

Abstract
The studies on the intrinsic structural determinants for export trafficking of G protein-coupled receptors (GPCRs) have been mainly focused on the C termini of the receptors. In this report we determined the role of the extracellular N termini of α2-adrenergic receptors (α2-ARs) in the anterograde transport from the endoplasmic reticulum (ER) through the Golgi to the cell surface. The N-terminal-truncated α2B-AR mutant is completely unable to target to the cell surface. A single Met-6 residue is essential for the export of α2B-AR from the ER, likely through modulating correct α2B-AR folding in the ER. The Tyr-Ser motif, highly conserved in the membrane-proximal N ter-mini of all α2-AR subtypes, is required for the exit of α2A-AR and α2B-AR from the Golgi apparatus, thus representing a novel Tyr-based motif modulating GPCR transport at the Golgi level. These data provide the first evidence indicating an essential role of the N termini of GPCRs in the export from distinct intracellular compartments along the secretory pathway.
G protein-coupled receptors (GPCRs)2 constitute a super-family of membrane proteins that respond to a vast array of sensory and chemical stimuli and regulate downstream effectors such as adenylyl cyclases, phospholipases, protein kinases, and ion channels through coupling to heterotrimeric G proteins (1, 2). GPCRs are synthesized in the ER. After being correctly folded in the ER, newly synthesized receptors are packaged into the ER-derived COPII transport vesicles and move to the ER-Golgi intermediate complex and the Golgi apparatus through which they are post-translationally modified. The receptors then move from the Golgi to the trans-Golgi network (TGN) to attain fully matured statuses (3) and are further targeted to their functional destination at the plasma membrane. GPCRs at the plasma membrane may undergo internalization upon stimulation with their agonists. The internalized receptors in the endosome may be sorted to target to the lysosome for degradation or to recycle back to the plasma membrane. Therefore, expression of an individual GPCR at the plasma membrane is determined by the overall balance of export from the ER to the cell surface, internalization, recycling, and degradation. However, compared with the extensive studies performed on the events of the endocytotic pathway (4-6), molecular mechanisms governing the export trafficking of GPCRs from the ER through the Golgi to the cell surface and their role in regulating receptor expression at the cell surface and function is relatively less well understood (3).
The progress achieved over the past few years indicates that export from the ER, the first step in intracellular trafficking of GPCRs, is a highly regulated process and influences the cell-surface expression level of GPCRs (3, 7). GPCR export from the ER is modulated by direct interactions with multiple regulatory proteins such as the ER chaperones and receptor activity modifying proteins (8, 9). GPCR dimerization also plays an important role in receptor folding and export from the ER. Several studies have indicated that some GPCR dimers are constitutively formed in the ER and that dimerization is required for their transport from the ER to the cell surface (10-13). Furthermore, the identification of conserved sequences in the membrane-proximal C termini essential for ER export indicates that GPCR export from the ER may be directed by specific motifs (14-17).
Protein transport from the Golgi may be mediated through constitutive or regulatory pathways (18). Recently, several studies have demonstrated that protein export from the Golgi is mediated through highly specified motifs. For example, vesicular stomatitis virus glycoprotein uses the Tyr-based di-acidic motif found in its cytoplasm tail to export from the TGN through recruiting adaptor protein complex 3 (19). The cytoplasmic N-terminal positively charged residues are necessary for the efficient export of inward rectifier potassium channels from the Golgi complex (20). However, the specific sequences for exit from the Golgi of the GPCR superfamily have not been identified.
As an initial approach to understanding the export pathways for different GPCRs, we focused on the adrenergic (AR) and angiotensin II type 1 receptor (AT1R) (14, 21-24). We have demonstrated that Rab1, a Ras-like small GTPase that coordinates protein transport specifically from the ER to the Golgi, selectively regulates the transport of AT1R and β2-AR. In contrast, the transport from the ER to the cell surface of α2B-AR is independent of Rab1 (21). These data demonstrated that different GPCRs may use distinct pathways for their transport from the ER to the cell surface. Most importantly, the transport of α2B-AR from the ER to the cell surface is mediated through a non-conventional Rab1-independent pathway. We then identified a motif consisting of a Phe and double Leu spaced by six residues (FX6LL), which is required for the export of AT1R and α2B-AR from the ER (14). This motif is highly conserved in the membrane-proximal C termini of GPCRs and, therefore, may provide a common signal in mediating export of the receptors from the ER. To further define the intrinsic structural determinants for GPCR export trafficking, in this manuscript we determined the role of the N termini of α2B-ARs in the transport from the ER to the cell surface. We demonstrated that the single Met-6 residue modulates α2B-AR export from the ER, and the Tyr-Ser motif, which is highly conserved in the membrane-proximal N termini of all α2-AR subtypes, regulates exit of α2A-AR and α2B-AR from the Golgi. These data provide strong evidence for the first time indicating that the N termini of GPCRs contain multiple signals modulating the export of the receptors from distinct intracellular compartments.
EXPERIMENTAL PROCEDURES
Materials
Rat α2B-AR in vector pcDNA3 was kindly provided by Dr. Stephen M. Lanier (Dept. of Pharmacology and Experimental Therapeutics, Louisiana State University Health Sciences Center). Human α2A-AR and α2B-AR tagged with three HAs were purchased from UMR cDNA Resource Center (Rolla, MO). The dominant negative arrestin-3 mutant Arr3-(201–409) and the dominant negative dynamin mutant DynK44A were kindly provided by Dr. Jeffery L. Benovic (Department of Microbiology and Immunology, Kimmel Cancer Center, Thomas Jefferson University). Antibodies against green fluorescent protein (GFP) and phospho-ERK1/2 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-ERK antibodies detecting total ERK1/2 expression were from Cell Signaling Technology, Inc. (Beverly, MA). Anti-HA monoclonal antibody 12CA5 was from Roche Applied Science. Antibodies against GM130 and p230 were from Transduction Laboratories (San Diego, CA). Brefeldin A (BFA), UK14304, rauwolscine, and dimethyl sulfoxide (Me2SO) were obtained from Sigma-Aldrich. Alexa Fluor 594-labeled secondary antibodies and 4,6-diamidino-2-phenylindole were from Molecular Probes, Inc. (Eugene, OR). The ER markers pDsRed2-ER and pECFP-ER were from BD Biosciences. Normal donkey serum was purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). [3H]RX821002 (45.0 Ci/mmol) was purchased from PerkinElmer Life Sciences. Penicillin/streptomycin, l-glutamine, trypsin/EDTA, and Lipofectamine 2000 reagent were from Invitrogen. Polyvinylidene difluoride membranes were obtained from Gelman Sciences (Ann Arbor, MI). All other materials were obtained as described elsewhere (14, 21).
Plasmid Constructions
α2B-AR tagged with GFP at its C terminus (α2B-AR-GFP) was generated as described previously (21). For generation of GFP-tagged α2B-AR-12 construct in which the N-terminal 12 amino acid residues (Ser-2—Ser-13) were deleted from α2B-AR, the full-length α2B-AR-GFP was amplified by PCR (forward primer, 5′- GATCAAGCTTATGGTGCAGGCCACCGCCGCCATCGCGTCG-3′; reverse primer, 5′-GATCGTCGACGCCCAGCCAGTCTGGGTC-3′) in which the truncated α2B-AR was in-frame with GFP, restricted with HindIII and SalI, and ligated into the pEGFP-N1 vector (Invitrogen). Similar strategies were used to generate C-terminal GFP-tagged α2A-AR and the α2A-AR mutant lacking the N-terminal 28 residues (Gly-2—Ser-29). For generation of the C-terminal YFP-tagged α2B-AR, α2B-AR was released from the pEGFP-N1 vector by digestion with HindIII and SalI and ligated into the pEYFP-N1 vectors (Invitrogen), which was cleaved with the same restriction enzymes. Receptor mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) using GFP-tagged receptors as templates. The sequence of each construct used in this study was verified by restriction mapping and nucleotide sequence analysis (Louisiana State University Health Sciences Center DNA Sequence Core).
