

Abstract
Two Artemis-deficient (mArt-/-) mouse models, independently generated on 129/SvJ backgrounds have the expected T-B-NK+SCID phenotype. However, they fail to mimic the human disease due to CD4+ T-cell leakiness. Moreover, immune reconstitution in these leaky mouse models following hematopoietic stem cell transplantation (HSCT) is more easily achieved than that seen in Artemis-deficient humans. To develop a more clinically relevant animal model we backcrossed the mArt-/- mutation onto the C57Bl/6 (B6) background (99.9%) resulting in virtually no CD4+ T-cell leakiness compared to 129/SvJ mArt-/- mice (0.3±0.25% vs 19.5±15.1%, p<0.001). The non-leaky mouse was also uniquely resistant to engraftment using allogeneic mismatched HSC, comparable to what is seen with human Artemis deficiency. The genetic background also influenced Artemis-associated radiation sensitivity with differing degrees of x-ray hypersensitivity evident in 129/SvJ and B6 backgrounds with both the mArt-/- and mArt-/+ genotypes. Our results indicate that immunogenic and DNA repair phenotypes associated with Artemis deficiency are significantly altered by genetic background, which has important implications for SCID diagnosis and treatment. Moreover, the B6 mArt-/- mouse is a more accurate model for the human disease, and a more appropriate system for studying human Artemis-deficiency and for developing improved transplant and gene therapy regimens for the treatment of SCID children.
Keywords: SCID, Hematopoietic Stem Cell Transplant (HSCT), Knockout Mouse, Artemis Deficiency, Athabascan
Introduction
Approximately 20% of patients with severe combined immunodeficiency disease (SCID) have a phenotype that is characterized by a profound absence of both T and B lymphocyte numbers and function, but includes normal NK cells, and is associated with a defect in the V(D)J recombination pathway [1]. Maturation of T and B lymphocytes requires successful V(D)J recombination by which the germline components, V(Variable), D(Diversity), and J(Joining) gene segments are assembled to encode for V regions of Ig and TCR [1]. The well-characterized factors in this pathway consist of Ku heterodimer complex (Ku70/86), DNA-dependent protein kinase catalytic subunit (DNA-PKcs), XRCC4, DNA ligase IV, and the more recently identified Artemis. The significance of this process has been well defined by analyses of animal models and some rare types of human T−B−NK+ SCID carrying germline mutations/deletions in these genes [2-9].
Artemis mutations were identified first in children with Radiation Sensitive-SCID (RS-SCID) and then in Athabascan-speaking children with SCID (SCIDA) [10-12]. Fibroblasts from patients with either SCIDA or RS-SCID show increased sensitivity to ionizing radiation and impaired coding joint formation [10,12,13]. Artemis is a substrate of DNA-PKcs having multiple phosphorylation sites in vivo and in vitro, and phosphorylation enables the nuclease activity of Artemis to open hairpin structures at the coding ends of V(D)J intermediates [13-15].
An Artemis-deficient mouse model was first reported in 129/SvJ mice [16]. These mice showed a significant degree of leakiness with respect to CD4+ T cells, especially in lymph nodes, in contrast to what has been seen in children with SCID and Artemis deficiency. Subsequently, we reported an Artemis deficient mouse with a mixture of 129/SvJ and B6 backgrounds (25% and 75% respectively), which had some degree of leakiness with respect to both T cell numbers and function, but more limited than previously reported [17]. We also found significant sensitivity to ionizing radiation in our mouse model as well as some resistance to allogeneic mismatched HSCT that could be overcome with non myeloablative doses (3 Gy) of total body irradiation [17].
To determine whether or not the lower level of leakiness in our model was due to the higher percent of B6 in the genetic background, we backcrossed our original mice onto the B6 background (99.9% B6). We also generated a 99.9% 129/SvJ with the same Artemis mutation. The 129/SvJ mArt-/- mouse exhibits levels of leakiness at least as high as previously reported [16]. In contrast, we found that the phenotype of the B6 mArt-/- mouse has virtually no leakiness and is highly resistant to allogeneic mismatched HSCT. In addition, our studies show that the Artemis deletion (-/-) and haploinsufficiency (+/-) in B6 and 129/SvJ embryonic fibroblasts are significantly different with respect to sensitivity to ionizing radiation, indicating a gene dosage effect that we describe here for the first time. Finally, unlike previously reported mArt-/- mice, the phenotype of our B6 mArt-/- mouse model closely resembles that seen in Artemis-deficient children and is therefore the appropriate model for the human disease.
Materials and Methods
Mice
The 129/SvJ-B6 mArt-/- chimeric mouse (25% 129/SvJ and 75% B6) has been previously described [17]. Heterozygote (mArt+/-) male chimeric mice were either crossed with wild type B6 or 129/SvJ females for an additional 7-8 generations to generate either N10 B6 (99.9%) or N9 129/SvJ (99.9%) mArt-/- mice. All references to mArt-/- are to these B6 or 129/SvJ mice carrying the mArt-/- mutation [17] with a 99.9% background unless otherwise indicated. C57Bl/6, 129x1/SVJ, BALB/c and B6.SJL (CD45.1) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animals were maintained in the Laboratory Animal Resource Center (LARC) Rodent Barrier Facility at UCSF. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of UCSF.
