

Abstract
DNA-templated organic synthesis enables the translation, selection, and amplification of DNA sequences encoding synthetic small-molecule libraries. Previously we described the DNA-templated multistep synthesis and model in vitro selection of a pilot library of 65 macrocycles. In this work we report several key developments that enable the DNA-templated synthesis of much larger (> 10,000-membered) small-molecule libraries. We developed and validated a capping-based approach to DNA-templated library synthesis that increases final product yields, simplifies the structure and preparation of reagents, and reduces the number of required manipulations. To expand the size and structural diversity of the macrocycle library, we augmented the number of building blocks in each DNA-templated step from four to 12, selected eight different starting scaffolds which result in four macrocycle ring sizes and two building-block orientations, and confirmed the ability of the 36 building blocks and eight scaffolds to generate DNA-templated macrocycle products. We computationally generated and experimentally validated an expanded set of codons sufficient to support 1,728 combinations of step 1, step 2, and step 3 building blocks. Finally, we developed new high-resolution LC/MS analysis methods to assess the quality of large DNA-templated small-molecule libraries. Integrating these four developments, we executed the translation of 13,824 DNA templates into their corresponding small-molecule macrocycles. Analysis of the resulting libraries is consistent with excellent (> 90%) representation of desired macrocycle products and a stringent test of sequence specificity suggests a high degree of sequence fidelity during translation. The quality and structural diversity of this expanded DNA-templated library provides a rich starting point for the discovery of functional synthetic small-molecule macrocycles.
Introduction
Functional molecules emerge in living systems through cycles of translation, selection, and amplification with mutation. Scientists have successfully adapted features of biological evolution to create DNA, RNA, and protein molecules with tailor-made binding or catalytic properties. The scope of this approach has traditionally been restricted to biopolymers that can be accessed through the use of DNA or RNA polymerases1-4 or the ribosome.5-8 The potential benefits of applying translation and selection-based methods to the discovery of functional synthetic molecules has inspired new approaches to addressing the problem of translating DNA sequences into structures not necessarily compatible with polymerase enzymes or ribosomal machinery. For example, our lab has developed a DNA-templated synthesis approach to translating DNA sequences into synthetic small molecules and synthetic polymers in which Watson-Crick base pairing to a DNA template recruits DNA-linked reagents to perform each synthetic step in a sequence-programmed manner.9-15 Harbury and co-workers have developed an elegant “DNA display” method in which resin-bound DNA hybridization directs split-and-pool combinatorial synthesis, and have used DNA display to generate libraries of linear peptides and peptoids.16-19 Neri and co-workers have used DNA base pairing to bring together noncovalent combinations of small pharmacophores that can suggest the synthesis of covalent small molecules with desired binding properties.20-23
These approaches to the central problem of translating nucleic acids into corresponding synthetic compounds are relatively recent developments, and their ability to provide molecules with desired functional properties will depend on their applicability to appropriately sized libraries of synthetic structures. Past efforts to synthesize and evaluate libraries of synthetic small molecules have resulted in the discovery of chemical genetic probes that perturb specific cellular functions in vivo in a temporally controlled, dose-dependent, and reversible manner,24-30 as well as the discovery of lead compounds for the development of new therapeutic agents.31-33
Macrocycles are of special interest for the development of biologically active small molecules.34-46 The increased rigidity of macrocyclic compounds can greatly decrease the entropic cost of their binding to biological targets, resulting in higher potential binding affinities and specificities than corresponding linear compounds.34,36,41,47-53 In addition, macrocyclic peptide-like structures can possess higher bioavailability, membrane permeability, and resistance to in vivo degradation than their linear counterparts.34,36,40,41,54-58 These advantages likely account for the significant representation of macrocyclic compounds among biologically active secondary metabolites.38,39
Despite these attractive features, the availability and use of small- and medium-sized macrocycles have been limited by the challenges associated with their synthesis.31,34,59-62 Macrocyclization reactions can be very sensitive to small structural variations within linear precursors,60,63 can be especially difficult for small peptide substrates that form medium-sized macrocycles,61,64-66 and can be prone to multimeric side-products arising from intermolecular bond formation.61,62 These challenges are magnified in a solid-phase library synthesis format in which the removal of truncated, multimeric, and acyclic byproducts from desired macrocycles (all of which are linked to the same bead) can be difficult.