Cell Culture and Transient Transfection
HEK293T cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Transient transfection of the HEK293T cells was carried out using Lipofectamine 2000 reagent as described previously (21). For measurement of cell-surface receptor expression and ERK1/2 activation, HEK293T cells were cultured on 6-well plates and transfected with 0.5 μg of GFP-tagged receptors. To determine the influence of the endocytotic pathway on α2B-AR expression at the cell surface, HEK293T cells were transfected with 0.25 μg of GFP-tagged α2B-AR or its mutants together with 0.75 μg of dominant negative mutants Arr3-(201–409), DynK44A, or Rab5S34N. For co-localization with pDsRed2-ER, an ER marker, HEK293T cells were co-transfected with 0.5 μg of pDsRed2-ER and equal amounts of receptors. The transfection efficiency was estimated to be about 80% based on the GFP fluorescence.
Measurement of α2-AR Expression at the Cell Surface
Cell-surface α2-AR expression was measured by [3H]RX821002 binding to intact cells. HEK293T cells were cultured on 6-well plates and transfected with α2-AR constructs for 6 h. The cells were then split onto poly-l-lysine-coated 12-well plates at a density of 4 × 105 cells/well and grown for 24 h. To determine whether the reduced temperature could rescue the transport of mutated α2B-AR, HEK293T cells were grown at 30 °C for 40 h after being split onto 12-well plates. To determine the effect of the chemical chaperone Me2SO on α2B-AR export, HEK293T cells were cultured at 37 °C for 24 h and then incubated with Me2SO at a concentration of 2% for another 24 h. The cells were incubated with Dulbecco's modified Eagle's medium containing 20 nM [3H]RX821002 for 90 min at room temperature with constant shaking (40 rpm). The nonspecific binding was determined in the presence of nonradioactive rauwolscine (10 μm). The cells were washed twice with 1 ml of ice-cold phosphate-buffered 3 saline (PBS), and the cell surface-bound [3H]RX821002 was extracted by 1 m NaOH treatment for 2 h at 37 °C. The radioactivity was counted by liquid scintillation spectrometry in 5 ml of Ecoscint A scintillation solution (National Diagnostics, Inc., Atlanta, GA).
Immunofluorescence Microscopy
HEK293T cells were grown on coverslips and fixed with a 4% paraformaldehyde, 4% sucrose mixture in PBS for 15 min. The cells were stained with 4,6-diamidino-2-phenylindole for 5 min, and the coverslips were mounted. For co-localization of the receptor with intra-cellular markers, HEK293T cells were permeabilized with PBS containing 0.2% Triton X-100 for 5 min and blocked with 5% normal donkey serum for 1 h. The cells were then incubated with antibodies against GM130 or p230 (1:50) for 1 h. After washing with PBS (3 × 5 min), the cells were incubated with Alexa Fluor 594-labeled secondary antibody (1:2000 dilution) for 1 h at room temperature. The fluorescence was detected with a Leica DMRA2 epifluorescent microscope.
For co-localization of YFP-tagged receptors with the ER marker pECFP-ER in live cells, HEK293T cells were plated on poly-l-lysine-precoated 35-mm glass bottom dishes and transiently transfected with 100 ng of YFP-tagged receptor and 100 ng of the ER marker pECFP-ER with Lipofectamine 2000 reagent for 24 h. One hour before imaging, culture medium was replaced with CO2-independent medium (Invitrogen). Fluorescence was detected with a Zeiss Axiovert microscope (200M). Images were deconvolved using SlideBook software and the nearest neighbors deconvolution algorithm (Intelligent Imaging Innovations, Denver, CO).
Measurement of ERK1/2 Activation
HEK293T cells were cultured on 6-well plates and transfected as described above. At 36 h after transient transfection, HEK293T cells were starved for at least 3 h and then stimulated with UK14304 at concentrations from 0.01 to 10 μm for 5 min. Stimulation was terminated by the addition of 1 × SDS gel loading buffer. After solubilizing the cells, 20 μl of total cell lysates was separated by 12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The ERK1/2 activation was determined by measuring the levels of phosphorylation of ERK1/2 with phosphospecific ERK1/2 antibodies (21). The signal was detected using ECL Plus (PerkinElmer Life Sciences) and a Fuji Film luminescent image analyzer (LAS-1000 Plus) and quantitated using the Image Gauge program (Version 3.4). The membranes were stripped and reprobed with anti-ERK1/2 antibodies to confirm equal protein loading.
Immunoprecipitation of α2B-AR
HEK293T cells cultured on 100-mm dishes were transfected with 4 μg of HA-tagged α2B-AR together with 4 μg of the pEGFP-N1 vector or GFP-tagged wild-type or mutated α2B-AR in the pEGFP-N1 vector for 28 h. The cells were washed twice with PBS and harvested. The cells were then lysed with 500 μl of lysis buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and Complete Mini protease inhibitor mixture. After gentle rotation for 1 h, samples were centrifuged for 15 min at 14,000 × g, and the supernatant was incubated with 50 μl of protein G-Sepharose for 1 h at 4 °C to remove nonspecific bound proteins. Samples were then incubated with 5 μg of anti-GFP antibodies overnight at 4 °C with gentle rotation followed by an incubation with 50 μl of protein G-Sepharose beads for 5 h. Resin was collected by centrifugation and washed 3 × 500 μl of lysis buffer. Immunoprecipitated receptors were eluted with 100 μl of 1 × SDS-PAGE loading buffer, separated by 8% SDS-PAGE, and visualized by immunoblotting using anti-HA antibodies.
Statistical Analysis
Differences were evaluated using Student's t test, and p < 0.05 was considered as statistically significant. Data are expressed as the mean ± S.E.
RESULTS
A Requirement of the N terminus of α2B-AR for the Transport to the Cell Surface
We have recently identified the FX6LL motif in the membrane-proximal C termini of α2B-AR and AT1R, which is required for their export from the ER (14). To further define the structural determinants for export trafficking of GPCRs, we determined the role of the N terminus of α2B-AR in its anterograde trafficking from the ER to the cell surface. We first determined the effect of deleting the entire N terminus on the transport of α2B-AR to the cell surface. α2B-AR and its mutant lacking N-terminal 12 amino acid residues (Ser-2—Ser-13) (α2B-AR-12) were conjugated with the GFP at their C termini and transiently expressed in HEK293T cells. The cell-surface expression of α2B-AR-12 was markedly reduced by 95% as compared with wild-type (WT) α2B-AR as measured by intact cell ligand binding using [3H]RX821002 (Fig. 1A). Consistent with the lack of α2B-AR-12 cell-surface expression as measured by ligand binding, ERK1/2 activation by the agonist UK14304 was completely lost in cells transfected with α2B-AR-12 (Fig. 1B). In contrast, ERK1/2 activation was stimulated dose-dependently by UK14304 in cells transiently transfected with α2B-AR (Fig. 1B). Subcellular localization of the receptors indicated that α2B-AR-12 was trapped in the perinuclear region, whereas α2B-AR was mainly localized at the cell surface (Fig. 1C), which was confirmed by co-localization with tetramethyl-rhodamine-conjugated concanavalin A, a plasma membrane marker (data not shown). These data indicate that the extracellular N-terminal portion is required for α2B-AR transport to the cell surface.
FIGURE 1. Effect of the deletion of extracellular N terminus on α2B-AR transport to the cell surface.