Phenotyping by Flow Cytometry
T, B and NK cell and granulocyte phenotyping and chimerism analyses were done by flow cytometry using a FACScalibur (Becton Dickinson) and data were processed by Cell Quest Pro software. BALB/c allogeneic (anti-H2Kd) donor and B6 (anti-H2Kb) recipient cells, or congenic (anti-CD45.1) donor and (anti-CD45.2) recipient cells were analyzed. Peripheral blood was obtained from the lateral saphenous vein every 4 weeks post transplant, and the cells stained by using fluorescently conjugated antibodies to CD3, CD4 and CD8 to detect T lymphocytes and anti-CD45R/B220 antibodies to detect B lymphocytes. To test for multilineage engraftment, cells were stained with Ly6G/C, a granulocyte marker. 7-8 week old leaky study mice or mice 3-6 months post transplant were euthanized and tissues collected. Single-cell suspensions were prepared from blood, spleen, thymus, lymph node and bone marrow and stained by using the same antibodies as above. In addition, bone marrow and spleen cells were stained with anti-CD45R/B220 and IgM to test for mature B-cell engraftment or leakiness. Blood and spleen cells were stained with CD49b/Pan-NK to detect NK cells in leaky study mice. Cells were incubated with Fc block and then antibodies for 30 minutes at 4°C, washed once, and fixed in PBS/1% formalin for analysis. All antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA) or e-Bioscience (San Diego, CA).
Cell Lines and Radiation Sensitivity Assays
Mouse embryonic fibroblast (MEF) lines established from early passage cultures were immortalized by infection with lentivirus constructs encoding genetic suppressor element GSE-22, a dominant negative p53 peptide, or by introduction of SV40 large T antigen as previously described [17,19]. Lentivirus infected MEF lines were selected with 1 μg/ml puromycin Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, penicillin (100U/ml), and streptomycin (100 μg/ml) at 37° C in a humidified incubator (5% CO2) for 7 days and all MEF cells were maintained in the same media without puromycin. For proliferation assays, cells were plated in triplicate on six-well dishes at 5×104cells per well and irradiated using a Pantak X-ray generator (East Haven, CT) operating at 320 kV/10 mA with 0.5 mm copper filtration. Cells recovered in the incubator for 24 hours after radiation and were then labeled by the addition of fresh medium containing 10 μM bromodeoxyuridine (BrdU) (Sigma) for an additional 24 hours, harvested, washed with PBS, and fixed in 70% ice-cold ethanol overnight at −20 °C. Cell cycle distribution and BrdU incorporation were analyzed and scored with a Guava Easy Cyte Plus System using Guava Cytosoft Version 3.6 Cell Acquisition and Analysis Software (Hayward, CA). Proliferating fraction was calculated as the percent of intact cells staining positive for BrdU and in G1-phase normalized to untreated controls of the same cell line (>10,000 events were scored for each point). Error bars are calculated from variation among the three biological replicates within each experiment.
Hematopoietic Stem Cell Preparation and Transplants
BALB/c allogeneic mismatched or B6 CD45.1 congenic mice (2-4 month-old) were used as donors. For allogeneic and congenic HSCT, purification of lin- c-kit+ HSC was done by negative selection of lineage-committed cells using the Lineage Cell Depletion Kit followed by positive selection using CD117 microbeads (Miltenyi Biotec) following the manufacturer’s instructions. The lin− c-kit+ HSC preparations were >90% pure by flow cytometric analysis.
Recipient B6 mArt-/- mice were 5–8 weeks of age at the time of transplantation, and 100,000 lin−c-kit+ congenic or allogeneic mismatched HSC were injected i.v. through the tail vein. Most of the mice received no prior conditioning therapy, but one group was pretreated with 3 Gy of TBI using a cesium137 animal irradiator (520A, Xetex Inc, Sunnyvale, CA). The transplanted mice were maintained in the LARC Barrier Facility with antibiotic-supplemented water. Controls included age-matched mArt-/-, WT, or heterozygotes that received a sham injection or HSCT.
In Vitro Lymphocyte Proliferation
At the tissue harvest, splenocytes were collected for in vitro lymphocyte proliferation studies. 4×106/ml post ficoll splenocytes were incubated in RPMI medium 1640 (Gibco, Carlsbad, CA) containing 10% FBS and supplemented with 1x penicillin and streptomycin (UCSF Cell Culture Facility), and stimulated with either lipopolysaccharide (LPS) (25 μg/ml; Sigma) or anti-CD3 (5 μg/ml; BD Pharmingen, San Diego, CA) in 96 well flat bottom plates. On day 1 (LPS) or day 2 (anti-CD3) radioisotope [H3]-Thymidine (Perkin Elmer, Waltham, MA) was added and plates were incubated an additional 24 hours, harvested, counted, and evaluated as described [17].
TCR Vβ Quantitative Immunoscope
RNA was obtained from T cells isolated from thymus or spleen using a Pan T cell positive selection kit (Miltenyi Biotec). TCR Vβ repertoire analysis was modified and performed as previously described [21,22]. Briefly, RNA was extracted and cDNA was amplified with each of the 24 TCR Vβ family member–specific primers together with a fluorescent (Rox) primer specific for the TCR Cβ segment (SuperScript First-strand Synthesis System, 11904-018, Invitrogen, Carlsbad, CA). Spectrotyping PCR was carried out on a GeneAmp PCR system 2400 (Perkin Elemer, Waltham, MA). The fluorescent products were mixed with Rox size standard (Gene Scan 500-40173, Applied Biosystems, Foster City, CA), 99.5% Formamide (Sigma, Saint Louis, MI), and capillary electrophoresis was run on a 3100 sequencer (ABI POP4 polymer) (Applied Biosystems, Foster City, CA). The size and intensity of each band were analyzed by using the Genemapper software (3.0) (TM) according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA).
