

1. Introduction
The opium poppy, Papaver somniferum, is one of several plants that have profoundly affected human history.1 It has provided an unmatched medicine for the relief of pain for centuries.2 More than 30 alkaloids have been identified in opium, and among the most relevant are morphine (1a) and codeine (1b) (Figure 1).3 These alkaloids are important for modern medicine as an analgesic and a cough suppressant, respectively. These and related opiates exert their pharmacological effects by interacting with opioid receptors.4
Figure 1.
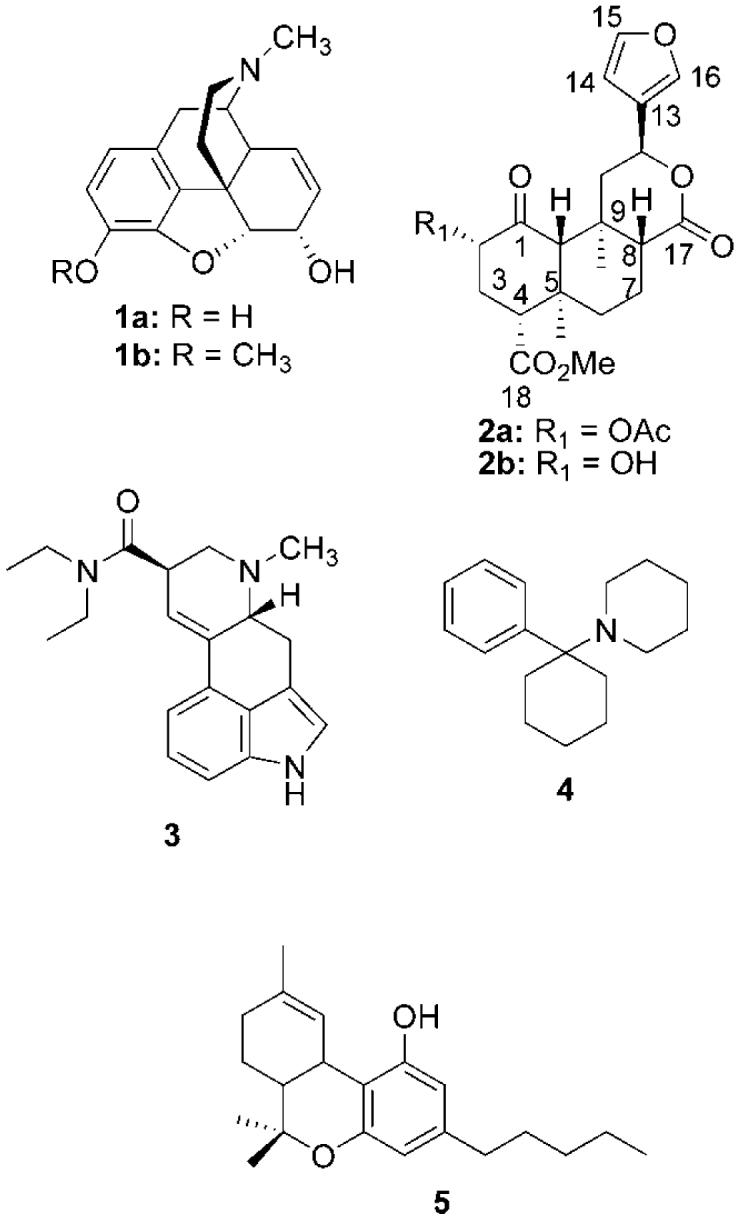
Structures of morphine (1a), codeine (1b), salvinorin A (2a), salvinorin B (2b), LSD (3), PCP (4), and Δ9-THC (5).
Research over the past 30 years has given great insight into the pharmacology, biochemistry, and biology of opioid receptors.5-7 Opioid receptors are members of the G-protein-coupled receptor (GPCR) superfamily of receptors and are divided into three types, μ (MOP), δ (DOP), and κ (KOP). In addition, there is a fourth member of the family, the nociception (NOP) receptor, which has low affinity for traditional opioids but has structural similarity with opioid receptors. Various pharmacological studies suggest the existence of additional opioid receptor subtypes.8-10 Recent data suggests that these subtypes may arise from the formation of receptor heterodimers.11 Each opioid receptor type plays a role in antinociception, as well as other biological responses.12 In addition to being important targets for medications used to treat pain, opioid receptors are potential targets for obesity,13 depression,14,15 and alcoholism.16
In 2002, opioid receptors were implicated in the actions of the psychoactive mint Salvia divinorum.17 The main active constituent isolated from the leaves of S. divinorum is the neoclerodane diterpene salvinorin A (2a).18,19 Diterpene 2a has been shown to produce short and very intense hallucinations in humans with a potency similar to lysergic acid diethyl amide (LSD) and 4-bromo-2,5-dimethoxyphenylisopropylamine (DOB).20,21 This activity was interesting given its lack of structural similarity to other psychotomimetic substances including LSD (3), phencyclidine (PCP, 4), or Δ9-tetrahydrocannabinol (Δ9-THC, 5). It came as little surprise that this molecule had a different mechanism of action given its lack of structural similarity to these substances.20 It was striking, however, that 2a was found to be a potent and selective agonist for κ opioid receptors over a battery of other receptors, including the 5-hydroxytryptamine (2a) (5-HT2A) receptor, which mediates the psychotomimetic effects of LSD, N-methyl-D-aspartate acid (NMDA) receptors, the target of PCP like agents, and cannabinoid receptors, which mediate the psychotomimetic effects of cannabinoids.17
As a neoclerodane diterpene, 2a is a truly unique opioid receptor ligand. Structurally it has little in common with other nonpeptidic opioid receptor ligands, such as 1a, metopon (6),22 bremazocine (7),23 sufentanil (8),24 SNC 80 (9),25 U50,488 (10),26 or etonitazene (11)27 (Figure 2).28,29 One common motif among these opioids is a basic nitrogen. Until the discovery of 2a, it had been assumed that the presence of a positively charged nitrogen atom in opioid compounds represented an absolute requirement for their interaction with opioid receptors.30 A general assumption was that the cationic amino charge of the opioid ligand would form a salt bridge with the side chain carboxyl group of an aspartate residue located in transmembrane (TM) III of the opioid receptor.31-33
Figure 2.

Structures of metopon (6), bremazocine (7), sufentanil (8), SNC 80 (9), U50,488H (10), and etonitazene (11).
Pharmacological and behavioral studies provide evidence that 2a acts as a KOP agonist. Diterpene 2a, for example, produces antinociception in mice that is blocked by a KOP antagonist.34,35 Importantly, the antinociceptive and hypothermic effects of 2a are not observed in KOP knockout mice.36 The KOP agonist U69,593 produces depressive-like behaviors in animal models, including increased mobility in the forced swim test, decreased extracellular dopamine in the nucleus accumbens,37 and increases in threshold for intracranial self-stimulation.38 A recent study showed that 2a produced the same effects as U69,593 in these models, confirming that 2a acts as a KOP agonist in vivo.39 Other notable findings include the finding that, as expected for a KOP agonist, 2a produced an aversive response in the conditioned place preference assay,40 blocked the locomotor-stimulant effects of cocaine,41 and did not exert DOM-like effects in nonhuman primates.42
This review describes the chemistry and structure-activity relationships of the growing number of nonpeptidic agonists and antagonists derived from 2a. It provides a review of the scientific literature up to the middle of 2007 without attempting to describe the detailed pharmacology of 2a. For this information, the reader is directed to several recent reviews on the pharmacology of 2a.43-45
2. Phytochemistry
S. divinorum is a relatively rare plant, and few chemical studies have characterized its components. The first compounds isolated from S. divinorum were the neoclerodane diterpenes salvinorin A (2a) and salvinorin B (2b).18,46 Prior to this report, Valdés was working on the isolation and characterization of the psychoactive substance from S. divinorum.47 Infusions of the plant had been shown to possess psychotropic activity,48 but the component responsible for this activity and its mechanism of action were not known.49 Having ascertained the active component to be a terpenoid, efforts were initiated by Valdés to identify the molecular target of this compound.47 These efforts were largely unsuccessful. A manuscript describing the isolation of the psychotropic terpenoid divinorin A and its congener divinorin B was then submitted to the Journal of Organic Chemistry.19 Comparison of the structures of divinorin A with 2a isolated by Ortega et al.18 found these compounds to be identical. Therefore, divinorin A and B are now called salvinorin A and B, respectively.
2.1. Chemical Constituents
Additional work by Valdés et al. on S. divinorum isolated a novel neoclerodane diterpene, salvinorin C (12) (Figure 3).50 Further phytochemical investigations have isolated salvinorinsD-I(13-18),51-54divinatorinsA-F(19-24),52,53,55salvinicins A and B (25 and 26),56 and salvidivins A-D (27-30).53 Eight additional constituents have been characterized from this plant: nepetoidin B, dehydrovomifoliol, harwickiic acid, isololiolide, methyl caffeate, methyl 3,4-dihydroxybenzoate, 3,4-dihydroxybenzaldehyde, and loliolide.53,57 To date, no alkaloids have been reported in S. divinorum.
Figure 3.

Naturally occurring analogs of salvinorin A.
2.2. Biosynthesis
Recently, the biosynthetic route of 2a was studied using the incorporation of [1-13C]glucose, [CH3-13C]methionine, and [1-13C;3,4-2H2]-1-deoxy-D-xylulose into its structure.58 Neoclerodane 2a, like other terpenoids, results from the assembly of isopentenylpyrophosphate and dimethylallyl pyrophosphate to form geranylgeranyl pyrophosphate (31) (Figure 4).59 Cyclization of 31 affords the labdanyl cation (32), which after several methyl shifts yields clerodane pyrophosphate (33). Further oxidation, acetylation, and methylation of 33 constructs 2a. Despite low incorporation, feeding experiments with [1-13C;3,4-2H2]-1-deoxy-D-xylulose (DOX) yielded a pattern of incorporation consistent with the deoxyxylulose phosphate pathway.58 Furthermore, enrichment of the C-23 methoxy group suggests the participation of an S-adenosyl-L-methionine dependent type III O-methyltransferase. However, the enzymes responsible for the oxygenation of 33 remain uncharacterized.
