

Abstract
A dual cycloaddition strategy for the synthesis of the hetisine alkaloids has been developed, illustrated by a concise asymmetric total synthesis of (+)-nominine (7). The approach relies on an early-stage intramolecular 1,3-dipolar cycloaddition of a 4-oxido-isoquinolinium betaine dipole with an ene–nitrile dipolarophile. Subsequent late-stage pyrrolidine-induced dienamine isomerization/Diels–Alder cascade allows for rapid construction of the carbon–nitrogen polycyclic skeleton within this class of C20-diterpenoid alkaloids.
Keywords: alkaloids, asymmetric synthesis, cycloaddition, hetisine, natural products
Introduction
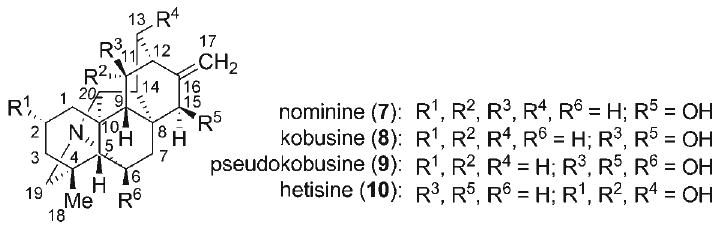
The genera of Aconitum and Delpinium, and to a lesser extent, Rumex, Consolida, and Spiraea, are rich sources of diterpenoid alkaloids.[1] The C20-diterpenoid alkaloids have been classified by Wang and Liang into two principal groups (Figure 1), the atisane class and the kaurane class, based on their respective carbon skeletons. The atisine (1), danudatine (2), hetidine (3), and hetisine (4) alkaloids comprise the atisane structural family, and the veatchine (5) and napeline (6) alkaloids make up the kaurane family. The hetisine natural products (4) are the largest subgroup of the atisane diterpenoid alkaloids, and are among the most structurally complex. Among the first of the hetisine alkaloids isolated (Figure 2) were nominine (7),[2,3] kobusine (8),[4,5] pseudokobusine (9),[6,7] and hetisine (10)[7–9] more than five decades ago. Subsequently, over 100 additional hetisine alkaloids have been isolated and characterized. The C20-diterpenoid alkaloids have long been recognized as active constituents within many traditional Eastern herbal medicines. Pharmacological investigations have revealed that the hetisine alkaloids exhibit a diverse range of biological activities,[1,10–12] exemplified by local anesthetic, anti-inflammatory, anti-arrhythmic activities of nominine (7), potent vasodilatory activities of kobusine (8) and pseudokobusine (9), and hypotensive activity of hetisine (10).
Figure 1.
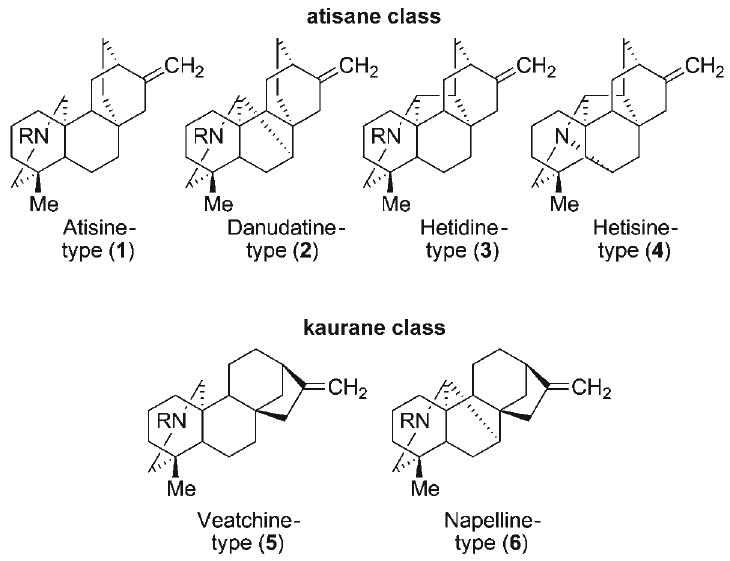
Figure 2.
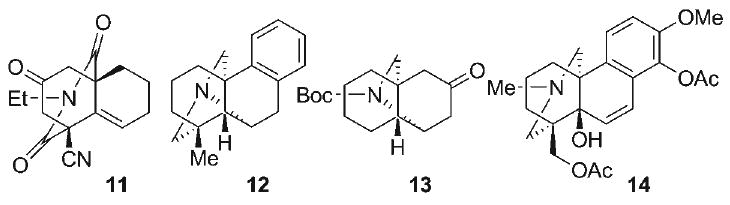
The C20-diterpenoid alkaloids have also served as classic targets in natural product synthesis, resulting in the total syntheses of natural products within the atisine (1),[13–18] veatchine (5),[19–23] and napelline (6)[24,25] subclasses. Despite this progress, synthetic efforts toward the hetisine subclass (4) have been comparatively sparse. Efforts by van der Baan and Bickelhaupt[26] led to the synthesis of the bridged aza-tricyclic core 11 (Figure 3) via intramolecular enolate alkylation, a strategy potentially amenable to multiple families of the C20-diterpenoid alkaloids. Shibanuma and Okamoto[27] were the first to disclose the synthesis of the fully-formed bridged N-heterocyclic array (i.e., 12) within the hetisine skeleton, involving the key sequence of intramolecular alkene aziridination and chloride-mediated ring opening, followed by reduction and Hofmann–Löffler–Freytag reaction to form the requisite C–N bonds. An elegant strategy was reported by Winkler and Kwak[28] to access the bridged pyrrolidine 13, employing a highly efficient intramolecular [2+2]-photocycloaddition of a vinylogous imide, followed by a retro-Mannich–Mannich cascade. More recently, Williams and Mander[29–31] applied a double Mannich protocol in conjunction with an intramolecular bridgehead arylation[32,33] as key elements in their synthesis of the advanced veatchine-like intermediate 14. The first completed synthesis of a hetisine natural product was reported by Muratake and Natsume in 2004 (Scheme 1),[34] more than 60 years after the initial discovery of this alkaloid family. This heroic synthesis of nominine (7)[35–38] involved several key transformations, including aldehyde α-arylation[39] to form the C9–C10 bond in 15, Lewis acid catalyzed acetal–ene reaction to construct the C14–C20 linkage in 16, reductive radical cyclization to form the C12–C16 bond in 17, and late-stage intramolecular N-alkylation to construct the N–C20 linkage, culminating in a 40-step racemic synthesis of nominine (7).
Figure 3.
Scheme 1.
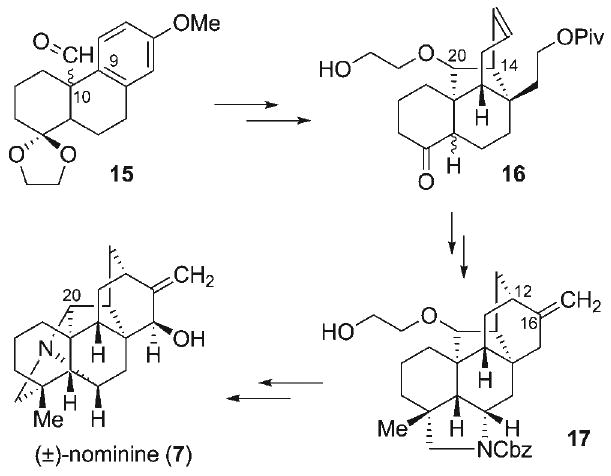
First completed synthesis of a hetisine natural product reported by Muratake and Natsume.[34]
In our development of a novel strategy to access the hetisine carbon–nitrogen scaffold, construction of the maximally bridging C–C bonds via multiple cycloaddition processes formed the basis of the synthesis plan (Scheme 2).[40] An edge-on conformational view of nominine (7) revealed two promising key elements for simplification, namely the bridging pyrrolidine embedded in the azabicyclo[3.2.1]octane core, as well as the cyclohexane moiety within the bicyclo[2.2.2]octane substructure of 7. The former could be constructed via an early-stage intramolecular 1,3-dipolar cycloaddition (i.e., 20) with an endocyclic 1,3-dipole, thereby rapidly establishing the entire N-heterocyclic array within the alkaloid target. Subsequent late-stage intramolecular Diels–Alder reaction of an appropriately positioned diene and dienophile (i.e., 19) would then complete the hetisine skeleton (i.e., 18), allowing for minimal additional functional group interconversions to realize the natural product 7. Taken together, these two cycloaddition transformations constitute a potentially expeditious entry into the hetisine framework from relatively simple precursors.
Scheme 2.
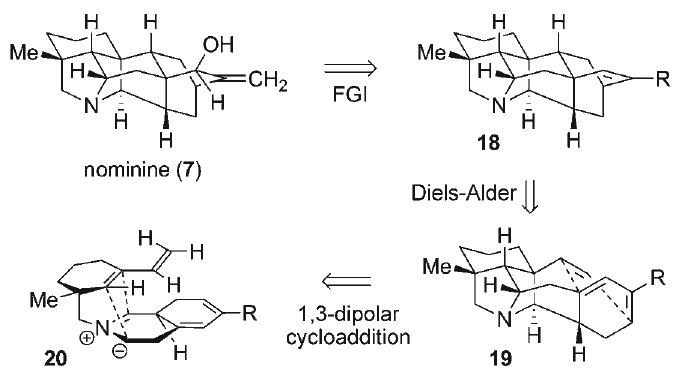
Retrosynthetic analysis.
Results and Discussion
1,3-Dipolar cycloaddition via non-stabilized endocyclic azomethine ylides
The use of azomethine ylides in the synthesis of pyrrolidine-containing complex alkaloids is well-established,[41–45] and formed the basis of our approach to construct the N-heterocyclic core within the hetisine alkaloids. A plethora of methods exist for the generation of these aza-1,3-dipoles, including, fluoride-mediated desilylation of α-iminium species, electrocyclic ring-opening of aziridines, and tautomerization of α-amino acid ester imines. Of these, the Vedejs strategy of desilylation of α-iminium intermediates is among the most widely used methods for the generation of non-stabilized azomethine ylides,[46] wherein a notable extension by Padwa has allowed access to this class of 1,3-dipoles from sequential N-alkylation and desilylation of vinylogous imidates.[47] A complementary variant to this approach has also been recently developed, involving the O-sulfonylation and desilylation of N-CH2TMS tertiary vinylogous amides.[48] In adapting this method to the construction of the pyrrolidine ring embedded in the azabicyclo[3.2.1]nonane core in the hetisine alkaloids, the generation of a six-membered endocyclic azomethine ylide to serve as a dipole in the presence of a tethered dipolarophile functionality (i.e., 22, Scheme 3) is required (for full experimental details, see Supporting Information). In this scenario, O-activation (ROTf) of an endocyclic vinylogous amide (i.e., the α-TMS-dihydropyridone 21), followed by subsequent desilylation of the resultant iminium salt, would furnish the azomethine ylide 22. If the torsional and steric constraints in 22 permit, intramolecular dipolar cycloaddition would ensue to provide the requisite bridged pyrrolidine core 23.
