

Abstract
Background
Gene expression microarray technologies are widely used across most areas of biological and medical research. Comparing and integrating microarray data from different experiments would be very useful, but is currently very challenging due to the experimental and hybridization conditions, as well as data preprocessing and normalization methods. Furthermore, even in the case of the widely-used, industry-standard Affymetrix oligonucleotide microarrays, the various array generations have different probe sets representing different genes, hindering the data integration.
Results
In this study our objective is to find systematic approaches to normalize the data emerging from different Affymetrix array generations and from different laboratories. We compare and assess the accuracy of five normalization methods for Affymetrix gene expression data using 6,926 Affymetrix experiments from five array generations. The methods that we compare include 1) standardization, 2) housekeeping gene based normalization, 3) equalized quantile normalization, 4) Weibull distribution based normalization and 5) array generation based gene centering. Our results indicate that the best results are achieved when the data is normalized first within a sample and then between-samples with Array Generation based gene Centering (AGC) normalization.
Conclusion
We conclude that with the AGC method integrating different Affymetrix datasets results in values that are significantly more comparable across the array generations than in the cases where no array generation based normalization is used. The AGC method was found to be the best method for normalizing the data from several different array generations, and achieve comparable gene values across thousands of samples.
Background
Microarray experiments have become an indispensable part of modern biological and biomedical research. As the number of studies using microarrays is growing all the time, it becomes increasingly important to compare and integrate data from multiple experiments and thereby improve the ability to make meaningful biological conclusions. Collections of microarray data from thousands of samples are emerging, but proper normalization methods are to a large extent lacking. To make optimal use of these datasets, improved methods for normalizing data from different studies in different laboratories are urgently required.
There are studies where gene expression data from different studies are systematically combined together. For example, computational models for defining modules in the transcriptional data [1-3] have been suggested. In addition, Oncomine, a database for gene expression data in cancer tissues including over 25,000 samples have been introduced [4,5]. Furthermore, a Celsius data warehousing system aggregates Affymetrix CEL-files and associated metadata [6]. These studies have included several thousands of samples from separate studies. Since different array types and normalization methods have typically been carried out for each study, the integration and direct comparison between the samples is difficult. Most of these meta-analyses are performed one-study-at-a-time, summing up the results together. There are also some publications describing the integration of data between different Affymetrix array generations. These methods are often based on the normalization of oligonucleotide microarray data using sequence overlaps between the individual oligos on the same slide [7-9]. However, the drawback of these approaches is that the non-overlapping probes need to be discarded. Therefore, particularly in the comparisons across multiple platforms, the number of informative genes is significantly reduced.
Here, our main objective was to test several known normalization methods for integrating gene expression values across thousands of experiments to be able to select a suitable method when combining datasets across Affymetrix array generations and experiment series. Even though the methods presented in this study are shown to work with the Affymetrix gene expression microarrays, they should be applicable also for integration experiments of other microarray platforms.
Results
We compared and assessed the accuracy of five normalization methods for Affymetrix gene expression data using 6,926 Affymetrix experiments from five array generations. The methods that we compared include 1) standardization (Z), 2) housekeeping gene based normalization (HK), 3) equalized quantile normalization (Q), 4) Weibull distribution based normalization (WBL) and 5) array generation based gene centering (AGC). These were tested in the following ten combinations: Pure preprocessed data (MAS) without any further normalization, Z-, HK-, Q-, WBL-normalizations, and all of these normalization methods combined with the AGC method: MASAGC, ZAGC, HKAGC, QAGC and WBLAGC. The MAS, Z, HK, Q and WBL methods normalize the data within the samples, while the AGC method normalizes the data gene-wise between the samples.
Goodness of normalization can be measured in many ways. Here, we applied five different ways to estimate the degree of comparability between data from different array generations, including: 1) correlation between technical replicates, 2) correlation between randomly selected genes, 3) classification of the samples based on the anatomical classes, 4) comparison of correlations between the samples computed based on the anatomical classes and array generations, 5) stability of the house-keeping genes.
The data collection used in this study contained samples from Affymetrix array generations Hu6800 (HuGeneFL), HG-U95A, HG-U95Av2, HG-U133A, HG-U133 Plus 2. These array generations were selected as there were more than 500 samples hybridized on each of them in the database by the time of the comparison. At least half of the genes were in common between the array generations.
Correlation between technical replicates
The first metric for comparing the goodness of the normalization methods is to study the correlation between technical replicates. We have utilized an experiment series from St. Jude University [10,11] with 132 replicated RNA samples, each analyzed with both HG-U95Av2 and HG-U1331A. We calculated the correlations between these samples with each normalization method. This comparison method has been used in several studies in which data from different generations of Affymetrix arrays are combined and compared [7,8,12]. Here, the results are identical for MAS, Z and HK methods, since the correlation is linearly invariant. When comparing the methods without the AGC correction, the WBL gave the best results. We calculated the significance of the results using one-way ANOVA and performed the multiple comparison with Tukey's HSD. When AGC was merged with any of the normalization methods, the correlations increased significantly as compared with the first level normalization alone, with a significance level of α = 0.01. The WBLAGC gave the best results from the AGC methods, but the difference with the other AGC normalization methods was not significant (Figure 1).