Figure 4.

Proposed biosynthetic pathway for salvinorin A. Green labels indicate incorporation pattern of [1-13C]-glucose isotopically labeled IPP derived from the DOXP pathway. Reprinted from Phytochemistry, 68, Lukasz Kutrzeba, Franck E. Dayan, J'Lynn Howell, Ju Feng, José-Luis Giner, and Jordan K. Zjawiony, Biosynthesis of salvinorin A proceeds via the deoxyxylulose phosphate pathway, pages 1872-1881, Copyright 2007, with permission from Elsevier.
3. Structure-Activity Relationships of Salvinorin A Analogs
Over the last 150 years, opioid receptor ligands have remained an active area in central nervous system (CNS) drug discovery. Opioid agonists are used clinically for the management of cancer pain, chronic pain, and cough and to treat diaherra.28 Opioid antagonists, such as naloxone and naltrexone, are used to treat opioid overdose, narcotic addiction, and alcohol dependence.60 Preclinical studies suggest that opioid agonists may have utility in treating stimulant dependence.61 Additional studies indicate that opioid antagonists may have utility in treating mood disorders,39,62 opioid abuse,63 stress-induced reinstatement of cocaine-seeking behavior,64 opioid-induced constipation,65 and gambling addiction.66 Thus, 2a, based on its novel structure, is a valuable lead in the development of opioid agents to treat all of these conditions.
Diterpene 2a possesses a rigid tricyclic core with seven chiral centers. This structural architecture opens various avenues for chemical investigation. Presently, there are a growing number of investigations into the activity and selectivity of 2a for κ opioid receptors. Interestingly, although 2a has low affinity for the MOP,5 2a has been shown to be an allosteric modulator of the MOP.67 To date, structure-activity relationships of 2a have focused on several main areas: (1) the 2-position acetoxy group, (2) the 4-position carbomethoxy group, (3) the 17-position carbonyl, and (4) the furan ring (Table 1). In addition, several attempts toward an asymmetric synthesis have been reported.
Table 1.
Opioid Receptor Affinity of Salvinorin A Analogs
| compd | MOP Ki ± SD | DOP Ki ± SD | KOP Ki ± SD | refs |
|---|---|---|---|---|
| 2a | >10000 nM | >10000 nM | 18.74 ± 3.38 nM | 17,70 |
| 2b | >10000 nM | >10000 nM | >10000 nM | 70 |
| 12 | >1 μM | >1 μM | 1022 ± 262 nM | 68 |
| 13 | >1 μM | >1 μM | >10000 nM | 68 |
| 14 | >1 μM | >1 μM | >10000 nM | 68 |
| 15 | a | a | 418 ± 117 nM | 52 |
| 16 | a | a | a | |
| 17 | a | a | a | |
| 18 | a | a | a | |
| 19 | a | a | a | |
| 20 | a | a | a | |
| 21 | a | a | >10000 nM | 52 |
| 22 | a | a | 230 ± 21 nM | 52 |
| 23 | a | a | >10000 nM | 52 |
| 24 | a | a | a | |
| 25 | >10000 nM | >10000 nM | 390 ± 30 nM | 69 |
| 26 | >10000 nM | >10000 nM | 7020 ± 750 nM | 69 |
| 27 | a | a | a | |
| 28 | a | a | a | |
| 29 | a | a | a | |
| 30 | a | a | a | |
| 34 | >10000 nM | >10000 nM | 32.63 ± 15.7 nM | 70 |
| 35 | >10000 nM | >10000 nM | 3199 ± 961.2 nM | 70 |
| 36 | >10000 nM | >10000 nM | >10000 nM | 70 |
| 37 | >10000 nM | >10000 nM | >10000 nM | 70 |
| 38 | >10000 nM | >10000 nM | >10000 nM | 70 |
| 10 ± 1 nM | 1410 ± 80 nM | 740 ± 40 nM | 72 | |
| 39 | >10000 nM | >10000 nM | >10000 nM | 70 |
| 40 | >10000 nM | >10000 nM | >10000 nM | 70 |
| 41 | >10000 nM | >10000 nM | >10000 nM | 70 |
| 42 | >10000 nM | >10000 nM | >10000 nM | 70 |
| >10000 nM | >10000 nM | 410 ± 40 nM | 77 | |
| 43 | >1 μM | >1 μM | 18 ± 2 nM | 68 |
| 44 | 520 ± 50 nM | 4030 ± 250 nM | 4 ± 1 nM | 72 |
| 45 | 310 ± 50 nM | 3970 ± 270 nM | 15 ± 2 nM | 72 |
| 46 | 520 ± 80 nM | 4240 ± 290 nM | 70 ± 4 nM | 72 |
| 47 | a | a | >10000 nM | 71 |
| 48 | a | a | >10000 nM | 71 |
| 49 | a | a | >10000 nM | 71 |
| 50 | 5660 ± 250 nM | >10000 nM | 90 ± 10 nM | 72 |
| 51 | 2980 ± 110 nM | >10000 nM | 19 ± 2 nM | 73 |
| 52 | 260 ± 6 nM | 8880 ± 390 nM | 42 ± 1 nM | 73 |
| 53 | >10000 nM | >17000 nM | 430 ± 10 nM | 73 |
| 54 | 12 ± 1 nM | 1170 ± 60 nM | 90 ± 2 nM | 73 |
| 55 | 73 ± 2 nM | 4820 ± 300 nM | 1930 ± 50 nM | 73 |
| 56 | 1090 ± 250 nM | >10000 nM | 290 ± 40 nM | 72 |
| 57 | 280 ± 40 nM | 9330 ± 1010 nM | 180 ± 10 nM | 72 |
| 58 | 110 ± 10 nM | >10000 nM | 90 ± 7 nM | 72 |
| 59 | 110 ± 10 nM | >10000 nM | 70 ± 7 nM | 72 |
| 60 | 10 ± 2 nM | 1380 ± 130 nM | 260 ± 20 nM | 72 |
| 61 | 1640 ± 90 nM | >10000 nM | 230 ± 20 nM | 77 |
| 62 | 7550 ± 970 nM | >10000 nM | 900 ± 50 nM | 77 |
| 63 | 30 ± 2 nM | 1140 ± 60 nM | 550 ± 30 nM | 77 |
| 64 | >10000 nM | >10000 nM | 800 ± 50 nM | 77 |
| 65 | 70 ± 4 nM | 1860 ± 140 nM | 540 ± 40 nM | 77 |
| 66 | 260 ± 210 nM | >10000 nM | 570 ± 40 nM | 77 |
| 67 | 180 ± 20 nM | >10000 nM | 5490 ± 640 nM | 77 |
| 68 | 10 ± 1 nM | 580 ± 30 nM | 70 ± 2 nM | 77 |
| 69 | 10 ± 1 nM | 690 ± 30 nM | 80 ± 3 nM | 77 |
| 70 | 1030 ± 80 nM | >10000 nM | 2010 ± 110 nM | 77 |
| 71 | a | a | 3.2 ± 0.2 nM | 71 |
| 72 | a | a | 83.0 ± 8.5 nM | 71 |
| 73 | a | a | 462 ± 20 nM | 71 |
| 74 | 640 ± 30 nM | 6460 ± 390 nM | 120 ± 4 nM | 73 |
| 75 | 16 ± 1 nM | 230 ± 10 nM | 93 ± 3 nM | 73 |
| 76 | a | a | >10000 nM | 86 |
| 77 | a | a | >10000 nM | 86 |
| 78 | a | a | 220 ± 12 nM | 71 |
| 79 | a | a | 7.9 ± 0.3 nM | 71 |
| 80 | a | a | 28.7 ± 3.0 nM | 71 |
| 81 | a | a | 35.8 ± 5.1 nM | 71 |
| 82 | a | a | 60.1 ± 9.1 nM | 71 |
| 83 | a | a | 75.7 ± 5.9 nM | 71 |
| 84 | >10000 nM | >10000 nM | 1610 ± 120 nM | 73 |
| 85 | a | a | 0.4 ± 0.02 nM | 86 |
| 86 | a | a | 328 ± 40 nM | 79 |
| 87 | a | a | 65.2 ± 24.6 nM | 79 |
| 88 | a | a | 17.6 ± 3.1 nM | 79 |
| 111 ± 49 nM | >10 μM | 4.5 ± 2.0 nM | 110 | |
| 89 | a | a | 168 ± 10 nM | 79 |
| 90 | a | a | 2.3 ± 0.6 nM | 79 |
| 91 | a | a | 149 ± 1 nM | 79 |
| 4180 ± 310 nM | >10000 nM | 30 ± 2 nM | 77 | |
| 92 | a | a | 374 ± 19 nM | 79 |
| 93 | a | a | 3.2 ± 0.1 nM | 79 |
| 135 ± 4 nM | 1690 ± 285 nM | 0.37 ± 0.30 nM | 110 | |
| 94 | a | a | 1.6 ± 0.1 nM | 79 |
| 15 ± 3 nM | 366 ± 38 nM | 0.11± 0.10 nM | 110 | |
| 95 | a | a | 27.6 ± 1.8 nM | 79 |
| 96 | a | a | 38.1 ± 1.9 nM | 79 |
| 97 | 3.1 ± 0.4 nM | 810 ± 30 nM | 7430 ± 880 nM | 77 |
| 98 | a | a | 38.1 ± 1.9 nM | 81 |
| 4370 ± 310 nM | 3990 ± 290 nM | 5.7 ± 0.4 nM | 77 | |
| 99 | a | a | 54.5 ± 25.7 nM | 81 |
| 100 | 290 ± 70 nM | 1930 ± 70 nM | 1410 ± 80 nM | 77 |
| 101 | 6820 ± 660 nM | >10000 nM | 2.3 ± 0.1 nM | 73 |
| 102 | >10000 nM | >10000 nM | 60 ± 6 nM | 72 |
| 103 | 220 ± 20 nM | 3720 ± 400 nM | 50 ± 5 nM | 72 |
| 104 | >10 μM | >10 μM | 2.9 ± 0.4 μM | 84 |
| 105 | >1 μM | >1 μM | 1125 ± 365 nM | 68 |
| 106 | >1 μM | >1 μM | 18 ± 2 nM | 68 |
| 107 | a | a | a | |
| 108 | a | a | a | |
| 109 | a | a | a | |
| 110 | a | a | a | |
| 111 | a | a | a | |
| 112 | a | a | a | |
| 113 | >10 μM | >10 μM | >10 μM | 84 |
| 114 | a | a | 1000 ± 269 nM | 79 |
| 115 | a | a | >10000 nM | 79 |
| 116 | a | a | >10000 nM | 79 |
| 117 | a | a | >10000 nM | 79 |
| 118 | a | a | >1000 nM | 86 |
| 119 | a | a | 28.5 ± 0.9 nM | 86 |
| 120 | a | a | >1000 nM | 86 |
| 121 | a | a | 201 ± 26 nM | 86 |
| 122 | a | a | 99.6 ± 15.9 nM | 86 |
| 123 | a | a | 613 ± 54.1 nM | 87 |
| 124 | a | a | >10000 nM | 79 |