ELISA for Serum IgM and IgG Antibody
To measure antibody responses, mice were injected i.p. with 100 μg of either NP41-Ficoll or NP24-KLH (Biosearch Technologies, Novato CA). Post-immune serum was collected via saphenous vein blood draw 11 days following NP-Ficoll immunization and stored at -20°C. Mice receiving NP-KLH immunizations were boosted with an additional 100 μg of NP-KLH five weeks following the initial injection, and post-immune serum was collected one week later. NP-specific antibody response was measured by sandwich ELISA. MaxiSorp plates (Nunc, Rochester, NY) were coated overnight with 5 μg/mL NP30-BSA (Biosearch Technologies) at 4°C. After washing, a blocking reagent (50mM TBS, 1% BSA, pH 8.0) was added to the wells for 1 hour followed by washing. Two-fold serial dilutions of serum from mice immunized with NP-Ficoll, or four-fold serial dilutions of serum from mice immunized with NP-KLH beginning at 1:100 were added to the plates and incubated for one hour. The plates were then washed and incubated with a goat-anti mouse HRP-conjugated detection antibody for one hour. After washing, horseradish peroxidase (Kirkegaard & Perry, Gaithersburg, MD) was added to the plates and incubated for 20 minutes, at which point the reaction was stopped with the addition of 4N H2SO4. Optical density was measured at 450 nm on a SpectraMax plate reader (Molecular Devices, Sunnyvale, CA) using SoftMaxPro software. Plates and ELISA reagents (with the exception of the coating antigen) were included in the “Mouse IgM ELISA Quantitation Kit” and the “Mouse IgG ELISA Quantitation Kit” from Bethyl Laboratories, Inc. (Montgomery, TX). O.D. values for sera were measured in duplicate and averaged, and the subsequent values of the pre-immune serum were subtracted from that of the post-immune. These values were then plotted against the corresponding dilution factor [20].
Statistical Analysis
Differences in leakiness between the two strains of mArt-/- mice were analyzed using either the independent samples T test or the Chi-square test of goodness of fit and independence.
Results
T, B and NK Cell Phenotypes in 129/SvJ and B6 mArt-/- Mice
CD4+ and CD8+ T cells in blood, spleen, thymus, and lymph node were evaluated in mArt-/- mice with either the 129/SvJ or B6 genetic background (Figure 1). The backcrossed 129/SvJ mArt-/- mice exhibited a degree of “leakiness” that was significantly greater than that previously observed in chimeric mArt-/- mice [17], with the most striking difference seen in the lymph nodes (leakiness defined as ≥ 1% CD4+ or CD8+ T cells). In contrast to a previous study of 129/SvJ Artemis-deficient mice in which only 40% of mice were leaky [16], all of the 129/SvJ mArt-/- mice that we studied (n=10) exhibited a leaky phenotype. CD45+ lymphocytes from 129/SvJ mArt-/- mice were 19.5±15.1% CD4+ (range 1.7-49%) while B6 mArt-/- T lymphocytes were 0.3±0.25% CD4+ (p<0.001). Absolute numbers of ficoll-purified CD4+ splenocytes were also compared between B6 and 129/SvJ mArt-/- mice (n=10 for each group). We found 3200±3700 CD4+ T cells per spleen in the B6 mArt-/- mice (0.07% of WT B6) versus 185,000±220,000 (3.9% of WT 129) in the 129/SvJ mArt-/- mice (p=0.013). A similar analysis of CD8+ T cells showed no remarkable CD8 T cell leakiness in either mArt-/- group and thus no significant difference between the two strains (p=0.496, Figure 1B).
Figure 1. CD4+(A) or CD8+(B) T-cells in 129/SvJ and B6 wt and mArt-/-mice.
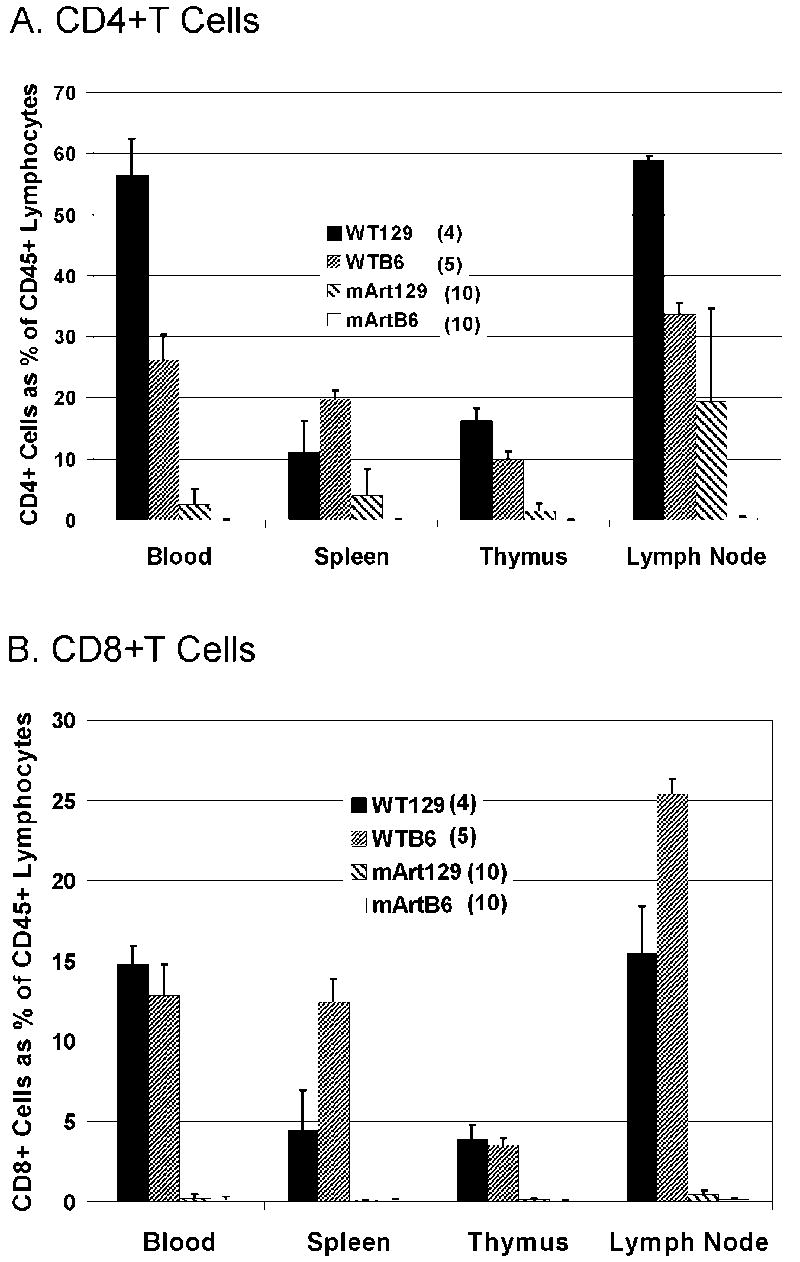
Each bar represents the mean percent ± standard deviation of CD4+ or CD8+ T cells gated on CD45+ CD3+ lymphocytes in blood, spleen, thymus, and lymph node.
