

Abstract
NOS is a key enzyme in the production of NO, a molecule that directly regulates vasorelaxation and blood supply. Diverse forms of muscle disease have been clinically associated with unusual fatigue after exercise. The localization of neuronal NOS (nNOS) at the plasma membrane of muscle has recently been shown to prevent muscle fatigue after exercise. In this issue of the JCI, Lai et al. show that dystrophin — the structural protein missing in individuals with Duchenne muscular dystrophy — anchors nNOS to the sarcolemma through a direct interaction with dystrophin spectrin-like repeats 16 and 17 (see the related article beginning on page 624). Furthermore, in another recently reported study of mouse models of muscular dystrophy, phosphodiesterase 5A inhibitors were used to treat the downstream ischemia that is associated with nNOS mislocalization. Collectively, these findings significantly advance our understanding of exercise-induced muscle fatigue and its role in muscle disease.
Dystrophin localizes neuronal NOS to the plasma membrane of muscle
The dystrophin-associated protein complex (Figure 1) is found at the plasma membrane of skeletal muscle, where it provides stability to the myofiber membrane during contraction (1). Genetic mutations that ablate dystrophin expression lead to Duchenne muscular dystrophy (DMD) in humans and muscular dystrophy in mdx mice. Dystrophin is a long cytoskeletal protein that contains an actin-binding site at its amino terminus, 23 spectrin-like repeats interrupted by four hinge points, and a carboxyl terminus that links dystrophin to dystroglycan and the other transmembrane and membrane-associated components of the larger protein complex. The syntrophins are cytosolic proteins. α-Syntrophin directly binds to neuronal NOS (nNOS or NOS1) by way of its PDZ (postsynaptic density 95, discs large, and zonula occludens–1) domains (2). Syntrophins bind directly to dystrobrevins and dystrophin, and this interaction was thought to be sufficient to localize nNOS to the plasma membrane (3). In the absence of dystrophin, nNOS is lost from the plasma membrane (2).
Figure 1. Mechanism for the localization of nNOS to the plasma membrane of muscle.
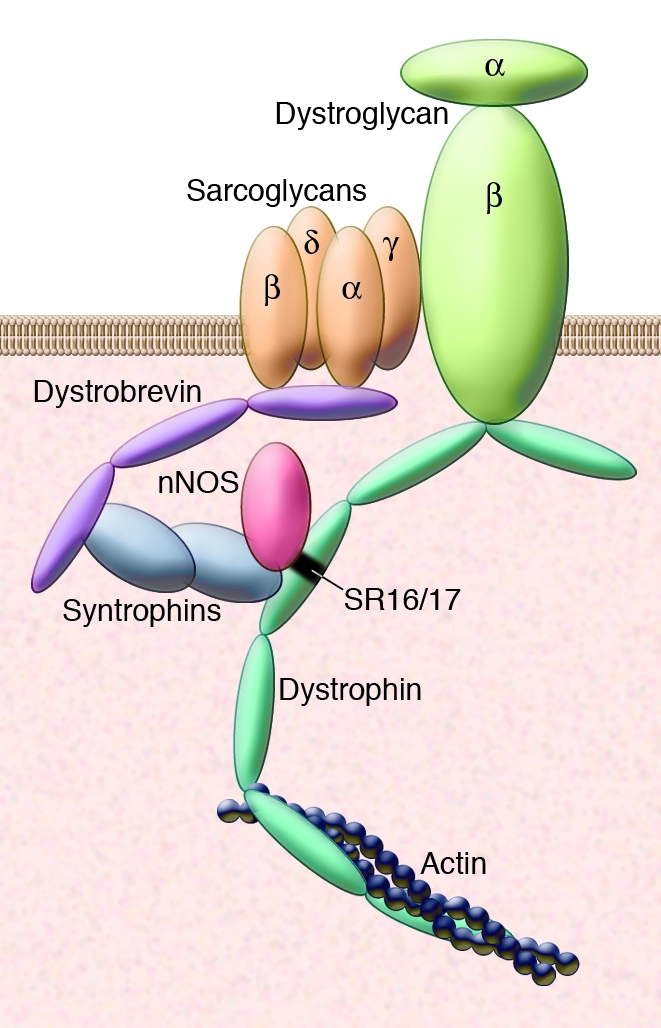
Sarcolemmal nNOS promotes vasodilation and improves blood flow into muscle. This blood flow is necessary to prevent early fatigue with exercise. In this issue of the JCI, Lai et al. show that a direct interaction between dystrophin and nNOS is mediated by spectrin-like repeats 16 and 17 in dystrophin (SR16/17) (14). It was previously shown that α-syntrophin could directly bind nNOS by way of its PDZ domain (15). Loss of dystrophin in humans with DMD or in mdx mice is associated with reduction in levels of dystrophin-associated proteins including α-syntrophin and nNOS (2). Work by Lai et al. (14) as well as recent reports by Kobayashi et al. (16) and Percival et al. (8) now clarify that loss of nNOS from the plasma membrane mediates muscle fatigue after minimal exercise and that this feature is present in diverse forms of muscle disease.