| 125 | a | a | >10000 nM | 79 |
| 126 | a | a | >10000 nM | 79 |
| 127 | a | a | 26.9 ± 1.8 nM | 86 |
| 128 | a | a | 470 ± 92 nM | 86 |
| 129 | a | a | 210 ± 32 nM | 86 |
| 130 | >1 μM | >1 μM | 59 ± 11 nM | 68 |
| 131 | 3670 ± 230 nM | >10000 nM | 7 ± 1 nM | |
| 132 | 2620 ± 150 nM | >10000 nM | 10 ± 1 nM | |
| 133 | >1 μM | >1 μM | 6 ± 1 nM | 68 |
| 134 | >1 μM | >1 μM | 6 ± 2 nM | 68 |
| 135 | 7240 ± 480 nM | >10000 nM | 14 ± 1 nM | 69 |
| S-135 | 9790 ± 1090 nM | >10000 nM | 3.7 ± 0.2 nM | 69 |
| 136 | 1450 ± 60 nM | 7620 ± 180 nM | 3.0 ± 0.2 nM | 69 |
| 137 | >10000 nM | >10000 nM | 300 ± 20 nM | 69 |
| 138 | >10000 nM | >10000 nM | 8530 ± 550 nM | 89 |
| 139 | >10000 nM | >10000 nM | 840 ± 90 nM | 89 |
| 140 | >10000 nM | >10000 nM | 410 ± 30 nM | 89 |
| 141 | >10000 nM | >10000 nM | 1620 ± 110 nM | 89 |
| 142 | >10000 nM | >10000 nM | 56 ± 3 nM | 69 |
| 143 | >10000 nM | >10000 nM | 25 ± 1 nM | 69 |
| 144 | >10000 nM | >10000 nM | 125 ± 1 nM | 69 |
| 145 | 3190 ± 230 nM | >10000 nM | 420 ± 20 nM | 69 |
| 146 | >10000 nM | >10000 nM | 125 ± 4 nM | 69 |
Not determined.
3.1. Other Salvinorins
Many naturally occurring analogues of 2a have been evaluated for affinity at κ opioid receptors.52,68,69 Generally, these compounds were found to have no affinity at KOPs (Ki > 10 000 nM).52,68,69 However, there are a few exceptions. Salvinorin C (12) was found to have 250-fold lower affinity compared with 2a (Ki = 1022 nM vs Ki = 4 nM).68 Divinatorins D (22) and E (23) also had reduced affinity at KOPs compared with 2a (Ki = 230 nM and Ki = 418 nM, respectively, vs Ki = 1.0 nM).52 More recently, salvinicin A (25) was identified as having affinity for KOPs (Ki = 390 nM), but salvidivin A (27) was identified as the first naturally occurring neoclerodane with κ antagonist activity (Ke = 440 nM).69 These findings suggest the possibility of additional naturally occurring analogues with opioid receptor affinity and activity.
3.2. Role of the 2-Position Acetyl Group
The most extensively studied substituent of 2a has been the C-2 acetoxy group. This is likely due to the ease of preparation from 2b and the early observation of 2b having little psychotropic activity.20 Modifications have been made in order to study (1) C-2 ester modifications, (2) preparation of carbamates or carbonates, (3) conversion to ethers or amines, (4) bioisosteric replacement to amides or thioesters, and (5) conversion to sulfonate esters.
3.2.1. Ester Modifications
Initially the role of the 2-acetyl group of 2a on affinity and selectivity for κ opioid receptors was investigated.70 Structural modification of this position was found to vary activity from full agonism to partial agonism for inhibition of forskolin-stimulated cAMP production. In particular, 2a was found to be a full agonist, while propionate 34 and heptanoate 35 were found to be partial agonists in this assay (Figure 5).70 Surprisingly, 2a was found to be more efficacious than the selective κ agonist U50,488 and similar in efficacy to the naturally occurring peptide ligand for κ receptors, dynorphin A. However, salvinorin B (2b), as well as, 36-42 were found to have no affinity at μ, δ, or κ receptors.70 Finally, the C-8 epimer of 2a was found to have 41-fold lower affinity for κ receptors compared with 2a (Ki = 163 nM vs Ki = 4nM).68
Figure 5.
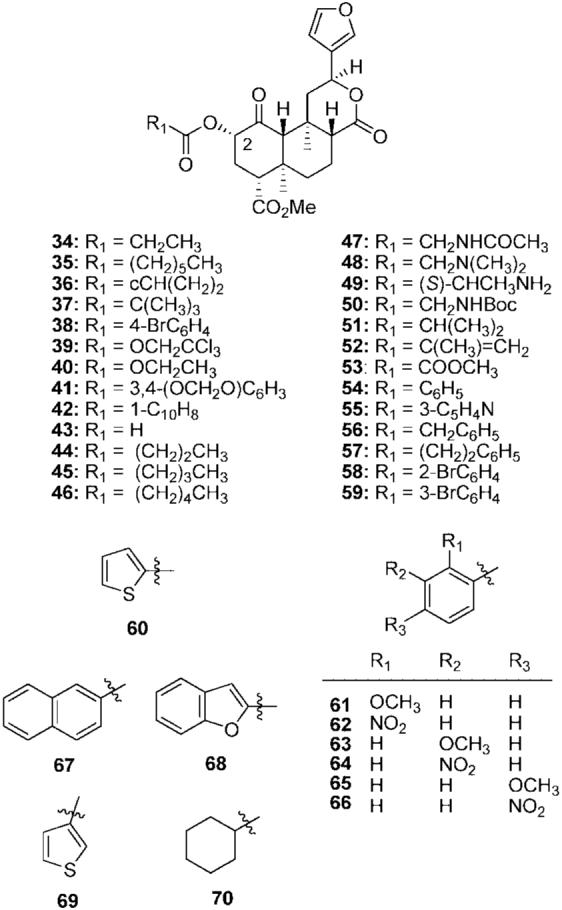
C-2 ester analogs of salvinorin A.
Replacement of the 2-acetyl group with a formate (43) decreased affinity and activity approximately 5-fold at KOPs compared with 2a.68 However, the introduction of a butyl group (44) was found to decrease affinity and activity approximately 2-fold.71 Additional studies found that 44 also had affinity at MOPs.72 Further extension of the carbon chain (45 and 46) decreased affinity for KOPs but had little effect on MOP binding.72 Introduction of an acetamido (47) or dimethylamino (48) to 2a abolished affinity at KOPs (Ki > 10 000 nM).71 Addition of an amino group to 34 (49) was also not tolerated. More recently, the addition of a tert-butoxycarbonylamino group (50) reduced affinity 47-fold at KOPs compared with 2a (Ki = 90 nM vs Ki =) 1.9 nM).72
The effect of branching and size of the 2-position has also been examined.73 Insertion of a methyl group to 34 (51) decreased affinity 10-fold at KOPs. Introduction of an alkene (52) decreased affinity 3-fold for KOPs but increased affinity 11-fold at MOPs. Replacement of the 2-methylacroyl group with a methyl glyoxyl group (53) decreased affinity 11-fold at KOPs. Introduction of a benzoyl group (54) resulted in 47-fold loss of affinity at KOPs compared with 2a. This change, however, increased affinity 25-fold at MOPs compared with 2a. Functional studies showed 54 to be a full agonist at MOPs and KOPs. This was the first example of a non-nitrogenous μ agonist.73
The pharmacological properties of 54, termed herkinorin, were examined in detail.74,75 Benzoate 54 did not promote recruitment of β-arrestin-2 to the MOP and internalization of the MOPs, even in cells that overexpress G-protein-coupled receptor kinase.75 In contrast, morphine, which does not normally recruit β-arrestin-2 and does not promote MOP internalization in HEK cells that express the MOP, will do so in the presence of G-protein-coupled receptor kinase overexpression. Thus, 54 provided a striking example of biased agonism.76 Recent work suggests that the ability of a μ agonist to produce MOP internalization contributes to its ability to produce tolerance and dependence. The availability of 54 provided a useful tool to test this hypothesis using CHO cells that express the MOP.74 The results showed that both noninternalizing (54) and internalizing (DAMGO) μ agonists produced tolerance, receptor desensitization, and upregulation of the cAMP system. The major difference between the two types of μ agonists was that chronic 54 induced the formation of constitutively active MOPs to a profound degree.
Given the unique characteristics of 54, additional studies were undertaken to more fully understand its preference for MOPs over KOPs.72 Replacement of the benzoyl group with a nicotinoyl group (55) decreased affinity 6-fold at MOPs and 20-fold at KOPs. Introduction of a one carbon spacer between the carbonyl and phenyl ring (56) decreased affinity at MOPs and KOPs. The addition of a second methylene unit (57) increased affinity at MOPs and KOPs compared with 56. Introduction of a bromo group in the 2-position (58) or 3-position (59) of the benzene ring had no effect on KOP affinity but decreased affinity for MOPs 9-fold compared with 54. The presence of a 4-bromo group (38) decreased affinity for KOPs 8-fold compared with 54 (Ki = 740 nM vs Ki = 90 nM). In contrast to a previous report, which indicated that 38 had no affinity for MOPs,70 this modification was found to retain high affinity for MOPs (Ki = 10 nM vs Ki = 12 nM).72 Finally, the bioisosteric replacement of the benzene ring with a 2-thiophene (60) reduced affinity for KOPs 3-fold but had no effect on affinity for MOPs or DOPs.