A, quantitation of the cell-surface expression of α2B-AR and α2B-AR-12 lacking the N-terminal 12 residues (Ser-2—Ser-13). HEK293T cells cultured on 6-well plates were transfected with 0.5 μg of GFP-conjugated α2B-AR or α2B-AR-12 and then split onto 12-well plates. The cell-surface expression of the receptors were measured by intact cell binding assays using [3H]RX821002 at a concentration of 20 nM in duplicate. The nonspecific binding was determined in the presence of nonradioactive rauwolscine (10 μm) as described under “Experimental Procedures.” The mean values of specific [3H]RX821002 binding were 16981 ± 956 and 849 ± 339 cpm (n = 3) from cells transfected with α2B-AR and α2B-AR-12, respectively. The data shown are percentages of the mean value obtained from cells transfected with α2B-AR and are presented as the mean ± S.E. of three experiments. *, p<0.05 versus cells transfected with α2B-AR. B, effect of the deletion of the N terminus of α2B-AR on ERK1/2 activation. HEK293T cells cultured on 6-well plates were transfected with α2B-AR (squares) or α2B-AR-12 (triangles). At 8–12 h after transfection, the cells were split and cultured for an additional 24 h. The cells were then stimulated with increasing concentrations of UK14304 (0.01–10 μm) for 5 min. ERK1/2 activation was determined by Western blot analysis using phosphospecific ERK1/2 antibodies. Left upper panel, a representative blot of ERK1/2 activation; left lower panel, total ERK1/2 expression; right panel, quantitative data expressed as percent of the ERK1/2 activation obtained in the cells transfected with α2B-AR and stimulated with 10 μM UK14304 (control). Similar results were obtained in at least three separate experiments. C, subcellular distribution of α2B-AR and α2B-AR-12. HEK293T cells cultured on coverslips were transfected with GFP-conjugated α2B-AR or α2B-AR-12, and subcellular distribution of the receptors was revealed by detecting GFP fluorescence as described under “Experimental Procedures.” The data are representative images of at least five independent experiments. Scalebar, 10 μm.
Identification of Amino Acid Residues in the N Terminus Required for the Cell-surface Expression of α2B-AR
To identify amino acid residues important for α2B-AR cell-surface expression, each amino acid residue (except Gly-3) in the N terminus (2SGPTMDHQEPYS13) (Fig. 2A) was substituted with Ala individually or in combination, and the cell-surface expression of each receptor mutant was determined by intact cell ligand binding. Mutation of Ser-2, Pro-4, Thr-5, Asp-7, His-8, Gln-9, Glu-10, and Pro-11 to Ala did not significantly influence the cell-surface expression of α2B-AR. In contrast, single substitution of Met-6 with Ala abolished α2B-AR transport to the cell surface, and mutation of Tyr-12 and Ser-13 significantly attenuated the cell-surface expression of α2B-AR by 65 and 55%, respectively, as compared with WT α2B-AR (Fig. 2B). Simultaneous mutation of Tyr-12 and Ser-13 (Y12A/S13A) to Ala more profoundly inhibited the cell-surface expression of α2B-AR than either mutant (Fig. 2B). Western blot analysis of total cell lysate using anti-GFP antibodies demonstrated that the reduction in the cell-surface expression of the mutant receptors was not due to the differences in total receptor expression as expression levels of these mutants were comparable with their WT (Fig. 2C). Consistent with the attenuated cell-surface expression, ERK1/2 activation in response to stimulation with UK14304 was also significantly inhibited in cells transfected with M6A, Y12A, S13A, or Y12A/S13A when compared with cells transfected with WT α2B-AR (Fig. 2D).
FIGURE 2. Site-directed mutagenesis identifies key residues at the N terminus required for the cell-surface expression of α2B-AR.

A, the sequence of the α2B-AR N terminus, in which each amino acid residue (except Gly-3) was mutated to Ala individually or in combination. B, specific [3H]RX821002 binding to intact HEK293T cells transfected with α2B-AR and its mutants. HEK293T cells were transiently transfected with α2B-AR and its mutants, and their expression at the cell surface was determined by intact cell ligand binding with [3H]RX821002 as described in the legend of Fig. 1. The data shown are the percentages of the mean value obtained from cells transfected with WT α2B-AR and are presented as the mean ± S.E. of three separate experiments. *, p < 0.05 versus cells transfected with WT α2B-AR. C, Western blot analysis of α2B-AR and its mutants M6A, Y12A, S13A, and Y12A/S13A (YS-AA). HEK293T cells were cultured on 6-well plates and transfected with 0.5 μg of GFP-tagged α2B-AR or its mutants. The cells were then solubilized in 300 μl of 1× SDS gel loading buffer. Five μl of total cell lysate were separated by 10% SDS-PGAE, receptor expression was visualized by immunoblotting using GFP antibodies (upperpanel), and the blots were stripped and then probed with β-actin antibodies (lower panel). D, ERK1/2 activation by UK14304 in HEK293T cells transiently transfected with α2B-AR or its mutants M6A, Y12A, S13A, and Y12A/S13A. HEK293T cells were transfected with α2B-AR (squares) and its mutants (triangles), and the cells were then treated with UK14304 (0.01–10 μm) for 5 min. P-, phosphorylated. Left panel, representative blots of ERK1/2 activation. Right panel, quantitative data expressed as percent of the ERK1/2 activation obtained in the cells transfected with α2B-AR and stimulated with 10 μM UK14304 (control).There is no significant difference in total ERK1/2 expression between samples (data not shown). Similar results were obtained in at least three separate experiments. E, subcellular localization of α2B-AR and its mutants. GFP-conjugated WT and mutated α2B-AR were transiently expressed in HEK293T cells, and their subcellular distribution was revealed by fluorescence microscopy detecting GFP as described under “Experimental Procedures.” The data shown are representative images of three independent experiments. Scale bar, 10 μm.
The subcellular localization of each α2B-AR mutant was then visualized. Consistent with quantitative measurement of receptor expression at the cell surface by intact cell ligand binding, M6A mutant was completely unable to transport to the cell surface, and Y12A and S13A mutants were partially trapped inside the cells. In contrast, S2A, P4A, T5A, D7A, H8A, Q9A, E10A, and P11A mutants exhibited clear cell-surface expression patterns (Fig. 2E). Ala substitution of Val-14, Gln-15, and Thr-17 at the beginning of the first transmembrane domain also did not significantly influence the cell-surface expression and subcellular localization of α2B-AR (data not shown). These data strongly indicate that three residues, Met-6, Tyr-12, and Ser-13, at the N terminus are critical for α2B-AR export to the cell surface.
Residues Met-6, Tyr-12, and Ser-13 Modulate α2B-AR Export atDistinct Organelles
The preceding data have demonstrated that residues Met-6, Tyr-12, and Ser-13 in the α2B-AR N terminus are required for cell-surface targeting. Interestingly, the subcellular localization pattern of M6A mutant is apparently different from those of Y12A, S13A, and Y12A/S13A mutants. To define the intracellular compartments in which the α2B-AR mutants M6A, Y12A, S13A, and Y12A/S13A were retained, each mutant was co-localized with markers of the ER, the Golgi, and the TGN. M6A mutant as well as α2B-AR-12 was extensively co-localized with the ER marker pDsRed2-ER (Fig. 3A) but not with the Golgi marker GM130 and the TGN marker p230 (data not shown) in fixed cells. To eliminate the possible nonspecific influence of cell fixation on the subcellular localization of α2B-AR, α2B-AR and its mutant M6A were tagged with YFP at their C termini, and their subcellular co-localization with the ER marker pECFP-ER was visualized by microscopic analysis in live cells. Similar to the results obtained from the fixed cells, M6A mutant was strongly co-localized with pECFP-ER in live HEK293T cells (Fig. 3B). These data demonstrate that M6A mutant was unable to export from the ER and indicate that Met-6 modulates α2B-AR export at the level of the ER.
FIGURE 3. Effect of mutation of Met-6 on the subcellular localization of α2B-AR.