The functional significance of nNOS displacement from the sarcolemma of muscle was previously shown to be associated with muscle ischemia in both mdx mice and boys with DMD (4, 5). However, mice engineered to lack nNOS itself lack the overt features of muscular dystrophy, such as muscle degeneration, reactive regeneration, and fibrofatty replacement of muscle (6, 7). In earlier studies, the phenotype of mice lacking both nNOS and dystrophin was no different from that observed in the mdx model itself (6, 7).
However, recent reevaluation of nNOS-null mice shows that male nNOS-null mice have smaller muscle mass and reduced force production compared with strain- and sex-matched WT mice (8). When normalized for the smaller muscle mass, force production in nNOS-null mice is normal. However, nNOS-null mice display a specific deficit in adapting to exercise and develop profound fatigue upon repeated muscle contraction. Thus, loss of nNOS or mislocalization of nNOS from its normal position at the plasma membrane causes fatigue with exercise. Interestingly, muscle expressing syntrophin that lacked an nNOS-binding site was also found to exhibit ischemia with exercise (9).
Minidystrophins only partially correct muscular dystrophy
DMD develops when dystrophin is absent, and gene replacement strategies have been pursued through both viral delivery and transgenesis. Internal truncations of dystrophin that leave the amino and carboxyl termini intact occur naturally with the milder muscle disease Becker muscular dystrophy (BMD). BMD patients display less muscle degeneration than DMD patients but frequently note exercise-induced fatigue (10). Gene replacement therapy for DMD is limited by vector capacity, as the dystrophin protein is large in size. Therefore, smaller dystrophins called mini- or microdystrophins have been engineered to mimic what is naturally found in BMD patients and have been extensively tested in the mdx mouse (11, 12). Gene replacement studies have shown that dystrophin lacking the middle portion of the protein can correct underlying pathology as long as the actin-binding domain and carboxyterminal domains are intact (13).
As now shown by Lai and colleagues in this issue of the JCI, this strategy, while successful at correcting histopathology, leaves the treated animals unable to exercise to the same extent as normal mice (14). To explain the exercise deficit, they noted that nNOS was not normally positioned at the plasma membrane in mdx mice expressing minidystrophins. Specifically, the authors correlated the absence of dystrophin’s spectrin-like repeats 16 and 17 with absent plasma membrane nNOS and an inability to undergo exercise conditioning. Moreover, these mice, referred to as ΔH2–R19 transgenic mdx mice, develop skeletal muscle ischemia with exercise. Ischemia and the absence of spectrin-like repeats 16 and 17 correlated with the inability to improve exercise capacity over time (summarized in Table 1). They concluded that association of nNOS with the dystrophin complex requires spectrin-like repeats 16 and 17 and is necessary to produce vasodilation and supply oxygen to exercising muscle. It was previously shown that the syntrophin PDZ domain was sufficient to anchor nNOS (15). Together, these findings indicate that nNOS requires both dystrophin and syntrophin for full localization and function at the plasma membrane.
Table 1 .
Genetic models of nNOS mislocalization

Muscle fatigue from nNOS mislocalization can be treated with phosphodiesterase 5A inhibitors
Recently, Kobayashi et al. studied the mdx mouse as well as another model, the Sgca–/– mouse, which lacks α-sarcoglycan and serves as a model of limb-girdle muscular dystrophy (16). When these animals were subjected to brief, mild exercise, they would not exercise further, unlike normal controls that continued to exercise. Moreover, in addition to being unable to sustain normal levels of exercise, the animals displayed arterial narrowing in skeletal muscle and evidence of ischemia. nNOS was displaced from the sarcolemma in these models. Treatment of the mutant animals with the phosphodiesterase 5A inhibitor sildenafil to enhance cyclic GMP signaling and promote vasodilation improved exercise ability and reduced evidence of edema within the musculature. These studies focused on skeletal muscle pathology and treatment. Interestingly, treatment of mdx mice with sildenafil was also recently shown to be successful in preventing cardiomyopathy and improving cardiac performance (17). Transgenic overexpression of cyclic GMP in the hearts of mdx mice was sufficient to ameliorate cardiomyopathy. Thus, phosphodiesterase 5A inhibition may be of benefit for both cardiopulmonary performance and skeletal muscle function in patients with muscular dystrophy.