More recently, the effects of additional modification to 54 were explored.77 The addition of a 2-methoxy group (61) or a 2-nitro group (62) decreased affinity for MOPs compared with 54. These results suggest that factors such as sterics are likely involved in the binding of 2-position analogues. However, this awaits further investigation. Introduction of a methoxy group in the 3-position of the benzene ring (63) also decreased affinity for MOPs and KOPs compared with 54. This modification, however, improved selectivity for MOPs over KOPs compared with 54. Substitution of a 3-nitro group (64) abolished affinity at MOPs (Ki > 10 000) and decreased affinity approximately 10-fold at KOPs compared with 54. The presence of 4-methoxy group (65) leads to an approximately 6-fold decrease in affinity and similar selectivity for MOPs compared with 54. The addition of a 4-nitro group (66) decreased affinity over 20-fold for MOPs and over 6-fold for KOPs compared with 54. These results indicate that factors other than electronics are likely involved in the binding of 4-position analogues to MOPs.
Replacement of the benzoyl group in 54 with a 1-naphthoyl group (42)70 decreased affinity roughly 1000-fold at MOPs, whereas substitution of a 2-naphthoyl group (67) reduced affinity at MOPs approximately 10-fold compared with 54. Introduction of a 2-benzofuran (68) or a 3-thiophene (69) was well tolerated as 68 and 69 showed equal affinity for MOPs compared with 54. Reduction of the benzene ring to a cyclohexane ring (70) reduced affinity for MOPs and KOPs compared with 54.
The effect of C-2 position stereochemistry on the affinity and activity of 2a has been probed.78,79 Inversion of the C-2 substituent of 2a decreased activity at KOPs 9-fold.78 A similar trend was seen when the C-2 position was inverted in 34 and 44.79
3.2.2. Carbamate and Carbonate Analogs
Replacement of the acetyl group in 2a with a carbamoyl group (71) was well tolerated at KOPs (Figure 6).71 Addition of methyl group (72) decreased affinity and activity. Extension of the methyl group to an ethyl group (73) was not well tolerated, and affinity and activity were further decreased. Exchange of the acetyl group in 2a with an allyl carbamoyl group (74) decreased affinity 63-fold at KOPs.73 Interestingly, this change resulted in moderate affinity at MOPs. Substitution of a phenylcarbamoyl group (75) for the allyl carbamoyl group in 74 had little effect at KOPs but increased affinity for MOPs and DOPs.73 Conversion of 72 and 73 to the corresponding carbonates, 76 and 77, respectively, was poorly tolerated at KOPs and affinity was abolished (Ki > 1000 nM).80
Figure 6.
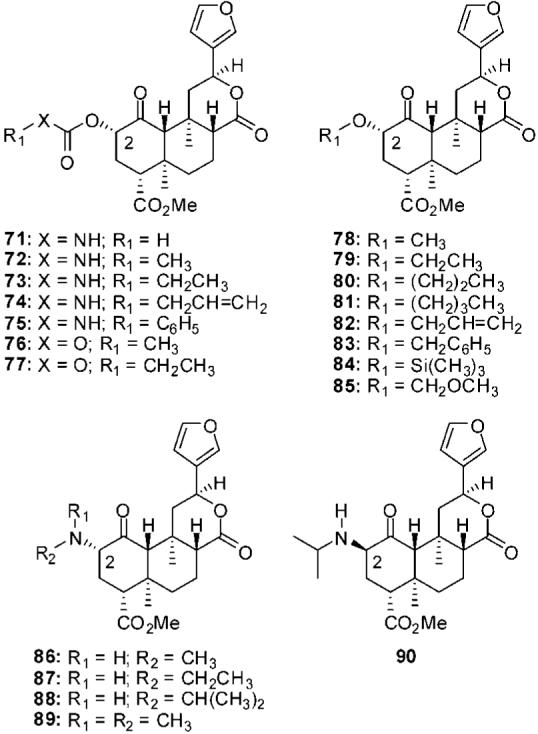
C-2 carbamate, carbonate, ether, and amine analogs of salvinorin A.
3.2.3. Ether and Amine Analogs
The conversion of 2a to various ethers has been studied.71,79,80 The addition of a methyl group to 2b (78) has little effect on affinity or efficacy at KOPs.71 Extending the chain to ethyl (79) increases affinity and activity 20-fold compared with 78. Further extension of the chain (80 and 81) decreased affinity and activity compared with 79. Allyl ether 82 and benzyl ether 83 were found to have similar activity at KOPs but were less potent than 79.71 Trimethylsilyl ether 84 was found to have reduced affinity compared with 2a.73 Finally, introduction of a methoxymethyl group (85) was found to increase affinity and activity at KOPs compared with 2a.80 This is the most potent salvinorin A derived κ agonist described to date.
The conversion of the methoxy group in 78 to a methylamino group (86) had little effect on affinity but decreased activity at KOPs.79 Extension of the chain to an ethylamino group (87) increased affinity and activity compared with 86. Substitution of an isopropylamino group (88) increased activity at KOPs compared with 87. Addition of an N-methyl group to 86 (89) also increased activity at KOPs. Generally, inversion of C-2 stereochemistry of these analogues was found to increase activity at KOPs. The most potent analogue (90) was found to be roughly equipotent with 2a (EC50 = 7.2 nM vs EC50 = 4.5 nM).79
3.2.4. Amides and Thioesters
Bioisosteric replacement of the acetoxy group in the 2-position of 2a with acetamido and thioacetoxy has also been investigated.71,77,79,81-83 The substitution of an acetamido group (91) for the acetoxy group in 2a decreases affinity and activity at KOPs (Figure 7).79 Extension of the carbon chain to a propionamido group (92) decreased affinity and activity at KOPs. The addition of an N-methyl group to 91 (93) increased affinity and activity at KOPs.79 A similar effect was seen when an N-methyl group was added to 92 (94) resulting in a derivative more potent than 2a (EC50 = 0.75 nM vs EC50 = 4.5 nM). Introduction of an N-ethyl group to 91 (95) and 92 (96) increased activity at KOPs, but these analogues were less potent than 94 and 95. Generally, inversion of the C-2 stereochemistry in 91-96 decreased affinity and activity.79 Conversion of the benzoyloxy group in 54 to a benzamido group (97) was found to increase affinity and selectivity for MOPs (Ki = 3.4 nM vs Ki = 12 nM).77 Amide 97 is the most potent μ agonist derived from 2a (EC50 = 360 nM) described to date.
Figure 7.

C-2 bioisosteric replacements of salvinorin A.
The substitution of a thioacetoxy group (98) for the acetoxy group in 2a decreased affinity and activity at KOPs (EC50 = 4.77 nM vs EC50 = 2.82 nM).77,81 Removal of the acetyl group in 98 (99) decreased affinity and activity at KOPs. Similarly, inversion of the C-2 stereochemistry in 87 and 88 decreased activity at KOPs. As shown in the amide and ester series, introduction of a benzene ring to 98 (100) increased affinity for KOPs. However, 100 had reduced affinity compared with 54 and 97.77
3.2.5. Sulfonyl Esters
An additional modification studied was the bioisosteric replacement of the acetyl group with a sulfonate ester.73 Substitution of a mesylate group (101) was well tolerated as this change had little effect on binding to KOPs. Mesylate 101 was also found to be slightly more potent than 2a as an agonist at KOPs (EC50 = 30 nM vs EC50 = 40 nM).73 Replacement of the mesylate in 101 with a benzenesulfonate (102) reduced affinity at KOPs compared with 101. This is in good agreement with previous SAR studies in the ester series where the replacement of a methyl group with a phenyl group (54) decreased affinity for KOPs.73 Surprisingly, 102 had no affinity for MOPs (Ki > 10 000 nM). An analogous replacement of methyl by phenyl (54) showed an increase in MOP affinity.73 Introduction of a 4-methyl group to 102 (103) had no effect on KOP affinity (Ki = 50 nM vs Ki = 60 nM) and increased affinity for DOPs (Ki = 3720 nM vs Ki > 10 000 nM) compared with 102. This change, however, increased affinity for MOPs compared with 102 (Ki = 220 nM vs Ki > 10 000 nM). These changes, however, are not parallel to the ester series suggesting that the sulfonate esters are not binding in an identical manner at either MOPs and KOPs.
3.3. Other A Ring Changes
There have been several other changes to the A ring studied.80,84,85 Ring-opened analogue 104 was found to have weak affinity at KOPs (Ki = 2.9 μM) (Figure 8).84 Reduction of the C-1 ketone to an α-alcohol (105) reduced affinity over 250-fold compared with 2a (Ki = 1125 nM vs Ki = 4 nM).68 This modification also changed the efficacy at KOPs from a full agonist (2a, Emax = 108%) to an antagonist (105, Ke = 240 nM).85 Removal of the ketone (106) resulted in a 5-fold loss of affinity compared with 2a (Ki = 18 nM vs Ki = 4 nM).68 Additional testing found that 106 was 3-fold less potent but more efficacious as a KOP agonist than 2a. A more recent study found that 106 was approximately as potent but less efficacious than 2a.85 In this study, 106 was found to also have antagonist activity at MOPs and DOPs. Replacement of the acetyl group in 106 with a benzoyl group (107) resulted in an antagonist at μ, δ, and κ receptors. This finding suggests that the C-1 deoxo analogues may be interacting at μORs in a nonidentical manner compared with C-1 keto analogues. Addition of a benzene ring to 105 (108) decreased activity 2-fold at KOPs (Ke = 450 nM vs Ke = 240 nM), whereas, the introduction of a mesylate group (109) resulted in a loss of antagonist activity at κ receptors.