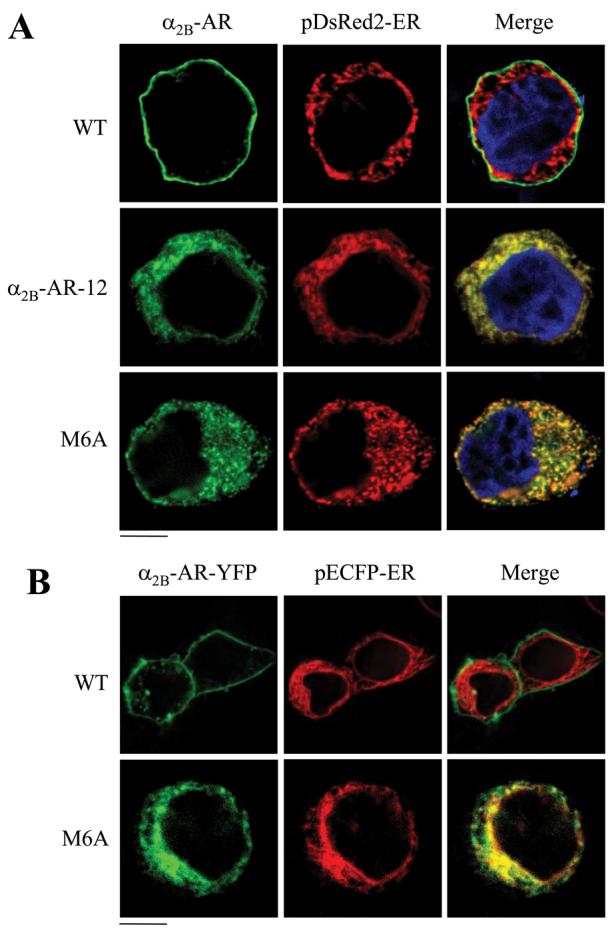
A, co-localization of α2B-AR, α2B-AR-12, and M6A with the ER marker pDsRed2-ER in fixed cells. HEK293T cells were transfected with GFP-tagged α2B-AR, α2b-AR-12, or M6A mutant together with pDsRed2-ER, and the subcellular distribution and co-localization of the receptors with pDsRed2-ER were revealed by fluorescence microscopy as described under “Experimental Procedures.” B, co-localization of α2B-AR and M6A mutant with the ER marker pECFP-ER in live cells. HEK293T cells plated on poly-l-lysine-precoated 35-mm glass-bottom dishes were transfected with YFP-tagged α2B-AR or M6A together with pECFP-ER, and the subcellular localization and co-localization of the receptors with pECFP-ER were obtained in living cells with a Zeiss Axiovert microscope. Green, α2B-AR tagged with GFP (A) or YFP (B); red, the ER markers pDsRed2-ER (A) and pECFP-ER (B); yellow, co-localization of the receptors with the ER; blue, DNA staining by 4,6-diamidino-2-phenylindole (nuclei). The data shown in A and B are representative images of at least three independent experiments. Scale bars, 10 μm.
In contrast to M6A retained in the ER, Y12A, S13A, and Y12A/S13A, mutants were strongly co-localized with GM130 (Fig. 4A) but not with pDsRed2-ER (data not shown) and p230 (Fig. 4B). Similar results were obtained from COS-7 cells (data not shown), suggesting the role of the Tyr-12—Ser-13 motif in modulating α2B-AR export is not cell-type specific. These data demonstrate that these mutants were able to exit from the ER and transport to the Golgi, but their abilities to export from the Golgi to the TGN were impaired. These data indicate that the Tyr-12—Ser-13 motif modulates α2B-AR export at the level of the Golgi.
FIGURE 4. Effect of mutation of Tyr-12 and Ser-13 on the subcellular localization of α2B-AR.
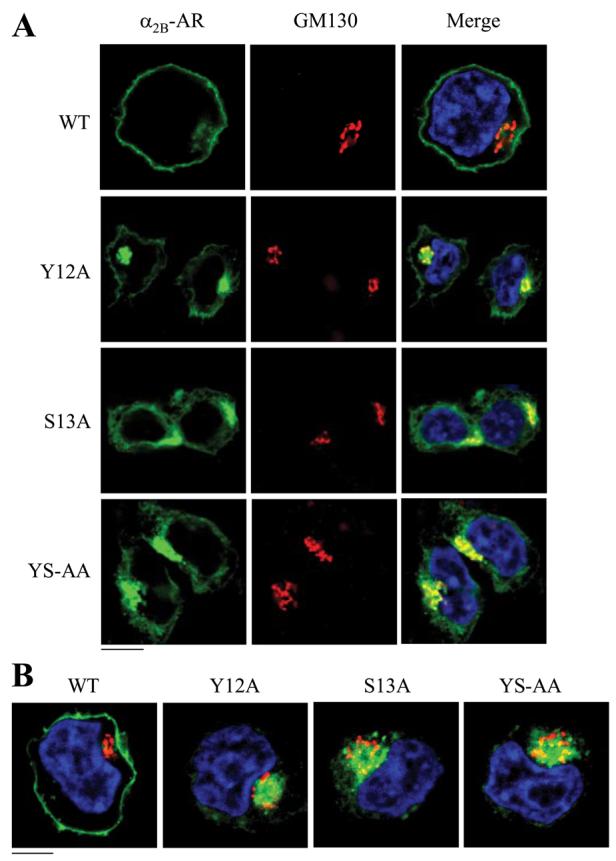
HEK293T cells were transfected with α2B-AR or its mutants Y12A, S13A, and Y12A/S13A and their co-localization with the Golgi (A) and theTGN (B) markers were revealed by fluorescence microscopy after staining with antibodies against GM130 (A) and p230 (B) (1:50 dilution), respectively, as described under “Experimental Procedures.” Green, α2B-AR tagged with GFP; red, the Golgi marker GM130 (A) or the TGN marker p230 (B); yellow, co-localization of the receptors with the Golgi (A) and the TGN markers (B); blue, DNA staining by 4,6-diamidino-2-phenylindole (nuclei). The data shown in A and B are representative images of at least five independent experiments. Scale bars, 10 μm. YS-AA, Y12A/S13A.
Effect of the Dominant Negative Mutants of Arrestin-3, Dynamin, and Rab5 and Treatment with BFA on α2B-AR Export
To eliminate the possibility that the accumulation of the Y12A, S13A, and Y12A/S13A mutants in the Golgi is caused by their constitutive internalization induced by the mutation, we determined the effect of transient expression of the dominant negative mutants Arr3-(201–409), DynK44A, and Rab5S34N on the cell-surface expression and subcellular localization of the mutated receptors. Arrestin-3 and dynamin modulate α2B-AR endocytotic trafficking, and Rab5 is involved in the transport from the plasma membrane to the endosome of many GPCRs (3, 25-27). Expression of Arr3-(201–409), DynK44A, and Rab5S34N did not have clear influence on the cell-surface expression (Fig. 5A) or subcellular localization (data not shown) of α2B-AR and its mutants Y12A, S13A, and Y12A/S13A.
FIGURE 5. Effect of blocking endocytotic and anterograde transport on the transport of the Tyr-Ser motif mutants.

A, specific [3H]RX821002 binding to intact HEK293T cells co-transfected with α2B-AR and internalization blockers. HEK293T cells were transfected with α2B-AR, Y12A, S13A, or Y12A/S13A (YS-AA) together with pcDNA3 vector (control) or the dominant negative mutants Arr3-(201–409), DynK44A, or Rab5S34N. Receptor expression at the cell surface was measured by intact cell ligand binding as described in the legend of Fig. 1. The data shown are percentages of the mean value obtained from cells transfected with WT α2B-AR and pcDNA3 and are presented as the mean ± S.E. of three separate experiments. Receptor cell-surface expression has no significant differ-ence in cells transfected with Arr3-(201–409), DynK44A, or Rab5S34N as compared with their respective controls. B, specific [3H]RX821002 binding to intact HEK293T cells transfected with α2B-AR and treated with BFA. HEK293 cells were transfected with α2B-AR or its mutants and then incubated with ethanol (control) or BFA at a concentration of 5 μg/ml for 8 h. The data shown are percentages of the mean value obtained from cells transfected with WT α2B-AR and treated with ethanol and are presented as the mean ± S.E. of three separate experiments. *p< 0.05 versus respective control. C, effect of BFA treatment on the subcellular distribution of α2B-ARand its mutants. HEK293T cells were transfected with GFP-conjugated α2B-AR or its mutants, and subcellular distribution of the receptors was revealed by detecting GFP fluorescence as described under “Experimental Procedures.” The data are representative images of at least three independent experiments. Scale bar, 10 μm.