Scheme 3.
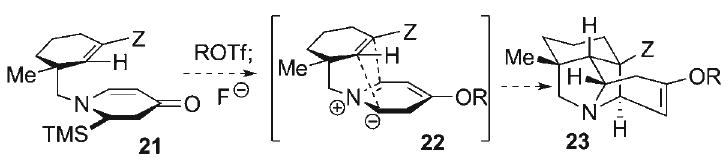
Approach to bridged pyrrolidine core 23.
Since examples of the generation and cycloaddition of endocyclic non-stabilized azomethine ylides are rare,[49–54] model investigations involving the dihydropyridone 28 (Scheme 4) were initiated to assess the feasibility of the key dipolar cycloaddition step in the synthesis. Attractive strategies to access 6-TMS-2,3-dihydro-4-pyridone moieties such as 28 have been reported, most notably involving the addition of silyl-Grignard nucleophiles to N-acyl-4-methoxypyridinium derivatives.[55,56] Unfortunately, the impracticality of late stage N-deprotection and subsequent neopentyl N-alkylation to form dihydropyridones such as 21 (i.e., Scheme 3) led us to consider an alternate, more flexible approach, incorporating an imine–hetero-Diels–Alder reaction employing Danishefsky's diene (27, Scheme 4).[57] To investigate this plan, the preparation of a model α-TMS-dihydropyridone 28 would require access to a C-silyl aldimine dienophile such as 26. Moffatt–Swern oxidation of trimethylsilyl-methanol (24) afforded an ether solution of highly reactive formyl silane 25,[58,59] which was immediately exposed to benzylamine to provide the transient C-silyl aldimine dienophile 26. Treatment of the unpurified aldimine 26 with Danishefsky's diene 27 and ZnCl2 yielded the dihydropyridone cycloadduct 28 (61 % from 24). This one-pot procedure provided a convenient protocol to access C-silyl aldimines, and established this class of imines to be competent dienophiles in the hetero-Diels–Alder reaction.
Scheme 4.
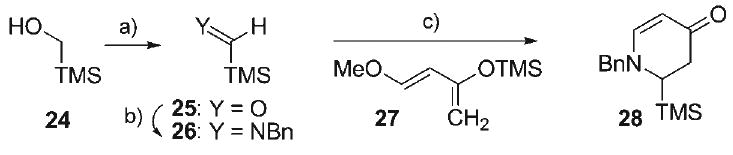
a) (COCl)2, DMSO, NEt3, Et2O, −78→0°C; b) BnNH2, solvent evaporation, 23°C; c) add 27, ZnCl2, CH2Cl2, 0→23°C, 61% overall.
The ready access to the endocyclic vinylogous amide 28 allowed for evaluation of its suitability to serve as an endocyclic non-stabilized azomethine ylide precursor. O-Activation of 28 (Scheme 5) with MeOTf or BzOTf followed by treatment with tetrabutylammonium triphenylsilyldifluorosilicate (TBAT) as a source of anhydrous fluoride[60] provided the putative azomethine ylide 29, which was trapped with several activated dipolarophiles. Dimethylacetylenedicarboxylate, phenylvinylsulfone and o-nitrobenzylacrylate all provided the desired azabicyclo[3.2.1]octane cycloadducts 30–33, albeit in low yields. While the efficiency of these intermolecular reactions is far from optimal, these experiments nonetheless demonstrate the feasibility of the dipolar cycloaddition, and signal the possibility of enhancing the efficiency of the process in an intramolecular mode, a variation required for the synthesis of the hetisine alkaloids.
Scheme 5.
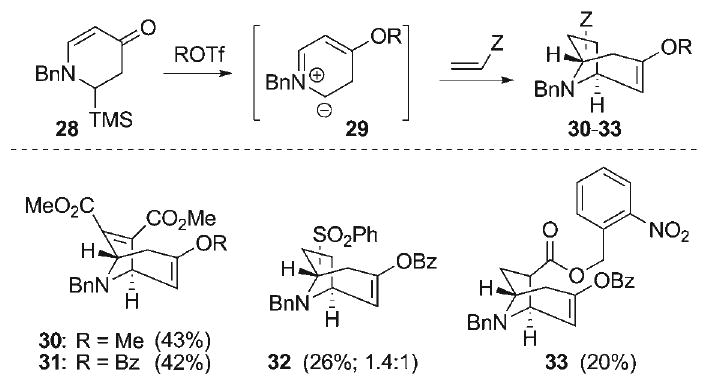
Synthesis of azabicyclo[3.2.1]octane cycloadducts 30–33.
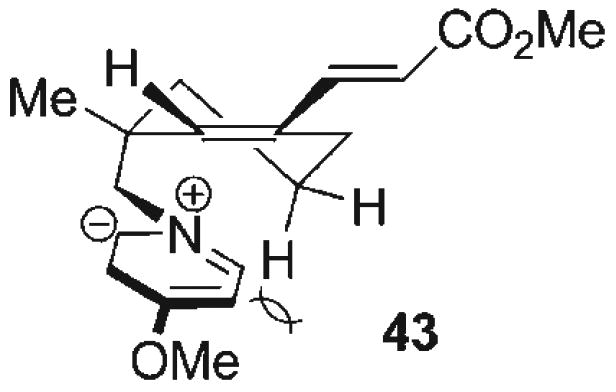
Efforts on this front commenced with 3-methylcyclohexenone (34, Scheme 6), employed as an electrophile for conjugate addition of cyanide with Al(CN)Et2[61] to afford the transient aluminum enolate. Activation of this enolate with anhydrous fluoride (TBAT) followed by treatment with Tf2O led to smooth O-triflation to provide the vinyl triflate 35 (81%). Reduction of the nitrile with DIBAL-H followed by NaBH4 produced the neopentyl amine 36, which served as the nitrogen source for an imine–hetero-Diels–Alder reaction. This proceeded with the conversion of amine 36 to its corresponding C-silyl aldimine by condensation with formyltrimethylsilane, generated in situ from Moffatt–Swern oxidation of trimethylsilylmethanol (see above). The unpurified imine was subsequently exposed to Danishefsky's diene 27 with ZnCl2 catalysis to provide the vinylogous amide 37 (55% from 36, 1.2:1 dr). Stille coupling[62] of vinyl triflate 37 with (E)-methyl 3-(tributylstannyl)acrylate afforded the dienoate 39 (87%), a substrate which incorporated both the dipolarophile group and the dipole precursor. Although the complex endocyclic vinylogous amide 39 was conveniently accessed, all attempts at azomethine ylide generation (via O-acylation or O-alkylation and subsequent desilylation) followed by intramolecular cycloaddition to prepare the advanced pyrrolidine intermediate 42 were unsuccessful, leading instead to complex mixtures of unidentifiable products. Nevertheless, several findings emerged from these studies. Interestingly, O-activation of dihydropyridone 39 with MeOTf proceeded with near quantitative conversion to the vinylogous iminium triflate 40 as evidenced by 1H NMR, even though this intermediate could not be successfully advanced to the cycloadduct 42. Furthermore, when the vinylogous amide 37, prior to installation of the pendant dienoate functionality, is activated with MeOTf and then exposed to TBAT, intermolecular cycloaddition could be performed with dimethyl acetylenedicarboxylate (DMAD) to provide the azabicyclo[3.2.1]octane 38 in 57% yield, thereby confirming at least the successful generation of the azomethine ylide dipole from 37. That the intramolecular cycloaddition could not be induced from 37 or 39 suggests prohibitive steric and/or torsional constraints in the desired transition state (i.e., 43, Figure 4) leading to competitive unproductive reaction manifolds arising from the highly reactive, short-lived 1,3-dipole. These results indicated that much of the difficulties may stem from the lack of stability of the azomethine ylide in 43, and this eventually led to re-evaluation of the cycloaddition strategy with the aim of employing a more stabilized 1,3-dipole.
Scheme 6.
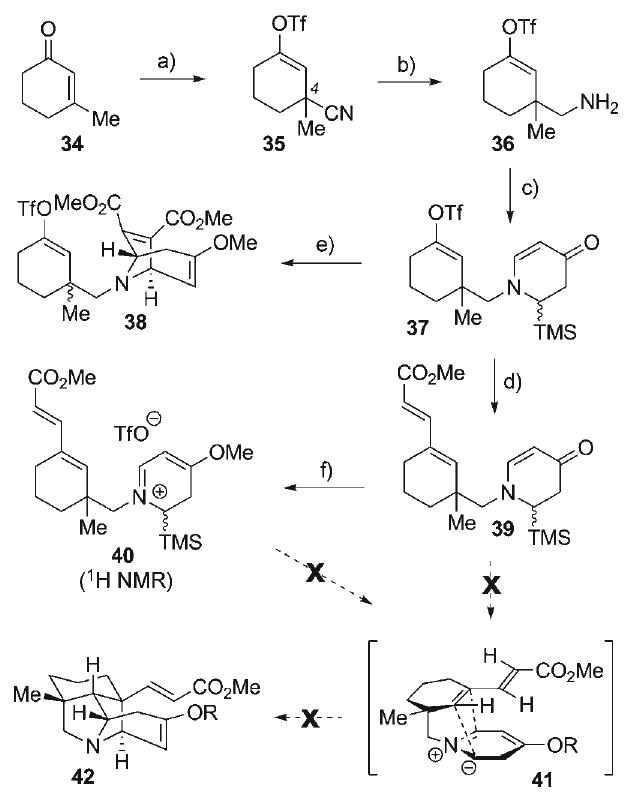
a) i) Al(CN)Et2, PhH, 23°C; ii) TBAT, then Tf2O, 81%; b) DIBAL-H, 0°C, toluene, NaBH4/MeOH quench, 88%; c) i) TMSCHO, Et2O, 0°C; ii) solvent evaporation, 0°C; iii) 27, ZnCl2, CH2Cl2, 0→23°C, 55%, 1.2:1 dr; d) methyl (E)-3-tributylstannylacrylate, [Pd(PPh3)4], LiCl, THF, reflux, 87%; e) MeOTf, CH2Cl2, 23°C; DMAD, TBAT, 57%, 1:1 dr; f) MeOTf, CDCl3, 23°C, >95%.
Figure 4.