Figure 1.
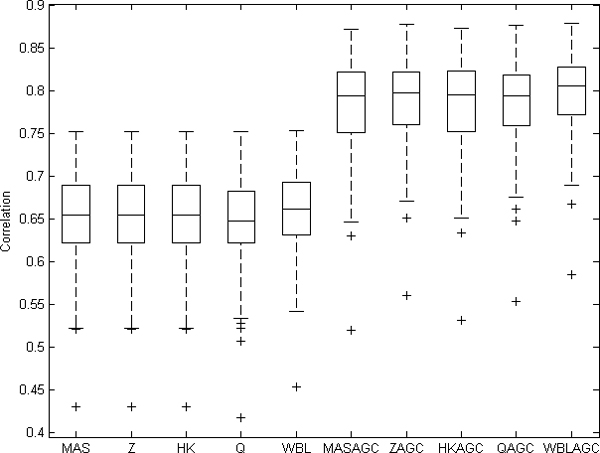
Correlations between technical replicates. Values from 132 technically replicated samples are normalized with different normalization methods and correlations are computed based on logarithmic values. Overall, the AGC improved the correlations.
Correlation of randomly selected gene pair
Another method for comparing the goodness of the normalization methods is the comparison between the correlations of randomly selected genes [13]. Since it is unlikely that two randomly chosen genes are correlated with each other, the expected value for their correlation is zero. Now, the hypothesis is that E(Corr(k1, k2)) = 0 for genes k1, k2 where k1 ≠ k2. The different array generations are known to induce some biases to the gene values that may further cause systematic errors in the data. These kinds of systematic array-wide variations may increase the correlation between randomly selected genes.
We selected randomly 500 genes that had values in each array generation and computed the correlations between each gene pair in the data normalized with the different methods. Further, we tested the mean values of the distributions of randomly selected correlations with one-way ANOVA and a utilized multiple testing procedure Tukey's HSD. The results showed that with significance level α = 0.01 the ZAGC, HKAGC, QAGC and WBLAGC had smaller mean values than the other methods. AGC correction was again found more robust than the other normalization methods, as these AGC-correction methods did not significantly differ from one another and were closer to zero (Figure 2).
Figure 2.
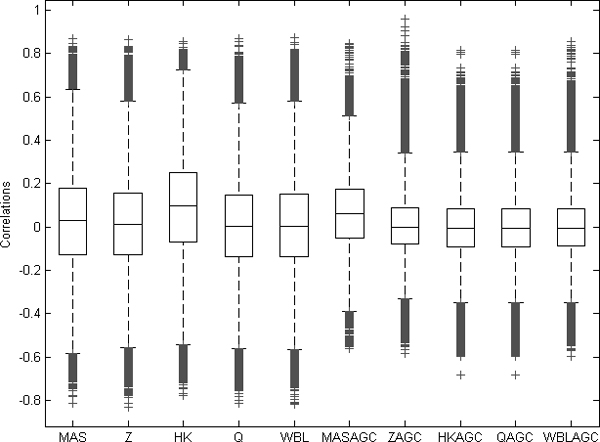
Correlation values between randomly selected gene pairs. The correlations were calculated between 500 randomly selected genes through 6,926 samples. The four AGC-normalized datasets ZAGC, HKAGC, QAGC, and WBLAGC have mean values closer to zero than the others (ANOVA with multiple testing procedure, α < 0.01).
Samples to profiles classification
Third way to estimate goodness of normalization was the use of anatomical classes with the eVOC Anatomical System ontology [14]. An anatomical profile is the mean value of the logarithmic values of healthy samples of each tissue type. The profiles were calculated independently between the array generations. To obtain the profiles we used 1,464 samples from healthy tissues and cells including 15,931 genes from 35 anatomical classes. All the samples were annotated based on the eVOC Anatomical System ontology and the profile for the anatomical classes was created only if there were more than ten samples from that tissue. The 1,464 healthy samples were classified to the anatomical profiles with the nearest neighbour algorithm.
We used Pearson correlation as the metric in the classification. We computed the distance d = 1-ri, j, where ri, j is the correlation between the logarithmic values of the sample i and the profile j. Each sample was classified to the profile with the smallest distance. With the AGC normalized data the number of correctly classified samples increased substantially (Table 1). Obviously, there will always be some biological variability within a tissue, as well as sampling errors, methodological variability, and lab-to-lab variability that will render 100% classification accuracy unattainable. Nevertheless, the significant improvement of classification accuracy again testifies for the value of the AGC-based normalization methods.
Table 1.
