

Summary
Because of advances in the high-throughput screening technology, identification of a hit that can bind to a target protein has become a relatively easy task; however, in the process of drug discovery, the following hit-to-lead and lead optimization still remain challenging. In a typical hit-to-lead and lead optimization process, the analogues of the most promising hits are synthesized for the development of structure-activity relationship (SAR) analysis, and in turn, in the effort of optimization of lead compounds, such analysis provides guidance for the further synthesis. The synthesis processes are usually long and labor-intensive. In silico searching has becoming an alternative approach to explore SAR especially with millions of compounds ready to be screened and most of them can be easily obtained. Here, we report our discovery of fifteen new Dishevelled PDZ domain inhibitors by using such an approach. In our studies, we first developed a pharmacophore model based on NSC668036, an inhibitor previously identified in our laboratory; based on the model, we then screened the ChemDiv database by using an algorithm that combines similarity search and docking procedures; finally, we selected potent inhibitors based on docking analysis and examined them by using NMR spectroscopy. NMR experiments showed that all the fifteen compounds we chose bound to the PDZ domain tighter than NSC668036.
Keywords: Chemical space, NMR, PDZ domain inhibitors, SAR, Virtual screening, Wnt signaling pathway
Introduction
With the advancement of technology in the field of drug discovery, hits of a potential therapeutic reagent can be identified in a comparatively straightforward fashion by using high-throughput screening [1; 2]. However the follow-up hit-to-lead process and lead optimization still remain as challenging problems in the drug discovery process [3; 4]. One of the most frequently taken approaches in the hit-to-lead process is hit evolution [4]. During hit evolution, analogues of the most promising hits are synthesized for the development of structure-activity relationship (SAR) data. The SAR is then used to guide the synthesis and optimization of lead compounds to improve their potencies and physico-chemical properties, and to reduce off-target activities. The synthesis processes are usually long and labor-intensive. Virtual screening of databases consisting of physically available compounds may help us to take advantage of the chemistry that has already been done and speed up projects, especially with the ever growing list of existing compounds. Indeed, the Zinc database has 4.6 million compounds [5] and the iResearch ™ Library (ChemNavigator®, San Diego, CA) has more than 50 million chemicals. Although the databases of available compounds are still under-sampled [6], the chemical space represented by those millions of compounds should never be neglected. We believed that the large chemical space of available compounds offers us with an opportunity to explore SAR of known hits; and as a proof of principle test, we searched the ChemDiv database for the Dishevelled (Dvl) PDZ domain inhibitors based on an inhibitor previously identified in our lab [7].
The Dvl PDZ (Post-synaptic density-95/Discs large/Zonula occludens-1) domain relays Wnt signaling and has been considered as a potential cancer therapeutic target [8; 9]. Different approaches have been taken to identify and develop PDZ domain inhibitors, including NMR-based screening and chemical synthesis [10; 11; 12]. Previously, using receptor-based virtual screening, we identified a PDZ domain inhibitor (NSC668036; 9,15-diisopropyl-2,2,6,12-tetramethyl-4,7,10,13-tetraoxo-3,8,14-trioxa-5,11-diazahexadec an-16-oic acid) from the NCI library. NSC668036 acted as a Wnt antagonist and inhibited the second axis formation in Xenopus induced by Wnt3A which is upstream of Dvl but not by β-cantenin which is downstream of Dvl [7]. Our results further suggest that the Dvl PDZ domain might be a suitable target for blocking Wnt signaling pathways at the Dvl level, and PDZ domain inhibitors may be used as inhibitors of Wnt signaling [13]. In order to develop more potent PDZ inhibitors and to understand the molecular determinants of PDZ-ligand binding, we carried out additional virtual screening to search for NSC668036 analogues and developed SAR models using experimentally verified inhibitors (Figure 1). First, we developed a pharmacophore model based on the complex structure of NSC668036 and the PDZ domain as well as the differences between NSC668036 and two other compounds [7]. These two compounds are similar to NSC668036 in structure but do not bind to the PDZ domain. We then used the pharmacophore to screen the ChemDiv database for potential inhibitors. Following virtual screening and docking, we selected fifteen compounds as potential inhibitors of the PDZ domain. By using NMR spectroscopy we showed that all the fifteen compounds bound to the PDZ domain. In fact, all the fifteen compounds bind to the PDZ domain much tighter than compound NSC668036, the starting compound in the virtual screening. Nevertheless, we think that these fifteen compounds will also allow us to develop SAR models of PDZ domain ligands, which should be very useful in the future hit optimizations.