Intramolecular 1,3-dipolar cycloadditions with 3-oxidopyridinium betaines
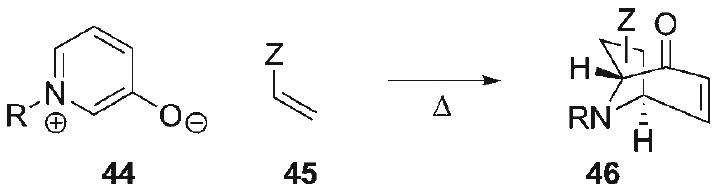
Given our lack of success in effecting intramolecular cycloaddition with non-stabilized azomethine ylides, a significantly more stable 1,3-dipole equivalent, namely a 3-oxidopyridinium betaine, was explored in the context of pyrrolidine construction for the hetisine alkaloids. Dipolar cycloaddition of 3-oxidopyridinium betaines, introduced by Katritzky,[63–69] are useful synthetic transformations for increasing structural complexity in the construction of bridged N-heterocycles (44+45→46, Scheme 7). This class of betaines constitutes moderately reactive aza-1,3-dipoles that undergo dipolar cycloadditions at the 2- and 6-positions with alkenes and alkynes to produce tropanone-like adducts of varying complexity, including a handful of examples that proceed in an intramolecular mode.[70–76]
Scheme 7.
3-Oxidopyridinium betaine cycloaddition.
In applying this approach to the synthesis of the hetisine alkaloids, the initial aim was to prepare the 3-oxidopyridinium betaine 50 (Scheme 8), incorporating a pendant ene–nitrile as an electronically activated, less sterically demanding dipolarophile. Although N-alkylation of substituted pyridines is the most direct and common method for the preparation of 3-oxipyridinium betaines,[63] concerns with nucleophilic substitution at a neopentyl carbon to form the N–C19 bond in 50 directed our efforts to pursuing an alternate approach involving the aza-Achmatowicz reaction.[77–80] This sequence relied on our previously prepared nitrile 35, which was reduced to the corresponding aldehyde 47 (DIBAL-H, 92%) and then subjected to reductive amination with furfurylamine to provide the secondary amine 48 (99%). To introduce an appropriate electron deficient dipolarophile activating group that would also serve as a convenient dienophile precursor, the vinyl triflate moiety in 48 was then converted to ene–nitrile 49 (75%) via Pd-catalyzed cyanation with KCN. Bromine-mediated aza-Achmatowicz reaction of 50 delivered the oxidopyridinium betaine 50 (65%), which could be purified by silica gel flash chromatography. Unfortunately, heating (up to 170°C) a solution of betaine 50 in a variety of solvents failed to effect the desired intramolecular dipolar cycloaddition (i.e., 50 → 51 → 52), as the tri-cyclic oxidopyridinium betaine 54 was the only discernable product (73%) after a prolonged reaction time (5 d). The formation of 54 was the result of direct conjugate addition of the betaine 53 into the ene-nitrile, followed by aromatization, being the dominant reaction pathway. While the formation of the indolizinium species 54 provides access to an interesting heterocyclic scaffold,[81] the unmet challenges of accessing the desired azabicyclo[3.2.1]octane skeleton of the hetisine alkaloids remained.
Scheme 8.
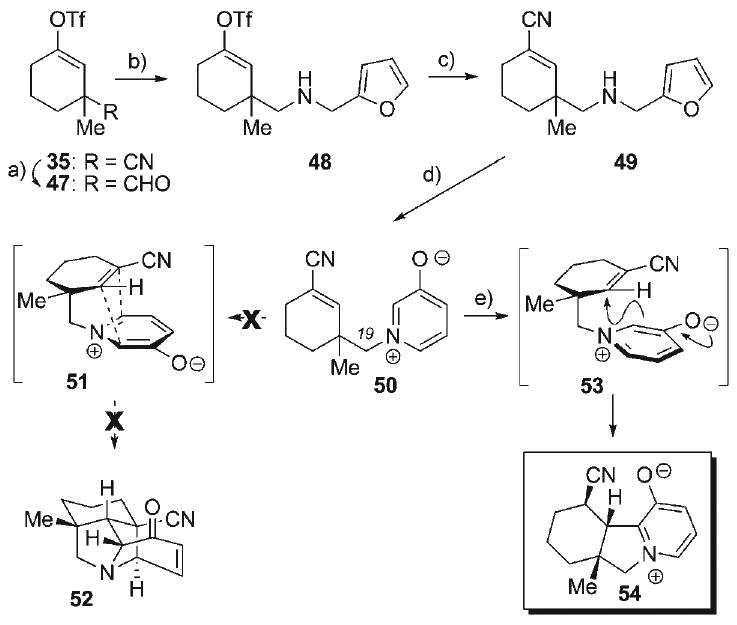
a) DIBAL-H, 0°C, PhMe, 92%; b) furfurylamine·HCl, NaBH4, MeOH, 3 Å MS, 23°C, 99%; c) KCN, [18]crown-6, [Pd(PPh3)4], PhH, 75°C, 75%; d) Br2, 1:1 AcOH/water, 0°C, 65%; e) [0.15 m] toluene, 170°C, 5 d, 73%.
Since the undesired conjugate addition pathway emerged as the principal obstacle at this juncture, a strategy was adopted in which a removable electron-deficient dipolarophile activating group (i.e., a sulfone) was installed at the vinyl C5-position in a cycloaddition substrate such as 59 (Scheme 9). It was anticipated that reversal of the polarization of the dipolarophile π-system would preclude a conjugate addition pathway (i.e., 59 → 62 → 63) that would necessarily proceed via a high energy bridged transition state. This may provide an opportunity for the favored cycloaddition process (i.e., 59 → 60 → 61) to surface as the dominant reaction pathway. This approach began with 2-cyano-2-methylcyclohexanone (55), which was readily prepared from 1,5-dicyanopentane via a previously reported three-step sequence.[82] Condensation of ketone 55 with thiophenol produced the vinyl sulfide 56 (84%). Subsequent reduction of the nitrile in 56 with DIBAL-H afforded the corresponding neopentyl aldehyde, which allowed for oxidation of the vinyl sulfide functionality to the vinylsulfone 57 (75%, 2 steps) with m-CPBA. Introduction of the oxidopyridinium betaine dipole proceeded with reductive amination of aldehyde 57 with furfurylamine to produce amine 58 (91%), which smoothly underwent aza-Achmatowicz oxidative rearrangement to provide the cycloaddition precursor 59 (77%). Heating of a dilute solution of 3-oxidopyridinium betaine 59 in refluxing toluene provided the desired cycloadduct 61 (70%), the structure of which was unambiguously verified by its X-ray crystal structure. That none of the isomeric conjugate addition product 63 was formed in this reaction reinforces the hypothesis of an energetically disfavored bridged transition state 62 giving way to the more favorable cycloaddition manifold (60). This promising result presented a highly efficient route to the entire N-heterocyclic array within the hetisine framework in the form of 61. Further advancement of this intermediate toward the hetisine alkaloids would involve installation of the diene–dienophile pair for late-stage intramolecular Diels–Alder reaction (i.e., 19, Scheme 2). While installation of the 1,3-diene moiety onto 61 (Scheme 9) can be envisioned to occur via a variety of six-membered annulation strategies at the C8–C14 positions, introduction of a dienophile moiety, required at the C10-position of 61, is considerably more challenging since a method for controlled C–H activation at this bridgehead position is not obvious.
Scheme 9.
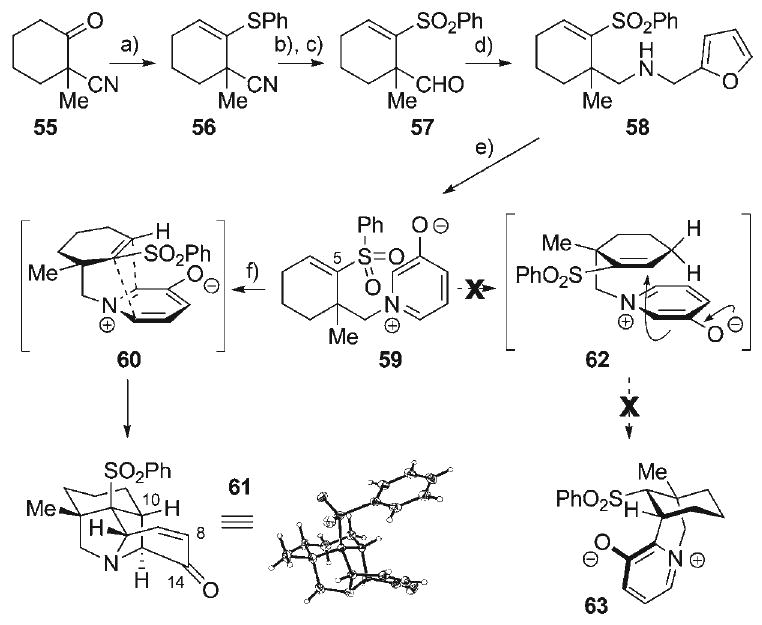
a) PhSH, P2O5, CH2Cl2, 23°C, 84%; b) DIBAL-H, toluene, 0°C, 82%; c) m-CPBA, CH2Cl2, 0°C, 91%; d) furfurylamine·HCl, NaBH3CN, 3 Å MS, NEt3, MeOH, 23°C, 91%; e) Br2, MeOH, H2O, 0°C, 77%; f) [0.05 m], PhMe, reflux, 70 %.
In light of this challenge, installation of an appropriate C10-dienophile precursor prior to the key dipolar cycloaddition event was pursued. This involved (Scheme 10) bromination of the alkene at C10 within our previously prepared vinyl sulfide 56 to form vinyl bromide 64 (92%), which allowed for metal–halogen exchange with iPrMgCl.[83] The resulting vinyl Grignard reagent was alkylated with BOMCl in the presence of CuCN to provide tetrasubstituted alkene 65 (81%). The nitrile within 65 was then reduced (DIBAL-H) to afford the corresponding aldehyde (74%), allowing for sulfide oxidation (m-CPBA) to provide the vinyl sulfone 66 (62%). Subsequent reductive amination with furfurylamine furnished amine 67 (78%), allowing for bromine-mediated oxidative rearrangement to provide the oxidopyridinium betaine 68 (67%). Investigation into the cycloaddition reaction of betaine 68 led to its heating in refluxing toluene with the hopes of acquiring pyrrolidine 70 via the anticipated transition state 69. However, none of the cycloadduct 70 was detected; rather, the isomeric intramolecular cycloadduct 73 was isolated (43%) as the primary product; its structure was initially delineated by a battery of spectroscopic data and ultimately unambiguously verified through single crystal X-ray analysis.
Scheme 10.