We then determined the effect of blocking anterograde protein transport by BFA treatment on the cell-surface expression and subcellular localization of the Tyr-Ser motif mutants. BFA is a fungal metabolite that disrupts the structures of the Golgi and blocks protein transport from the ER to the Golgi. Treatment with BFA dramatically inhibited the cell-surface expression of α2B-AR and almost abolished the cell-surface expression of Y12A, S13A, and Y12A/S13A mutants (Fig. 5B). BFA treatment arrested α2B-AR in the perinuclear regions of the transfected cells, presumably in the ER. Y12A, S13A, and Y12A/S13A mutants were redistributed from the Golgi to the ER in the presence of BFA (Fig. 5C). These data suggest that the Golgi accumulation of the Tyr-Ser motif mutants is likely caused by defective export from the Golgi rather than the constitutive endocytotic transport from the plasma membrane.
Hydrophobic Properties of Met-6 and Tyr-12 Are Important for Their Function in α2B-AR Export
To characterize physiochemical properties required for the function of Met-6, Tyr-12, and Ser-13 in α2B-AR transport to the cell surface, we first determined if hydrophobicity of Met-6 and Tyr-12 played a role in their function by mutating them to hydrophobic residues (M6L and Y12F) and non-hydrophobic residue (M6Q and Y12Q). The cell-surface expression of M6L and Y12F mutants was markedly enhanced as compared with M6A and Y12A mutants, respectively. In contrast, the cell-surface expression of M6Q and Y12Q mutants was the same as their respective Ala mutants (Fig. 6, A and C). Consistently, subcellular localization analysis showed that M6L and Y12F mutants were able to transport to the cell surface, whereas M6Q and Y12Q mutants were trapped inside the cell (Fig. 6, B and D). These data indicate that the hydrophobic characteristics of the residues Met-6 and Tyr-12 are crucial for their function in α2B-AR export.
FIGURE 6. Characterization of Met-6, Tyr-12, and Ser-13.

A, effect of mutating Met-6 to hydrophobic Leu (M6L) and non-hydrophobic Gln (M6Q) residues on the cell-surface expression of α2B-AR. B, subcellular localization of α2B-AR and its mutants M6L and M6Q. C, effect of mutating Tyr-12 to Leu (Y12L) and Gln (Y12Q) on the cell-surface expression of α2B-AR. D, subcellular localization of α2B-AR and its mutants Y12L and Y12Q. E, effect of mutating Tyr-12 to Asp (Y12D) and Ser-13 to Asp (S13D) and Thr (S13T) on the cell-surface expression of α2B-AR. HEK293T cells were transfected with α2B-AR or its mutants, their expression at the cell surface was determined by intact cell ligand binding with [3H]RX821002, and subcellular localization was revealed by fluorescence microscopy detecting GFP signal as described in the legend of Fig. 1. The data shown in A, C, and E are percentages of the mean value obtained from cells transfected with WT α2B-AR and are presented as the mean ± S.E. ofthree separate experiments. *,p< 0.05 versus cells transfected with WT α2B-AR; **, p < 0.05 versus cells transfected with M6A (A) and Y12A (C). The data shown in B and D are representative images of three independent experiments. Scalebars, 10 μm.
We next determined if the function of Tyr-12 and Ser-13 residues in α2B-AR export was regulated by their potential phosphorylation. Similar to the mutation to Ala, substitution of Tyr-12 and Ser-13 with Asp, which will mimic the status of phosphorylation, significantly attenuated α2B-AR export to the cell surface (Fig. 6E). Mutation of Ser-13 to Thr also inhibited receptor expression at the cell surface (Fig. 6E). These data suggest that phosphorylation of Tyr-12 and Ser-13 residues is unlikely involved in regulating α2B-AR export.
Rescue ofM6A and Y12A/S13A Transport by Low Temperature and Me2SO Treatment
To determine if Met-6 and the Tyr-Ser motif are involved in the proper α2B-AR folding, we determined the effect of low temperature culture and treatment with Me2SO, a chemical chaperone, on the cell-surface expression of the M6A and Y12A/S13A mutants. HEK293T cells cultured at a reduced temperature (30 °C) significantly enhanced the cell-surface expression of M6A mutant without influencing WT α2B-AR transport as measured by intact cell ligand binding (Fig. 7A) and subcellular distribution (Fig. 7C), suggesting that 30 °C is a permissive temperature for proper M6A folding to achieve a status competent for export from the ER. Similar to M6A, the transport of the Y12A/S13A mutant was also rescued by reducing culture temperature. However, the magnitude of rescue by low temperature was greater for M6AthanY12A/S13A.
FIGURE 7. Effect of reduced temperature and Me2SO treatment on the cell-surface expression of M6A and Y12A/S13A mutants.

A, specific [3H]RX821002 binding to intact HEK293Tcells transfected with α2B-AR, M6A, or Y12A/S13A (YS-AA) and cultured at 37 and 30 °C for 40 h. Left panel, data shown are percentages of the mean value obtained from cells transfected with WT α2B-AR and cultured at 37 °C and are presented as the mean ± S.E. of three separate experiments. Right panel,-fold increase in receptor cell-surface expression at 30 °C over 37 °C. *, p < 0.05 versus cells cultured at 37 °C (left panel) and WT (right panel). B, specific [3H]RX821002 binding to intact HEK293T cells transfected with α2B-AR, M6A or Y12A/S13A and treated with Me2SO (DMSO). HEK293T cells transfected with α2B-AR, M6A, or Y12A/S13A were incubated with 2% Me2SO for 24 h. Left panel, the data shown are percentages of the mean value obtained from cells transfected with WT α2B-AR (control) and are presented as the mean ± S.E. of three separate experiments. Right panel, -fold increase in receptor cell-surface expression after Me2SO treatment. *, p < 0.05 versus control (left panel) and WT (right panel). Con, control. C, the effect of low temperature culture and Me2SO treatment on the subcellular localization of α2B-AR, M6A, and Y12A/S13A. The data shown are representative images of three independent experiments. Arrows indicate α2B-AR expressed at the plasma membrane. Scale bar, 10 μm.
Me2SO treatment facilitated the transport to the cell surface of α2B-AR and M6A mutant as determined by intact cell ligand binding (Fig. 7B) and subcellular localization (Fig. 7C). Further-more, the Me2SO-enhanced cell-surface expression was much greater for M6A mutant compared with WT α2B-AR (Fig. 7B). In contrast, Me2SO treatment did not produce a significant effect on the transport of Y12A/S13A mutant (Fig. 7,B and C). These data indicate that both low temperature culture and the chemical chaperone Me2SO could rescue at least in part the cell-surface delivery of the ER-retained M6A mutant and sug-gest that Met-6 residue may be involved in the regulation of correct α2B-AR folding in the ER.
Effect of Mutation of Met-6 and the Tyr-Ser Motif on Dimerization of α2B-AR
We previously demonstrated that α2B-AR constitutively forms dimers in the ER, and dimerization of α2B-AR plays a crucial role in modulating its transport from the ER to the cell surface (23). To test if the influence of Met-6 and the Tyr-Ser motif on export trafficking of α2B-AR was caused by disrupting its dimerization in the ER, we determined whether GFP-tagged M6A or Y12A/S13A could form heterodimers with HA-tagged WT α2B-AR when co-expressed in HEK293T cells. HA-α2B-AR was found in the anti-GFP immunoprecipitate from cells transfected with either GFP-tagged M6A (Fig. 8A) or Y12A/S13A (Fig, 8B), but not from cells expressing GFP. To determine specific interaction of α2B-AR and its mutants, HEK293T cells were separately transfected with α2B-AR or its mutants, mixed, and immunoprecipitated with anti-GFP antibodies. Anti-GFP antibodies did not immunoprecipitate HA-tagged α2B-AR from the mixture (data not shown). These data indicate that α2B-AR forms heterodimers with M6A and Y12A/ S13A when co-expressed in same cell populations.
FIGURE 8. Western blot (IB) analysis of heterodimerization of α2B-AR and M6A or Y12A/S13A mutants.