We hypothesized that several features of DNA-templated synthesis could facilitate access to macrocycles. DNA-templated synthesis is compatible with aqueous solvent,67 extremely low (nM) reactant concentrations,9 and selection-based purification methods for bond formation or bond cleavage that are not available to solid-phase synthesis10— all factors that could promote macrocycle formation or facilitate macrocycle isolation. We also anticipated that the ability of base pairing to hold together relevant reactive groups at elevated effective molarities during the macrocyclization step would further assist the ring closure reaction. Indeed, these features enabled the successful DNA-templated synthesis and model in vitro selection of a pilot library of 65 macrocycles.15
Here, we report the DNA-templated synthesis of a large library of synthetic macrocycles suitable for in vitro selection. Achieving this goal required several significant methodological advances. We developed a more robust and efficient library synthesis route to these compounds and conducted a thorough study of the compatibility of 36 building blocks and eight scaffolds in DNA-templated macrocycle synthesis. Guided by our previous studies on template secondary structure,68 we designed a new set of DNA codons to support large-scale library synthesis. We confirmed the efficacy, quality, and sequence fidelity of our library synthesis method by examining a series of small-molecule sublibraries with PAGE and high resolution LC/MS analysis. Integrating these findings, we completed the DNA-templated synthesis of the full library of 13,824 macrocycles and generated sufficient material for hundreds of in vitro selections against biological targets of interest.
Materials and Methods
All chemicals, unless otherwise specified, were purchased from Sigma-Aldrich. All reagents for DNA synthesis, including modified phosphoramidites and CPG resins, were purchased from Glen Research. All reactions were performed at 25 °C unless otherwise noted. All buffers were prepared at 25 °C to match reaction conditions.
DNA Synthesis
DNA oligonucleotides were synthesized with a PerSeptive Biosystems Expedite 8090 DNA synthesizer or with a Bioautomation Corporation MerMade 12 DNA synthesizer, using standard phosphoramidite protocols. Oligonucleotides were purified by reverse-phase HPLC (Agilent) using a gradient of acetonitrile (8% to 80%) in 100 mM triethylammonium acetate (TEAA), pH 7.0. DNA was synthesized on standard CPG (1000 angstrom) beads, unless otherwise noted. The oligonucleotides were quantitated by UV spectroscopy (except where otherwise noted) using a NanoDrop ND-1000 Spectrophotometer.
Secondary Structure Prediction for Codon Screening (see also Reference 68)
Secondary structure prediction was performed using the Oligonucleotide Modeling Platform (OMP, DNA Software, Inc.) with the following parameters: 25 °C in 1.0 M NaCl with 100 nM template. Reagent-template hybridization was simulated at 150 nM reagent and 100 nM template under the same conditions.
DNA Templates
DNA templates were made by the enzymatic ligation of two oligonucleotides mediated by a complementary splint oligonucleotide. The 3′ halves of the templates (24, 27 or 33 bases each) were synthesized with a 5′-phosphate group using the Chemical Phosphorylation Reagent II (CPR-II) phosphoramidite. The 5′ halves of the templates (21 or 24 bases each) were prepared using the 5′-Amino-5 phosphoramidite; solid-phase synthesis protocols were used (see Supporting Information) to append the scaffold to the 5′ template halves following DNA synthesis. Ligation reactions contained a 1:1:1 mixture of 5′-half:3′-half:splint, T4 DNA ligase and T4 DNA Ligase Buffer (New England Biolabs). Ligation reactions were maintained at 16 °C for 3 h. Ligation products were precipitated with ethanol, 0.1 volumes of 3 M NaOAc (pH 5.0), and 50 μg of glycogen/mL (Roche) and purified by PAGE. DNA was recovered by excising the desired acrylamide gel band, and crushing, freezing, and thawing the excised gel bands in 400 μL 10 mM Tris (pH 7.5) with 1 mM EDTA. Tubes containing the resulting material were shaken for 3 h at 37 °C, and the desired DNA was recovered by removal of the polyacrylamide using an Ultrafree-MC centrifuge filter (Millipore) and precipitation with ethanol as described above.