Results of the samples to profiles classification. 1,464 samples from healthy tissues and cells including 15,931 genes from 35 anatomical classes were classified. Anatomical classes, number of samples and number of different array generations within each of the classes are listed in the table. The percentages of correctly classified samples are calculated with each normalization method.
| # samples | # array gens | MAS | Z | HK | Q | WBL | MASAGC | ZAGC | HKAGC | QAGC | WBLAGC | |
| TOTAL | 1464 | 5 | 74.2% | 77.9% | 75.5% | 77.6% | 76.1% | 86.7% | 89.6% | 89.7% | 89.8% | 89.3% |
| Aorta | 24 | 1 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| Bronchus | 94 | 2 | 81.9% | 81.9% | 81.9% | 81.9% | 81.9% | 86.2% | 86.2% | 86.2% | 86.2% | 86.2% |
| Lung | 117 | 4 | 47.9% | 46.2% | 46.2% | 46.2% | 46.2% | 91.5% | 88.0% | 89.7% | 86.3% | 83.8% |
| Alveolus | 34 | 1 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| Bone marrow | 104 | 3 | 53.8% | 53.8% | 53.8% | 52.9% | 52.9% | 86.5% | 84.6% | 86.5% | 85.6% | 86.5% |
| Peripheral blood | 260 | 3 | 53.1% | 70.8% | 61.2% | 70.8% | 61.2% | 62.7% | 82.3% | 82.3% | 83.5% | 82.3% |
| Tonsil | 16 | 2 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| Colon | 18 | 3 | 88.9% | 88.9% | 88.9% | 88.9% | 88.9% | 88.9% | 94.4% | 88.9% | 94.4% | 88.9% |
| Liver | 12 | 3 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| Pancreas | 21 | 4 | 76.2% | 81.0% | 81.0% | 76.2% | 81.0% | 95.2% | 95.2% | 95.2% | 95.2% | 95.2% |
| Kidney | 49 | 3 | 73.5% | 75.5% | 73.5% | 73.5% | 75.5% | 89.8% | 87.8% | 89.8% | 87.8% | 87.8% |
| Testis | 13 | 3 | 92.3% | 92.3% | 92.3% | 92.3% | 92.3% | 92.3% | 92.3% | 92.3% | 92.3% | 92.3% |
| Prostate | 70 | 4 | 80.0% | 75.7% | 80.0% | 75.7% | 75.7% | 98.6% | 95.7% | 98.6% | 95.7% | 95.7% |
| Foreskin | 30 | 2 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| Ovary | 14 | 2 | 57.1% | 64.3% | 57.1% | 64.3% | 57.1% | 78.6% | 78.6% | 78.6% | 78.6% | 78.6% |
| Uterus | 12 | 3 | 83.3% | 75.0% | 83.3% | 75.0% | 83.3% | 100% | 91.7% | 100% | 91.7% | 91.7% |
| Endometrium | 12 | 1 | 50.0% | 83.3% | 50.0% | 66.7% | 83.3% | 100% | 100% | 100% | 100% | 100% |
| Placenta | 28 | 3 | 67.9% | 67.9% | 64.3% | 67.9% | 67.9% | 82.1% | 75.0% | 75.0% | 75.0% | 75.0% |
| Breast | 25 | 4 | 64.0% | 64.0% | 64.0% | 64.0% | 64.0% | 84.0% | 96.0% | 84.0% | 96.0% | 100% |
| Thyroid | 16 | 2 | 87.5% | 100% | 88% | 100% | 100% | 100% | 93.8% | 93.8% | 93.8% | 87.5% |
| Thymus | 12 | 2 | 66.7% | 75.0% | 75.0% | 75.0% | 75.0% | 83.3% | 75.0% | 83.3% | 83.3% | 83.3% |
| Muscle | 11 | 2 | 90.9% | 100% | 90.9% | 100% | 100% | 54.5% | 54.5% | 54.5% | 54.5% | 54.5% |
| Skin cuticle | 85 | 3 | 91.8% | 91.8% | 91.8% | 91.8% | 91.8% | 96.5% | 97.6% | 97.6% | 97.6% | 97.6% |
| Brain | 35 | 3 | 54.3% | 60.0% | 54.3% | 60.0% | 60.0% | 88.6% | 88.6% | 85.7% | 85.7% | 85.7% |
| Cerebral cortex | 34 | 1 | 55.9% | 61.8% | 55.9% | 61.8% | 58.8% | 58.8% | 61.8% | 52.9% | 61.8% | 58.8% |
| Frontal lobe | 33 | 1 | 93.9% | 93.9% | 93.9% | 93.9% | 93.9% | 93.9% | 93.9% | 93.9% | 93.9% | 93.9% |
| Hypothalamus | 25 | 1 | 100% | 100% | 100% | 100% | 100% | 100% | 96.0% | 96.0% | 96.0% | 96.0% |
| Cerebellum | 33 | 2 | 84.8% | 84.8% | 84.8% | 84.8% | 84.8% | 97.0% | 97.0% | 97.0% | 97.0% | 97.0% |
| Lens | 11 | 1 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 90.9% | 100% | 100% |
| Optic nerve | 44 | 1 | 93.2% | 93.2% | 93.2% | 93.2% | 93.2% | 93.2% | 93.2% | 93.2% | 93.2% | 93.2% |
| Striated skeletal muscle | 92 | 2 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| Umbilical vein | 38 | 1 | 97.4% | 97.4% | 97.4% | 97.4% | 97.4% | 97.4% | 97.4% | 97.4% | 97.4% | 97.4% |
| Intervertebral disc | 11 | 1 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| Ventricle myocardium | 18 | 2 | 61.1% | 61.1% | 61.1% | 61.1% | 61.1% | 100% | 100% | 100% | 100% | 100% |
| Atrium myocardium | 13 | 1 | 100% | 100% | 100% | 100% | 100% | 76.9% | 76.9% | 84.6% | 76.9% | 76.9% |
Correlations between samples from the same anatomical class
The correlation between samples from same anatomical class should be high indicating the similarity of samples in question. However, often the experimental conditions, preprocessing and array generation may cause high correlation, even if samples have very little in common based on the anatomy. This causes problems if the technical details of the experiments have more effect on the final data than the biological properties of the samples.