A, dimerization of α2B-ARand M6A. HEK293T cells were transfected with HA-tagged α2B-AR plus the pEGFP-N1 vector (control), GFP-tagged α2B-AR (WT) or GFP-tagged M6A. IP, immunoprecipitation. B, dimerization of α2B-AR and Y12A/S13A. HEK293T cells were transfected with HA-α2B-AR together with the pEGFP-N1 vector (control), GFP-tagged α2B-AR (WT) or GFP-tagged Y12A/S13A. The cells were solubilized and immu-noprecipitated with anti-GFP antibodies. The immunoprecipitate was sepa-rated by SDS-PAGE, and immunoprecipitated receptors were revealed by Western blotting with anti-HAantibodies (upper panels).The blots were then stripedand reprobed with anti-GFP antibodies (lower panels).The data shown in A and B are representative of three independent experiments.
The Tyr-Ser Motif Modulates α2A-AR Export from the Golgi
We then searched the GPCR data base to see if the Tyr-Ser motif is conserved in the superfamily of GPCRs. The Tyr-Ser motif is highly conserved in the membrane-proximal N termini of α2A-, α2B-, and α2C-AR subtypes in different species (Fig. 9A). To test if the Tyr-Ser motif is also important for the export of other α2-ARs, we investigated the effect of mutating Tyr-28 and Ser-29 residues on the cell-surface expression and subcellular localization of α2A-AR, as the majority of α2C-AR is normally expressed intracellularly (28, 29). Similar to α2B-AR, mutation of Tyr-28 and Ser-29 significantly inhibited α2A-AR transport to the cell surface by 57 and 46%, respectively (Fig. 9B). Simultaneous mutation of Tyr-28 and Ser-29 (Y28A/S29A) blocked α2A-AR expression at the cell surface by 75%. Consistent with the attenuated cell-surface expression, the α2A-AR mutants Y28A, S29A (data not shown) and Y28A/S29A were accumulated in the perinuclear regions of the transfected cells (Fig. 9C) and were extensively co-localized with the Golgi marker GM130 (data not shown). These data indicate that Tyr-28 and Ser-29 are important for α2A-AR transport from the Golgi to the cell surface. These results together with the data obtained from α2B-AR suggest that the conserved Tyr-Ser motif may function as a common signal specifically modulating Golgi export of the α2-AR subfamily.
FIGURE 9. Effect of mutation of theTyr-Ser motif on the cell-surface expression and subcellular localiza-tion of α2A-AR.

A, the conserved Tyr-Ser motif in the N termini of α2A-, α2B-, and α2C-AR subtypes. The entire N-terminal sequencesof α2A-, α2B-, and α2C-ARs from human, rat, and mouse are shown. The Tyr-Ser motifs are underlined. B, specific [3H]RX821002 binding to intact HEK293T cells transfected with α2A-AR and its mutants Y28A, S29A, and Y28A/S29A (YS-AA) as described in the legend of Fig. 1. The mean values of specific [3H]RX821002 binding were 18873 ± 1432,8115 ± 2264,10568 ± 1321,and 4718 ± 755 cpm (n = 3)fromcells transfected with α2A-AR, Y28A, S29A, and Y28A/S29A, respectively. The data shown are percentages of the mean value obtained from cells transfected with WT α2A-AR and are presented as the mean ± S.E. of three experiments. *, p < 0.05 versus cells transfected with α2A-AR. C, subcellular localization of α2A-AR and Y28A/ S29A. GFP-conjugated WT and mutated α2A-AR were transiently expressed in HEK293T cells, and their subcellular distribution was revealed by fluorescence microscopy as described under “Experimental Procedures.” The data shown are representative images of at lease three independent experiments. Scale bar, 10 μm.
DISCUSSION
The studies on the intrinsic structural determinants for GPCR export trafficking have been mainly focused on the C termini of the receptors. A number of sequences essential for exit from the ER have been identified in the membrane-proximal C termini of GPCRs including the EX3LL motif in vasopres-sin V2 receptor, the FX3FX3F motif in dopamine D1 receptor, and the FNX2LLX3L motif in vasopressin V1b/V3 receptor (15-17). We have recently identified the FX6LL motif, which is necessary for export of α2B-AR and AT1R from the ER (14). In contrast to the C termini, the roles of the N termini in regulating GPCR export trafficking have been much less investigated and remain controversial. For instance, whereas proteolytic cleavage of the N-terminal 64 amino acid residues reduces the expression of the endothelin B receptor at the cell surface, removal of the N termini facilitates the cell-surface transport of α1D-AR and cannabinoid receptor 1 (CB1), and deletion of the N terminus does not influence α1B-AR transport to the cell surface (30-32). In the present study we determined the role of N termini in the export of α2-ARs from the ER through the Golgi to the cell surface. Our data demonstrated that, similar to the intracellular C terminus, the extracellular N terminus is essential for cell-surface targeting of α2B-AR. First, the α2B-AR-12 mutant lacking the N-terminal 12 residues was unable to transport to the cell surface, as quantified by intact cell radioli-gand binding. Second, consistent with the lack of receptor cell-surface expression, α2B-AR-12 was unable to activate ERK1/2. Third, subcellular localization analysis revealed that α2B-AR-12 was extensively trapped in the ER. These results together with our previous data (14) strongly indicate that both extracellular and intracellular terminal tails of α2B-AR contain structural determinants for its targeting to the cell surface.
The most important finding in this manuscript is the identification of three specific amino acid residues, Met-6, Tyr-12, and Ser-13, within the N terminus by an alanine-scanning mutagenesis approach, which are essential for proper α2B-AR export at discrete intracellular compartments. M6A mutant was completely unable to transport to the cell surface, whereas Y12A and S13A transport to the cell surface was markedly attenuated. Double mutation of Tyr-12 and Ser-13 (Y12A/S13A) more profoundly inhibited α2B-AR transport to the cell surface. Consistently, ERK1/2 activation by M6A, Y12A, S13A, and Y12A/S13A mutants was significantly blocked. More importantly, subcellular co-localization of the mutated receptors with intracellular organelle markers demonstrated that M6A, Y12A, S13A, and Y12A/S13A mutants were arrested in distinct subcellular compartments. M6A mutant was extensively co-localized with the ER marker pDsRed2-ER and pECFP-ER, indicating M6A mutant was unable to export from the ER. In contrast to M6A mutant, Y12A, S13A, and Y12A/S13A mutants were markedly co-localized with the Golgi marker GM130 but not the ER marker pDsRed2-ER and the TGN marker p230, indicating that they were able to exit from the ER and were further transported to the Golgi compartment but could not export from the Golgi to reach the TGN. These data indicate that the N terminus of α2B-AR possesses multiple signals that are required for the export from the ER and the Golgi.
There are at least two possibilities regarding the accumulation of α2B-AR in the Golgi induced by mutation of the Tyr-Ser motif. It is possible that the Tyr-Ser motif regulates α2B-AR export from the Golgi, and mutation of this motif results in defective export of the receptor from the Golgi. It is also possible that mutation of the Tyr-Ser motif does not alter α2B-AR export trafficking, but the mutated receptors are constitutively internalized from the plasma membrane to the intracellular compartments in the absence of agonists. To dissect these possibilities, we determined the effect of blocking endocytotic and anterograde transport pathways on the cell-surface expression of the Tyr-Ser motif mutants. Our data demonstrated that the internalization blockers Arr3-(201–409), DynK44A, and Rab5S34N did not influence the cell-surface expression and subcellular localization of the Tyr-Ser motif mutants. In contrast, inhibition of the anterograde ER-to-Golgi transport by BFA treat-ment abolished cell-surface expression of the motif mutants and arrested them in the ER. These data indicate that the accumulation of the Tyr-Ser motif mutants in the Golgi is due to defective export rather than constitutive internalization.