Displacement of nNOS from the sarcolemma is not unique to DMD, and Kobayashi et al. showed that nNOS levels were reduced in many genetically distinct forms of human muscle disease, including those resulting from loss of the genes encoding extracellular matrix proteins laminin α2 and collagen VI (16). Mutations in dysferlin are associated with defective muscle repair, and this distinct form of muscular dystrophy was also shown to be characterized by reduced levels of nNOS (16). Whether neurogenic muscle diseases share this nNOS-mediated ischemia has not yet been determined. These observations raise interesting questions since deconditioning itself is a component of most neuromuscular disease. Thus, it is possible that in the absence of genetic disease, deconditioning alone may be associated with relative ischemia and potential mislocalization of nNOS from the plasma membrane. Therefore, it is possible that exercise conditioning could reverse nNOS mislocalization and enhance blood flow to the conditioned muscle. Whether this mechanism of nNOS recruitment occurs in normal muscle and is impaired in diseased muscle are areas for future study.
In mdx muscle, repetitive muscle contraction leads to a reduction of force production (18, 19). It was previously assumed that this force reduction arose from contraction-induced membrane damage. While this mechanism may still be at play, muscle fatigue from nNOS-mediated ischemia is also likely to be a significant contributor. Recently, it was shown that activation of the serine-threonine kinase Akt prevents contraction-induced force drop in mdx muscle (20). Akt or protein interactions at the plasma membrane may influence the local activity of NOS and thereby regulate further its ability to promote blood flow and counteract muscle fatigue. As gene therapy approaches for DMD evolve, nNOS restoration mediated by dystrophin spectrin-like repeats 16 and 17 will need to be incorporated in order to correct muscle fatigue.
Acknowledgments
We acknowledge the support of NIH grant HL61322 and the Doris Duke Charitable Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: BMD, Becker muscular dystrophy; DMD, Duchenne muscular dystrophy; nNOS, neuronal NOS.
Citation for this article: J. Clin. Invest. 119:448–450 (2009). doi:10.1172/JCI38618.
See the related article beginning on page 624.
References
- 1.Lapidos K.A., Kakkar R., McNally E.M. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 2.Brenman J.E., Chao D.S., Xia H., Aldape K., Bredt D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 3.Ahn A.H., et al. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J. Biol. Chem. 1996;271:2724–2730. doi: 10.1074/jbc.271.5.2724. [DOI] [PubMed] [Google Scholar]
- 4.Sander M., et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. . Proc. Natl. Acad. Sci. U. S. A. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas G.D., et al. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. U. S. A. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chao D.S., Silvagno F., Bredt D.S. Muscular dystrophy in mdx mice despite lack of neuronal nitric oxide synthase. J. Neurochem. 1998;71:784–789. doi: 10.1046/j.1471-4159.1998.71020784.x. [DOI] [PubMed] [Google Scholar]
- 7.Crosbie R.H., et al. mdx muscle pathology is independent of nNOS perturbation. Hum. Mol. Genet. 1998;7:823–829. doi: 10.1093/hmg/7.5.823. [DOI] [PubMed] [Google Scholar]
- 8.Percival J.M., Anderson K.N., Gregorevic P., Chamberlain J.S., Froehner S.C. Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS ONE. 2008;3:e3387. doi: 10.1371/journal.pone.0003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas G.D., Shaul P.W., Yuhanna I.S., Froehner S.C., Adams M.E. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ. Res. 2003;92:554–560. doi: 10.1161/01.RES.0000061570.83105.52. [DOI] [PubMed] [Google Scholar]
- 10.Phillips B.A., Mastaglia F.L. Exercise therapy in patients with myopathy. Curr. Opin. Neurol. 2000;13:547–552. doi: 10.1097/00019052-200010000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Duan D. Challenges and opportunities in dystrophin-deficient cardiomyopathy gene therapy. . Hum. Mol. Genet. 2006;15:R253–R261. doi: 10.1093/hmg/ddl180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapsa R., Kornberg A.J., Byrne E. Novel therapies for Duchenne muscular dystrophy. Lancet Neurol. 2003;2:299–310. doi: 10.1016/S1474-4422(03)00382-X. [DOI] [PubMed] [Google Scholar]
- 13.Banks G.B., Gregorevic P., Allen J.M., Finn E.E., Chamberlain J.S. Functional capacity of dystrophins carrying deletions in the N-terminal actin-binding domain. Hum. Mol. Genet. 2007;16:2105–2113. doi: 10.1093/hmg/ddm158. [DOI] [PubMed] [Google Scholar]
- 14.Lai Y., et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenman J.E., et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/S0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi Y.M., et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456:511–515. doi: 10.1038/nature07414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khairallah M., et al. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7028–7033. doi: 10.1073/pnas.0710595105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox G.A., et al. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature. 1993;364:725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- 19.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U. S. A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaauw B., et al. Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum. Mol. Genet. 2008;17:3686–3696. doi: 10.1093/hmg/ddn264. [DOI] [PubMed] [Google Scholar]
- 21.Grady R.M., et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat. Cell. Biol. 1999;1:215–220. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]