Fig 1.
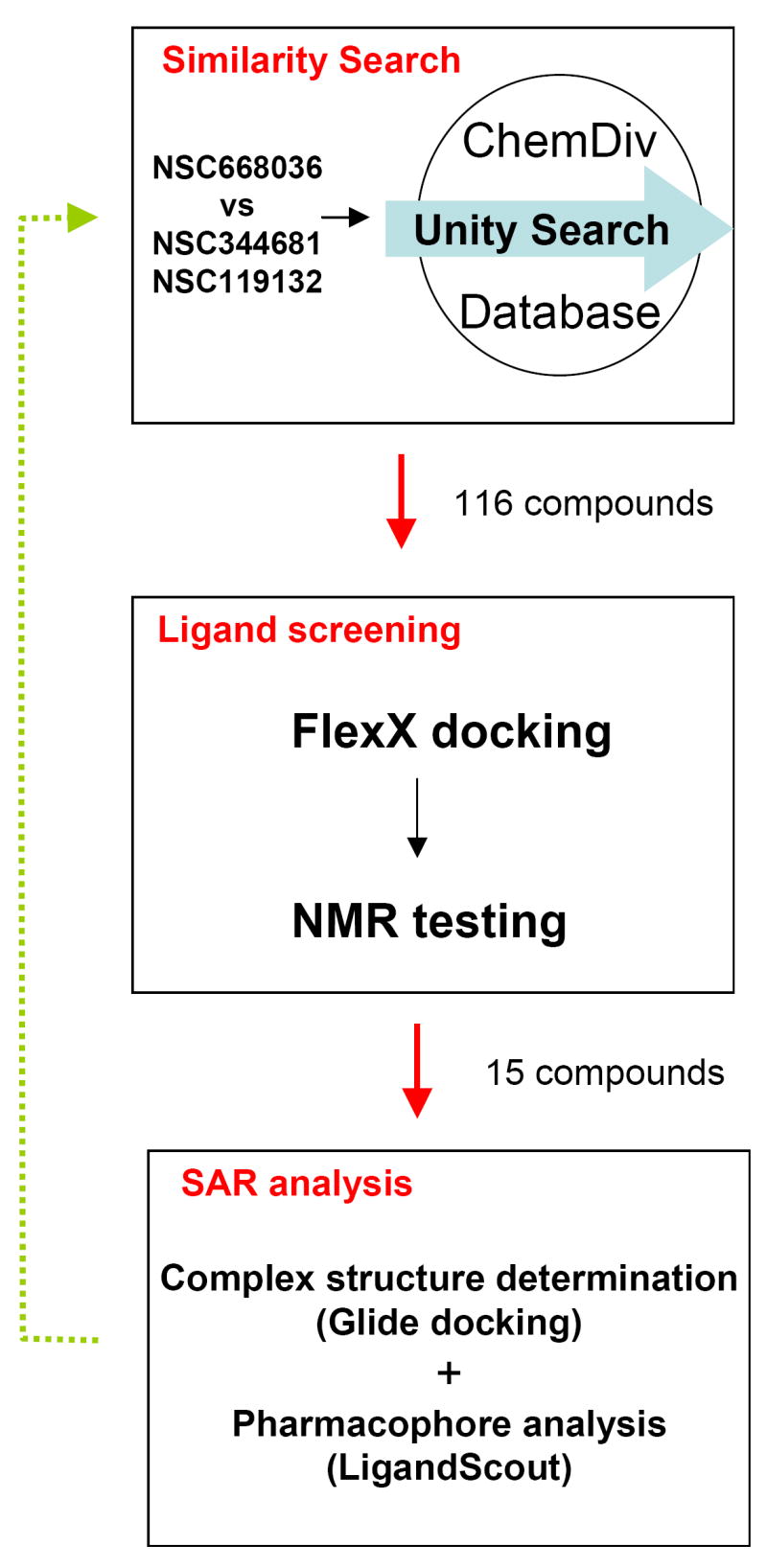
The workflow of optimizing PDZ domain inhibitors by exploring chemical space. Queries designed based on a PDZ-NSC668036 complex was used to search ChemDiv database for PDZ domain inhibitors by using Unity 2D/2D similarity searches. Returned 116 hits were docked using FlexX and 15 compounds were selected for NMR testing and their docking conformations were refined by Glide (Schrödinger Inc.). Combining NMR testing, Glide optimization of docking poses and LigandScout (Inte:Ligand, Austria) pharmacophore analysis, we derived SAR for the 15 new PDZ domain inhibitors.
Materials and Methods
Chemicals
The fifteen compounds identified by virtual screening were purchased from ChemDiv Inc. (San Diego, CA).
Pharmacophore generation
Pharmacophores were generated with LigandScout (Inte:Ligand, Austria). LigandScout extracts 3-D pharmacophores based on complex structures [14]. The complex structure of NSC668036 and the PDZ domain was generated by docking and extensive molecular dynamics simulations [7]. Complex structures of compounds 4, 5 and 7 were modeled using Glide (Schrödinger Inc., Portland, OR). Complex structures of compounds 9 and 10 were modeled by superimposing them onto docked compound 1 followed by ligand minimization in the ligand binding pocket of the PDZ domain with LigandScout.
Similarity search
The UNITY module in the SYBYL® software package (Tripos, Inc.) was used to screen the ChemDiv database for potential PDZ domain inhibitors.
FlexX docking
After screening, the candidate compounds were docked into the binding site of the Dvl PDZ domain (PDB entry 1L6O) [15] by using the FlexX module of SYBYL® (Tripos, Inc.) [16] as previously reported [7]. Default docking parameters were used.
Glide docking
The docking models of the fifteen PDZ ligands were refined by using Glide (Schrödinger Inc.). During Glide docking, compounds’ amide bonds were kept rigid; hydrogen-bond pharmacophores were designed on the protein to induce ligands to form hydrogen-bonds with the βA-βB loop and the βB strand of the PDZ domain. Other than fixed amide bonds and H-bond pharmacophores, default docking parameters were used. All ligand binding poses generated by Glide have reasonable Glide scores, suggesting that they are likely very close to the true binding modes. For example, the Glide score of compound 1 is -7.57. According to Schrödinger Inc., low-micromolar inhibitors should have scores around -7. Glide 2.5 predicted binding affinities of a set of 125 crystallized complexes with an RMSD of 2.2 kcal/mol against experimental data [17]. Based on the facts that compound 1 binds to the PDZ domain with a moderate binding affinity and its glide score fits with the experimental data, we infer that this docking conformation is close to the true binding mode. This conformation was then used to generate complex structures for compounds 9 and 10 which have different binding poses than those of the rest of thirteen compounds.
NMR
The 15N-labeled mouse Dvl1 PDZ domain (residues 247-341 of mouse Dvl1) was prepared as described previously [18; 19] by the protein production facility at St. Jude Children’s Research Hospital. NMR 15N HSQC experiments were performed by using a Varian Inova 600 MHz NMR spectrometer at 25 °C. Samples consisted of the mouse Dvl1 PDZ domain (0.2 - 0.3 mM) in 100 mM potassium phosphate buffer (pH 7.5), 10% D2O, and 0.5 mM EDTA. Compounds were dissolved in the same buffer but with 5% DMSO, which did not change the spectra of the PDZ domain (data not shown). NMR spectra were processed with NMRpipe [20] and analyzed by using Sparky [21].
Calculating KD by using HSQC titration spectra
The binding affinities (KD) of PDZ ligands were calculated using HSQC spectra by following the method described by Worrall et al. [22]. The mean chemical-shift perturbation changes caused by the binding of ligands were calculated using Eq. (1). KD was then calculated using Eq. (2) and (3) by applying a one-site binding model with corrections for dilutions, where R was the ligand to protein molar ratio, P was the protein concentration before titration, C was the ligand stock concentration, and KD was the dissociation constant. Two-parameter nonlinear least-squares fitting was performed with program Prism (GraphPad Software, La Jolla, CA).
| (1) |
| (2) |
| (3) |
Results
Deduction of Pharmacophore based on a PDZ-inhibitor complex structure and two non-binders
A pharmacophore is composed of functional groups essential and necessary to receptor-ligand binding; without those groups, ligands will no longer bind to receptors and lose their activities [23]. Pharmacophore-based approaches have been widely used and shown successes in the field of computer-aided drug design [24; 25]. In order to identify more PDZ domain inhibitors, we took such an approach, derived the pharmacophore of PDZ ligands and used it to screen for PDZ domain inhibitors in the ChemDiv database. The pharmacophore was derived from the PDZ-NSC668036 complex structure. The structure-based pharmacophore was then examined and essential components were selected based on the differences between NSC668036 and two similar compounds that did not bind to the PDZ domain [7], which were then used to screen for more PDZ domain inhibitors.