1a: 5′-(scaffold)-GCATGTCTACCACGTTCTGAGCACACTGACTCCACTGTACACCTCGAG
5′-segment for 1a: 5′-(scaffold)-GCATGTCTACCACGTTCTGAGCAC
3′-segment for 1a: 5′-(P)-ACTGACTCCACTGTACACCTCGAG
splint for 1a : 5′-GTGGAGTCAGTGTGCTCAGAAC
1b: 5′-(scaffold)-CCCTGTACACTTCCTCAAGTTGCTGAAATGATGGCTTTCTACCCACTC
5′-segment for 1b: 5′-(scaffold)- CCCTGTACACTTCCTCAAGTT
3′-segment for 1b: 5′-(P)- GCTGAAATGATGGCTTTCTACCCACTC
splint for 1b: 5′- ATCATTTCAGCAACTTGAGGAA
The general template structure for libraries 21 (12×1×1), 22 (1×12×1), and 23 (1×1×12) is shown below; each codon is six bases in length:
5′-(scaffold)-CCCTGTACAC[codon 3]AAGTT[codon 2]ATGAT[codon 1]CTACCCACTC
The 12-membered DNA template libraries that produced macrocycle libraries 21, 22, and 23 were assembled by ligating the appropriate oligonucleotide 5′-halves and 3′-halves in a one-pot mixture using the corresponding splint oligonucleotides, as described above. Templates were then purified by PAGE, as described above. The sequences for 21 (12×1×1), 22 (1×12×1), and 23 (1×1×12) are as follows:
21 - 5′-(scaffold)-CCCTGTACACTTCCTCAAGTTGCTGAAATGAT[codon 1]CTACCCACTC
5′-segment for 21 - 5′-(scaffold)- CCCTGTACACTTCCTCAAGTT
3′-segments for 21 (12 in total): - 5′-(P)-GCTGAAATGAT[codon 1]CTACCCACTC
Splint for 21 – 5′- ATCATTTCAGCAACTTGAGGAA
22 - 5′-(scaffold)-CCCTGTACACTTCCTCAAGTT[codon 2]ATGATGGCTTTCTACCCACTC
5′-segment for 22 - 5′-(scaffold)- CCCTGTACACTTCCTCAAGTT
3′-segments for 22 (12 in total): - 5′-(P)- [codon 2]ATGATGGCTTTCTACCCACTC
Splints for 22 (12 in total) - 5′- ATCAT[anticodon 2]AACTTGAGGAA
23- 5′-(scaffold)-CCCTGTACAC[codon 3]AAGTTGCTGAAATGATGGCTTTCTACCCACTC
5′-segments for 23 (12 in total) - 5′-(scaffold)- CCCTGTACAC[codon 3]AAGTT
3′-segment for 23 - 5′-(P)- GCTGAAATGATGGCTTTCTACCCACTC
Splints for 23 (12 in total) - 5′- ATCATTTCAGCAACTT[anticodon 3]
The general template structure for template libraries 24, 26, and 28 is as follows; each building-block codon is six nucleotides, while the scaffold codon is three nucleotides (scaffold codons are listed in the Supporting Information, Figure S2):
5′-(scaffold)-CCCTGTACAC[codon 3]AAGTT[codon 2]ATGAT[codon 1]CTA[scaffold codon]-CATCCCACTC
Template libraries 24, 26, and 28 were assembled by ligation of the appropriate oligonucleotide 5′-halves and 3′-halves in a one-pot mixture using the corresponding splint oligonucleotides, as described above. Templates were then purified by PAGE, as described above. The sequences for 24, 26, and 28 are as follows:
5′-segments of 24, 26, and 28 - 5′-(scaffold)-CCCTGTACAC[codon 3]AAGTT
3′-segments of 24, 26, and 28 - 5′-(P)- [codon 2]ATGAT[codon 1]CTA[scaffold codon]-CATCCCACTC
Splints for 24, 26, and 28 - 5′- ATCAT[anticodon 2]AACTT[anticodon 3]
12-membered template library 24 was synthesized by performing 12 separate splint ligations (3A + [2A-1A], 3B + [2B-1B],… 3L + [2L-1L]); each ligation used one 5′-half, one 3′-half, and the corresponding splint. The resulting 12 templates were purified by PAGE, as described above. The templates were pooled together to form a final 12-membered template library 24.
Template libraries 26 and 28 were produced by one-pot mixed ligations. The 3′-segments were assembled by split/pool oligonucleotide synthesis: the first round of synthesis (5′-ATGAT[codon 1]CTA[scaffold codon]CATCCCACTC) was performed in 12 separate columns; the CPG was then removed from the columns and pooled into a single container using acetonitrile. After agitation by vortexing, the beads were redistributed to the columns evenly, and subjected to a second round of oligonucleotide synthesis (5′-[codon 2]). The CPG was pooled again, shaken, and transferred to a column to add the 5′-CPR-II phosphoramidite. The oligonucleotides were cleaved from CPG, prepared, and purified following standard protocols for use in the ligation reaction with their corresponding 5′-segments. This produced a mixture containing 144 different 3′-segments (12×12) per split/pool synthesis.
The splints for template libraries 26 and 28 were also generated by split/pool oligonucleotide synthesis: the first round of synthesis (5′- AACTT[anticodon 3]) was performed in 12 separate columns; the CPG was then removed from the columns and pooled into a single container using acetonitrile. After agitation by vortexing, the beads were redistributed to the columns evenly, and subjected to a second round of oligonucleotide synthesis (5′- ATCAT[anticodon 2]). The CPG was pooled, and the oligonucleotides were cleaved from CPG and purified as above. This produced a mixture containing 144 different splint oligonucleotides (12×12) per split/pool synthesis.