There was variation in the amount of leakiness among individual 129/SvJ mArt-/- mice and a notable variation between tissues, with lymph nodes expressing the greatest degree of leakiness (Figure 1A). Leakiness was only seen in the CD4+ T cell subset in the 129/SvJ mArt-/- mice. We observed the expected staining pattern of CD4+CD8- T cells for various tissues from “leaky” 129/SvJ mArt-/- mice as compared to wild type (Figure 2). Similar to what we have found in Artemis-deficient children with a null mutation [26] and unlike the 129/SvJ mArt-/- mice, we detected virtually no CD4+ T cells in the “non leaky” B6 mArt-/- mice (Figure 2).
Figure 2. Flow cytometric analysis of CD4+ and CD8+ lymphocytes in a representative leaky 129/SvJ N9 mArt-/- mouse and non leaky B6 N10 mArt-/- mouse.

WT B6 LN (lymph node) was used as a control. Percent expressed as number of CD4+, CD8+, or CD4+CD8+ cells gated on CD45+CD3+ lymphocytes.
NK cells were measured in the blood and spleens of the B6 and 129/SvJ mArt-/- mouse strains (n=3 for each group). CD45+ NK lymphocytes in blood were 84.2±1.7% in B6 mArt-/- mice and 54.2±12.1% in 129/SvJ mArt-/- mice. In the spleen the NK levels were 51.9±13.4% in B6 mArt-/- mice and 32.1±6.8% in 129/SvJ mArt-/- mice. Interestingly, the higher percent of NK cells in the B6 mArt-/- mice is consistent with what is seen in human SCIDA patients [26].
Leakiness is Proportional to 129/SvJ Genetic Contribution
We evaluated lymphocyte phenotypes in several stages of backcrossing including N2, N4, N10 B6 and N4, N9 129/SvJ (Table 1). As the percent of the 129/SvJ background in a mouse declined so did the percent of mice that were leaky. This trend in leakiness correlates well with the percentage of 129/SvJ genetic background in all cases. The effect of 129/SvJ genetic modifiers on leakiness was most dramatically evident when comparing animals >99% 129/SvJ with mice harboring identical Artemis mutations but having ≤0.1% 129/SvJ genetic content (Table 1 top and bottom rows respectively). Statistically significant differences in leakiness were evident in the spleen and lymph node when all groups were analyzed (p≤0.001).
Table 1.
Leaky mice as a function of percent of 129/SvJ in B6 genetic background.
| Percent 129/SvJ | CD4+ Blood* | CD4+ Spleen* | CD4+ Thymus* | CD4+ Lymph node* | |
|---|---|---|---|---|---|
| N9 129/SvJ mArt-/- (n=10) |
99.9† | 6/10 | 7/10 | 7/10 | 10/10 |
| N4 129/SvJ mArt-/- (n=10) |
95.3 | 1/10‡ | 4/10‡ | 2/10 | 10/10‡ |
| N2 B6 mArt-/- (n=8) |
25 | 1/8 | 1/8 | 2/8 | not available |
| N4 B6 mArt-/- (n=10) |
6.3 | 0/10 | 0/10 | 0/10 | 1/10 |
| N10 B6 mArt-/- (n=10) |
0.1 | 0/10 | 0/10 | 0/10 | 0/10 |
Leakiness defined as ≥1% CD4+ T cells. When compared by Chi-square analysis for differences among groups 99.9%→0.1%, p=0.001 for spleen and 0.0001 for lymph node.
Number of mice exhibiting leakiness in that tissue /total evaluated.
Represents 99.9% 129/SvJ and 0.1% B6.
Leaky number includes one mouse leaky in the CD8 subset.
The Mouse Genetic Background Alters Artemis-Associated Radiation Sensitivity
To assess whether the DNA repair function of Artemis is similarly dependent on the mouse strain, we evaluated the effect of Artemis deletion on the radiation sensitivity of mouse embryonic fibroblast (MEF) cultures from both 129/SvJ and B6 backgrounds (Figure 3). Because mechanisms of cellular immortalization may influence radiation sensitivity, we also carried out radiation sensitivity assays with cells immortalized with either GSE-22 or SV40-Large T antigen as well as in cells derived from several different animals to control for animal-to-animal variations. We found that cells from B6 mice appear to be less sensitive to radiation effects in this assay than cells from 129/SvJ mice. As expected, the homozygous Artemis mutants were the most sensitive to ionizing radiation relative to the other genotypes tested in both the 129/SvJ and B6 backgrounds. However, the increase in radiation sensitivity in the Artemis null mutation relative to the wild type was more pronounced in the 129/SvJ background than in the B6 background (Figure 3, compare A, B, and C). This greater sensitivity in 129/SvJ was consistently evident in multiple cell lines when 129/SvJ and B6 mArt-/- were compared directly in both GSE and SV40 immortalized cells (Figure 3D). When we examined the effects of radiation on heterozygous cells from each strain, we saw a response that was intermediate between wild type and null. As in the case of the null mutants, Artemis haploinsufficiency in the 129/SvJ background conferred increased radiation sensitivity relative to the wild type, while the identical heterozygous mutation in B6 was not significantly different from wild type (compare Figures 3A and B). The increased sensitivity of the heterozygous Artemis mutant was also evident in a second set of 129/SvJ cell lines from different animals immortalized with SV40 rather than GSE-22 (Figure 3C).
Figure 3. Radiation sensitivity assays.