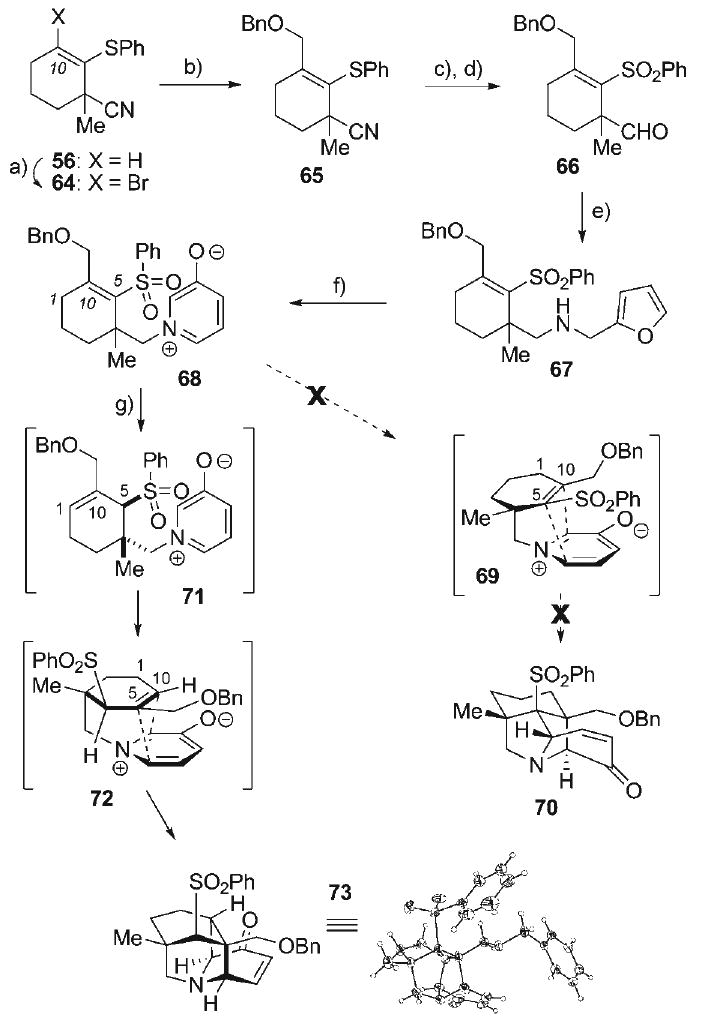
a) Br2, MeCN, 80°C, 92%; b) iPr2MgCl, CuCN, THF, 0°C, then BOMCl, 81%; c) DIBAL-H, toluene, 0°C, 74%; d) m-CPBA, CH2Cl2, 0°C, 62%; e) furfurylamine·HCl, NaBH4, 3 Å MS, NEt3, MeOH, 23°C, 78%; f) Br2, MeOH, H2O, 0°C, 41%; g) [0.01 m], PhMe, 120°C, 67%.
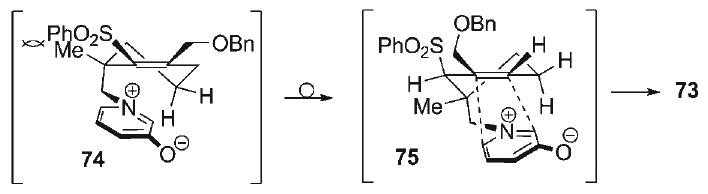
This outcome arose from initial isomerization of the tetrasubstituted C5–C10 alkene in 68 to its trisubstituted C1–C10 alkene counterpart 71, eventually leading to intramolecular dipolar cycloaddition of this unactivated dipolarophile to generate the observed cycloadduct 73. This initial “deconjugation” event (68 → 71) is likely driven by release of steric strain to allow for access to a lower energy pathway for intramolecular cycloaddition. Qualitative conformational analysis of the initially anticipated cycloaddition transition state of the tetrasubstituted dipolarophile (74, Scheme 11) reveals that the pendant oxidopyridinium dipole must extend from a pseudo-axial orientation. This would constrain three neighboring exocyclic substituents, namely the vinyl benzyloxymethyl, the vinyl sulfone, and pseudoequatorial allylic methyl group, to all be nearly coplanar in an early transition state model 74. By contrast, significant relief of steric strain would result from alkene isomerization to the C1–C10 position to form the trisubstituted alkene 75, in which the same three exocyclic substituents would approximate mutual pseudo-gauche relative orientations. While the mechanism of the alkene isomerization of 68 to 71 is not known, the process is not surprising in retrospect given that electronic influences of the sulfone moiety on α,β-alkenes are primarily inductive in nature. It has been shown that the sulfone moiety imparts little electronic stabilization to attached alkenes through conjugation, since ground state d-orbital resonance does little to stabilize α,β-2p-π-systems.[84] Indeed, this property of vinyl sulfones has on occasion been synthetically exploited.[85–87] Although the formation of cycloadduct 73 constitutes entry into a novel bridged pyrrolidine scaffold, its formation could not be abated in favor of the desired cycloadduct 70, as all attempts to preclude alkene isomerization through either rigorous exclusion of adventitious proton sources, or decreasing substrate concentration, met with no success.
Scheme 11.
The unavoidable alkene isomerization of vinyl sulfone 68 led to the exploration of a cycloaddition substrate incorporating a C10-nitroalkene dipolarophile functionality (i.e., 81, Scheme 12). It was anticipated that use of the nitroalkene would preclude alkene isomerization in the cycloaddition due to an increased degree of conjugative stabilization, even though the propensity of 81 to undergo unproductive conjugate addition might be enhanced. These efforts were initiated with the formation of the thermodynamic TMS-enol ether of 2-methylcyclohexanone (76, Scheme 12), followed by trityl-ClO4-catalyzed α-alkylation with 2-methoxy-1,3-dioxolane to afford the keto-dioxolane 77 (81%).[88] Nitration at the α′-position of 77 was accomplished with iBuONO2 and K-OtBu to provide the α′-nitroketone 78 (91%) as a diastereomeric/tautomeric mixture, which could be converted to the corresponding nitroalkene (69%) under the Luche reduction protocol.[89] After acetal hydrolysis to form aldehyde 79 (78%), the familiar sequence (unoptimized) of reductive amination with furfurylamine and aza-Achmatowicz rearrangement was executed to form amine 80 (34%) and oxidopyridinium betaine 81 (64%), respectively. Microwave heating of oxidopyridinium 81 at 170°C provided the cycloadduct 83. While this experiment afforded, for the first time, a C10-derivatized cycloadduct comprising the hetisine N-heterocyclic substructure, the transformation advanced to no further than ≈14% conversion. Moreover, the structure of pyrrolidine 83, verified by spectroscopic scrutiny, was that derived from a cycloaddition which proceeded opposite to what would be expected from a dipole HOMO-controlled process, implying contra-kinetic electronic selectivity. These data suggest that the cycloaddition outcome might be the result of thermodynamic selection, a fact that was verified when purified cycloadduct 83 was re-subjected to the reaction conditions, leading to re-establishment of a ≈6:1 ratio of betaine 81 and cycloadduct 83, respectively. From this critical finding, a more efficient synthetic approach to the hetisine alkaloids was pursued based on a thermodynamic (as opposed to a kinetic) analysis of the key 1,3-dipolar cycloaddition event.
Scheme 12.
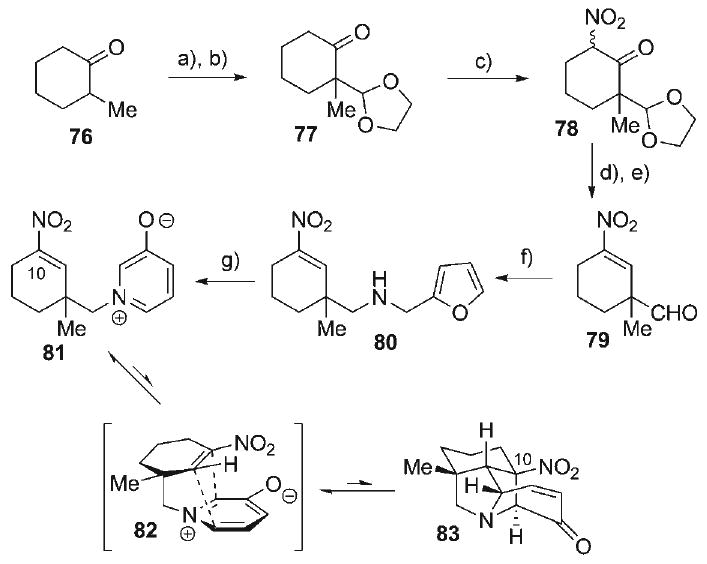
a) iPr2NMgBr; TMSCl, NEt3, HMPA, Et2O, 23°C, 77%; b) TrClO4, 2-methoxy-1,3-dioxolane, CH2Cl2, −78°C, 81%; c) iBuONO2, tBuOK, 23°C, 91 %; d) CeCl3·7H2O, NaBH4, EtOH, 23°C, 69%; e) TFA/H2O 4:1, 23°C, 78%; f) i) furfurylamine·HCl, NEt3, 4 Å MS, MeOH; ii) NaBH(OAc)3, CH2Cl2, 23°C, 34%; g) Br2, 1:1 AcOH/H2O, 0°C, 64%; h) [0.01 m], PhMe, 170°C (MW wave), 14% (plus 81 % rec. 81).
Intramolecular 1,3-dipolar cycloadditions with 4-oxidoisoquinolinium betaines
The unfavorable equilibrium observed in the conversion of 3-oxidopyridinium 81 to its pyrrolidine cycloadduct 83 (Scheme 12) led to an overhaul in the design of the cycloaddition substrate. As a result, the exploration of 4-oxidoisoquinolinium betaine dipoles was initiated with the goal of lowering the energetic cost of breaking aromaticity of the dipole in the cycloaddition event to the point where the transformation might be rendered exothermic. Calculations by Bird regarding resonance stabilization energies (RSE) of various aromatic and heteroaromatic compounds provide a useful framework in this context.[90] In these estimates, the sum of the individual RSEs (Figure 5) of pyridine (84) and benzene (85) exceeds the RSE (by ≈8 kcal mol−1) of isoquinoline (86), the molecular fusion of the two individual aromatic systems. This in turn suggests that the aromatic resonance stabilization of the isolated heterocyclic “pyridinyl subunit” within isoquinoline (86) is less than that of pyridine (84) itself, perhaps by a few kcal mol−1. If this analogy can be reasonably transferred to aromatic betaine substrates, one can infer that the aromatic stabilization of the “oxidopyridinium subunit” (i.e., the dipole component) within 4-oxidoisoquinolinium intermediate 88 is also less (i.e., more destabilized) compared to that of 3-oxidopyridinium substrate 87 itself. This proposed net relative ground state destabilization of a 4-oxidoisoquinilinium dipole highlights its potential to serve in a more thermodynamically favorable dipolar cycloaddition.
Figure 5.