We assume that the expected value of correlation between the samples from same anatomical class is higher than the expected value of correlation of samples from different anatomical class, even if the samples were from the same experiment series or from same array generation. We calculated the correlations of gene expression levels between all the 1,464 healthy samples in the dataset and analyzed the values of them. Based on the array generation and the anatomy of samples, we divided these correlations into two groups: 1) Correlations from healthy samples from the same array generation but from different anatomical class, and 2) Correlations of healthy samples from the same anatomical class done with different array generations.
When AGC was not used, the array generation was superior to biological origin in defining the identity of the sample. In such cases the correlation between samples from the same array generations was significantly higher than the correlations between samples from the same anatomical class. When the data were AGC normalized, the correlations from the same anatomical origin were significantly higher than the correlations from different anatomical classes within the same array generation. The significance was tested with one-way ANOVA and multiple comparisons performed with Tukey's HSD with α < 0.01. As evidenced by the significance analysis, the AGC-normalization method reduced noise due to different array generations (Figure 3).
Figure 3.
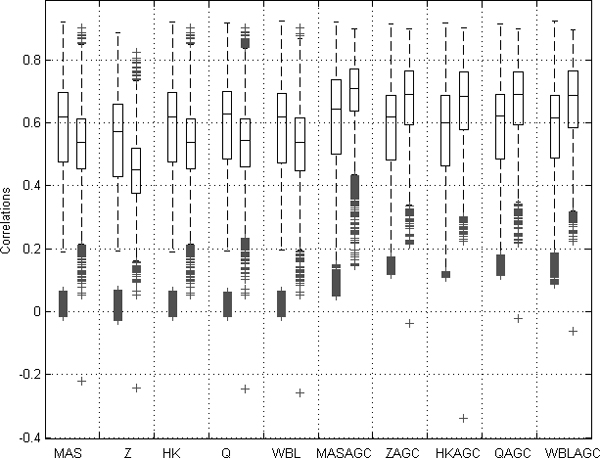
Boxplot of distributions for two sets of correlations from data normalized with each normalization method. The left values are correlations between samples from same array generation within different anatomical class. The right values are correlations between samples within the same anatomical class but with different array generation. All the 1,464 healthy samples were used in this comparison. When the AGC method was used, the mean value of the correlations between samples from the same anatomical class but different array generations were significantly higher than the mean of the correlations between the samples from same array generation but different anatomical class.
Stability of housekeeping genes
If the data are normalized properly, the housekeeping gene values should be stable between experiments. This is based on the assumption that the housekeeping genes are expressed similarly in all samples across the array generations and tissues. However, it is known that the array generation can impact also on values for housekeeping genes, and the expression values of housekeeping genes in our material also seemed to differ based on the array generation.
We investigated the effects of different normalization methods on similarity of distributions of housekeeping genes from different array generations. The housekeeping genes under consideration were the same ones than used in the HK-normalization. The similarities were quantified with the Kullback-Leibler measure. We assumed that the values for each housekeeping gene from one array generation should be distributed similarly with the distribution of the gene across all the array generations. We divided the range of the gene value into 50 bins so that within each bin there are 2% of the gene values of the gene:
| aj = f([0, 0.02, 0.04,..., 0.98, 1]), |
where f is the empirical cumulative density function of the gene. We define D to be a set of all expression values measuring the expression of gene k and array generations to A1,..., Ap. Now, for every array generation i, Di ⊆ D is a set of all expression values of the housekeeping gene that are measured with the array generation Ai. The probability density function of distribution Q for each array generation i is constant:
where #(D) is the number of values in the data set D. We assume that the data values within each percentile group are distributed along this constant distribution. We compute the discrete distribution of the gene values from all array generations within each of these percentiles of the data:
where i is the array generation and j the percentile group. Thus, it is assumed that the distribution P is similar with the distribution Q. The distance between these distributions for each array generation i is calculated with the Kullback-Leibler distance:
where j goes through the percentiles. The smaller the distance is between the distributions, the closer the distributions are to each other.
We calculated these distances for each of the 126 housekeeping genes [15] from each array generation. The AGC method greatly reduced the distance between gene values from one array generation and gene values from all array generations (Figure 4).
Figure 4.