Mouse embryonic fibroblasts (MEFs) were scored for proliferation 48 hours after x-ray exposure based on BrdU containing G1 cells relative to untreated controls. Cell line details are denoted in the legend by cell line number, mouse strain, and Artemis genotype. (A) Radiation sensitivity of GSE-22 transformed mArt+/+, mArt+/- and mArt-/- on 129/SvJ background. (B) Radiation sensitivity of GSE-22 transformed mArt+/+, mArt+/- and mArt-/- on B6 background. (C) Radiation sensitivity of SV40 transformed mArt+/+, mArt+/- and mArt-/- on 129/SvJ background, derived from different animals. (D) Radiation sensitivity of additional mArt-/- lines from additional animals of noted backgrounds immortalized with GSE-22 (top graph) or SV40 (lower graph).
Congenic and Allogeneic HSCT in Non-leaky B6 mArt-/- Mice
Transplant studies were carried out to assess the impact of the B6 background on the outcome of various BMT regimens. HSCT using 1×105 lin-c-kit+ congenic donor cells (CD45.1) in young adult B6 mArt-/- recipients (CD45.2) resulted in T but not B cell reconstitution with T cell numbers increasing over time (Table 2 and Figure 4). However, when mice were pretreated with a non-myeloablative dose of radiation (3 Gy TBI), significant donor B cells and granulocytes in addition to T cells were seen following a congenic transplant (Table 2). Donor engraftment of these cells persisted for more than 5 months post transplant (Table 2). Further phenotyping analysis revealed that congenic transplant recipients with and without TBI pre-treatment developed CD4+ and CD8+ T cells in the peripheral blood, spleen, lymph nodes, and thymus, as well as CD4+CD8+ T cells in the thymus, comparable to wild type controls (Figure 4). With respect to B cell reconstitution, IgM+B220+ cells were only present in the bone marrow of congenic recipients that received TBI (Figure 4). HSCT using 1×105 allogeneic mismatched (BALB/c) donor HSC resulted in no donor cell engraftment (Table 2). When the B6 mArt-/- recipient mice were pretreated with low dose TBI, there was a degree of T and B cell engraftment, but at a much lower level than with congenic donor cells.
Table 2.
T cell, B cell, and granulocyte engraftment in blood in B6 mArt-/- mice following congenic and allogeneic transplants.
| BMT | Marrow Prep and Pre-condition | 1 Month Post-HSCT | 2 Months Post-HSCT | At least 3 Months Post-HSCT | ||||
|---|---|---|---|---|---|---|---|---|
| T cells, %* | B cells, %* | T cells, %* | B cells, %* | T cells, %* | B cells, %* | Granulocytes, %† | ||
| Congenic (CD45.1) | HSC‡ (n=6) | 4.0±2.7 | 0 | 16.3±4.1 | 0 | 64.4±8.2 | 0 | N/A |
| Congenic (CD45.1) | HSC‡/TBI (n=4) | 44.6±3.5 | 18.4±2.8 | 56.6±4.5 | 23.5±7.0 | 46.1±2.2 | 35.2±2.1 | 64.1±11.2 |
| Allogeneic (BALB/c) | HSC‡ (n=5) | 0 | 0 | 0 | 0 | not available | not available | not available |
| Allogeneic (BALB/c) | HSC‡/TBI (n=4) | 1.8±1.2(3)£ | 3.3±1.5(3)£ | 4.4±3.4(2)£ | 3.4±0.7(2)£ | not available | not available | not available |
WT mice used as controls had a mean percent of 28.5±17.7 CD3+ (T cells) and a mean percent of 37.5 ± 2.1 CD45R/B220+ (B cells) in the CD45+ lymphocyte gate.
Mean percent of CD3+ (T cells) or CD45R/B220+ (B cells) in CD45+ lymphocyte gate ± standard deviation.
Mean percent of donor granulocytes of total granulocytes ± standard deviation.
For HSC (lin−c-kit+), 1 × 105 cells were injected. Mice treated with TBI (Total Body Irradiation) received 3Gy.
Mean±SD of engrafted animals. ( ) = the number of transplanted mice that engrafted (≥0.75% donor cells).
Figure 4. Immune reconstitution in a representative B6 WT mouse, mArt-/- mouse, congenic transplanted mArt-/- mouse, and a congenic transplanted mArt-/- mouse treated with TBI (3 Gy).

T cell phenotyping was performed on tissues (peripheral blood (PB), spleen, lymph node (LN), and thymus) collected ≥3 months post transplant. B cell phenotyping was done on bone marrow (BM). Percent expressed as number of CD4+, CD8+, or CD4+CD8+ cells gated on CD3+ lymphocytes; mature B cells (IgM+ B220+) and immature B cells (IgM- B220+) were gated on lymphocytes.
T and B Cell Proliferative Responses in Leaky and HSC Transplanted Mice
We measured the proliferative responses of 129/SvJ mArt-/- leaky T and B cells to either anti-CD3 MoAb or to LPS, a B cell mitogen (Figure 5A). The response to anti-CD3 MoAb in the 129/SvJ mArt-/- mice was an average of 9.5% (13617±9379 cpm) of normal wild type litter mate controls, and all nine mice tested had a response ranging from 0.9% to 19.4% of control. The proliferative response of cells from leaky mice to lipopolysaccharide (LPS) was less apparent, averaging only 1.0% (1619±2325 cpm) of that seen in normal controls. Only three of seven mice tested showed any B cell response, with the highest response among the three being 3.5% of normal. The non leaky B6 mArt-/- mice exhibited virtually no response to either anti-CD3 or LPS. Lymphocyte proliferative responses to anti-CD3 and LPS were tested in the two congenic HSC transplant groups (± 300cGy TBI). Nearly normal proliferative responses to anti-CD3 stimulation were seen in the recipients of congenic HSCT while the responses to LPS were absent (Figure 5A). When 3 Gy TBI was added to the preparative regimen the proliferative responses to both anti-CD3 and LPS stimulation were normal (Figure 5A).