The 90-residue PDZ domain consisting of six β-sheets flanked by two α-helices is one of the protein interaction module domains that organize superamolecular complexes which are essential for biological functions. PDZ domains have been considered as potential therapeutic targets [8; 9]. Previously, using receptor-based virtual screening, we identified a PDZ domain inhibitor (NSC668036) after testing nine compounds from the NCI library. In the complex structure of the PDZ domain with NSC668036 (Figure 2), the carboxyl group of the ligand forms hydrogen-bonds with the βA-βB loop in the PDZ domain and the isopropyl group (attached to carbon “c1” in Figure 2b) occupies a hydrophobic cavity between the βB strand and the αB helix [7]. We used this complex structure to build a pharmacophore by using LigandScout (Inte:Ligand, Austria). The LigandScout pharmacophore model of NSC668036 is consistent with our structural analysis. The carboxyl group contributes three hydrogen-bond acceptors and the isopropyl group contributes a hydrophobic interaction to the pharmacophore (Figure 3a, 3b). We then compared NSC668036 with other compounds that did not bind to the PDZ domain as verified by our pervious virtual screening. When we aligned two non-binders, NSC344681 (((((2-amino-3-hydroxybutanoyl)amino)acetyl)amino)acetic acid) and NSC119132 (methyl 3-((2-amino-3-(aminooxy)propanoyl)amino)-2-((aminooxy)methyl)-3-oxopropanoate), against NSC668036, we identified the differences among these structures that might render NSC344681 and NSC119132 inactive. NSC334681 does not have a hydrophobic group at the 2-position; and NSC119132 is an ester instead of a free acid (Figure 3c, 3d and 3e). This finding strongly suggested that both the carboxyl group and the hydrophobic group next to it were important to the binding and might compose the pharmacophore of PDZ ligands since compounds without them did not bind to the protein. To test this hypothesis, we decided to search for compounds with 2-(3-methylbutanoic acid) groups.
Fig 2.
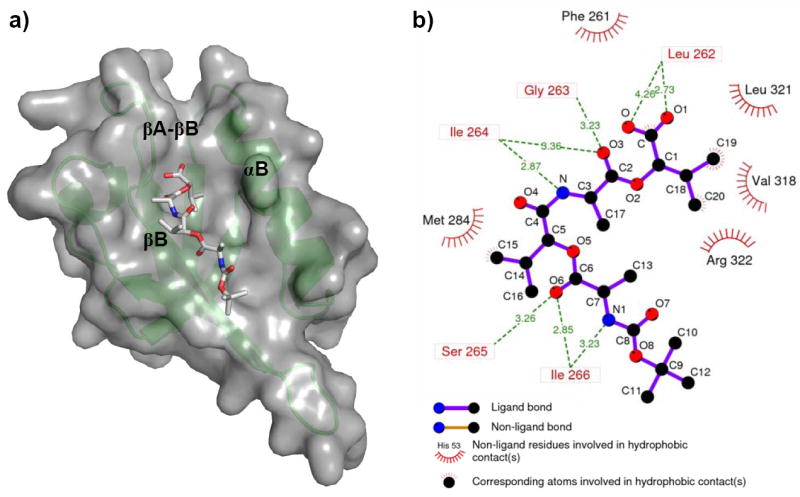
The binding mode of PDZ domain inhibitor NSC668036. (a) Compound NSC668036 lies in a grove formed by the αB helix and the βB strand, which is the binding site for PDZ ligands. The PDZ domain is in green cartoon representation and grey surface. The compound is in stick representation with carbons in white, nitrogens in blue and oxygens in red. The figure was prepared with PyMol (Delano Scientific LLC, San Carlos, CA). (b) Detailed interactions between NSC668036 and the PDZ domain. The carboxyl group of NSC668036 forms hydrogen-bond with Leu262 in the βA-βB loop while the isopropyl group directly contacts with Val318, Leu321 and Arg322 (hydrophobic side chains) in the αB helix of the PDZ domain. The figure was prepared with LIGPLOT [28].
Fig 3.
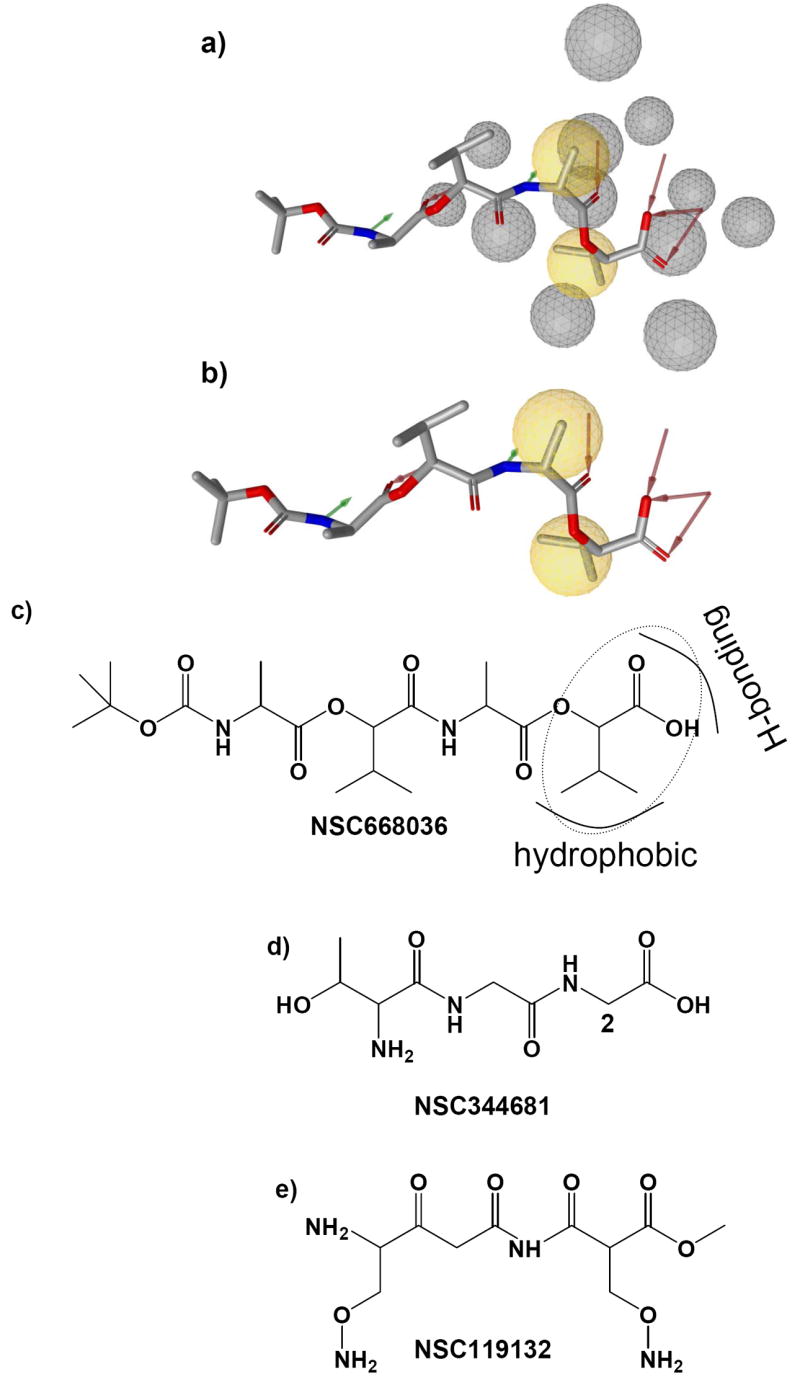
Structure-based pharmacophore of NSC668036 generated by LigandScout [14]. The pharmacophore of NSC668036 generated from its complex structure with the PDZ domain [7] with (a) and without (b) excluded volume spheres (grey). The pharmacophore has two hydrophobic interactions (yellow), five hydrogen-bond acceptors (red) and two hydrogen-bond donors (green). (c) The 2-D structure of PDZ domain inhibitor NSC668036. The substructure 2-(3-methylbutanoic acid) important to PDZ binding is in dotted circle. (d, e) 2-D structures of compounds that do not bind to the PDZ domain. Compared with NSC668036, NSC344681 lacks a hydrophobic group next to the carboxyl group while NSC119132 is an ester instead of an acid.