Figure 8.
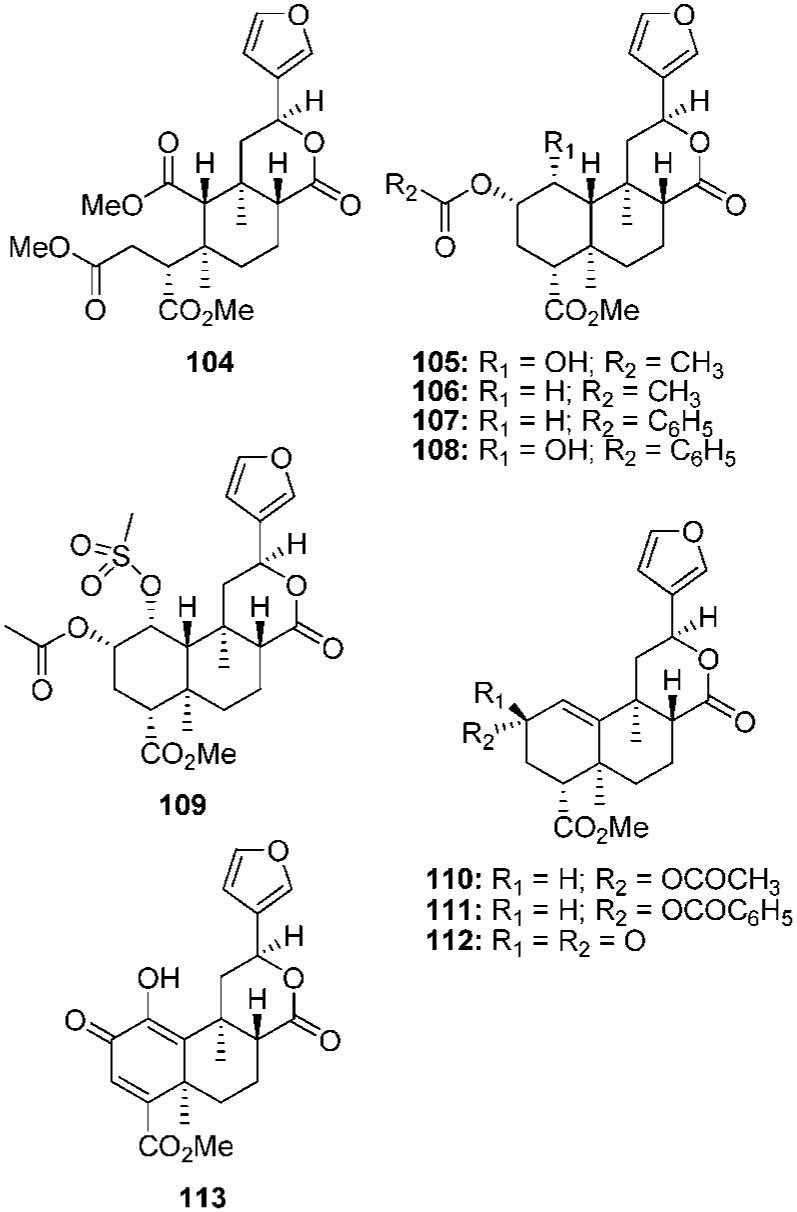
Ring A modified salvinorin A analogs.
The presence of the 1,10-alkene has also been investigated.85 Introduction of a 1,10-alkene to 106 (110) resulted in a switch of efficacy from partial agonist to antagonist at KOPs. Furthermore, 110 had higher antagonist activity at MOPs and DOPs than at KOPs. When compared with 106, 110 had similar antagonist activity at MOPs and reduced activity at DOPs. Replacement of the acetyl of 110 with benzoyl group (111) decreased activity 9-fold at MOPs and 6-fold at DOPs but had little effect on KOPs. This also suggests that the 1,10-dehydro analogues are not interacting in a similar manner to analogues that contain a C-1 keto group. Several additional α,β-unsaturated ketones have been investigated. Ketone 112 was found to have similar antagonist activity as 110 at KOPs but reduced activity at MOPs and DOPs, whereas, 113 was found to have no affinity for KOPs (Ki > 10 μM).80,84,85
3.4. Role of the 4-Position Carbomethoxy Group
Several reports have investigated the role of the 4-position carbomethoxy group.68,79,86,87 Reduction of the carbomethoxy group to the primary alcohol (114) reduced affinity 89-fold at KOPs (Figure 9).68 Bioisosteric replacement of the alcohol with a primary amine (115) was not tolerated as affinity for KOPs was lost (Ki > 10 000 nM).79 N-Alkylation of 115 (116 and 117) did not lead to an enhancement of affinity. Similarly, epimerization of the C-8 position of 115-117 did not lead to an increase in affinity for KOPs.79
Figure 9.

C-4 modified analogs of salvinorin A.
Hydrolysis of the methyl ester in 2a to the corresponding acid (118) resulted in a loss of affinity at KOPs (Ki > 1000 nM).86 Extension of the carbon chain in 2a to an ethyl group (119) also resulted in a loss of affinity and activity at KOPs. Further extension of the alkyl chain, such as 120, was not tolerated.86 However, incorporation of an alkyne (121) did result in modest affinity at KOPs (Ki = 201 nM). Addition of a methoxymethyl (MOM) group to 118 (122) resulted in a loss in affinity and activity at KOPs. Interestingly, the C-8 epimer of 122 had similar affinity for KOPs but was 3-fold less potent as an agonist.86 Replacement of the MOM group with a MEM group (123) decreased affinity and activity at KOPs.87
Conversion of the 4-position methyl ester into various amides has also been accomplished.79,86,87 Bioisosteric replacement of the methyl ester with a methyl amide (124) resulted in over a 500-fold loss of affinity at KOPs.79 Extension of the carbon chain (125) or addition of an N-methyl group (126) were not tolerated as affinity at KOPs was lost (Ki > 10 000 nM). Substitution of other alkyl chains was also not tolerated.86 However, several amino acid derivatives (127-129) were found with affinity and activity less than 2a.86 The most potent of these analogues was alanine derivative 127 (EC50 = 46.7 nM).
3.5. Role of the 17-Position Carbonyl
Reduction of the lactone carbonyl to a lactol (130) was found to reduce affinity 14-fold and activity 2-fold at KOPs (Figure 10).68 Addition of a methyl group to 130 creates 131 and 132. This change was well tolerated, and C-17 stereochemistry was found to have little effect on binding as 131 and 132 had similar affinities at μ, δ, and κ receptors (131, μ Ki = 3670 ± 230 nM; δ Ki > 10 000 nM; κ Ki = 7 ± 1 nM vs 132, μ Ki = 2620 ± 150 nM; δ Ki > 10 000 nM; κ Ki = 10 ± 1 nM).88 Removal of the carbonyl (133) was found to have little effect on affinity at KOPs compared with 2a (Ki = 6 nM vs Ki = 4 nM), but activity was reduced 5-fold.68 Similarly, introduction of C-8-C-17 alkene (134) had little effect on binding, but activity was reduced 14-fold compared with 2a (EC50 = 624 nM vs EC50 = 46 nM).68
Figure 10.
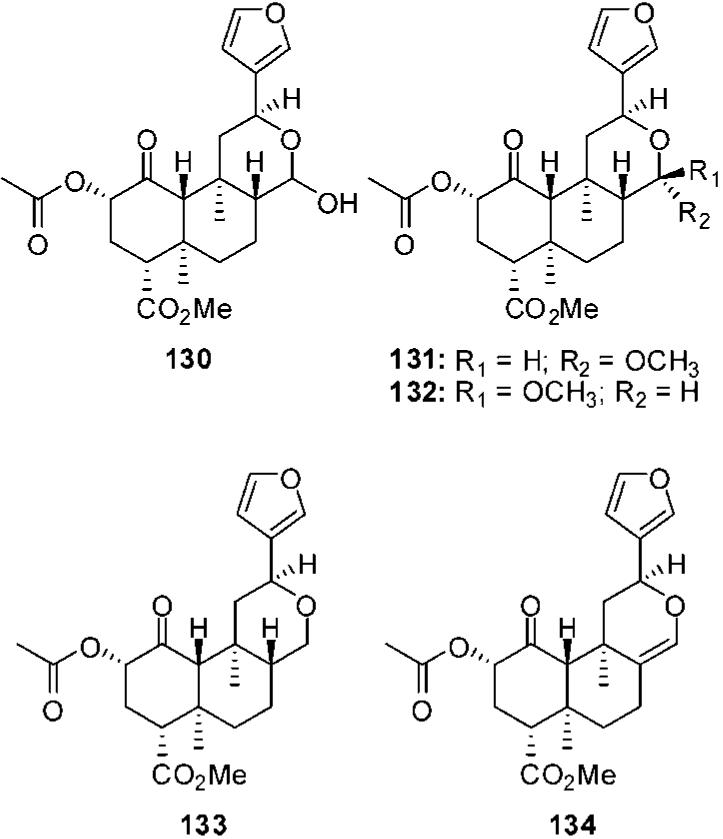
C-17 modified analogs of salvinorin A.
3.6. Role of the Furan Ring
Additional work has focused on the role of the furan ring.68,69,89 Reduction of the furan ring to a mixture of C-13 epimers (135) reduced affinity for κ receptors compared with 2a (Ki = 156 nM vs Ki = 4 nM) (Figure 11).68 Additional testing found 135 to possess high affinity at KOPs (Ki = 14 nM).69 The R epimer (R-135) was found to have similar affinity for KOPs as 2a but was 17-fold less active than 2a. Bromination of the furan ring is well tolerated as 136 retained high affinity and activity at KOPs. Replacement of the furan ring with a 2-oxazoline ring (137) or a 4-carbomethoxyoxazole (138) decreased affinity for KOPs.69,89 A series of N-sulfonylpyrroles (139-141) were also found to have reduced affinity for KOPs compared with 2a.89 Interestingly, these modifications resulted in partial agonism at KOPs. Substitution of the furan ring with a 4-methyl-1,3,5-oxadiazole (142) resulted in a 29-fold loss in affinity at KOPs compared with 2a. However, this change resulted in antagonist activity at MOPs and KOPs.69 The addition of 2,5-dimethoxy groups to R-135 (143 and 144) was also probed and found to decrease affinity at KOPs. Incorporation of a C-13-C-14 alkene to 143 (145) and 144 (146) did not enhance affinity at KOP.