The absolute requirement of the single Met-6 residue for the exit of α2B-AR from the ER is quite surprising. The role of the Met residue in GPCR trafficking has been sug-gested in opioid receptors as mutation of a Met residue in the third intracellular loop altered internalization and lysosomal targeting of the receptors (33). We demonstrated that the cell-surface expression of M6A mutant was significantly restored by reduced temperature during culture and the chemical chaper-one Me2SO. These data suggest that the function of Met-6 in modulating α2B-AR export from the ER is likely mediated through regulating proper receptor folding and that Met-6 is not involved in the insertion of the receptor into the ER membrane. We previously demonstrated that α2B-AR constitutively forms dimers in the ER, and dimerization of α2B-AR plays an important role in modulating its export from the ER. Our data showed that M6A was able to form dimers with WT α2B-AR, indicating that Met-6 is not involved in the dimerization of α2B-AR in the ER. Furthermore, we demonstrated that the function of Met-6 in modulating α2B-AR export depends on its hydrophobic prop-erty. It is possible that the hydrophobic characteristic of Met-6 is important for proper α2B-AR folding to achieve a conformation competent for passing the ER quality control mechanism.
Structural determinants for exit from the Golgi of the GPCR superfamily have not been identified. Olfactory and chemokine receptors have been reported to be released from the ER but accumulated in the Golgi (34, 35), suggesting that multiple trafficking steps are involved in the regulation of GPCR export. Our studies identified the first motif consisting of a Tyr and a Ser in the GPCR superfamily, which is crucial for export from the Golgi. Similar to Met-6, the function of Tyr-12 in α2-AR export is dependent on its hydrophobic property. However, the function of the Tyr-Ser motif is unlikely modulated by their phos-phorylation, as mutation of Tyr-12 and Ser-13 to Asp and Ser-13 to Thr produced an inhibitory effect on the α2B-AR export similar to their mutation to Ala. By searching the GPCR data base, we found that the Tyr-Ser motif is highly conserved in the membrane-proximal N termini of α2A-, α2B-, and α2C-AR subtypes in different species. Mutation of Tyr-28 and/or Ser-29 residues attenuated α2A-AR transport to the cell surface and induced α2A-AR accumulation in the Golgi. These data suggest that the Tyr-Ser motif in the N-terminal portion may provide a common mechanism for the Golgi export of this subgroup of adrenergic receptors.
The Tyr-Ser motif identified in the membrane-proximal N termini of α2-ARs is apparently different from other Tyr-based signals, which have been demonstrated to modulate intracellular trafficking at distinct steps for a variety of proteins including GPCRs (36). For example, the NPXY motif functions as an internalization signal for non-GPCRs (37-39), and the NPXXY motif, which is highly conserved in many GPCRs within the putative seventh transmembrane domain near the cytoplasmic face of the plasma membrane, is involved in the endocytotic trafficking of the receptors (40, 41). In contrast, the YXXQ motif (where Xis any amino acid, and Φ has a bulky hydrophobic side chain) modulates protein transport from the TGN to the lysosome or basolateral membranes in polarized cells (42-45). Therefore, the Tyr-Ser motif may represent a novel Tyr-based motif that is required for the transport of α2-ARs from the Golgi to the cell surface.
The molecular mechanism underlying the function of the Tyr-Ser motif in regulating α2-AR trafficking remains largely unknown. Our data show that the Tyr-Ser motif mutants were exported from the ER and transported to the Golgi. These data suggest that these mutants were correctly inserted into the ER membrane for export and properly folded to pass through the ER quality control mechanism. In addition, mutation of the Tyr-Ser motif did not alter α2B-AR ability to dimerize in the ER. It has been well demonstrated that ER export motifs such as DXE and FF motifs are decoded by physical interaction with components of the ER-derived COPII-coated vesicles, which exclusively transport cargo proteins from the ER to the ER-Golgi intermediate complex (46-48). Interestingly, the function of the FF motif can be functionally substituted by other dihydrophobic motifs such as YY, LL, II, and VV. The DXE motif also interacts with adaptor protein 3 to coordinate vesic-ular stomatitis virus glycoprotein export from the TGN (24). The function of the C-terminal motifs in the ER export of GPCRs, as discussed above, may be mediated through direct interaction with the transport machinery or by regulating correct receptor folding in the ER. However, the Tyr-Ser motif within the N terminus of α2-AR are positioned toward the lumen of the ER or the Golgi during the export process and are unable to directly interact with components of the transport machinery or other proteins in the cytoplasm. It is unlikely that the Tyr-Ser motif would function as linear independent export motifs directing α2-AR exit from the Golgi. Therefore, the molecular mechanism underlying the function of the Tyr-Ser motif in mediating α2-AR export is different from those pro-posed for the export motifs identified in the C termini of receptors and other membrane proteins.
The physiological functions of GPCRs are dependent on their precise localization in the cell, and defective GPCR transport from the ER through the Golgi to the cell surface is asso-ciated with the pathogenesis of a variety of human diseases. These diseases may result from defects in ER export (resulting in ER retention) and/or in the machinery for transport after ER exit. For example, numerous naturally occurring mutations in the GPCRs themselves prevent proper folding and lead to ER retention. Such mutations have been implicated in the inher-ited diseases such as nephrogenic diabetes insipidus, retinitis pigmentosa, and male pseudohermaphroditism (49-51). Further characterization of the molecular mechanisms of the motif-facilitated traffic of GPCRs may be used as an important foundation for developing new therapeutic strategies in treat-ing diseases by targeting GPCR export trafficking.
Acknowledgments
We are grateful to Stephen M. Lanier and Jeffery L. Benovic for sharing reagents. We also thank Fuguo Zhou and Erin Fugetta for superb technical assistance.
Footnotes
This work was supported by National Institutes of Health Grants R01GM076167 (to G. W.) and P20RR018766 (to G. W. and Stephen M. Lanier) and by the American Heart Association, Southeast affiliate (to C. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: GPCR, G protein-coupled receptor; AR, adrenergic receptor; AT1R, angiotensin II type 1 receptor; ER, endoplasmic reticulum; TGN, trans-Golgi network; GFP, green fluorescent protein; YFP, yellow fluorescent protein; ERK1/2, extracellular signal-regulated kinase 1/2; WT, wild type; BFA, brefeldin A; PBS, phosphate-buffered saline; HA, hemagglutinin.