Pharmacophore-based virtual screening
Based on the pharmacophore proposed above, we carried out a 2-D search to retrieve all the compounds with 2-(3-methylbutanoic acid) groups in the ChemDiv database by using UNITY in SYBYL® (Tripos, Inc.). The search returned 116 hits. To reduce the number of compounds needed to test experimentally and to select ligands with higher potentials to bind to the PDZ domain, we filtered the hits by using FlexX docking and “Cscore” ranking following a procedure described previously [7]. After manually inspecting top-ranked compounds, we chose fifteen compounds (Table 1) for further examinations based on the following four criteria: (a) the compounds were docked into the designated binding site, (b) their docking conformations were complementary to that of the PDZ domain, and (c) the docked compounds formed hydrogen-bonds with the βA-βB loop as well as (d) additional hydrogen-bonds with the PDZ domain.
Table 1.
Structures and binding affinities of the fifteen compounds
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound ChemDiv ID | R1 | R2 | Exchange regimeb | Δδc | KDd (mM) | KD’e (μM) | ||
| 1 | 5435-0027 | Aa | -ph-4-CH3 | -ph | ++++ | 0.411 | 0.98 | 8.6 |
| 2 | 3865-0097 | A | -ph | -CH3 | +++ | 0.285 | 3.10 | 27.2 |
| 3 | 0277-0075 | A | -ph-4-N(CH2CH2Cl)2 | -CH3 | + | 0.063 | 8.98 | 78.8 |
| 4 | 3865-0011 | B1 | -ph | -ph | ++ | 0.204 | 5.67 | 49.8 |
| 5 | 3379-2711 | B1 | -ph-4-Cl | -ph | n/a | 0.245 | 1.16 | 10.2 |
| 6 | 3802-0264 | B1 | -ph | -ph-4-(OCH3) | + | 0.187 | 3.48 | 30.5 |
| 7 | 3865-0047 | B1 | -ph-4-F | -ph | + | 0.142 | 5.09 | 44.7 |
| 8 | 5613-0298 | B1 | -ph-4-(OCH3) | -ph-4-CH3 | + | 0.167 | 2.33 | 20.5 |
| 9 | 2509-0036 | B1 | -ph-4-Br | -ph-3,4,5-3(OCH3) | ++ | 0.239 | 2.34 | 20.5 |
| 10 | 2509-0040 | B1 | -ph-3,5-2(OCH3) | -ph-3,4,5-3(OCH3) | +++ | 0.188 | 2.46 | 21.6 |
| 11 | 5613-0343 | B1 | -5-benzo[d][1,3]dioxol | -ph-4-CH3 | + | 0.208 | 1.59 | 14.0 |
| 12 | 3631-0149 | B2 | -ph | -2-furan | ++++ | 0.206 | 2.58 | 22.6 |
| 13 | 3379-2681 | B2 | -ph-4-Cl | -2-furan | + | 0.192 | 2.99 | 26.2 |
| 14 | 3379-2909 | B2 | -ph-4-F | -2-furan | + | 0.170 | 4.28 | 37.6 |
| 15 | 3802-0137 | B3 | -2-(furan-3-Br) | -2-furan | + | 0.175 | 4.47 | 39.2 |
| NSC668036 | + | 0.035 | 27 | 237 | ||||
Compounds labeled A have scaffold A and compounds labeled B1, B2, and B3 have scaffold B.
Compounds from being in intermediate exchange (++++) to fast exchange (+) with the PDZ domain. Compounds with “++++” caused some peaks disappear and reappear; compounds with “+++” caused peaks disappear only; compounds with “++” caused peaks become weak only; and compounds with “+” did not cause any peak intensity changes.
The chemical shift perturbation changes of Arg322 caused by 10 equivalents of compounds except for 10.8 equivalents of compound 1 and 10.4 equivalents of NSC668036.
KD (in mM) measured by NMR titration experiments at 25 °C.
KD (in μM) values were obtained by normalizing the KD values obtained in the NMR experiments with the difference of the KD values of NSC668036 measured by NMR and fluorescence methods respectively.