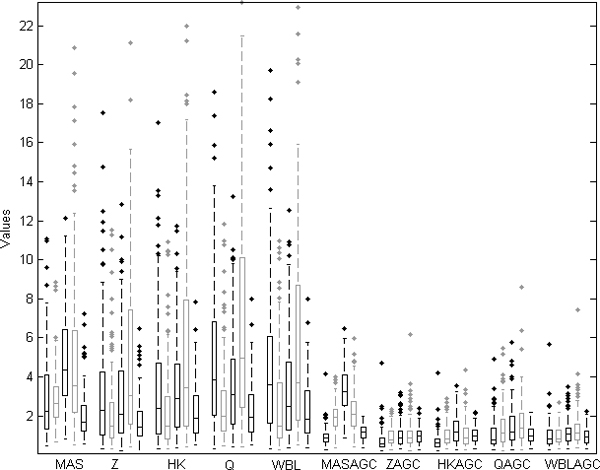
The boxplot of the Kullback-Leibler distances with each array generation and normalization. The KL-distances of 126 housekeeping genes from each array generation from all 6,926 samples were computed. Each boxplot includes the KL-distances of the 126 genes. The array generations are ordered within each of the normalization methods in chronological order of array generations; Hu6800, HG-U95A, HG-U95Av2, HG-U133A and HG-U133 Plus 2.0.
Discussion
An important step to integrate Affymetrix data is to develop methods that result in comparable values for a wide spectrum of array generations. Further, it is crucial to use different measures for goodness of the normalization as the objectives for the normalization may vary between studies. The methods we have developed will significantly facilitate data comparisons across thousands of samples, with minimal loss of informative genes, which was a serious limitation in earlier studies.
We have applied five different normalization methods for Affymetrix gene expression data. The array-generation based gene centering method (AGC) [16] can be merged together with any within-slide normalization method. Here, we tested the values normalized with the AGC method combined with five different normalization methods and observed significantly improved results. All the normalization methods compared here are based on different assumptions and therefore also the effect on normalization strategies may vary. The traits of the normalization methods are collected into Table 2.
Table 2.
The properties of the normalization methods. MAS, Z, HK, Q, and WBL are sample-wise normalization and the gene-wise method AGC can be merged to each of them. This table lists the properties of each method.
| MAS | Z | HK | Q | WBL | AGC | |
| Sample-wise normalization | Yes | Yes | Yes | Yes | Yes | No |
| Gene-wise normalization | No | No | No | No | No | Yes |
| Considers the array generation | No | No | No | No | Yes | Yes |
| Includes scaling | Yes | Yes | Yes | No | Yes | Yes |
| Based on distribution | No | No | No | Yes | Yes | No |
| May change the order of the values within a sample | No | No | No | No | No | Yes |
We have employed five different criteria to measure goodness of the normalizations. The results showed that the AGC method improved the results systematically and that the AGC normalized data became comparable across the array generations, as suggested by the classification accuracy of different anatomical samples, and the improved correlation of the data from the same samples analyzed on two different array generations. The AGC method combined with the Q normalization is used in for almost 10,000 samples in GeneSapiens database [16,17].
Conclusion
The gene expression data from 6,926 samples were analyzed together in order to find computationally effective and well-performing method to normalize a large number of the data samples to be directly comparable with each other. All the samples were measured with Affymetrix microarrays, but the various array generations hinder the comparability. Ten different combinations of five normalization method were utilized. The array generation based centering of gene values was found to perform the best, especially if utilized together with the equalized quantile normalization or WBL-normalization.
Methods
Data preprocessing
The data set of 6,926 Affymetrix arrays includes several different array generations with different probe sets. Further, the probe set values need to be converted to gene values. We took median of the normalized values from different probe sets that linked to the same ENSEMBL gene identifier [18] in order to have only one expression value for each gene.
As different preprocessing methods often complicate the data integration, we used data from which the raw data (CEL-files) were available. For all these experiments we used MAS5 preprocessing method with default parameters [19].
The selection of preprocessing method for Affymetrix gene expression data is a controversial topic, and although different opinions exist for optimal preprocessing method [20] in recent comparison studies MAS5 provided the most faithful cellular network construction [21] and optimal identification of differentially expressed genes [22]. In addition, in several studies [13,21,23] it has been stated that other preprocessing methods may also create false correlation between the samples.
Expression value standardization (Z)
Gene value standardization is widely used method for normalizing the gene expression values. In standardization the logarithmic signal values of genes are normalized to have zero as mean or median and one as standard deviation:
where the vector x consists of the logarithmic values of a sample, μ is the mean or median and σ the standard deviation of the sample. The standardized values are often called as Z-scores or median Z-scores. Here, Z refers to median Z-scores.
Housekeeping gene centering (HK)
The housekeeping gene centering (HK-centering) scales the data using a scaling factor that is defined based on the housekeeping genes common with most popular array generations. The assumption behind the HK-centering is that the set of housekeeping genes is expressed identically across the samples. This assumption is found to be unrealistic in several settings [24]. However, even though the assumption that a gene set is constantly expressed across a wide spectrum of tissues may be unrealistic, there are genes that are relatively constant in one tissue type. Consequently, we have included the HK-centering in this study. The scaling factor is defined based on a limited set of housekeeping genes that are found from the most common array generations. First, a suitable set of HK-genes needs to be selected. We selected a set of 126 genes that were found from most of the Affymetrix array generations [15]. Next, the target intensity (TI) value for the gene set is selected. Here, the target intensity value is computed as
where xj, HK are the values of housekeeping genes in sample j. The gene values of each sample can be calculated with
where x are the expression values of the sample.