Figure 11.

C-12 modified analogs of salvinorin A.
3.7. Total Synthesis Efforts
The first studies toward the total synthesis of 2a were reported by Lingham et al.90 The proposed synthetic route dissected 2a into cyclohexanone 147 and lactone 148 (Figure 12). It was envisioned that Michael addition followed by olefin metathesis and hydrogenation would afford 2a. This work, however, did not report a completed synthesis but rather an enantioselective route to ring A and a model route to ring C.90
Figure 12.

Synthetic targets and intermediates used in synthesis efforts of salvinorin A. Adapted from ref 91.
More recently, an asymmetric synthesis of 2a in 33 steps was reported.91 The construction of the tricyclic core of 2a (149) was accomplished by a transannular Michael reaction cascade of macrocycle 150. Bisenone 150 was prepared in a convergent assembly from vinyl iodide 151 and aldehyde 152. However, this synthetic route produces predominantly the C-8 epimer of 2a but epimerization studies produced 2a identical to natural material. This synthesis demonstrates the utility of a transannular reaction cascade in constructing polycyclic structures and offers a new method of preparing analogues of 2a with modified C-12 functionality.91
4. Studies on the Mode of Binding of Salvinorin A
A number of models have been proposed to explain the selectivity and binding of opioid receptor ligands. Current thinking is that KOP-selective opiate antagonists recognize three elements within the KOP: (1) a highly conserved aspartate in TM III; (2) an aromatic pocket formed by TMs V, VI, and VII; (3) a KOP specific selectivity site in TM VI.92-97 This model, however, does not readily apply to other classes of KOP ligands, such as 10. However, several models of κ agonist binding have been reported.98-103 It has been proposed that the Asp138 carboxylate in TM III forms a salt bridge with the protonated nitrogen of arylacetamides and benzomorphans, a hydrophobic pocket consisting of Tyr312, Leu224, Leu295, and Ala298 side chains hosts the phenyl ring of arylacetamides such as 10, and His291 makes a hydrogen bond with the phenolic group of the benzomorphans such as 7.98 Another model was developed to study the mode of binding of dynorphin A(1-8).102 This model found that amino acid residues in EL2, TM III, TM IV, and TM V determine the selectivity of peptide agonists for KOPs.
There have been several studies directed at elucidating the mode of binding of 2a at the KOP.17,83,104,105 Initially, molecular modeling studies suggested four interactions that might explain the binding of 2a: (1) Gln115 forming a hydrogen bond with the furan oxygen; (2) Tyr139 interacting with the C-17 carbonyl; (3) Tyr312 interacting with the 4-carbomethoxy group; (4) Tyr313 interacting with the C-2 acetyl group.17 This model, however, was revised based on structure-activity relationship studies. A more recent model83 proposed three points of interaction: (1) the furan oxygen interacting with Tyr119 and Tyr320; (2) the 4-carbomethoxy group interacting with Glu297 and Ile294; (3) the 2-acetyl group interacting in a hydrophobic manner with Tyr313. This model was developed out of site-directed mutagenesis studies using thiol 99. For the Tyr313 mutant, the affinity of 99 was enhanced compared with 2a suggesting that this residue in helix 7 is close to the C-2 position of 2a.83 Collectively, the mutagenesis studies imply that 2a utilizes unique residues within the commonly shared binding pocket of the KOP.
An additional model has been proposed by Kane et al. through chimeras and single-point mutations.104 This work proposes that 2a recognizes the KOP through a unique binding epitope involving interactions in TM II, TM IV, and EL-2.104 In the qualitative binding-site model, 2a vertically spans residues Tyr119, Tyr320, Gln115, Tyr313, and Tyr312. As in the model proposed by Yan et al., 2a is in close proximity to EL-2. Overall, this model begins to elucidate how 2a binds using hydrophobic interactions rather than saltlink interactions at the KOP.
Recently, an approach utilizing chimeras, site-directed mutagenesis, and the substituted cysteine accessibility method was used to investigate the selectivity of 2a for KOPs.105 It was found that helix 2 is required for the binding of 2a to the KOP and two residues, Val108 and Val118, are responsible for the selectivity for the KOP. These two residues result in a differential pattern of amino acids that have access to the binding pocket. This finding supports 2a having a novel mode of binding that imparts subtype selectivity for GPCR ligands.
5. Summary and Outlook
The body of knowledge of the chemistry, pharmacology, and neurobiology of opioid receptors continues to grow rapidly. Despite these advances, many aspects of the three-dimensional structure of μ, δ, and κ opioid receptors remain elusive. Salvia divinorum and its major active component 2a have the potential to identify novel opioid receptor probes that will aid in addressing these aspects, as well as opening additional areas for chemical investigation. Opioid agonists based on 2a have the potential to treat pain, cough, diarrhea, stimulant dependence, and mood disorders. Antagonists derived from 2a have potential utility in treating a number of conditions including drug dependence, depression, opioid-induced constipation, and obesity. Thus, analogues of 2a may prove to be excellent research tools and give greater insight into opioid receptor mediated phenomena.
Medicinal chemistry efforts have begun to explore the structure-activity relationship studies of 2a. To date, these have mainly focused on its high affinity and selectivity for the KOP. Future work is likely to identify additional analogues of 2a with altered selectivity for MOPs and DOPs, as well as novel allosteric modulators of opioid receptors.67 The SAR known at this time is summarized in Figure 13. At the C-1 position, (1) reduction or removal of the carbonyl is tolerated, and (2) introduction of a 1,10-alkene increases the likelihood of antagonist activity. At the C-2 position, (1) small alkyl esters favor binding to KOPs, whereas aromatic esters favor binding to MOPs, (2) replacement of the acetoxy with ethers and amines is tolerated, (3) generally α-substituents are preferred over the corresponding β-substituents, and (4) bioisosteric replacement of the acetoxy group with amide, thioester, and sulfonate esters is tolerated. At the C-4 position, (1) small alkyl chains are preferential for binding to KOPs, (2) hydrolysis or reduction of the carbomethoxy group leads to reduced affinity at KOPs, and (3) generally conversion to an amide is not tolerated. At the C-17 position, (1) reduction or removal of the carbonyl is tolerated, and (2) introduction of an 8,17-alkene is also tolerated. Finally at C-12, the furan ring may be reduced or replaced, but this leads to a reduction in affinity. Several models suggest that the affinity and selectivity of 2a for KOPs is the result of interactions with unique residues compared with other KOP ligands.
Figure 13.

Summary of SAR for salvinorin A analogs.
In 1929, the eminent pharmacologist Reid Hunt articulated a guiding premise for the development of a program at the NIH to address drug abuse problems. He stated that, “A thorough study of the morphine molecule might show a possibility of separating the analgesic from the habit forming property . . . work along these lines would involve cooperation between the highest type of organic chemists and pharmacologists.”106-108 Studies under this directive lead to the identification of metopon (6).22 Early preliminary pharmacological studies of 6 in animals and humans showed this compound to retain morphine-like analgesia but cause fewer undesirable side effects than morphine. These observations provided the initial “proof of principle” that permeates contemporary opioid research.108 Continued research into chemistry and pharmacology of opioid receptor ligands, such as 2a or other natural products, may yet yield the holy grail of opioids.109
6. Acknowledgments
The authors thank the National Institute on Drug Abuse, NIH, and the Universities of Iowa and Kansas for financial support of ongoing research programs. The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
Biographies

Thomas E. Prisinzano received his Ph.D. degree in Medicinal Chemistry from Virginia Commonwealth University, Richmond, VA, in 2000. After postdoctoral studies at the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (2000-2003), Dr. Prisinzano joined the Division of Medicinal and Natural Products Chemistry at the University of Iowa as an Assistant Professor. Dr. Prisinzano has received the 2005 D. John Faulkner Travel Award from the American Society of Pharmacognosy, the 2006 Jack L. Beal Award from the Journal of Natural Products, and the 2008 Matt Suffness Award from the American Society of Pharmacognosy. Currently, he is an Associate Professor in the Department of Medicinal Chemistry at the University of Kansas. His research interests include the discovery, design, and synthesis of novel opioids with the potential to treat drug abuse and pain.

Richard B. Rothman received an M.D. and a Ph.D. degree in pharmacology from the University of Virginia, Charlottesville, VA, in 1982. After postdoctoral studies at the National Institute of Mental Health (NIMH) (1982-1984), Dr. Rothman served a residency in psychiatry at St Elizabeth's Hospital, Washington, DC (1984-1987) and later a 2-year fellowship in psychiatry at the NIMH. In 1991, Dr. Rothman joined the Intramural Research Program of the National Institute on Drug Abuse (NIDA-IRP) in 1991. He has coauthored more than 340 original articles focused mostly on the pharmacology of opioids and stimulants and is an inventor on several U.S. patents. Dr. Rothman is board certified in psychiatry and currently serves as Chief of the Clinical Psychopharmacolgy Section, Chemical Biology Research Branch.