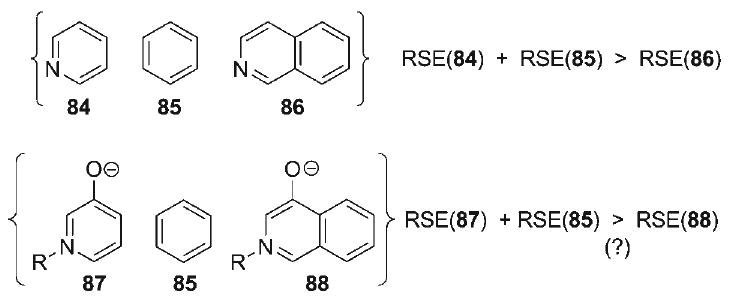
Notably, the earlier cycloaddition employing the nitroalkene–oxidopyridinium substrate 81 (Scheme 13a, also Scheme 12) was found to be endothermic by ≈1.6 kcal mol−1 (i.e., ≈14% conversion at 170°C). Thus, only a relatively small perturbation in the relative ground state energies of the reagent and product need be achieved to reverse the thermodynamic outcome of the reaction. If this could be accomplished with the use of a moderately destabilized 4-oxidoisoquinolinium dipole such as 89 (Scheme 13b), rapid access to a highly advanced synthetic intermediate such as 90 would be possible. Indeed, the allure of this strategy is heightened by the likelihood of the pendant benzo-ring within the isoquinolinium dipole 89 to eventually serve as the structural foundation for a Diels–Alder diene in the later stages of the synthesis, a prospect contingent, of course, on an acceptable level of regioselectivity in the dipolar cycloaddition.
Scheme 13.
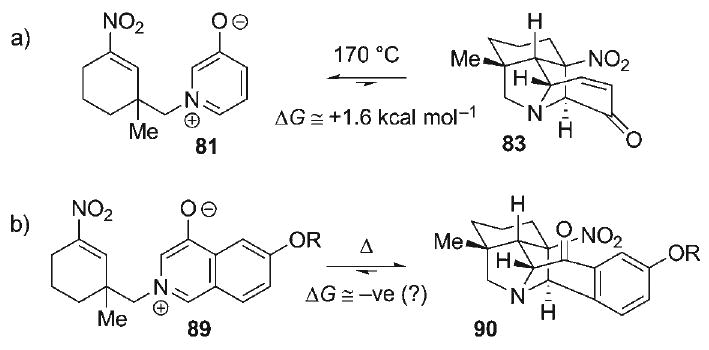
The use of 4-oxidoisoquinolinium betaines in 1,3-dipolar cycloadditions was first reported by Katritzky.[91] Despite the synthetic potential of this transformation, only a handful of these cycloadditions have appeared,[92–99] wherein the paucity of intramolecular examples is notable.[100] Several methods exist for the preparation of 4-oxidoisoquinolinium betaines, including N-alkylation of hydroxyisoquinolines, fragmentation of benzo-keto aziridines,[92] cyclocondensation–oxidation of diesters,[98] and RhII-catalyzed annulation of α-diazocarbonyl groups onto imines.[96] However, direct application of any of these strategies toward the appropriately substituted 4-oxidoisoquinolinium 89 was not obvious, and so an alternative strategy was pursued based on a late-stage cycloaromatization–condensation event, similar to that in the second phase of the classic aza-Achmatowicz reaction. Preparation of the 4-oxidoisoquinolinium betaine commenced with commercially available 4-methoxybenzaldehyde dimethyl acetal (91, Scheme 14) in which directed ortho-lithiation[101] and acylation with α-chloro-N-methoxy-N-methyl-acetamide provided the chloroacetophenone 92 (52%). Nucleophilic substitution of the chloride in 92 with NaN3 proceeded smoothly (95%), allowing for subsequent acid-catalyzed acetal isomerization in MeOH to produce the bicyclic diacetal 93 (99%) as a 3:2 mixture of diastereomers.
Scheme 14.
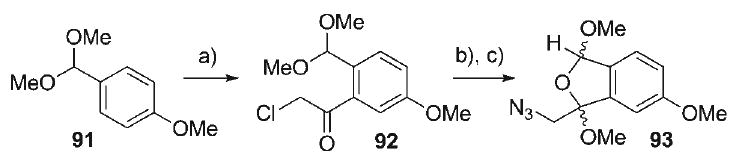
a) tBuLi, Et2O, −23°C; ClCH2C(O)N(OMe)Me, 52%; b) NaN3, acetone, 23°C, 95%; c) AcCl, MeOH, 99%, 3:2 dr.
Convergent attachment of the dipole precursor 93 with the nitroalkene dipolarophile component 79 was accomplished (Scheme 15) with a Staudinger–aza-Wittig reaction, in which azide 93 was initially exposed to Bu3P, followed by sequential treatment with the nitroalkene–aldehyde 79 and NaBH(OAc)3. The resultant amine 94 (74%) was isolated as an inconsequential 3:3:2:2 mixture of diastereomers, all of which were converged to the 4-oxidoisoquinolinium betaine 95 via TFA-catalyzed extrusion of MeOH (98%). Investigations into the intramolecular 1,3-dipolar cycloaddition of 95 involved its heating in a dilute solution of 5% MeCN in toluene at 90°C in a microwave reactor. After 30 min at this temperature, 4-oxidoisoquinolinium betaine 95 was quantitatively converted to three isomeric products, namely the bridged pyrrolidine cycloadducts 98 and 100, as well as the tetracyclic betaine 96 (arising from intramolecular conjugate addition of 95) in a 4:2:3 isomeric ratio, respectively. Upon increasing the reaction time, the proportion of the tetracyclic betaine 96 also increased, ultimately to the point of its exclusive formation. These data suggest that the cycloaddition of 95 to form the bridged pyrrolidines 98 and 100 is relatively rapid compared with that of conjugate addition of 95 to form betaine 96. Additionally, the cycloaddition of 95 is reversible, in which reversion of cycloadducts 98/100 to a small quantity of bicyclic betaine 95 allows for its eventual irreversible funneling via conjugate addition to the tetracyclic betaine 96, the thermodynamic sink in the reaction. Despite the unavoidable diminution of the desired cycloadducts in this process, the presence of only a small (undetectable) amount of the intermediary betaine 95 in the eventual accumulation of 96 from 98/100 suggests that the partial equilibrium of the cycloaddition (95 ⇄ 98/100) indeed represents an exothermic process.[102] This validates the strategy of using oxidoisoquinolinium betaines over oxidopyridinium betaines to effect a more favorable cycloaddition equilibrium, yet there remained the specific issue of avoiding irreversible conjugate addition of 95 for application to the synthesis of the hetisine alkaloids.
Scheme 15.
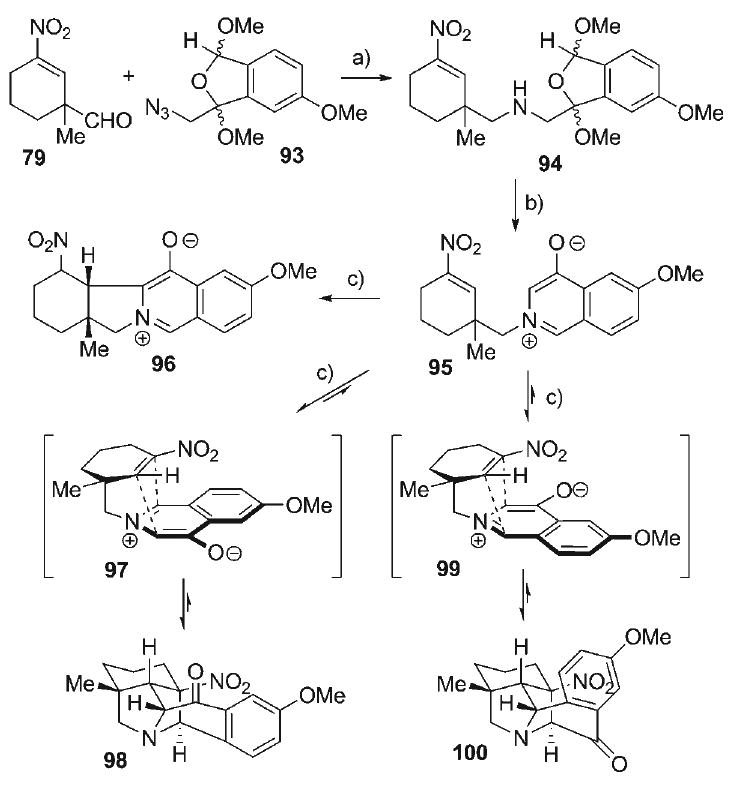
a) 93, PBu3, CH2Cl2, 23°C; add 79; add NaBH(OAc)3, 74% (3:3:2:2 dr); b) 1:10 TFA/CH2Cl2, 0°C, 98%; c) [0.008 m], PhMe, 90°C (MW wave), 30 min, → 98/100/96 with 4:2:3 isomeric ratio.
This challenge was addressed with a final modification of the dipolarophile component of the 4-oxidoisoquinilinium betaine cycloaddition precursor, wherein the use of the less activated ene–nitrile dipolarophile was re-investigated. Although previous studies with this class of dipolarophile failed to effect intramolecular cycloaddition with 3-oxidopyridinium dipoles (i.e., 50, Scheme 8), the conjugate addition manifold was also extremely slow (170°C, 5 d). Thus, the use of a more reactive isoquinolinium betaine dipole with an ene–nitrile dipolarophile may result in the cycloaddition emerging as the preferred reaction pathway. The ene–nitrile was readily prepared from the vinyl triflate 47 (Scheme 16), beginning with Pd-catalyzed cyanation with Zn(CN)2[103] to provide the nitrile–alkene 101 (85%). Application of the previously established Staudinger–aza-Wittig sequence in the coupling of aldehyde 101 and azide 92 provided the amine 102 (79%) as a mixture of four diastereomers. Treatment of this mixture with TFA effected cycloaromatization to afford the 4-oxidoisoquinolinium betaine 103 (93%). When a dilute solution of this cycloaddition precursor was heated to 180°C in THF (sealed tube) for 15 h, a mixture of the betaine 103 and the cycloadducts 104 and 105 was isolated in a 03:21:76 ratio, respectively. Importantly, this ratio was invariant to extended reaction times, and contained no trace of conjugate addition by-products, indicating that the cycloaddition was indeed exothermic (97% conversion), and that the unproductive conjugate addition pathway had been completely suppressed. However, the 1:3.6 isomeric ratio of the cycloadducts favored the undesired isomer 105. Nevertheless, the cycloaddition process was verified to be under thermodynamic control, as isolation and re-subjection of the undesired cycloadduct 105 to the reaction conditions led to re-establishment of the equilibrium ratio of isomers. Given this near quantitative interconversion of isomers from 103, a procedure for iterative thermal re-equilibration in the recycling of 105 was established, allowing for accumulation of the desired cycloadduct 104 at ≈20% per equilibration with minimal loss of material.
Scheme 16.