Equalized quantile normalization (Q)
In several cases it is desirable to scale the samples so that the minimum and the maximum values are the same order of magnitude. Further, often down-stream analysis methods assume that at least standard deviations or means of the values are equal. Therefore, we have utilized equalized quantile normalization (Q) algorithm to normalize the data [25].
In the basic quantile normalization all samples are normalized to have the same distribution [26-28]. This distribution is the mean distribution of all samples in analysis. Therefore, the quantile normalization requires the same number of values in each sample and hence, the quantile normalization is not directly usable for normalizing expression data from different array generations.
We utilized the equalized quantile normalization (Q) that constructs a data set having the desired distribution that has been determined prior to transformation. In the Q-normalization the assumptions are the same as in quantile normalization; the sorted order of the data values should not be changed by the normalization method and the logarithmic signal values can be presented with a predefined distribution. This distribution is the same for every sample through all array generations.
In this study, we evaluated the distribution of the logarithmic MAS5 probe set values for every Affymetrix array generation and found that we can approximate the signals by equalizing the signal-log-values to normal distribution. The distribution of logarithmic values from all samples (N = 6,926) was very near to the normal distribution with mean value 8 and standard deviation 2 (Figure 5). Accordingly, we use N(8,2) normal distribution as the target distribution for the probe set values of the samples.
Figure 5.
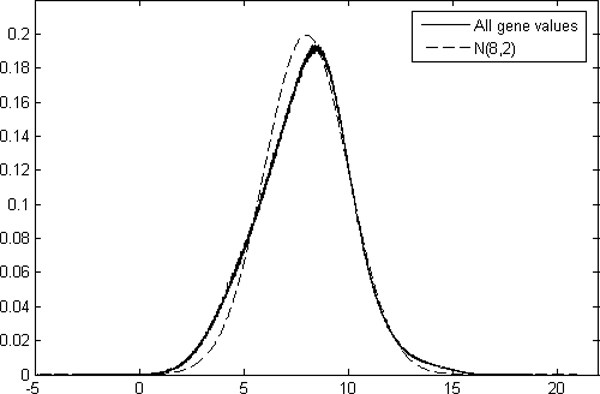
Distribution of the gene values and the normal distribution. The normal distribution with mean 8 and standard variation 2 is near to the distribution of logarithmic values of all 6,926 samples from five different array generations.
Weibull distribution based normalization (WBL)
We have also used the Weibull distribution based normalization (WBL), a way to normalize and correct Affymetrix microarray probe set data [29]. In the Weibull distribution based normalization method it is assumed that the logarithmic probe set values can be adjusted based on the parameters of the Weibull distribution. In order to obtain comparable data, each sample is corrected to have the same shape and scale parameters as the corresponding array generation has. Depending on the array generation, the scale and shape parameters of the Weibull distribution may still differ a bit after normalization, but they are the same for every sample within the same array generation.
For each array generation we collected the array-generation data that are a group of samples analyzed using the MAS5 with default parameters [19,29]. These samples were selected since they represent the distribution of all the samples done with the same array generation. These were used as comparison material and the parameters of these data were set to be the default parameters for each array generation. Based on these data we computed the Maximum Likelihood (ML) estimates [30] for the shape parameter βi and the scale parameter ηi for each array generation i. The scale parameter varied from 8.19 to 8.57, while the shape parameter varied from 3.11 to 3.85. The ML-estimates were calculated for each sample and probe set values were adjusted in order to have same parameter estimates for each of the samples within an array generation. For the sample j this normalization can be done with the formula:
where ηi and βi are the ML-estimates for scale and shape parameters in the array generation i, and ηj and βj are the ML-estimates for the scale and shape parameters in the sample j. Finally, the WBL-normalized gene values were set to be the median values of the probe sets linked to each gene.
Array generation based gene centering (AGC)
In the AGC method we assume that the mean of expression values of one gene in each array generation should be the same. If the mean value of some of the array generations differs substantially from the others, the shift is assumed to be caused by the array generation based variation. The AGC method aims to correct this variation.
The AGC method requires the collection of samples to be relatively large so that one can assume the distribution of logarithmic values of each gene k to represent the total distribution of all potential expression values across all tissues in that array generation i. Therefore, the AGC normalization method normalizes the data to have the mean values μi, k = μk for all array generations i, where μk is the mean of all logarithmic values of the gene k. We assume that the minimum and the maximum estimates for the gene value are reached and the range of the gene k should approximately be [ak, bk], where ak is the lowest 2% value and bk is the largest 2% value of gene k. After array-generation based centering none of the values should go over this range. However, if the new centered value exceeds the range, the difference is diminished towards the range limits with coefficient c, 0 ≤ c ≤ 1. Here, the coefficient is set to c = 1/5 in order to diminish the greatest and smallest values. The centered values can now be obtained with
where xk, j is the value of gene k in sample j measured with the array generation i, μk, iis the mean of the logarithmic values of gene k across array generation i and μk is the mean of the logarithmic values of gene k across all array generations. Further, the resulted AGC values are adjusted based on the equation
Finally, the values are converted back to the original scale by
The AGC method can be used together with any of the within slide methods presented above. After the AGC normalization the mean values of distributions of array generations are centered to have the same mean (Figure 6).