7. References
- (1).Spinella M. The Psychopharmacology of Herbal Medicine: Plant Drugs that Alter Mind, Brain, and Behavior. MIT Press; Cambridge, MA: 2001. [Google Scholar]
- (2).Casy AF, Parfitt RT. Opioid Analgesics: Chemistry and Receptors. Plenum Press; New York: 1986. [Google Scholar]
- (3).Robbers JE, Speedie MK, Tyler VE. Pharmacognosy and Pharmacobiotechnology. Williams and Wilkins; Baltimore, MD: 1996. [Google Scholar]
- (4).Pert CB, Snyder SH. Science. 1973;179:1011. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- (5).Waldhoer M, Bartlett SE, Whistler JL. Annu. Rev. Biochem. 2004;73:953. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- (6).Reisine T. Neuropharmacology. 1995;34:463. doi: 10.1016/0028-3908(95)00025-2. [DOI] [PubMed] [Google Scholar]
- (7).Snyder SH, Pasternak GW. Trends Pharmacol. Sci. 2003;24:198. doi: 10.1016/S0165-6147(03)00066-X. [DOI] [PubMed] [Google Scholar]
- (8).Pasternak GW. Neuropharmacology. 2004;47(Suppl 1):312. doi: 10.1016/j.neuropharm.2004.07.004. [DOI] [PubMed] [Google Scholar]
- (9).Zaki PA, Bilsky EJ, Vanderah TW, Lai J, Evans CJ, Porreca F. Annu. Rev. Pharmacol. Toxicol. 1996;36:379. doi: 10.1146/annurev.pa.36.040196.002115. [DOI] [PubMed] [Google Scholar]
- (10).Rothman RB. Analgesia. 1994;1:27. [Google Scholar]
- (11).Rios CD, Jordan BA, Gomes I, Devi LA. Pharmacol. Ther. 2001;92:71. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- (12).Kieffer BL, Gaveriaux-Ruff C. Prog. Neurobiol. 2002;66:285. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- (13).Cota D, Tschop MH, Horvath TL, Levine AS. Brain Res. Rev. 2006;51:85. doi: 10.1016/j.brainresrev.2005.10.004. [DOI] [PubMed] [Google Scholar]
- (14).Zhang H, Shi YG, Woods JH, Watson SJ, Ko MC. Eur. J. Pharmacol. 2007;570:89. doi: 10.1016/j.ejphar.2007.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jutkiewicz EM. Mol. Interventions. 2006;6:162. doi: 10.1124/mi.6.3.7. [DOI] [PubMed] [Google Scholar]
- (16).Koob GF, Roberts AJ, Kieffer BL, Heyser CJ, Katner SN, Ciccocioppo R, Weiss F. Recent Dev. Alcohol. 2003;16:263. doi: 10.1007/0-306-47939-7_19. [DOI] [PubMed] [Google Scholar]
- (17).Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ortega A, Blount JF, Manchand PS. J. Chem. Soc., Perkin Trans. 1. 1982:2505. [Google Scholar]
- (19).Valdes LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda M. J. Org. Chem. 1984;49:4716. [Google Scholar]
- (20).Siebert DJ. J. Ethnopharmacol. 1994;43:53. doi: 10.1016/0378-8741(94)90116-3. [DOI] [PubMed] [Google Scholar]
- (21).Sheffler DJ, Roth BL. Trends Pharmacol. Sci. 2003;24:107. doi: 10.1016/S0165-6147(03)00027-0. [DOI] [PubMed] [Google Scholar]
- (22).Small L, Fitch HM, Smith WE. J. Am. Chem. Soc. 1936;58:1457. [Google Scholar]
- (23).Romer D, Buscher H, Hill RC, Maurer R, Petcher TJ, Welle HBA, Bakel HCCK, Akkerman AM. Life Sci. 1980;27:971. doi: 10.1016/0024-3205(80)90107-1. [DOI] [PubMed] [Google Scholar]
- (24).Niemegeers CJ, Schellekens KH, Van Bever WF, Janssen PA. Arzneim.-Forsch. 1976;26:1551. [PubMed] [Google Scholar]
- (25).Calderon SN, Rothman RB, Porreca F, Flippen-Anderson JL, McNutt RW, Xu H, Smith LE, Bilsky EJ, Davis P, Rice KC. J. Med. Chem. 1994;37:2125. doi: 10.1021/jm00040a002. [DOI] [PubMed] [Google Scholar]
- (26).Von Voigtlander PF, Lewis RA. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 1982;6:467. doi: 10.1016/s0278-5846(82)80130-9. [DOI] [PubMed] [Google Scholar]
- (27).Hunger A, Kebrle J, Rossi A, Hoffmann K. Experientia. 1957;13:400. doi: 10.1007/BF02161116. [DOI] [PubMed] [Google Scholar]
- (28).Aldrich JV, Vigil-Cruz SC. In: Burger's Medicinal Chemistry and Drug Discovery. 6th ed. Abraham DA, editor. Vol. 6. John Wiley; New York: 2003. p. 329. [Google Scholar]
- (29).Eguchi M. Med. Res. Rev. 2004;24:182. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- (30).Rees DC, Hunter JC. In: Comprehensive Medicinal Chemistry. Emmet JC, editor. Pergammon; New York: 1990. p. 805. [Google Scholar]
- (31).Surratt C, Johnson P, Moriwaki A, Seidleck B, Blaschak C, Wang J, Uhl G. J. Biol. Chem. 1994;269:20548. [PubMed] [Google Scholar]
- (32).Lu Y, Weltrowska G, Lemieux C, Chung NN, Schiller PW. Bioorg. Med. Chem. Lett. 2001;11:323. doi: 10.1016/s0960-894x(00)00660-0. [DOI] [PubMed] [Google Scholar]
- (33).Mansour A, Taylor LP, Fine JL, Thompson RC, Hoversten MT, Mosberg HI, Watson SJ, Akil H. J. Neurochem. 1997;68:344. doi: 10.1046/j.1471-4159.1997.68010344.x. [DOI] [PubMed] [Google Scholar]
- (34).John TF, French LG, Erlichman JS. Eur. J. Pharmacol. 2006;545:129. doi: 10.1016/j.ejphar.2006.06.077. [DOI] [PubMed] [Google Scholar]
- (35).McCurdy CR, Sufka KJ, Smith GH, Warnick JE, Nieto MJ. Pharmacol., Biochem. Behav. 2006;83:109. doi: 10.1016/j.pbb.2005.12.011. [DOI] [PubMed] [Google Scholar]
- (36).Ansonoff MA, Zhang J, Czyzyk T, Rothman RB, Stewart J, Xu H, Zjwiony J, Siebert DJ, Yang F, Roth BL, Pintar JE. J. Pharmacol. Exp. Ther. 2006;318:641. doi: 10.1124/jpet.106.101998. [DOI] [PubMed] [Google Scholar]
- (37).Spanagel R, Herz A, Shippenberg TS. Proc. Natl. Acad. Sci. U.S.A. 1992;89:2046. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA., Jr. Psychopharmacology (Berlin) 2004;172:463. doi: 10.1007/s00213-003-1680-y. [DOI] [PubMed] [Google Scholar]
- (39).Carlezon WA, Jr., Beguin C, Dinieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. J. Pharmacol. Exp. Ther. 2006;316:440. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- (40).Zhang Y, Butelman ER, Schlussman SD, Ho A, Kreek MJ. Psychopharmacology (Berlin) 2005;179:551. doi: 10.1007/s00213-004-2087-0. [DOI] [PubMed] [Google Scholar]
- (41).Chartoff EH, Potter D, Damez-Werno D, Cohen BM, Carlezon WA., Jr. Neuropsychopharmacology. 2008 doi: 10.1038/sj.npp.1301659. doi: 10.1038/sj.npp.1301659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Li J-X, Rice KC, France CP. J. Pharmacol. Exp. Ther. 2008;324:827. doi: 10.1124/jpet.107.130625. [DOI] [PubMed] [Google Scholar]
- (43).Prisinzano TE. Life Sci. 2005;78:527. doi: 10.1016/j.lfs.2005.09.008. [DOI] [PubMed] [Google Scholar]
- (44).Grundmann O, Phipps SM, Zadezensky I, Butterweck V. Planta Med. 2007;73:1039. doi: 10.1055/s-2007-981566. [DOI] [PubMed] [Google Scholar]
- (45).Vortherms TA, Roth BL. Mol. Interventions. 2006;6:257. doi: 10.1124/mi.6.5.7. [DOI] [PubMed] [Google Scholar]
- (46).Valdes LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda M. J. Org. Chem. 1984;49:4716. [Google Scholar]
- (47).Valdes LJ., III . The Pharmacognosy of Salvia Divinorum (Epling and Jativa-M): An Investigation of Ska Maria Pastora, Ph.D. Thesis. University of Michigan; Ann Arbor, MI: 1983. [Google Scholar]
- (48).Valdes LJ, III, Diaz JL, Paul AG. J. Ethnopharmacol. 1983;7:287. doi: 10.1016/0378-8741(83)90004-1. [DOI] [PubMed] [Google Scholar]
- (49).Tyler VE., Jr. Lloydia. 1966;29:275. [Google Scholar]
- (50).Valdes LJ, III, Chang HM, Visger DC, Koreeda M. Org. Lett. 2001;3:3935. doi: 10.1021/ol016820d. [DOI] [PubMed] [Google Scholar]
- (51).Munro TA, Rizzacasa MA. J. Nat. Prod. 2003;66:703. doi: 10.1021/np0205699. [DOI] [PubMed] [Google Scholar]
- (52).Lee DY, Ma Z, Liu-Chen LY, Wang Y, Chen Y, Carlezon WA, Jr., Cohen B. Bioorg. Med. Chem. 2005;13:5635. doi: 10.1016/j.bmc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- (53).Shirota O, Nagamatsu K, Sekita S. J. Nat. Prod. 2006;69:1782. doi: 10.1021/np060456f. [DOI] [PubMed] [Google Scholar]
- (54).Ma Z, Lee DYW. Tetrahedron Lett. 2007;48:5461. doi: 10.1016/j.tetlet.2007.05.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bigham AK, Munro TA, Rizzacasa MA, Robins-Browne RM. J. Nat. Prod. 2003;66:1242. doi: 10.1021/np030313i. [DOI] [PubMed] [Google Scholar]
- (56).Harding WW, Tidgewell K, Schmidt M, Shah K, Dersch CM, Snyder J, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Org. Lett. 2005;7:3017. doi: 10.1021/ol0510522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Valdes LJ., III J. Nat. Prod. 1986;49:171. doi: 10.1021/np50043a031. [DOI] [PubMed] [Google Scholar]