Figure 5. T and B cell proliferative responses, T cell receptor diversity, and specific IgM response post transplantation.

(A) Proliferative responses of lymphocytes to anti-CD3 antibody and LPS. B6 wild type mice (positive controls) and mArt-/- mice (negative controls) were compared to recipients of congenic HSCT with and without TBI at ≥3 months post transplant. Results (stimulated minus resting) are expressed as counts per minute (cpm) on a log scale. (B) TCR Vβ repertoire analysis. T cells were enriched from either thymus or spleen and prepared as in Materials and Methods. Four representative Vβ regions are shown from a representative B6 WT mouse, a leaky 129/SvJ mArt-/- mouse, two mArt-/- recipients of congenic HSCT (451 and 454), and two mArt-/- recipients of congenic HSCT with TBI (658, 660). (C) Serum IgM-specific antibody response to NP-Ficoll after immunization (minus pre-immune response). The figure shows the average response from recipients (n=4) of congenic HSCT at ≥3 months post transplant. B6 mArt+/- (n=7) and B6 mArt-/- (n=4) mice were used as positive and negative controls, respectively. (D) Serum IgM-specific antibody response to NP-KLH after immunization (minus pre-immune response). Average response from recipients (n=3) of congenic HSCT with TBI (3Gy) are compared to B6 mArt+/- (n=3) and B6 mArt-/- (n=3) controls.
TCR Vβ Repertoire in Leaky and HSCT Recipient Mice
To roughly approximate the baseline frequency of successful V(D)J recombination in the leaky mice, we assessed the TCR Vβ repertoire in the T cells from these animals. In 129/SvJ mArt-/- mice, clonal patterns were observed in all four of the regions analyzed, unlike the normal Gaussian distribution of the different CDR3 lengths in wild type mice (Figure 5B). A comparative TCR Vβ repertoire analysis for B6 mArt-/- mice was not possible due to the absence of T cells.
The TCR Vβ repertoire was also analyzed in engrafted circulating T cells of congenic HSCT mice. Although these mice exhibited T cell numbers in the normal range by 3 months post transplant (see Table 2), the T cell repertoire had skewed profiles or clonal patterns in the immunoscope analysis, indicating preferential and restricted clonal expansion of the engrafted T cells whether or not low dose TBI was used (Figure 5B).
Serum IgM and IgG Responses in HSCT Recipient Mice
B cell function was assayed in the congenic recipients by measuring serum IgM-specific antibody production. In congenic recipients that were not treated with TBI, immunization with the T cell independent antigen NP-Ficoll did not elicit a humoral response (Figure 5C). In the congenic group treated with TBI, we detected serum IgM-specific antibody in response to immunization with the T cell dependent antigen, NP-KLH (Figure 5D). Specific IgG antibody to NP-KLH was also detected in these recipients (data not shown).
Discussion
Leakiness vs Non-leakiness
Two independently generated mArt-/- mouse models [16,17] failed to faithfully replicate the phenotypes seen in Artemis-deficient humans, thereby limiting the utility of these mouse models in developing improved clinical regimens. We found that by backcrossing the mArt-/- mutation [17] onto the B6 mouse (99.9%), we were able to generate a non-leaky mouse model of Artemis-deficient SCID with respect to both T cell numbers and function that also expresses significant graft resistance in response to allogeneic mismatched HSCT. The leakiness in our Artemis-deficient 129/SvJ mice is similar to that reported by Alt et al [16] except that in their model the deficient mice were shown to have significant leakiness with respect to CD4+ cells in the lymph nodes from only 4 of 10 mice studied. In contrast, we found CD4+ T cells in the lymph nodes of all ten of the backcrossed 129/SvJ mArt-/- mice that we studied. Our results unambiguously show that the mouse genetic background proportionally alters the phenotypes associated with Artemis-deficiency. Spectrotyping patterns revealed a very limited TCR repertoire in the leaky T cells, indicating impaired T cell development and clonal outgrowth. Finally, while the lymphocyte function in the spleens from our 129/SvJ mArt-/- mice was ~10% of that seen in the WT animals and comparable to what we had previously reported in the 129/B6 chimeric mArt-/- mice [17], there was no lymphocyte function in our B6 Artemis-deficient mice, similar to what is found in human Artemis deficiency.
While the leakiness in this model is principally reflected in the CD4 subset, of ten 129/SvJ N4 mArt-/- (95.3 % 129/SvJ) animals studied, we found one animal that expressed primarily CD8+ T cells and three that had mature B cells in the bone marrow (data not shown). This indicates that the mechanism(s) that allows the escape of lymphocytes past the block in maturation affects all subsets, although the greatest effect is clearly on CD4 cells.
Consistent with this idea of a general escape mechanism, we found differences in the radiation sensitivity of cell lines from 129/SvJ and B6 backgrounds. Most notably, cells from heterozygote 129/SvJ mArt+/- animals show a gene dosage effect, having radiation sensitivity intermediate between wild type and null, while the same haploinsufficiency in the B6 background lacks the distinction compared to wild type. Moreover, like the differences in leakiness in these backgrounds, the Artemis null cells are more radiation sensitive on 129/SvJ than the B6 background. Together these data show that the genetic background significantly modifies both immunological and DNA repair phenotypes associated with Artemis deficiencies although in somewhat different ways. Curiously, relative to the B6 background, Artemis deficiency in the 129/SvJ better repairs V(D)J intermediates, manifesting as leakiness, yet less efficiently repairs radiation induced damage, manifesting as more pronounced radiation sensitivity. While the mechanism for this disparity is not clear, we propose several plausible reasons for this apparent contradiction: 1) Failed V(D)J recombination involves one double-strand DNA break per cell, with one type of terminus, while radiation damage causes far more breaks and a wide spectrum of termini, 2) Repair and survival processes in fibroblasts and lymphocytes are not equivalent, 3) Repair processes other than NHEJ may account for the leakiness (i.e., homologous recombination) but these may not compensate for Artemis deficiency in repairing random breaks introduced by radiation. Interestingly, we did not find tumor formation in any of the animals with or without radiation exposure, although it is possible that we did not follow them long enough to detect this side effect.