Figure 6.
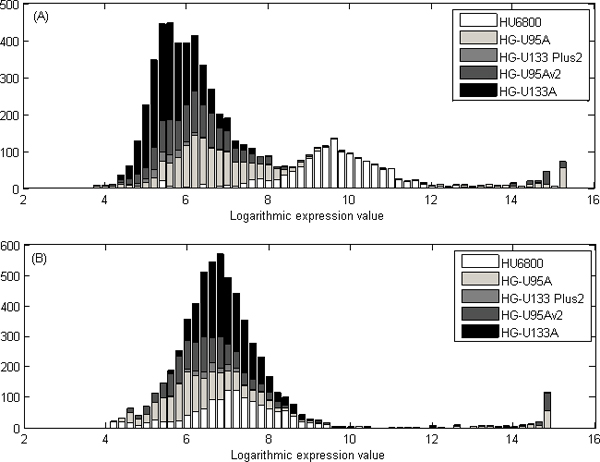
Illustration of the effect of AGC normalization for a prostate specific gene KLK3. (A) The logarithmic expression values of the KLK3 gene before the AGC normalization are distributed based on the array generation. (B) The values are normalized with the AGC method, and there is no significant difference between the values from different array generations. The figure illustrates prostate specific gene, KLK3, and gene values in prostate samples have great values both before and after normalization. After AGC-normalization, the large values not caused by anatomy but array generation HU6800 are diminished.
We have utilized equalized quantile normalization Q combined with the AGC method in the GeneSapiens database [16] with almost 10,000 samples in [17]. There are also few other methods [31-33] used to combine different datasets. However, these are computationally demanding and therefore impractical to use for a dataset including thousands of samples.
Competing interests
A patent application of the AGC normalization protocol has been filed.
Authors' contributions
RA, SK and MS contributed to the development and testing of the normalization. SH contributed to the development of normalization and supervised the comparison and validation of the normalization methods. JA and OK supervised the project. RA wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
Authors want to thank Elmar Bucher, Rolf Skotheim, Mari Björkman, Kristiina Iljin, Matthias Nees, Saija Haapa-Paananen, Henrik Edgren and Maija Wolf for excellent assistance in collecting and annotating the data, as well as the biological evaluation of the normalized data and Dr Outi Monni for providing facilities in Biomedicum Biochip Center.
The work was supported in part by the Academy of Finland, the Finnish Foundation for Economic and Technology Sciences (KAUTE), the Foundation of Technology, the Emil Aaltonen Foundation, the Otto A. Malm Foundation and the Finnish Konkordia Fund.
This article has been published as part of BMC Bioinformatics Volume 10 Supplement 1, 2009: Proceedings of The Seventh Asia Pacific Bioinformatics Conference (APBC) 2009. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/10?issue=S1
Contributor Information
Reija Autio, Email: reija.autio@tut.fi.
Sami Kilpinen, Email: sami.k.kilpinen@helsinki.fi.
Matti Saarela, Email: matti.saarela@tut.fi.
Olli Kallioniemi, Email: olli.kallioniemi@vtt.fi.
Sampsa Hautaniemi, Email: sampsa.hautaniemi@helsinki.fi.
Jaakko Astola, Email: jaakko.astola@tut.fi.
References
- Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–76. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Segal E, Yelensky R, Koller D. Genome-wide discovery of transcriptional modules from DNA sequence and gene expression. Bioinformatics. 2003;19:i273–282. doi: 10.1093/bioinformatics/btg1038. [DOI] [PubMed] [Google Scholar]
- Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan A. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (New York) 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Chinnaiyan A. Mining for regulatory programs in the cancer transcriptome. Nat Genet. 2005;37:579–583. doi: 10.1038/ng1578. [DOI] [PubMed] [Google Scholar]
- Day A, Carlson MR, Dong J, O'Connor BD, Nelson SF. Celsius: a community resource for Affymetrix microarray data. Genome Biol. 2007;8:R112. doi: 10.1186/gb-2007-8-6-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang KB, Kong S, Greenberg S, Park P. Combining gene expression data from different generations of oligonucleotide arrays. BMC Bioinformatics. 2004;5:159. doi: 10.1186/1471-2105-5-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elo LL, Lahti L, Skottman H, Kyläniemi M, Lahesmaa R, Aittokallio T. Integrating probe-level expression changes across generations of Affymetrix arrays. Nucleic Acids Res. 2005;33:e193. doi: 10.1093/nar/gni193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Mariani TJ. Transformation of expression intensities across generations of Affymetrix microarrays using sequence matching and regression modeling. Nucleic Acids Res. 2005;33:e157. doi: 10.1093/nar/gni159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoh EJ, Ross M, Shurtleff S, Williams W, Patel D, Mahfouz R, Behm F, Raimondi S, Relling M, Patel A, Cheng C, Campana D, Wilkins D, Zhou X, Li J, Liu H, Pui C, Evans W, Naeve C, Wong L, Downing J. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–143. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- Ross ME, Zhou X, Song G, Shurtleff S, Girtman K, Williams W, Liu H, Mahfouz R, Raimondi S, Lenny N, Patel A, Downing J. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102:2951–2959. doi: 10.1182/blood-2003-01-0338. [DOI] [PubMed] [Google Scholar]