Confirmation of the pharmacophore by using NMR spectroscopy
We then examined the interactions between the fifteen compounds we chose and the Dvl PDZ domain experimentally by using NMR chemical shift perturbation experiments. In the studies, we titrated the compounds individually to the solution of the 15N-labled Dvl PDZ domain and monitored the 15N-HSQC spectra of the protein. The experiments showed that all the compounds perturbed similar residues on the PDZ domain as NSC668036 did, suggesting that all the selected compounds bound to the PDZ domain at the same binding site as the one occupied by NSC668036 (Figure 4). Furthermore, the amounts of chemical shift perturbation changes caused by these compounds were all much larger than those caused by NSC668036 (Table 1), indicating that they all bind to the PDZ domain with higher affinities than NSC668036. In addition, unlike NSC668036 which was in the fast exchange regime when it bound to the PDZ domain, six compounds (1, 2, 4, 9, 10 and 12) (Figure 4, Table 1) were in the intermediate exchange regime; for example, when compound 1 was titrated into the 15N-labled PDZ domain, the amide cross peak of Arg322 became weaker, disappeared, reappeared and became stronger again during titrations (Figure 4a), which is a classical slow/intermediate exchange pattern [26]. Ligands in intermediate exchange with proteins usually have low-micromolar binding affinities. The fact that all these compounds with 2-(3-methylbutanoic acid) groups bound to the PDZ domain supported the hypothesis that group 2-(3-methylbutanoic acid) might be important to PDZ domain-ligand binding.
Fig 4.
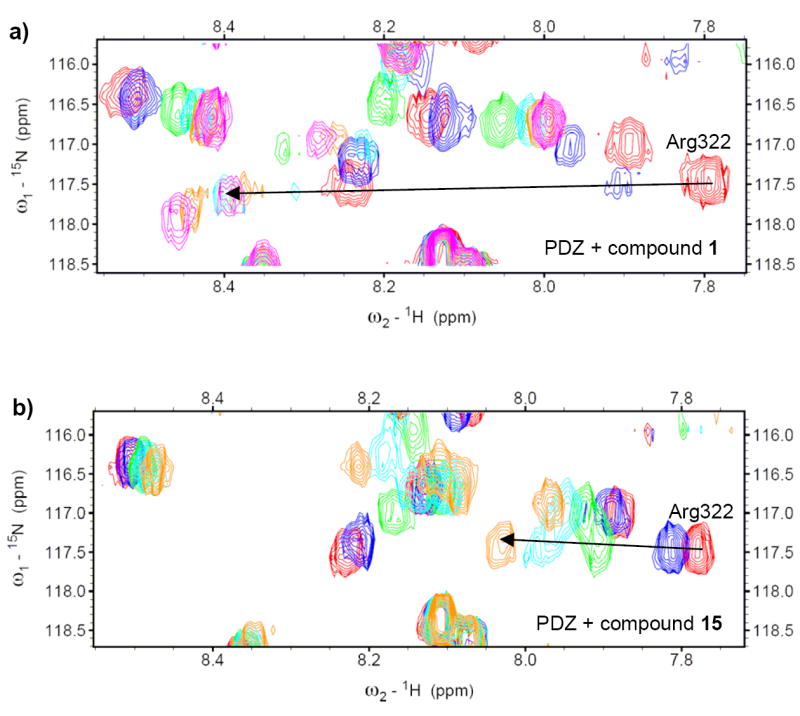
Example NMR spectra of PDZ domain titrated with compounds in the intermediate or in the fast exchange regime. (a) NMR spectra of PDZ domain titrated with compound 1, which is in intermediate exchange with the PDZ domain with a low-micromolar binding affinity. Spectra are colored by the protein to ligand ratios: red (1:0), blue (1:0.72), green (1:3.6), cyan (1:7.2), orange (1:10.8) and magenta (1:14.4). (b) NMR spectra of PDZ domain titrated with compound 15, which is in fast exchange with the PDZ domain with a relative weak binding affinity. Spectra are colored by the protein to ligand ratios: red (1:0), blue (1:1), green (1:6), cyan (1:10) and orange (1:15).
Calculating binding affinities by using chemical-shift perturbation changes
After verifying that all the fifteen compounds bound to the PDZ domain by using NMR spectroscopy, we calculated their binding affinities by using the method published by Worrall et al. [22] in order to compare their binding strengths quantitatively. In detail, we monitored chemical shift perturbation changes of Arg322 when the 15N-labeled proteins were titrated with compounds. Residue Arg322 is at the ligand binding site of the PDZ domain. For most of the titration experiments, five titration points were taken with the ligand-to-protein ratios ranging from 1 to 15. The calculated KD values of these fifteen compounds ranged from 0.98 mM to 8.98 mM. However, as showed in Figure 4a, NMR titration data clearly indicated that at least some of the fifteen compounds were in the intermediate exchange range when they interacted with the PDZ domain, suggesting the true binding affinities between those compounds and the PDZ domain should be around micromolar. Such appeared differences may be due to a systemic error in calculating KD values by using NMR chemical shift perturbation data; such error is likely caused by the averaging effect during the NMR measurement. To further address this issue, we compared the KD values of the interaction between the compound NSC668036 and the PDZ domain measured by two different methods, fluorescence anisotropy and NMR chemical shift perturbation. In our earlier studies, using fluorescence spectroscopy, we measured the KD value of NSC668036 as 237 μM [7]. However, using the NMR chemical shift perturbation data, this value was calculated as 27 mM, which is about 100 times higher than that was measured by fluorescence spectroscopy. Nevertheless, the KD values calculated by the NMR chemical shift perturbation data can still be used to compare the strengths of the interactions between those compounds and the PDZ domain since the relative values are still correctly reflected (Table 1). To get an idea what their KDs would be if measured by fluorescence, we calibrated their NMR KDs by using the differences between KDs of NSC668036 by NMR and by fluorescence (114 fold). The results were included as the last column in Table 1. Table 1 illustrates that compounds in the intermediate exchange regime have low-micromolar affinities while compounds in the fast exchange regime have affinities ranging from low-millimolar to sub-millimolar. This is consistent with the general understanding of NMR. Both the magnitudes of chemical-shift perturbation changes and compounds’ exchange rates with the PDZ domain suggest that all the fifteen new compounds were potent than NSC668036.