In any case, it is clear that both the immunological and DNA repair/radiation survival functions of Artemis are significantly impacted by the genetic background. These findings have implications for diagnosis and treatment of Artemis defects in humans. Specifically, it is not unlikely that some carriers of Artemis mutation may have increased sensitivities to radiation and chemotherapy with alkylating agents, and/or increased cancer risks. In addition, the leakiness in Artemis-deficient mice may also become evident in some affected humans, potentially confounding the correct diagnosis. Finally, the leakiness in the mArt-/- mice closely corresponds with the degree of 129/SvJ in the background. These results suggest that genetic modifiers exist in the 129/SvJ genetic background that permit low level resolution of V(D)J intermediates, resulting in at least some T and B lymphocyte differentiation, despite Artemis deficiency.
Graft Resistance and Disparate T and B cell Reconstitution Following HSCT
We previously reported that there was a significant T cell and some B cell engraftment in the leaky 129/SvJ mArt-/- mice following congenic HSCT with or without low-dose TBI conditioning. These results were significantly different from our findings in the non-leaky B6 mArt-/- mice in which no B cell reconstitution occurred.. Moreover, we found that in contrast to our previous chimeric (25% 129/SvJ) model [17] in which 4×104 donor BALB/c HSC (lin-Sca1+) resulted in T but not B cell engraftment, the non-leaky B6 mArt-/- mice were completely resistant to engraftment with 1×105 allogeneic mismatched BALB/c HSC. Even when low dose TBI was used, the non-leaky mice only recovered T and B cell immunity to a limited extent, much less than what we saw in the chimeric mice [17]. The different results in engraftment that we found following allogeneic HSCT between the B6 mArt-/- mouse and our previous 129/SvJ-B6 mArt-/- chimeric mouse model (25% 129/SvJ) likely reflect the impact of the genetic backgrounds [17]. Sequence comparison of conserved genes indicates that Ly49 receptors in 129/SvJ and BALB/c are more closely related to each other than to the corresponding B6 Ly49 receptor [23,24]. NK cells use inhibitory Ly49 receptors to differentiate self from foreign cells based on interactions with major histocompatibility (MHC) class I molecules. Thus, BALB/c donors are potentially less likely to be rejected in leaky 129/SvJ mArt-/- mice than in B6 mArt-/- mice. It is interesting that the T cells in the leaky animals that we previously studied [17] did not appear to increase graft resistance. We assessed the TCR Vβ repertoire in the T cells from leaky 129/SvJ mArt-/- mice. Clonal patterns were observed in all four of the regions analyzed, unlike the normal Gaussian distribution of the different CDR3 lengths in wild type mice, indicating that the leaky CD4+ T cells are not fully functional. Therefore, we assume that the NK cells are the critical reason for graft resistance in these animals and that the leaky T cells play little to no role.
B6 mArt-/- SCID Mice Are Comparable to Children with Artemis Deficiency
There is a very high incidence of T-B-NK+ SCID in Athabascan-speaking children that is associated with a founder mutation in Artemis [11,25]. We have previously reported our experience with transplantation in a large number of Athabascan-speaking Navajo children with this mutation [26]. Not only do these children have virtual absence of T and B cells in both numbers and function, but their fibroblasts show increased sensitivity to ionizing radiation and they appear to have clinical manifestations of increased sensitivity to alkylating agents and radiation. In addition, transplants from HLA matched siblings (differing in minor HLA antigens) usually result in T but not B cell reconstitution [26], which is similar to congenic transplants in the B6 mArt-/- mice model as CD45.1 congenic marrow is MHC-matched to CD45.2 mice except for a minor H antigen–mismatch. When allogeneic mismatched (BALB/c) HSC are used the B6 mArt-/- mice are resistant, comparable to children with Artemis-deficiency who typically reject haplocompatible stem cell enriched, T cell-depleted grafts unless some kind of conditioning therapy is used. Those who receive marrow ablative therapy and/or TBI reconstitute both T and B cell immunity. However, relatively few patients survive the harsh conditioning treatments, presumably due to their increased sensitivity to alkylating agents and ionizing radiation. Not surprisingly, when we exposed the B6 mArt-/- mice to higher doses of non-ablative radiation (6 Gy TBI) followed by HSCT the mortality was 100% (data not shown).
In summary, our results demonstrate that the immunological leakiness as well as the radiation sensitivity of mArt-/- mice is strain-dependent. We saw a dose-dependency of leakiness in terms of the amount of 129/SvJ background in the mice suggesting gene dosage effects with these Artemis genetic modifiers. We found that Artemis haploinsufficiency increases radiation sensitivity, but that the effect is less evident in the B6 background. In addition, the B6 mArt-/- mice showed significant resistance to allogeneic HSCT: there was no engraftment without conditioning and limited engraftment when low dose TBI was used as conditioning, significantly different from what we saw in the leaky mouse model [17]. Even with congenic HSCT in the B6 mArt-/- mice there was only T cell reconstitution while in the leaky mouse, T and B cells reconstituted. In all phenotypes assessed, the B6 mArt-/- mouse reported here more closely mimics the phenotypes that we see in Athabascan-speaking children with Artemis deficiency [26]. This B6 mArt-/- mouse will therefore be extremely useful in developing more efficacious treatment regimens including gene therapy to reconstitute the immune system of Artemis-deficient children.
Acknowledgments
This work was supported by NIH R01grant (5 R01 HL058842-07) and MOD grant (6-FY05-84) and work at LBNL was supported by the U.S. Department of Energy Office of Science, under contract no. DE-AC02-05CH11231 (SMY) and US National Institutes of Health grant CA104660 (SMY). We thank Randa Ibeid, Jerry Chen, Jennifer Sasaki, and Shirley Lee for their valuable technical support and Dr. Lee Ann Baxter-Lowe, Liang Peng, and Charlyn Dames for helping us with the immunoscope analysis.