- Nimgaonkar A, Sanoudou D, Butte A, Haslett J, Kunkel L, Beggs A, Kohane I. Reproducibility of gene expression across generations of Affymetrix microarrays. BMC Bioinformatics. 2003;4:27. doi: 10.1186/1471-2105-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploner A, Miller L, Hall P, Bergh J, Pawitan Y. Correlation test to assess low-level processing of high-density oligonucleotide microarray data. BMC Bioinformatics. 2005;6:80. doi: 10.1186/1471-2105-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso J, Visagie J, Theiler G, Christoffels A, Bardien S, Smedley D, Otgaar D, Greyling G, Jongeneel CV, McCarthy MI, Hide T, Hide W. eVOC: a controlled vocabulary for unifying gene expression data. Genome Res. 2003;13:1222–1230. doi: 10.1101/gr.985203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. Human housekeeping genes are compact. Trends Genet. 2003;19:362–365. doi: 10.1016/S0168-9525(03)00140-9. [DOI] [PubMed] [Google Scholar]
- Kilpinen S, Autio R, Ojala K, Iljin K, Bucher E, Sara H, Pisto T, Saarela M, Skotheim RI, Björkman M, Mpindi J-P, Haapa-Paananen S, Vainio P, Edgren H, Wolf M, Astola J, Nees M, Hautaniemi S, Kallioniemi O. Systematic bioinformatic analysis of expression levels of 17330 human genes across 9783 samples from 175 types of healthy and pathological tissues. Genome Biol. 2008;9:R139. doi: 10.1186/gb-2008-9-9-r139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneSapiens http://www.genesapiens.org
- Hubbard T, Andrews D, Caccamo M, Cameron G, Chen Y, Clamp M, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, Cutts T, Down T, Durbin R, Fernandez-Suarez XM, Gilbert J, Hammond M, Herrero J, Hotz H, Howe K, Iyer V, Jekosch K, Kahari A, Kasprzyk A, Keefe D, Keenan S, Kokocinsci F, London D, Longden I, McVicker G, Melsopp C, Meidl P, Potter S, Proctor G, Rae M, Rios D, Schuster M, Searle S, Severin J, Slater G, Smedley D, Smith J, Spooner W, Stabenau A, Stalker J, Storey R, Trevanion S, Ureta-Vidal A, Vogel J, White S, Woodwark C, Birney E. Ensembl 2005. Nucleic Acids Res. 2005;33:D447–453. doi: 10.1093/nar/gki138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statistical algorithms description document http://www.affymetrix.com/support/technical/whitepapers/sadd_whitepaper.pdf
- Seo J, Hoffman EP. Probe set algorithms: is there a rational best bet? BMC Bioinformatics. 2006;7:395. doi: 10.1186/1471-2105-7-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WK, Wang K, Lefebvre C, Califano A. Comparative analysis of microarray normalization procedures: effects on reverse engineering gene networks. Bioinformatics. 2007;23:i282–288. doi: 10.1093/bioinformatics/btm201. [DOI] [PubMed] [Google Scholar]
- Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS. Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 2005;6:R16. doi: 10.1186/gb-2005-6-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr B, Schlotterer C. Comparison of algorithms for the analysis of Affymetrix microarray data as evaluated by co-expression of genes in known operons. Nucleic Acids Res. 2006;34:e8. doi: 10.1093/nar/gnj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauraniemi P, Hautaniemi S, Autio R, Astola J, Monni O, Elkahloun A, Kallioniemi A. Effects of Herceptin treatment on global gene expression patterns in HER2-amplified and nonamplified breast cancer cell lines. Oncogene. 2004;23:1010–1013. doi: 10.1038/sj.onc.1207200. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay Y, Antonellis K, Scherf U, Speed T. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad B, Collin F, Cope L, Hobbs B, Speed T. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry R, Astrand M, Speed T. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Autio R, Kilpinen S, Saarela M, Hautaniemi S, Kallioniemi O, Astola J. Proceedings of the 2006 IEEE International Workshop on Genomic Signal Processing and Statistics (Gensips 2006), 28–30 May 2006. College Station, Texas, USA; 2006. The Weibull distribution based normalization method for Affymetrix gene expression microarray data; pp. 9–10. [Google Scholar]
- Johnson R, Wichern D. Applied multivariate statistical analysis. 4. Prentice-Hall; 1998. [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Benito M, Parker J, Du Q, Wu J, Xiang D, Perou CM, Marron JS. Adjustment of systematic microarray data biases. Bioinformatics. 2004;20:105–114. doi: 10.1093/bioinformatics/btg385. [DOI] [PubMed] [Google Scholar]
- Gilks WR, Tom BD, Brazma A. Fusing microarray experiments with multivariate regression. Bioinformatics. 2005;21:ii137–143. doi: 10.1093/bioinformatics/bti1123. [DOI] [PubMed] [Google Scholar]