- (58).Kutrzeba L, Dayan FE, Howell JL, Feng J, Giner J-L, Zjawiony JK. Phytochemistry. 2007;68:1872. doi: 10.1016/j.phytochem.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Eisenreich W, Bacher A, Arigoni D, Rohdich F. Cell. Mol. Life Sci. 2004;61:1401. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Cunningham CW, Coop A. Chim. Oggi. 2006;24:54. [Google Scholar]
- (61).Prisinzano TE, Tidgewell K, Harding WW. AAPS J. 2005;7:E592. doi: 10.1208/aapsj070361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr., Jones RM, Portoghese PS, Carlezon WA., Jr. J. Pharmacol. Exp. Ther. 2003;305:323. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- (63).Carroll FI, Harris LS, Aceto MD. Eur. J. Pharmacol. 2005;524:89. doi: 10.1016/j.ejphar.2005.09.013. [DOI] [PubMed] [Google Scholar]
- (64).Beardsley PM, Howard JL, Shelton KL, Carroll FI. Psychopharmacology (Berlin) 2005;183:118. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- (65).Bohn LM, Raehal KM. Curr. Opin. Pharmacol. 2006;6:559. doi: 10.1016/j.coph.2006.06.007. [DOI] [PubMed] [Google Scholar]
- (66).Hollander E, Sood E, Pallanti S, Baldini-Rossi N, Baker B. J. Gambl. Stud. 2005;21:99. doi: 10.1007/s10899-004-1932-8. [DOI] [PubMed] [Google Scholar]
- (67).Rothman RB, Murphy DL, Xu H, Godin JA, Dersch CM, Partilla JS, Tidgewell K, Schmidt M, Prisinzano TE. J. Pharmacol. Exp. Ther. 2007;320:801. doi: 10.1124/jpet.106.113167. [DOI] [PubMed] [Google Scholar]
- (68).Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J. Med. Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Simpson DS, Katavic PL, Lozama A, Harding WW, Parrish D, Deschamps JR, Dersch CM, Partilla JS, Rothman RB, Navarro H, Prisinzano TE. J. Med. Chem. 2007;50:3596. doi: 10.1021/jm070393d. [DOI] [PubMed] [Google Scholar]
- (70).Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. J. Pharmacol. Exp. Ther. 2004;308:1197. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- (71).Beguin C, Richards MR, Wang Y, Chen Y, Liu-Chen LY, Ma Z, Lee DY, Carlezon WA, Jr., Cohen BM. Bioorg. Med. Chem. Lett. 2005;15:2761. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- (72).Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J. Nat. Prod. 2006;69:914. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- (73).Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. J. Med. Chem. 2005;48:4765. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- (74).Xu H, Partilla JS, Wang X, Rutherford JM, Tidgewell K, Prisinzano TE, Bohn LM, Rothman RB. Synapse. 2007;61:166. doi: 10.1002/syn.20356. [DOI] [PubMed] [Google Scholar]
- (75).Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. Mol. Pharmacol. 2007;71:549. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. J. Pharmacol. Exp. Ther. 2007;320:1. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- (77).Tidgewell K, Groer CE, Harding WW, Lozama A, Schmidt M, Marquam A, Hiemstra J, Partilla JS, Dersch CM, Rothman RB, Bohn LM, Prisinzano TE. J. Med. Chem. 2008 doi: 10.1021/jm701162g. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Gilmour B, Navarro H, Rothman RB, Prisinzano TE. J. Nat. Prod. 2006;69:107. doi: 10.1021/np050398i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Beguin C, Richards MR, Li J-G, Wang Y, Xu W, Liu-Chen L-Y, Carlezon WA, Jr., Cohen BM. Bioorg. Med. Chem. Lett. 2006;16:4679. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- (80).Lee DYW, Karnati VVR, He M, Liu-Chen L-Y, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C. Bioorg. Med. Chem. Lett. 2005;15:3744. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- (81).Bikbulatov RV, Yan F, Roth BL, Zjawiony JK. Bioorg. Med. Chem. Lett. 2007;17:2229. doi: 10.1016/j.bmcl.2007.01.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Stewart DJ, Fahmy H, Roth BL, Yan F, Zjawiony JK. Arzneim.-Forsch. 2006;56:269. doi: 10.1055/s-0031-1296720. [DOI] [PubMed] [Google Scholar]
- (83).Yan F, Mosier PD, Westkaemper RB, Stewart J, Zjawiony JK, Vortherms TA, Sheffler DJ, Roth BL. Biochemistry. 2005;44:8643. doi: 10.1021/bi050490d. [DOI] [PubMed] [Google Scholar]
- (84).Munro TA, Goetchius GW, Roth BL, Vortherms TA, Rizzacasa MA. J. Org. Chem. 2005;70:10057. doi: 10.1021/jo051813e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Holden KG, Tidgewell K, Marquam A, Rothman RB, Navarro H, Prisinzano TE. Bioorg. Med. Chem. Lett. 2007;17:6111. doi: 10.1016/j.bmcl.2007.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Lee DY, He M, Kondaveti L, Liu-Chen LY, Ma Z, Wang Y, Chen Y, Li JG, Beguin C, Carlezon WA, Jr., Cohen B. Bioorg. Med. Chem. Lett. 2005;15:4169. doi: 10.1016/j.bmcl.2005.06.092. [DOI] [PubMed] [Google Scholar]
- (87).Lee DY, He M, Liu-Chen LY, Wang Y, Li JG, Xu W, Ma Z, Carlezon WA, Jr., Cohen B. Bioorg. Med. Chem. Lett. 2006;16:5498. doi: 10.1016/j.bmcl.2006.08.051. [DOI] [PubMed] [Google Scholar]
- (88).Prisinzano TE, Rothman RB. Unpublished results.
- (89).Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Dersch CM, Rothman RB, Prisinzano TE. Bioorg. Med. Chem. Lett. 2006;16:3170. doi: 10.1016/j.bmcl.2006.03.062. [DOI] [PubMed] [Google Scholar]
- (90).Lingham AR, Hogel HM, Rook TJ. Aust. J. Chem. 2006;59:340. [Google Scholar]
- (91).Scheerer JR, Lawrence JF, Wang GC, Evans DA. J. Am. Chem. Soc. 2007;129:8968. doi: 10.1021/ja073590a. [DOI] [PubMed] [Google Scholar]
- (92).Lin CE, Takemori AE, Portoghese PS. J. Med. Chem. 1993;36:2412. doi: 10.1021/jm00068a020. [DOI] [PubMed] [Google Scholar]
- (93).Hjorth S, Thirstrup K, Grandy D, Schwartz T. Mol. Pharmacol. 1995;47:1089. [PubMed] [Google Scholar]
- (94).Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. J. Med. Chem. 1998;41:4911. doi: 10.1021/jm9805182. [DOI] [PubMed] [Google Scholar]
- (95).Larson DL, Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. J. Med. Chem. 2000;43:1573. doi: 10.1021/jm000059g. [DOI] [PubMed] [Google Scholar]
- (96).Stevens WC, Jr., Jones RM, Subramanian G, Metzger TG, Ferguson DM, Portoghese PS. J. Med. Chem. 2000;43:2759. doi: 10.1021/jm0000665. [DOI] [PubMed] [Google Scholar]
- (97).Metzger TG, Paterlini MG, Ferguson DM, Portoghese PS. J. Med. Chem. 2001;44:857. doi: 10.1021/jm000381r. [DOI] [PubMed] [Google Scholar]
- (98).Lavecchia A, Greco G, Novellino E, Vittorio F, Ronsisvalle G. J. Med. Chem. 2000;43:2124. doi: 10.1021/jm991161k. [DOI] [PubMed] [Google Scholar]
- (99).Metzger TG, Paterlini MG, Portoghese PS, Ferguson DM. Neurochem. Res. 1996;21:1287. doi: 10.1007/BF02532369. [DOI] [PubMed] [Google Scholar]
- (100).Iadanza M, Holtje M, Ronsisvalle G, Holtje HD. J. Med. Chem. 2002;45:4838. doi: 10.1021/jm0209127. [DOI] [PubMed] [Google Scholar]
- (101).Subramanian G, Paterlini MG, Larson DL, Portoghese PS, Ferguson DM. J. Med. Chem. 1998;41:4777. doi: 10.1021/jm9803166. [DOI] [PubMed] [Google Scholar]
- (102).Wan XH, Huang XQ, Zhou DH, Jiang HL, Chen KX, Chi ZQ. Acta Pharmacol. Sin. 2000;21:701. [PubMed] [Google Scholar]
- (103).Holzgrabe U, Brandt W. J. Med. Chem. 2003;46:1383. doi: 10.1021/jm0210360. [DOI] [PubMed] [Google Scholar]
- (104).Kane BE, Nieto MJ, McCurdy CR, Ferguson DM. FEBS J. 2006;273:1966. doi: 10.1111/j.1742-4658.2006.05212.x. [DOI] [PubMed] [Google Scholar]
- (105).Vortherms TA, Mosier PD, Westkaemper RB, Roth BL. J. Biol. Chem. 2007;282:3146. doi: 10.1074/jbc.M609264200. [DOI] [PubMed] [Google Scholar]
- (106).White WC. Report of the Committee on Drug Addiction, 1929-1941. National Research Council; 1941. [DOI] [PubMed] [Google Scholar]
- (107).Eddy NB. The National Research Council Involvement in the Opiate Problem 1928-1971. National Academy of Sciences; Washington, DC: 1973. [Google Scholar]
- (108).Rice KC. In: Opioids and Pain Relief: A Historical Perspective, Progress in Pain Research and Management. Meldrum ML, editor. IASP Press; Seattle, WA: 2003. [Google Scholar]
- (109).Corbett AD, Henderson G, McKnight AT, Paterson SJ. Br. J. Pharmacol. 2006;147(Suppl 1):S153. doi: 10.1038/sj.bjp.0706435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Beguin C, Potter DN, DiNieri JA, Munro TA, Richards MR, Paine TA, Berry L, Zhao Z, Roth BL, Xu W, Liu-Chen L-Y, Carlezon WA, Jr., Cohen BM. J. Pharmacol. Exp. Ther. 2008;324:188. doi: 10.1124/jpet.107.129023. [DOI] [PubMed] [Google Scholar]