Footnotes
Authorship and Conflit of Interest Statements
Contribution: Z.X. performed transplant experiments, analyzed, interpreted data and drafted the manuscript; E.D. performed leaky mouse experiments, analyzed data, helped to draft the manuscript; K.S. performed ELISA, helped to draft the manuscript; I.K. performed the radiation sensitivity assay; S.Y. analyzed, interpreted and discussed data, helped to draft the manuscript; M.C. designed research, analyzed and interpreted data, and drafted the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lewis SM. The mechanism of V(D)J joining: lessons from molecular, immunological, and comparative analyses. Adv Immunol. 1994;56:27–150. doi: 10.1016/s0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- 2.Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol. 2000;18:495–527. doi: 10.1146/annurev.immunol.18.1.495. [DOI] [PubMed] [Google Scholar]
- 3.Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109 Suppl:S45–55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 4.Grawunder U, West RB, Lieber MR. Antigen receptor gene rearrangement. Curr Opin Immunol. 1998;10:172–180. doi: 10.1016/s0952-7915(98)80246-x. [DOI] [PubMed] [Google Scholar]
- 5.Zhu C, Bogue MA, Lim DS, Hasty P, Roth DB. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell. 1996;86:379–389. doi: 10.1016/s0092-8674(00)80111-7. [DOI] [PubMed] [Google Scholar]
- 6.Taccioli GE, Amatucci AG, Beamish HJ, et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity. 1998;9:355–366. doi: 10.1016/s1074-7613(00)80618-4. [DOI] [PubMed] [Google Scholar]
- 7.Sawchuk DJ, Mansilla-Soto J, Alarcon C, et al. Ku70/Ku80 and DNA-dependent protein kinase catalytic subunit modulate RAG-mediated cleavage: implications for the enforcement of the 12/23 rule. J Biol Chem. 2004;279:29821–29831. doi: 10.1074/jbc.M403706200. [DOI] [PubMed] [Google Scholar]
- 8.Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 9.Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 10.Moshous D, Callebaut I, de Chasseval R, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Moshous D, Zhou Y, et al. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol. 2002;168:6323–6329. doi: 10.4049/jimmunol.168.12.6323. [DOI] [PubMed] [Google Scholar]
- 12.Moshous D, Li L, Chasseval R, et al. A new gene involved in DNA double-strand break repair and V(D)J recombination is located on human chromosome 10p. Hum Mol Genet. 2000;9:583–588. doi: 10.1093/hmg/9.4.583. [DOI] [PubMed] [Google Scholar]
- 13.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 14.Ma Y, Pannicke U, Lu H, Niewolik D, Schwarz K, Lieber MR. The DNA-dependent protein kinase catalytic subunit phosphorylation sites in human Artemis. J Biol Chem. 2005;280:33839–33846. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Pluth JM, Cooper PK, Cowan MJ, Chen DJ, Yannone SM. Artemis deficiency confers a DNA double-strand break repair defect and Artemis phosphorylation status is altered by DNA damage and cell cycle progression. DNA Repair (Amst) 2005;4:556–570. doi: 10.1016/j.dnarep.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Rooney S, Sekiguchi J, Zhu C, et al. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol Cell. 2002;10:1379–1390. doi: 10.1016/s1097-2765(02)00755-4. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Salido E, Zhou Y, et al. Targeted disruption of the Artemis murine counterpart results in SCID and defective V(D)J recombination that is partially corrected with bone marrow transplantation. J Immunol. 2005;174:2420–2428. doi: 10.4049/jimmunol.174.4.2420. [DOI] [PubMed] [Google Scholar]
- 18.Bhattacharyya S, Cowan MJ. B7.2-/- mature dendritic cells generate T-helper 2 and regulatory T donor cells in fetal mice after in utero allogeneic bone marrow transplantation. Biol Blood Marrow Transplant. 2005;11:657–671. doi: 10.1016/j.bbmt.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Itahana K, Zou Y, Itahana Y, et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23:389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Obukhanych TV, Nussenzweig MC. T-independent type II immune responses generate memory B cells. J Exp Med. 2006;203:305–310. doi: 10.1084/jem.20052036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hori S, Collette A, Demengeot J, Stewart J. A new statistical method for quantitative analyses: application to the precise quantification of T cell receptor repertoires. J Immunol Methods. 2002;268:159–170. doi: 10.1016/s0022-1759(02)00187-4. [DOI] [PubMed] [Google Scholar]
- 22.Pannetier C, Cochet M, Darche S, Casrouge A, Zoller M, Kourilsky P. The sizes of the CDR3 hypervariable regions of the murine T-cell receptor beta chains vary as a function of the recombined germ-line segments. Proc Natl Acad Sci U S A. 1993;90:4319–4323. doi: 10.1073/pnas.90.9.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makrigiannis AP, Etzler J, Winkler-Pickett R, Mason A, Ortaldo JR, Anderson SK. Identification of the Ly49L protein: evidence for activating counterparts to inhibitory Ly49 proteins. J Leukoc Biol. 2000;68:765–771. [PubMed] [Google Scholar]
- 24.Proteau MF, Rousselle E, Makrigiannis AP. Mapping of the BALB/c Ly49 cluster defines a minimal natural killer cell receptor gene repertoire. Genomics. 2004;84:669–677. doi: 10.1016/j.ygeno.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Drayna D, Hu D, et al. The gene for severe combined immunodeficiency disease in Athabascan-speaking Native Americans is located on chromosome 10p. Am J Hum Genet. 1998;62:136–144. doi: 10.1086/301688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Marcaigh AS, DeSantes K, Hu D, et al. Bone marrow transplantation for T-B- severe combined immunodeficiency disease in Athabascan-speaking native Americans. Bone Marrow Transplant. 2001;27:703–709. doi: 10.1038/sj.bmt.1702831. [DOI] [PubMed] [Google Scholar]