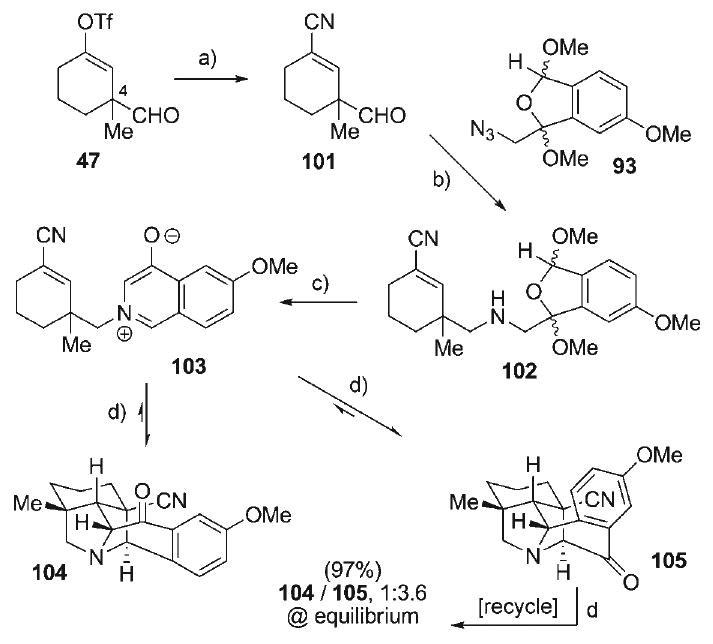
a) Zn(CN)2, [Pd(PPh3)4], DMF, 60°C, 85%; b) 93, PBu3, CH2Cl2, 23°C; add 101; add NaBH(OAc)3, 79% (3:3:2:2 dr); c) 1:10 TFA/CH2Cl2, 0°C, 93%; d) [1.5 m], 180°C, THF, 97%, 104/105 1:3.6 at equilibrium.
Intramolecular Diels–Alder reaction and the synthesis of (±)-nominine (7)
Advancement of the pyrrolidine cycloadduct 104 (Scheme 17) toward nominine (7), the simplest of the hetisine alkaloid family, began with benzylic ketone-to-methylene reduction. This was performed with the sequential operations of: 1) reduction to the benzylic alcohol (5:1 dr); 2) chlorination of the resultant benzylic alcohol (6:1 dr); and 3) free radical benzylic de-chlorination to provide the bridged pyrrolidine skeleton 106 (68%, three steps). Conversion of the nitrile in 106 to an alkene dienophile was straightforward, initially involving its reduction to the corresponding aldehyde (85%) with DIBAL-H, followed by Wittig methylenation to afford the alkene 107 (96%). In securing the diene component for the proposed Diels–Alder reaction, the arene in 107 was reduced under the Birch protocol (Na, THF, iPr2CHOH)[104] to afford the transient 1,4-dienol ether, which was hydrolyzed upon acidic work-up to afford the γ,δ-unsaturated cyclohexenone derivative 108 (97%).
Scheme 17.
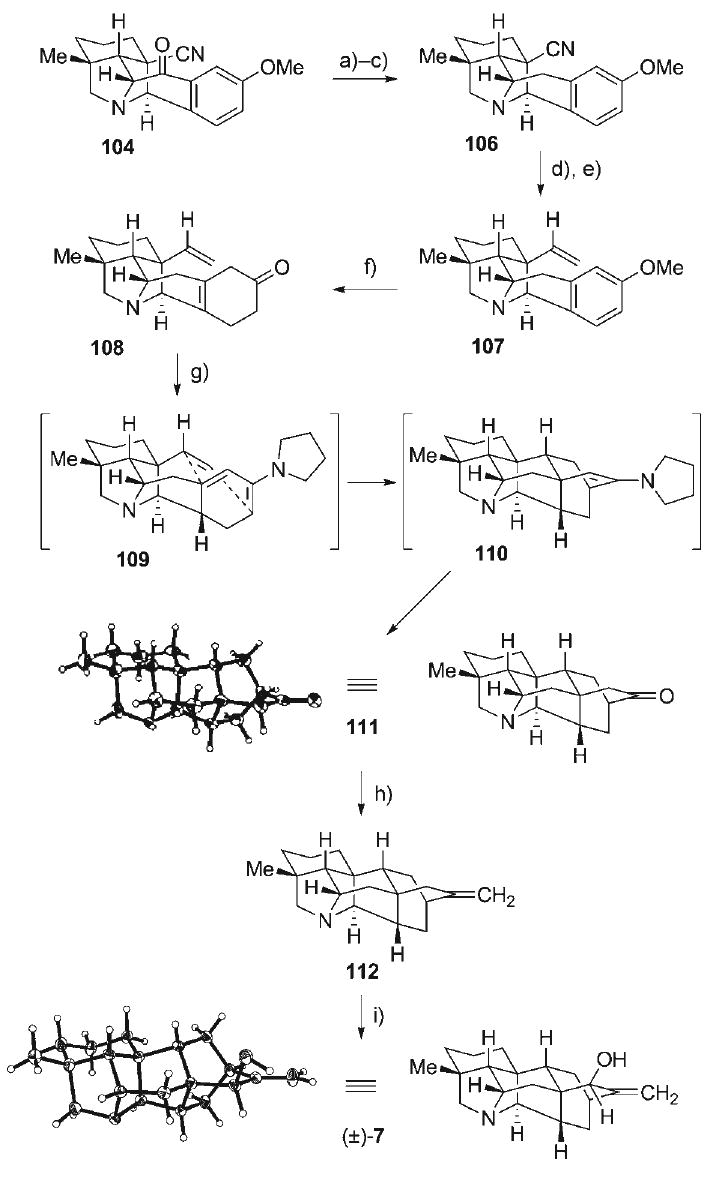
a) NaBH4, EtOH, 23°C; b) SOCl2, CH2Cl2, reflux; c) Bu3SnH, AIBN, PhH, reflux, 68% (3 steps); d) DIBAL-H, PhMe, 0°C, 85%; e) Ph3P=CH2, THF, 23°C, 96%; f) Na0, Me2CHOH, THF, NH3, −78°C; HCl(aq), 97%; g) 9:1 MeOH/pyrrolidine, 60°C, 78%; h) Ph3P=CH2, THF, 70°C, 77%; i) SeO2, tBuOOH, CH2Cl2, 23°C, 66%, 7:1 dr.
With efficient access to γ,δ-unsaturated cyclohexenone 108, it was envisioned that isomerization of the endocyclic alkene to provide the conjugated enone functionality would provide a key intermediate for generation of the required 1,3-diene for intramolecular [4 + 2] cycloaddition. Unfortunately, all efforts to convert 108 to is α,β-enone isomer met with no success. Exposure of 108 to a variety of either acidic or basic reagents/catalysts under mild conditions resulted in no reaction, while more forcing conditions led only to its decomposition. This suggests that the de-conjugated enone moiety in 108 may already be in its thermodynamically favored form. However, a significant advance emerged during attempts at generating conjugated diene functionalities through the formation of iminium/enamine intermediates. When enone 108 was treated with pyrrolidine in refluxing MeOH, the fully intact polycyclic hetisine skeleton 111 (verified by X-ray analysis) could be isolated in 78%. This intriguing outcome is likely the result of initial reversible iminium formation via dehydrative condensation of pyrrolidine and enone 108. In a polar protic solvent (MeOH) at elevated temperature, isomerization of the transient iminium species to several distinct dienamine constitutional- and stereo-isomeric forms would occur, with one of them being the dienamine 109. Among all of the possible isomers in dynamic equilibration, only dienamine 109 is conducive to a favorable intramolecular Diels–Alder transition state, resulting in Curtin-Hammett kinetics that funnel the equilibrating intermediates irreversibly to the Diels–Alder adduct 110, and ultimately to ketone 111 after enamine hydrolysis. Having secured the critical [4+2]-cycloaddition reaction in the synthesis, the final functional group interconversions involved Wittig olefination of the ketone in 111 to form alkene 112 (77%), followed by diastereoselective allylic hydroxylation of with SeO2[105,106] to afford (±)-nominine (7, 66%, 7:1 dr), whose structure was verified by single crystal X-ray analysis. Overall, the total synthesis of (±)-7 was accomplished in a 15-step sequence with only a single protective group manipulation.
Enantioselective total synthesis of (+)-nominine (7)
Adaptation of our racemic synthesis of nominine (7) to a non-racemic route focused on asymmetric induction at an early stage in the synthetic route to establish the absolute stereochemical course of the synthesis. Although nominine (7) incorporates ten chiral carbon centers, initial establishment of the absolute configuration of the C4-quaternary carbon would allow for effective diastereoselective stereochemical relay to the remaining stereocenters in the natural product. In the racemic synthesis of nominine (7), the enol triflate intermediate 47 (Scheme 16) served as a key early chiral racemic intermediate. The C–C bond-forming event to establish the quaternary C4 center in 47 was accomplished earlier by conjugate addition of cyanide onto an enone electrophile (34 → 35, Scheme 6); unfortunately, an enantioselective variant of this cyanide conjugate addition has not been reported.
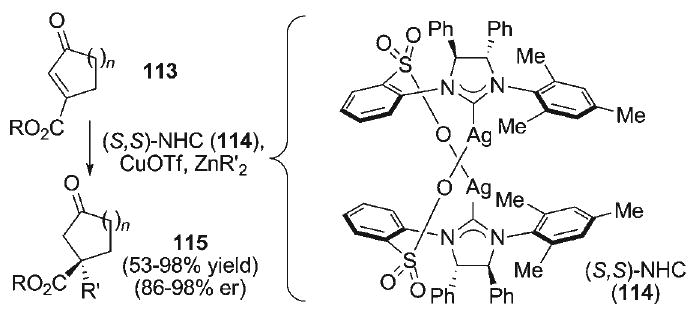
Recently, Hoveyda reported the enantioselective conjugate addition of alkylzinc and arylzinc nucleophiles for the formation of quaternary carbon stereogenic centers.[107] In this method (Scheme 18), treatment of cyclic γ-keto enoates 113 with dialkyl or diarylzinc reagents in the presence of CuOTf and the chiral N-heterocyclic carbene ligand complex 114 [(S,S)-NHC] provided the conjugate addition products 115 with good to excellent levels of asymmetric induction. Moreover, in related conjugate additions with 3-alkyl-substituted α,β-enones, the initial Zn–enolate product of nucleophilic addition could be trapped at oxygen with Tf2O to furnish enol triflate products.[108]
Scheme 18.