Ligand binding modes
Interestingly, all these fifteen compounds are similar in structure that they are all peptidomimetics. This observation might be explained by two possible reasons: one is that compounds with 2-(3-methylbutanoic acid) groups in the ChemDiv database were all peptidomimetics, the other is that this class of compounds are favored by the PDZ domain and stand out after the docking procedure. To address this issue, we re-examined the 115 hits returned by the initial 2-D search and found that these hits had various structures instead of all being peptidomimetics. Thus the first possibility is excluded and it is very likely that peptidomimetics are energetically favorable PDZ ligands comparing with other hits. To understand why this class of compounds were energetically favored by the PDZ domain, we then further studied the docked conformations of these compounds. The docking models generated by program Glide (Schrödinger Inc.) showed that these compounds bound to the PDZ domain in extended conformations (Figure 5) which are similar to that of β strand peptides and fit well with the binding pocket lined by the αB helix and the βB strand of the PDZ domain. For example, compound 1 forms hydrogen-bonds with residues Leu265 and Gly266 in the βA-βB loop of the PDZ domain as well as residue Ile267 in the βB strand of the protein. The conformation of compound 1 is very similar to that of the last three amino acids at the C-terminus of the Dapper peptide complexed with Xenopus Dvl PDZ domain [15]. Therefore, we conclude that β strand peptidomimetics are energetically favorable PDZ ligands. This observation is consistent with what was reported previously that β strand peptidomimetics were potent PDZ domain inhibitors [27].
Fig 5.
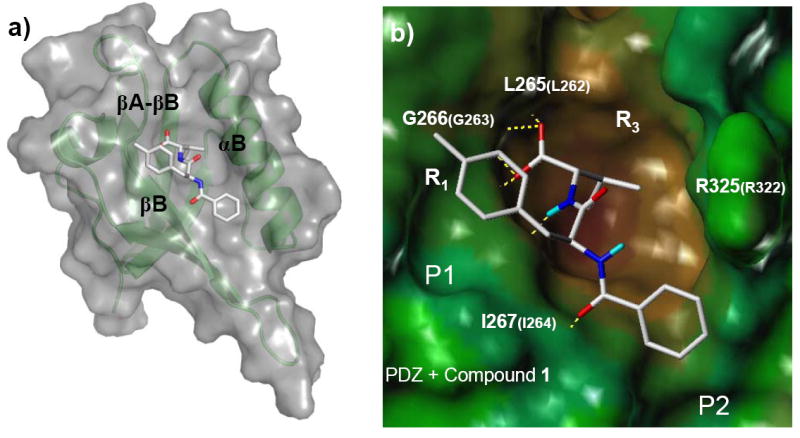
Compound 1 bound to the PDZ domain in an extended conformation as β-peptidomimetics. Compound 1 was docked to the PDZ domain by using Glide (Schrödinger Inc.). The othercompounds adopt similar conformations as that of 1 except 9 and 10. (a) Representations and colors are the same as in Figure 2a. (b) PDZ domain is in surface representation colored by hydrophobicity from brown (hydrophobic) to blue (hydrophilic). Compound 1 is in stick representation. Polar hydrogen atoms are in cyan. hydrogen-bonds between PDZ domain and 1 are in yellow dash lines. The amino acid numbers in bigger fonts are those in Xenopus Dvl PDZ domain and those in smaller fonts correspond to residue numbers in the mouse Dvl1 PDZ domain. The figure was prepared with SYBYL® 7.0 (Tripos, Inc.).
SAR of PDZ domain inhibitors
In order to gain further insights into the molecular determinants of PDZ-ligand binding which might facilitate further ligand optimization, we studied SAR of these fifteen compounds by comparing their structures and binding strengths in the context of docking complex structures presented above. These fifteen compounds have similar scaffolds except that some of them have an extra double bond in their scaffold (scaffold B) (Table 1). According to their scaffolds compounds 1 to 3 were grouped together (group A) and the rest 12 compounds with an extra double bond were placed in group B for SAR analysis.
Among the three compounds in group A, compound 1 is the most potent one and binds to the PDZ domain in the slow/intermediate change regime; compound 2 is less potent although it is also in the intermediate-exchange regime and 3 is the weakest binder in fast exchange with the PDZ domain (Figure 4, Table 1). The relative binding affinities of these three compounds suggest that the R1 position prefers groups with moderate sizes such as a phenyl ring (2) instead of a bigger −ph-4-N(CH2CH2Cl)2 group (3). The SAR of R1 substitution can be explained by docking models (Figure 5), in which the P1 pocket of the PDZ domain interacting with the ligands’ R1 groups is relatively shallow and rigid and may not be able to accommodate large groups such as −ph-4-N(CH2CH2Cl)2.
Group B has twelve compounds and should give us more understanding of the SAR. These 12 compounds were further clustered into three sub-groups based on the heterogeneities of their R1 and R2 substituents (Table 1). Subgroup B1 has eight compounds (4 to 11) with phenyl substituents at both the R1 and R2 positions; subgroup B2 has three compounds (12 to 14) with one phenyl and one -2-furan substituents; subgroup B3 has only one compound (15) with two -2-furan substituents. SAR derived from compounds in group B is discussed in the following paragraphs.
Among the eight compounds in subgroup B1, four compounds are (4, 5, 9 and 10) in the intermediate exchange regime; and the remaining four compounds are in the fast exchange regime; compound 5 is the strongest binder in group B1 (Table 1). For the compounds in subgroup B1, we analyzed the SAR for R1 and R2, respectively. Compounds 4, 5, and 7 have the same R2 substitutions, and thus provided a good data set for analysis of the SAR at R1. Among the three compounds, a fluorine substitution at the para-position of the phenyl ring decreases the binding affinity and a chlorine substitution further decreases the binding affinity (KD: 5 < 7 < 4). Pharmacophore analysis by LigandScout suggested that both fluorine and chlorine can form hydrophobic interactions with the PDZ domain (Figure 6a, 6b and 6c). Taken together, the data suggest that the increased potency is possibly due to the increased size of the hydrophobic group of R1. Similarly, comparing compound 9 and 10, substitution of a 3, 5-2(O-CH3) group by a 4-Br makes the ligand a stronger binder (KD: 9 < 10). And pharmacophore analysis by LigandScout showed that the 4-Br group on 9 can form hydrophobic interactions with the PDZ domain while the 3, 5-2(O-CH3) group on 10 can not (Figure 6d, 6e). This is consistent with the SAR derived from compounds 4, 5 and 7 that R1 prefers phenyl rings with small hydrophobic substituents such as Cl and Br. This might also explain why compound 1 was stronger than compound 2 in group A. For the SAR at the R2 position among compounds in subgroup B1, a methoxy substitution at the para-position of phenyl ring at R2 decreases the binding affinity (KD: 6 < 4). This might be due to that the methyl group in methoxy extended into the hydrophobic P2 pocket and formed more favorable hydrophobic interactions with the PDZ domain (Figure 7a). The same SAR might also be true for compounds in group A (KD: 1 <2).
Fig 6.