REFERENCES
- 1.Premont RT, Inglese J, Lefkowitz RJ. FASEB J. 1995;9:175–182. doi: 10.1096/fasebj.9.2.7781920. [DOI] [PubMed] [Google Scholar]
- 2.Wess J. Pharmacol. Ther. 1998;80:231–264. doi: 10.1016/s0163-7258(98)00030-8. [DOI] [PubMed] [Google Scholar]
- 3.Duvernay MT, Filipeanu CM, Wu G. Cell. Signal. 2005;17:1457–1465. doi: 10.1016/j.cellsig.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 4.von Zastrow M. Life Sci. 2003;74:217–224. doi: 10.1016/j.lfs.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Marchese A, Chen C, Kim YM, Benovic JL. Trends Biochem. Sci. 2003;28:369–376. doi: 10.1016/S0968-0004(03)00134-8. [DOI] [PubMed] [Google Scholar]
- 6.Tan CM, Brady AE, Nickols HH, Wang Q, Limbird LE. Annu. Rev. Pharmacol. Toxicol. 2004;44:559–609. doi: 10.1146/annurev.pharmtox.44.101802.121558. [DOI] [PubMed] [Google Scholar]
- 7.Petaja-Repo UE, Hogue M, Laperriere A, Walker P, Bouvier M. J. Biol. Chem. 2000;275:13727–13736. doi: 10.1074/jbc.275.18.13727. [DOI] [PubMed] [Google Scholar]
- 8.Tai AW, Chuang JZ, Bode C, Wolfrum U, Sung CH. Cell. 1999;97:877–887. doi: 10.1016/s0092-8674(00)80800-4. [DOI] [PubMed] [Google Scholar]
- 9.McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- 10.Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- 11.Hague C, Uberti MA, Chen Z, Hall RA, Minneman KP. J. Biol. Chem. 2004;279:15541–15549. doi: 10.1074/jbc.M314014200. [DOI] [PubMed] [Google Scholar]
- 12.Salahpour A, Angers S, Mercier JF, Lagace M, Marullo S, Bouvier M. J. Biol. Chem. 2004;279:33390–33397. doi: 10.1074/jbc.M403363200. [DOI] [PubMed] [Google Scholar]
- 13.Overton MC, Chinault SL, Blumer KJ. J. Biol. Chem. 2003;278:49369–49377. doi: 10.1074/jbc.M308654200. [DOI] [PubMed] [Google Scholar]
- 14.Duvernay MT, Zhou F, Wu G. J. Biol. Chem. 2004;279:30741–30750. doi: 10.1074/jbc.M313881200. [DOI] [PubMed] [Google Scholar]
- 15.Schulein R, Hermosilla R, Oksche A, Dehe M, Wiesner B, Krause G, Rosenthal W. Mol. Pharmacol. 1998;54:525–535. doi: 10.1124/mol.54.3.525. [DOI] [PubMed] [Google Scholar]
- 16.Bermak JC, Li M, Bullock C, Zhou QY. Nat. Cell Biol. 2001;3:492–498. doi: 10.1038/35074561. [DOI] [PubMed] [Google Scholar]
- 17.Robert J, Clauser E, Petit PX, Ventura MA. J. Biol. Chem. 2005;280:2300–2308. doi: 10.1074/jbc.M410655200. [DOI] [PubMed] [Google Scholar]
- 18.Traub LM, Kornfeld S. Curr. Opin. Cell Biol. 1997;9:527–533. doi: 10.1016/s0955-0674(97)80029-4. [DOI] [PubMed] [Google Scholar]
- 19.Nishimura N, Plutner H, Hahn K, Balch WE. Proc. Natl. Acad. Sci. U. S. A. 2002;99:6755–6760. doi: 10.1073/pnas.092150699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stockklausner C, Klocker N. J. Biol. Chem. 2003;278:17000–17005. doi: 10.1074/jbc.M212243200. [DOI] [PubMed] [Google Scholar]
- 21.Wu G, Zhao G, He Y. J. Biol. Chem. 2003;278:47062–47069. doi: 10.1074/jbc.M305707200. [DOI] [PubMed] [Google Scholar]
- 22.Filipeanu CM, Zhou F, Claycomb WC, Wu G. J. Biol. Chem. 2004;279:41077–41084. doi: 10.1074/jbc.M405988200. [DOI] [PubMed] [Google Scholar]
- 23.Zhou F, Filipeanu CM, Duvernay MT, Wu G. Cell. Signal. 2006;18:318–327. doi: 10.1016/j.cellsig.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filipeanu CM, Zhou F, Fugetta EK, Wu G. Mol. Pharmacol. 2006;69:1571–1578. doi: 10.1124/mol.105.019984. [DOI] [PubMed] [Google Scholar]
- 25.DeGraff JL, Gagnon AW, Benovic JL, Orsini MJ. J. Biol. Chem. 1999;274:11253–11259. doi: 10.1074/jbc.274.16.11253. [DOI] [PubMed] [Google Scholar]
- 26.Wu G, Krupnick JG, Benovic JL, Lanier SM. J. Biol. Chem. 1997;272:17836–17842. doi: 10.1074/jbc.272.28.17836. [DOI] [PubMed] [Google Scholar]
- 27.DeGraff JL, Gurevich VV, Benovic JL. J. Biol. Chem. 2002;277:43247–43252. doi: 10.1074/jbc.M207495200. [DOI] [PubMed] [Google Scholar]
- 28.Wozniak M, Limbird LE. J. Biol. Chem. 1996;271:5017–5024. doi: 10.1074/jbc.271.9.5017. [DOI] [PubMed] [Google Scholar]
- 29.von Zastrow M, Link R, Daunt D, Barsh G, Kobilka B. J. Biol. Chem. 1999;268:763–766. [PubMed] [Google Scholar]
- 30.Grantcharova E, Furkert J, Reusch HP, Krell HW, Papsdorf G, Beyermann M, Schulein R, Rosenthal W, Oksche A. J. Biol. Chem. 2002;277:43933–43941. doi: 10.1074/jbc.M208407200. [DOI] [PubMed] [Google Scholar]
- 31.Andersson H, D'Antona AM, Kendall DA, von Heijne G, Chin CN. Mol. Pharmacol. 2003;64:570–577. doi: 10.1124/mol.64.3.570. [DOI] [PubMed] [Google Scholar]
- 32.Hague C, Chen Z, Pupo AS, Schulte NA, Toews ML, Minneman KP. J. Pharmacol. Exp. Ther. 2004;309:388–397. doi: 10.1124/jpet.103.060509. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Loh HH, Law PY. J. Biol. Chem. 2003;278:36848–58368. doi: 10.1074/jbc.M301540200. [DOI] [PubMed] [Google Scholar]
- 34.Gimelbrant AA, Haley SL, McClintock TS. J. Biol. Chem. 2001;276:7285–7290. doi: 10.1074/jbc.M005433200. [DOI] [PubMed] [Google Scholar]
- 35.Venkatesan S, Petrovic A, Van Ryk DI, Locati M, Weissman D, Murphy PM. J. Biol. Chem. 2002;277:2287–2301. doi: 10.1074/jbc.M108321200. [DOI] [PubMed] [Google Scholar]
- 36.Bonifacino JS, Traub LM. Annu. Rev. Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 37.Chen WJ, Goldstein JL, Brown MS. J. Biol. Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- 38.Collawn JF, Kuhn LA, Liu LF, Tainer JA, Trowbridge IS. EMBO J. 1991;10:3247–3253. doi: 10.1002/j.1460-2075.1991.tb04888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouley R, Sun TX, Chenard M, McLaughlin M, McKee M, Lin HY, Brown D, Ausiello D. Am. J. Physiol. Cell Physiol. 2003;285:750–762. doi: 10.1152/ajpcell.00477.2002. [DOI] [PubMed] [Google Scholar]
- 40.Kalatskaya I, Schussler S, Blaukat A, Muller-Esterl W, Jochum M, Proud D, Faussner A. J. Biol. Chem. 2004;279:31268–31276. doi: 10.1074/jbc.M401796200. [DOI] [PubMed] [Google Scholar]
- 41.Lee TH, Linstedt AD. Mol. Biol. Cell. 2000;11:2577–2590. doi: 10.1091/mbc.11.8.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams MA, Fukuda M. J. Cell Biol. 1990;111:955–966. doi: 10.1083/jcb.111.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harter C, Mellman I. J. Cell Biol. 1992;117:311–325. doi: 10.1083/jcb.117.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunziker W, Harter C, Matter K, Mellman I. Cell. 1991;66:907–920. doi: 10.1016/0092-8674(91)90437-4. [DOI] [PubMed] [Google Scholar]
- 45.Rajasekaran AK, Humphrey JS, Wagner M, Miesenbock G, Le Bivic A, Bonifacino JS, Rodriguez-Boulan E. Mol. Biol. Cell. 1994;5:1093–1103. doi: 10.1091/mbc.5.10.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishimura N, Balch WE. Science. 1997;277:556–558. doi: 10.1126/science.277.5325.556. [DOI] [PubMed] [Google Scholar]
- 47.Fiedler K, Veit M, Stamnes MA, Rothman JE. Science. 1996;273:1396–1399. doi: 10.1126/science.273.5280.1396. [DOI] [PubMed] [Google Scholar]
- 48.Nufer O, Guldbrandsen S, Degen M, Kappeler F, Paccaud JP, Tani K, Hauri HP. J. Cell Sci. 2002;115:619–628. doi: 10.1242/jcs.115.3.619. [DOI] [PubMed] [Google Scholar]
- 49.Stojanovic A, Hwa J. Receptors Channels. 2002;8:33–50. [PubMed] [Google Scholar]
- 50.Morello JP, Bichet DG. Annu. Rev. Physiol. 2001;63:607–630. doi: 10.1146/annurev.physiol.63.1.607. [DOI] [PubMed] [Google Scholar]
- 51.Themmen AP, Brunner HG. Eur. J. Endocrinol. 1996;134:533–540. doi: 10.1530/eje.0.1340533. [DOI] [PubMed] [Google Scholar]