Application of this methodology to the asymmetric synthesis of (+)-nominine (7) thus necessitated several changes in the early stages of the sequence (Scheme 19), commencing with allylic oxidation of methyl 1-cyclohexene-1-carboxylate (116) with CrO3. The resultant γ-keto enoates 117 (56%) was then used as the electrophile for asymmetric conjugate addition with the Hoveyda protocol, employing the (S,S)-NHC catalyst 114 in the presence of CuOTf and ZnMe2. The corresponding zinc enolate intermediate was then trapped with Tf2O to provide the enol triflate (+)-118 (91%) with good enantioselectivity (92:8 er) as determined by chiral HPLC. The enantio-enriched vinyl triflate 118 was then subjected to low-temperature DIBAL-H reduction of the ester to provide the corresponding aldehyde (+)-47 (92%), an enantio-enriched intermediate that directly intercepts its racemic counterpart in our earlier synthesis (i.e., 47, Scheme 16). Palladium-catalyzed cyanation of enol triflate 47 then furnished the ene–nitrile (+)-101 (82%). Aldehyde (+)-101 was then coupled to the cyclic ketal azide 93 via the Staudinger–aza-Wittig protocol to afford amine 102 (82%) as a diastereomeric mixture, which was readily converted to the oxidoisoquinolinium betaine 103 upon TFA-mediated cycloaromatization (98%). The intramolecular dipolar cycloaddition of enantio-enriched 103 proceeded as expected, providing the cycloadducts 105 and 104 in a 3.6:1 isomeric ratio, respectively. Reiterative thermal re-equilibration of the isolated cycloadduct 105 allowed for accumulation of the desired cycloadduct (+)-104, which could be recrystallized to enantiopurity (>99:1 er). The remaining steps in the asymmetric synthesis mirrored that of the above-mentioned racemic synthesis (Scheme 17), whereby the identical nine-step sequence was executed in comparable yields, providing (+)-nominine (7, [α]D = + 50.7, c = 1.09, MeOH).[2,3] These efforts mark the first enantioselective synthesis of (+)-nominine (7), comprising a 16-step sequence.
Scheme 19.
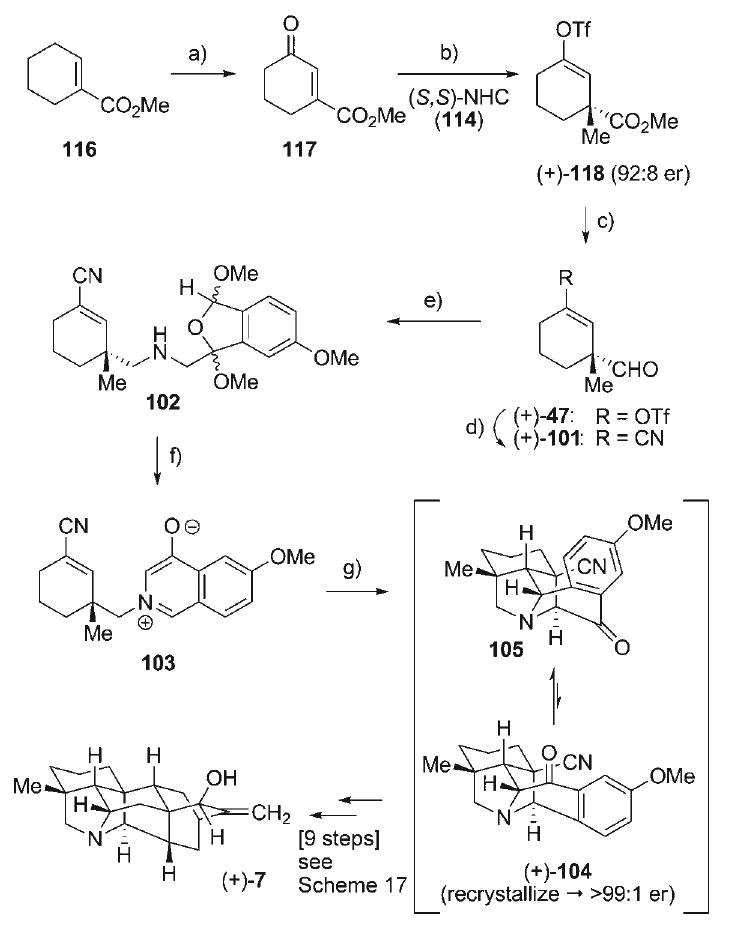
a) CrO3, Ac2O, AcOH, CH2Cl2, 23°C, 56%; b) 114, (CuOTf)2·PhH, ZnMe2, tBuOMe, 0°C; add 117, −30°C; add Tf2O, 0°C, 88%, 92:08 er; c) DIBAL-H, pentane, −105°C, 92%; d) Zn(CN)2, [Pd-(PPh3)4], DMF, 60°C, 82%; e) 93, PBu3, CH2Cl2, 23°C; add (+)-101; add NaBH(OAc)3, 82% (3:2 dr); f) 1:10 TFA/CH2Cl2, 0°C, 98%; g) [0.32 m], 180°C, THF, 97%, (+)-104/105 1:3.6 at equilibrium; recrystallized from iPrOH, 104 92:8 → > 99:01 er.
Conclusion
A concise synthetic entry to the hetisine class of diterpenoid alkaloids is described. The synthesis was predicated on the successful development of a dual cycloaddition strategy including an intramolecular 1,3-dipolar cycloaddition with a 4-oxidoisoquinilinium betaine dipole as well as a pyrrolidine-induced dienamine isomerization/Diels–Alder cascade. Early investigations demonstrated the facility with which 3-oxidopyridinium betaines could be prepared and applied to complex intramolecular cycloadditions in the preparation of the N-heterocyclic subunit of the hetisine alkaloids. Later studies extended the approach to the use of 4-oxidoisoquinolinium betaine dipoles, prepared by a novel cycloaromatization strategy, ultimately enabling an expedient racemic synthesis of (±)-nominine (7) in a 15-step sequence. The synthetic route also provided an attractive setting with which to apply novel catalytic asymmetric conjugate addition methodology recently disclosed by Hoveyda. Early-stage modification in the racemic synthetic route permitted successful application of this enantioselective transformation in the context of multi-step synthesis, culminating in the first asymmetric synthesis of (+)-nominine (7). This total synthesis of (+)-7 signals a novel strategic approach to the hetisine class of C20-diterpenoid alkaloids that will likely enable ready access to the other, more highly oxidized members of this natural product family.
Supplementary Material
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author: Experimental Section.
Acknowledgments
This research was supported by the NIH (GM67659), Abbott, Eli Lilly, Johnson & Johnson, Merck, and Pfizer. A Pharmacia (Pfizer) predoctoral fellowship to K.M.P is acknowledged. We thank Dr. H. Muratake for supplying spectral data for 7, and Professor A. Hoveyda for a generous gift of catalyst 114 to initiate model investigations into asymmetric conjugate addition.
References
- 1.Wang FP, Liang XT. Alkaloids. 2002;59:1–280. doi: 10.1016/s0099-9598(02)59008-8. [DOI] [PubMed] [Google Scholar]
- 2.Ochiai E, Okamoto T, Sakai S, Saito A. Yakugaku Zasshi. 1956;76:1414–1418. [Google Scholar]
- 3.Sakai S, Yamamoto I, Yamaguchi K, Takayama H, Ito M, Okamoto T. Chem Pharm Bull. 1982;30:4579–4582. [Google Scholar]
- 4.Jacobs WA, Craig LC. J Biol Chem. 1942;143:605–609. [Google Scholar]
- 5.Przybylska M. Can J Chem. 1962;40:566–568. [Google Scholar]
- 6.Suginome H, Kakimoto S, Sonoda J, Noguchi S. Proc Jpn Acad. 1946;22:122. [Google Scholar]
- 7.Pelletier SW, Wright LH, Newton MG, Wright HE. J Chem Soc Chem Commun. 1970:98–99. [Google Scholar]
- 8.Suginome H, Simamouti H. Liebigs Ann. 1940;545:220–228. [Google Scholar]
- 9.Okamoto T. Chem Pharm Bull. 1959;7:44–49. [Google Scholar]
- 10.Wang XW, Xie H. Drugs Future. 1999;24:877–882. [Google Scholar]
- 11.Jacyno JM. Chem Tox Diverse Class Alkal. 1996:301–336. [Google Scholar]
- 12.Wada K, Ishizuki S, Mori T, Fujihira E, Kawahara N. Biol Pharm Bull. 2000;23:607–615. doi: 10.1248/bpb.23.607. [DOI] [PubMed] [Google Scholar]
- 13.Nagata W, Sugasawa T, Narisada M, Wakabayashi T, Hayase Y. J Am Chem Soc. 1963;85:2342–2343. [Google Scholar]
- 14.Nagata W, Sugasawa T, Narisada M, Wakabayashi T, Hayase Y. J Am Chem Soc. 1967;89:1483–1499. [Google Scholar]
- 15.Masamune S. J Am Chem Soc. 1964;86:291–292. [Google Scholar]
- 16.Guthrie RW, Valenta Z, Wiesner K. Tetrahedron Lett. 1966:4645–4654. doi: 10.1016/s0040-4039(01)89239-x. [DOI] [PubMed] [Google Scholar]
- 17.Ihara M, Suzuki M, Fukumoto K, Kametani T, Kabuto C. J Am Chem Soc. 1988;110:1963–1964. [Google Scholar]
- 18.Ihara M, Suzuki M, Fukumoto K, Kabuto C. J Am Chem Soc. 1990;112:1164–1171. [Google Scholar]
- 19.Nagata W, Narisada M, Wakabayashi T, Sugasawa T. J Am Chem Soc. 1964;86:929–930. [Google Scholar]
- 20.Nagata W, Narisada M, Wakabayashi T, Sugasawa T. J Am Chem Soc. 1967;89:1499–1504. [Google Scholar]
- 21.Valenta Z, Wiesner K, Wong CM. Tetrahedron Lett. 1964;5:2437–2442. [Google Scholar]
- 22.Wiesner K, Uyeo S, Philipp A, Valenta Z. Tetrahedron Lett. 1968;9:6279–6282. doi: 10.1016/s0040-4039(00)75452-9. [DOI] [PubMed] [Google Scholar]
- 23.Masamune S. J Am Chem Soc. 1964;86:290–291. [Google Scholar]
- 24.Wiesner K, Ho PT, Tsai CSJ, Lam YK. Can J Chem. 1974;52:2355–2357. [Google Scholar]
- 25.Sethi SP, Atwal KS, Marini-Bettolo RM, Tsai TYR, Wiesner K. Can J Chem. 1980;58:1889–1891. [Google Scholar]
- 26.van der Baan JL, Bickelhaupt F. Recl Trav Chim Pays-Bas. 1975;94:109–112. [Google Scholar]
- 27.Shibanuma Y, Okamoto T. Chem Pharm Bull. 1985;33:3187–3194. [Google Scholar]
- 28.Kwak YS, Winkler JD. J Am Chem Soc. 2001;123:7429–7430. doi: 10.1021/ja010542w. [DOI] [PubMed] [Google Scholar]
- 29.Williams CM, Mander LN. Org Lett. 2003;5:3499–3502. doi: 10.1021/ol0353060. [DOI] [PubMed] [Google Scholar]
- 30.Williams CM, Mander LN, Bernhardt PV, Willis AC. Tetrahedron. 2005;61:3759–3769. [Google Scholar]
- 31.Hutt OE, Mander LN, Willis AC. Tetrahedron Lett. 2005;46:4569–4572. [Google Scholar]