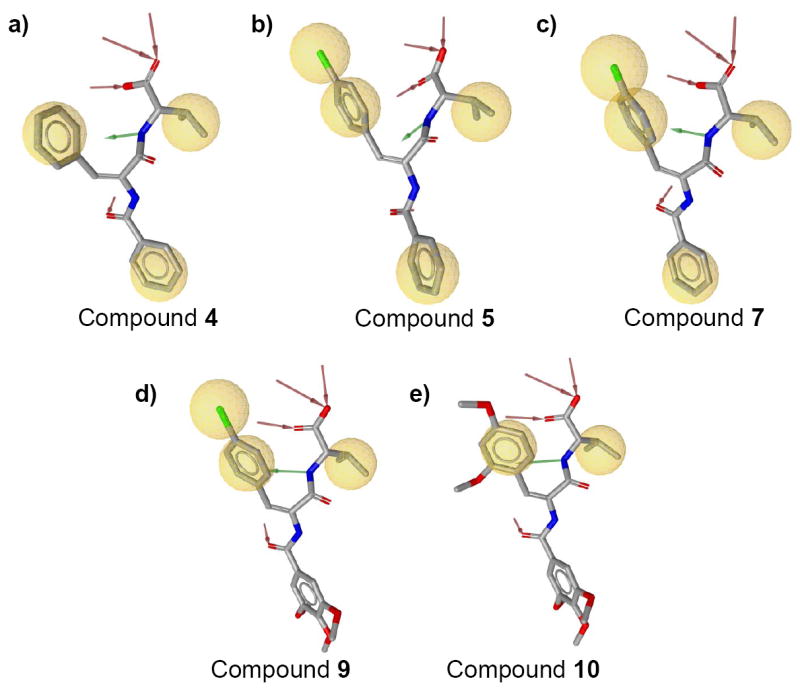
Complex-structure-based pharmacophores of compounds 4 (a), 5 (b), 7 (c), 9 (d), and 10 (e). Pharmacophores were generated from complex structures by using LigandScout [14]. All of them contain two or three hydrophobic interactions (yellow), four hydrogen-bond acceptors (red) and one hydrogen-bond donors (green). Complex structures for 4, 5 and 7 were generated by Glide, and those for 9 or 10 were generated by superimposing ligands onto the docking conformation of 1 and minimizing ligands in the binding pocket.
Fig 7.
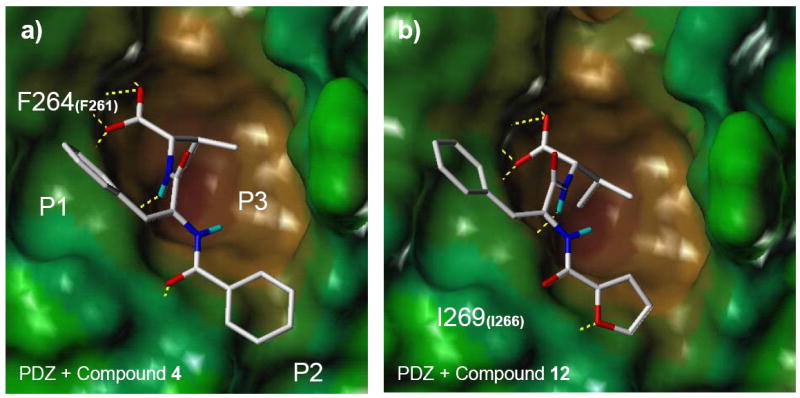
Group -2-furan at R2 forms an additional hydrogen-bond with the PDZ domain. Docking models of compound 4 (a) and 12 (b) generated by Glide (Schrödinger Inc.). Representations and colors are the same as in Figure 5b. Compound 12 forms an extra hydrogen-bond with Ile269 in the βB sheet of the PDZ domain.
Among the three compounds in subgroup B2, 12 is in intermediate exchange with the PDZ domain and the most potent one; the other two compounds 13 and 14 are in fast exchange with the PDZ domain. The SAR at the phenyl ring of compounds in this subgroup is slightly different than that of subgroup B1: F and Cl substitutions at the para-position of the phenyl ring increase the binding affinities (14 and 13 bind to the PDZ domain weaker than 12). Perhaps this is due to that the furan ring in scaffold B2 can form an hydrogen-bond with Ile269 which slightly changes the binding conformations of this class of compounds and/or it changes compounds’ electrostatic potential distributions.
Comparing compounds in group B, R1 position favors a phenyl ring (or a phenyl ring with 4-F, Cl substitutions) over a furan ring since compound 12, 13 and 14 bind to the PDZ domain tighter than 15 does. This is likely due to the limited size or the hydrophobic nature of the P1 pocket which is lined by Phe264 of the PDZ domain (Figure 7b). However, at the R2 position, a -2-furan ring is preferred than a phenyl ring (12 binds tighter to the PDZ domain than 4), which can be explained by the ability of the -2-furan ring to form an extra hydrogen-bond with the amide nitrogen of Ile269.
To summarize the SAR (Table 2), R1 and R2 positions favors hydrophobic groups such as phenyls in both scaffolds A and B. As to the R1 position, hydrophobic -ph is preferred than less hydrophobic -2-furan. And additions of F or Cl on phenyl rings increase ligand potencies while large groups (e.g., -N(CH2CH2Cl)2) weaken binding strengths. However, additions of F or Cl on compounds in subgroup B2 which have -2-furan rings at R2 are not favorable. One of the reasons is that the binding modes of compounds in B2 might be a little different from compounds in other groups. As to the R2 position, a -2-furan is better than -ph because the -2-furan can form an additional hydrogen-bond with the backbone of the PDZ domain. And the addition of methoxy group at the 4-position of phenyl increases the binding potency because it may extend into the P2 hydrophobic cavity of the PDZ domain and form more hydrophobic interactions.
Table 2.