- 32.Shimizu B, Ogiso A, Iwai I. Chem Pharm Bull. 1963;11:333–336. doi: 10.1248/cpb.11.770. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu B, Ogiso A, Iwai I. Chem Pharm Bull. 1963;11:766–769. doi: 10.1248/cpb.11.766. [DOI] [PubMed] [Google Scholar]
- 34.Muratake H, Natsume M. Angew Chem. 2004;116:4746–4749. doi: 10.1002/anie.200460332. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:4646–4649. doi: 10.1002/anie.200460332. [DOI] [PubMed] [Google Scholar]
- 35.Muratake H, Natsume M. Tetrahedron Lett. 2002;43:2913–2917. [Google Scholar]
- 36.Muratake H, Natsume M. Tetrahedron. 2006;62:7056–7070. [Google Scholar]
- 37.Muratake H, Natsume M. Tetrahedron. 2006;62:7071–7092. [Google Scholar]
- 38.Muratake H, Natsume M, Nakai H. Tetrahedron. 2006;62:7093–7112. [Google Scholar]
- 39.Muratake H, Nakai H. Tetrahedron Lett. 1999;40:2355–2358. [Google Scholar]
- 40.Peese KM, Gin DY. J Am Chem Soc. 2006;128:8734–8735. doi: 10.1021/ja0625430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grigg R. Chem Soc Rev. 1987;16:89–121. [Google Scholar]
- 42.Harwood LM, Vickers RJ. Chem Heter Comp. 2002;59:169–252. [Google Scholar]
- 43.Najera C, Sansano JM. Curr Org Chem. 2003;7:1105–1150. [Google Scholar]
- 44.Coldham I, Hufton R. Chem Rev. 2005;105:2765–2809. doi: 10.1021/cr040004c. [DOI] [PubMed] [Google Scholar]
- 45.Pandey G, Banerjee P, Gadre SR. Chem Rev. 2006;106:4484–4517. doi: 10.1021/cr050011g. [DOI] [PubMed] [Google Scholar]
- 46.Vedejs E, West FG. Chem Rev. 1986;86:941–955. [Google Scholar]
- 47.Padwa A, Haffmanns G, Tomas M. Tetrahedron Lett. 1983;24:4303–4306. [Google Scholar]
- 48.Epperson MT, Gin DY. Angew Chem. 2002;114:1856–1858. doi: 10.1002/1521-3773(20020517)41:10<1778::aid-anie1778>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 2002;41:1778–1780. [Google Scholar]
- 49.Fowler FW. Angew Chem. 1971;83:148–149. [Google Scholar]; Angew Chem Int Ed Engl. 1971;10:135–136. [Google Scholar]
- 50.Pandey G, Lakshmaiah G, Ghatak A. Tetrahedron Lett. 1993;34:7301–7304. [Google Scholar]
- 51.Pandey G, Bagul TD, Sahoo AK. J Org Chem. 1998;63:760–768. doi: 10.1021/jo971728+. [DOI] [PubMed] [Google Scholar]
- 52.Pandey G, Laha JK, Mohanakrishnan AK. Tetrahedron Lett. 1999;40:6065–6068. [Google Scholar]
- 53.Pandey G, Sahoo AK, Bagul TD. Org Lett. 2000;2:2299–2301. doi: 10.1021/ol006070s. [DOI] [PubMed] [Google Scholar]
- 54.Pandey G, Laha JK, Lakshmaiah G. Tetrahedron. 2002;58:3525–3534. [Google Scholar]
- 55.Comins DL, Joseph SP, Goehring RR. J Am Chem Soc. 1994;116:4719–4728. [Google Scholar]
- 56.Streith J, Boiron A, Sifferlen T, Strehler C, Tschamber T. Tetrahedron Lett. 1994;35:3927–3930. [Google Scholar]
- 57.Kerwin JF, Jr, Danishefsky S. Tetrahedron Lett. 1982;23:3739–3742. [Google Scholar]
- 58.Ireland RE, Norbeck DW. J Org Chem. 1985;50:2198–2200. [Google Scholar]
- 59.Linderman RJ, Suhr Y. J Org Chem. 1988;53:1569–1572. [Google Scholar]
- 60.Pilcher AS, Ammon HL, Deshong P. J Am Chem Soc. 1995;117:5166–5167. [Google Scholar]
- 61.Nagata W, Yoshioka M, Hirai S. J Am Chem Soc. 1972;94:4635–4643. [Google Scholar]
- 62.Pena MR, Stille JK. J Am Chem Soc. 1989;111:5417–5424. [Google Scholar]
- 63.Katritzky AR, Takeuchi Y. J Am Chem Soc. 1970;92:4134–4136. [Google Scholar]
- 64.Katritzky AR, Dennis N. Chem Rev. 1989;89:827–861. [Google Scholar]
- 65.Jung ME, Zeng LM, Peng TS, Zeng HY, Le Y, Su JY. J Org Chem. 1992;57:3528–3530. [Google Scholar]
- 66.Pham VC, Charlton JL. J Org Chem. 1995;60:8051–8055. [Google Scholar]
- 67.Sliwa W. Heterocycles. 1996;43:2005–2029. [Google Scholar]
- 68.Sung MJ, Lee HI, Chong Y, Cha JK. Org Lett. 1999;1:2017–2019. doi: 10.1021/ol9911932. [DOI] [PubMed] [Google Scholar]
- 69.Lee HI, Sung MJ, Lee HB, Cha JK. Heterocycles. 2004;62:407–422. [Google Scholar]
- 70.Rumbo A, Mourino A, Castedo L, Mascarenas JL. J Org Chem. 1996;61:6114–6120. doi: 10.1021/jo960854v. [DOI] [PubMed] [Google Scholar]
- 71.Sammes PG, Watt RA. J Chem Soc Chem Commun. 1976:367–368. [Google Scholar]
- 72.Joshi RA, Ravindranathan T. Ind J Chem B. 1984;23:300–302. [Google Scholar]
- 73.Bromidge SM, Archer DA, Sammes PG. J Chem Soc Perkin Trans 1. 1990:353–359. [Google Scholar]
- 74.Orlek BS, Sammes PG, Weller DJ. J Chem Soc Chem Commun. 1993:1412–1413. [Google Scholar]
- 75.Orlek BS, Sammes PG, Weller DJ. Tetrahedron. 1993;49:8179–8194. [Google Scholar]
- 76.Smith MP, George C, Kozikowski AP. Tetrahedron Lett. 1998;39:197–200. [Google Scholar]
- 77.Petersen JB, Norris K, Clauson-Kaas N, Svanholt K. Acta Chem Scand. 1969;23:1785–1790. [Google Scholar]
- 78.Shono T, Matsumura Y, Tsubata K, Inoue K, Nishida R. Chem Lett. 1983:21–24. [Google Scholar]
- 79.Ciufolini MA, Hermann CYW, Dong Q, Shimizu T, Swaminathan S, Xi N. Synlett. 1998:105–114. [Google Scholar]
- 80.Müller C, Diehl V, Lichtenthaler FW. Tetrahedron. 1998;54:10703–10712. [Google Scholar]
- 81.Peese KM, Gin DY. Org Lett. 2005;7:3323–3325. doi: 10.1021/ol051184v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dehli JR, Gotor V. J Org Chem. 2002;67:1716–1718. doi: 10.1021/jo011092t. [DOI] [PubMed] [Google Scholar]
- 83.Abarbri M, Dehmel F, Knochel P. Tetrahedron Lett. 1999;40:7449–7453. [Google Scholar]
- 84.O'Connor DE, Lyness WI. J Am Chem Soc. 1964;86:3840–3846. [Google Scholar]
- 85.Hamann PR, Toth JE, Fuchs PL. J Org Chem. 1984;49:3865–3867. [Google Scholar]
- 86.Lee SW, Fuchs PL. Tetrahedron Lett. 1991;32:2861–2864. [Google Scholar]
- 87.Nickel A, Maruyama T, Tang H, Murphy PD, Greene B, Yusuff N, Wood JL. J Am Chem Soc. 2004;126:16300–16301. doi: 10.1021/ja044123l. [DOI] [PubMed] [Google Scholar]
- 88.Meulemans TM, Stork GA, Jansen BJM, De Groot A. Tetrahedron Lett. 1998;39:6565–6568. [Google Scholar]
- 89.Stork G, Clark G, Weller T. Tetrahedron Lett. 1984;25:5367–5370. [Google Scholar]
- 90.Bird CW. Tetrahedron. 1992;48:335–340. [Google Scholar]
- 91.Dennis N, Katritzky AR, Takeuchi Y. J Chem Soc Perkin Trans 1. 1972:2054–2057. [Google Scholar]
- 92.Garling DL, Cromwell NH. J Org Chem. 1973;38:654–658. [Google Scholar]
- 93.Dennis N, Katritzky AR, Parton SK. Chem Pharm Bull. 1975;23:2899–2903. [Google Scholar]
- 94.Dennis N, Katritzky AR, Parton SK. J Chem Soc Perkin Trans 1. 1976:2285–2288. [Google Scholar]
- 95.Hanaoka M, Wada A, Yasuda S, Mukai C, Imanishi T. Heterocycles. 1979;12:511–514. [Google Scholar]
- 96.Padwa A, Dean DC, Osterhout MH, Precedo L, Semones MA. J Org Chem. 1994;59:5347–5357. [Google Scholar]
- 97.DiCesare JC, Burgess JP, Mascarella SW, Carroll FI, Rothman RB. J Heterocycl Chem. 1994;31:187–192. [Google Scholar]
- 98.Edmunds JJ, Cheng XM, Tobias B. J Chem Soc Perkin Trans 1. 1996:2005–2008. [Google Scholar]
- 99.Constable KP, Blough BE, Carroll FI. Chem Commun. 1996:717–718. [Google Scholar]
- 100.Cushman M, Patrick DA, Toma PH, Byrn SR. Tetrahedron Lett. 1989;30:7161–7164. [Google Scholar]
- 101.Plaumann HP, Keay BA, Rodrigo R. Tetrahedron Lett. 1979;20:4921–4924. [Google Scholar]
- 102.The formation of 96 from a retro-Mannich reaction of 98 cannot be discounted.
- 103.Tschaen DM, Desmond R, King AO, Fortin MC, Pipik B, King S, Verhoeven TR. Synth Commun. 1994;24:887–890. [Google Scholar]
- 104.Rabideau PW, Marcinow Z. Org React. 1992;42:1–334. [Google Scholar]
- 105.Umbreit MA, Sharpless KB. J Am Chem Soc. 1977;99:5526–5528. [Google Scholar]
- 106.Furber M, Mander LN. J Am Chem Soc. 1987;109:6389–6396. [Google Scholar]
- 107.Brown MK, May TL, Baxter CA, Hoveyda AH. Angew Chem. 2007;119:1115–1118. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]
- 108.Lee KS, Brown MK, Hird AW, Hoveyda AH. J Am Chem Soc. 2006;128:7182–7184. doi: 10.1021/ja062061o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author: Experimental Section.