SAR of the fifteen new PDZ domain inhibitors
| Sidechain | SAR |
|---|---|
| R1 | -ph > -2-furan |
| -ph > -ph-4-N(CH2CH2Cl)2 | |
| -ph-4-Cl > -ph-4-F > ph | |
| -ph-4-Br > -ph-3,5-2(OCH3) | |
| -5-benzo[d][1,2]dioxol > ph-4-OCH3 | |
| Exception: ph> ph-4-Cl > ph-4-F (B2) | |
| R2 | -2-furan > -ph |
| -ph-4(OCH3) > -ph | |
Discussion
In summary, we designed a query based on a previously identified inhibitor, the complex structure of this inhibitor bound to the protein as well as two non-binders, and discovered fifteen new compounds with improved binding affinities. These fifteen compounds are similar in structure but have considerable differences in binding affinities, and thus provide an opportunity to study their SAR. The SAR model helps us to get a better understanding of the PDZ-ligand interaction which presents us with further opportunities to optimize PDZ domain inhibitors. For example, the isopropyl group neighbor to the important carboxyl group is a potential modification site (R3). We can explore physical databases for compounds with various R3 groups to determine its optimal size, shape and charge. Furthermore, although substituents R1 and R2 have been studied in this article, their SAR can still be further optimized. In addition, besides optimizing side chains, the scaffolds of this class of compounds can also be optimized using similar methods. Finally, chemical synthesis may also be carried out to complement the chemical space of physically available compounds.
Conclusions
In conclusion, we present here the discovery of fifteen novel inhibitors of the Dvl PDZ domain by virtually exploring the existing chemical space and the development of SAR models of PDZ ligands. The fact that the fifteen compounds selected all bind to the PDZ domain indicates that group 2-(3-methylbutanoic acid) might be essential for PDZ-ligand binding. Indeed, these fifteen analogues with decent variances in binding affinities ranging from weak binding to moderate binding offered an opportunity to develop SAR models and to explore the molecular determinants of PDZ-ligand binding. We think that our SAR model can be used to guide furthering optimization of PDZ ligands. In turn, those developments should contribute to furthering our understanding of Wnt signaling and developing therapeutic agents for Wnt related diseases.
Acknowledgments
We thank the Protein Production Facility at St. Jude Children’s Research Hospital and Dr. Ho-Jin Lee, Youming Shao for producing proteins, Dr. Weixing Zhang for his assistance with NMR experiments, Dr. Charles Ross and Scott Malone for their computer support. This work is supported by grants CA21765 and GM061739. We are grateful to the American Heart Association for a Predoctoral Fellowship to J. Shan.
Abbreviations
- Dvl
Dishevelled
- HSQC
Heteronuclear Single Quantum Coherence
- NCI
National Cancer Institute
- NMR
Nuclear Magnetic Resonance
- PDZ
Post-synaptic density-95/Discs large/Zonula occludens-1
- SAR
structure-activity relationship
References
- 1.Burbaum J, Sigal N. Curr Opin Chem Biol. 1997;1:72. doi: 10.1016/s1367-5931(97)80111-1. [DOI] [PubMed] [Google Scholar]
- 2.Liu B, Li S, Hu J. Am J Pharmacogenomics. 2004;4:263. doi: 10.2165/00129785-200404040-00006. [DOI] [PubMed] [Google Scholar]
- 3.Bleicher K, Böhm H, Müller K, Alanine A. Nat Rev Drug Discov. 2003;2:369. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 4.Keseru G, Makara G. Drug Discov Today. 2006;11:741. doi: 10.1016/j.drudis.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 5.Irwin J, Shoichet B. J Chem Inf Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hann M, Oprea T. Curr Opin Chem Biol. 2004;8:255. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Shan J, Shi D, Wang J, Zheng J. Biochemistry. 2005;44:15495. doi: 10.1021/bi0512602. [DOI] [PubMed] [Google Scholar]
- 8.Dev K. Nat Rev Drug Discov. 2004;3:1047. doi: 10.1038/nrd1578. [DOI] [PubMed] [Google Scholar]
- 9.Fry D, Vassilev L. J Mol Med. 2005;83:955. doi: 10.1007/s00109-005-0705-x. [DOI] [PubMed] [Google Scholar]
- 10.Fujii N, Haresco J, Novak K, Stokoe D, Kuntz I, Guy R. J Am Chem Soc. 2003;125:12074. doi: 10.1021/ja035540l. [DOI] [PubMed] [Google Scholar]
- 11.Fujii N, You L, Xu Z, Uematsu K, Shan J, He B, Mikami I, Edmondson L, Neale G, Zheng J, Guy R, Jablons D. Cancer Res. 2007;67:573. doi: 10.1158/0008-5472.CAN-06-2726. [DOI] [PubMed] [Google Scholar]
- 12.Joshi M, Vargas C, Boisguerin P, Diehl A, Krause G, Schmieder P, Moelling K, Hagen V, Schade M, Oschkinat H. Angew Chem Int Ed Engl. 2006;45:3790. doi: 10.1002/anie.200503965. [DOI] [PubMed] [Google Scholar]
- 13.Barker N, Clevers H. Nat Rev Drug Discov. 2006;5:997. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 14.Wolber G, Langer T. J Chem Inf Model. 2005;45:160. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 15.Cheyette B, Waxman J, Miller J, Takemaru K, Sheldahl L, Khlebtsova N, Fox E, Earnest T, Moon R. Dev Cell. 2002;2:449. doi: 10.1016/s1534-5807(02)00140-5. [DOI] [PubMed] [Google Scholar]
- 16.Kramer B, Rarey M, Lengauer T. Proteins. 1999;37:228. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Friesner R, Banks J, Murphy R, Halgren T, Klicic J, Mainz D, Repasky M, Knoll E, Shelley M, Perry J, Shaw D, Francis P, Shenkin P. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 18.London T, Lee H, Shao Y, Zheng J. Biochem Biophys Res Commun. 2004;322:326. doi: 10.1016/j.bbrc.2004.07.113. [DOI] [PubMed] [Google Scholar]
- 19.Wong H, Bourdelas A, Krauss A, Lee H, Shao Y, Wu D, Mlodzik M, Shi D, Zheng J. Mol Cell. 2003;12:1251. doi: 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 21.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 22.Worrall J, Reinle W, Bernhardt R, Ubbink M. Biochemistry. 2003;42:7068. doi: 10.1021/bi0342968. [DOI] [PubMed] [Google Scholar]
- 23.Gund P. Progress in molecular and subcellular biology. 1977;5:17. [Google Scholar]
- 24.Guner OF. Pharmacophore perception, development, and use in drug design. International University Line; 2000. [Google Scholar]
- 25.Langer T, Hoffmann RD. Pharmacophores and pharmacophore searches. Wiley-VCH; 2006. [Google Scholar]
- 26.Zerbe O. BioNMR in drug research (methods and principles in medicinal chemistry, V 16) Wiley-VCH; 2003. [Google Scholar]
- 27.Hammond M, Harris B, Lim W, Bartlett P. Chem Biol. 2006;13:1247. doi: 10.1016/j.chembiol.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Wallace A, Laskowski R, Thornton J. Protein Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]