Template library 26 was produced by using the Lys-s scaffold codon (5′-AAC) for the 3′-segments; the Lys-s 5-scaffold (see Supporting Information) was attached to the 5′-segments. The ensuing ligation, using 144 different 3′-segments, 12 different 5′-halves, and 144 splints, produced library 26 with 1,728 different templates (12×144).
Template library 28 was produced by performing the synthesis of library 26 for each corresponding 5′-scaffold/3′-scaffold codon pair. The eight different libraries (1,728 different templates each) were then pooled together to form the complete template library 28 consisting of 13,824 different templates.
The identities of all appended 5′ scaffolds were confirmed by S1 nuclease digestion of the templates (see below), followed by high-resolution LC/MS analysis (Supporting Information, Figure S3).
Preparation of Step 1 and Step 2 DNA–Linked Reagents
The commercial sources for the amino acids are listed in the Supporting Information, Figures S5, S6, and S7.
For hydrophobic amino acids
The corresponding reagent oligonucletides were synthesized using 3′-Amino-C7-CPG beads (500 Å), and prepared using standard protocols. To attach the amino acid, 50 μL of a ∼2 mM solution of the appropriate 3′-amine-terminated DNA oligonucleotide was mixed with 50 μL of 100 mM amino acid in 1 M sodium phosphate buffer (pH 7.0). To the resulting solution 25 uL of 100 mg/mL bis-[2-(succinimidooxycarbonyloxy)-ethyl]sulfone (BSOCOES, Pierce) in DMF was added. The resulting mixture was sonicated to homogeneity and agitated for 1 hr at 25 °C. The product was diluted to 200 μL total, desalted by gel filtration (Nap-5, Amersham Biosciences), and purified by reverse-phase HPLC using a gradient of acetonitrile (8%-80%) in 100 mM TEAA buffer (pH 7.0).
For hydrophilic amino acids
When coupled to DNA, certain amino acids (e.g., glycine) generate desired oligonucleotide conjugates that co-elute on the HPLC with undesired BSOCOES-linked oligonucleotide byproducts lacking the amino acid. To avoid this problem, the concentration of amino acid was increased from 100 mM to 250 mM. The remainder of the synthesis and purification was performed as described above.
Preparation of Step 3 DNA-Linked Reagents
Preparation of 3′-phosphine reagents
The corresponding oligonucleotides were synthesized on 3′-Amino-C7-CPG beads (500 Å). The 3′ FMOC group on the beads was removed using three consecutive washes with 20% piperidine in DMF (5 min agitation at 25 °C for each wash). The beads were then washed with DMF and acetonitrile. In a separate container, 4-(diphenylphosphino)-benzoic acid was activated by reaction with N,N′-dicyclohexyl carbodiimide (DCC) (100 μmol), 1-hydroxybenzotriazole (HOBt) (100 μmol), and N,N-diisopropylethylamine (DIPEA) (100 μmol) in 600 μL dry DMF for 1 hr. The crude product was filtered through a glass frit to remove the N,N′-dicyclohexylurea (DCU) and added to the oligonucleotide beads with agitation for 4 h. The beads were washed with DMF and acetonitrile. Treatment for 10 min with a solution of 50:50 40% aqueous ammonium hydroxide:methylamine (v:v, AMA) and 1 mg/mL tris-(2-carboxyethyl)phosphine hydrochloride (TCEP) at 65 °C deprotected the modified oligonucleotide and cleaved it from the resin. Products were dried in vacuo and purified by reverse-phase HPLC using a gradient of acentonitrile (8%-80%) in 100 mM TEAA (pH 7.0). After lyophilization, the samples were resuspended in 10 mM sodium phosphate buffer (pH 7.0), divided into 100 uL aliquots (∼100 nmol of DNA each), and immediately frozen for later use.
For hydrophilic amino acids
50 μL of 500 mM amino acid in 1 M sodium phosphate buffer (pH 7.0) was combined with 50 μL of 50 mg/mL SIA (N-succinimidyl-iodoacetamide, Pierce) in DMF and agitated for 5 min. The 3′-4-(diphenylphosphino)-benzoic acid amide-linked oligonucleotide solution (∼100 uL of 1 mM DNA in 10 mM sodium phosphate buffer, pH 7.0) was then added and the resulting mixture was agitated for 2 h. The mixture was then diluted to 300 μL total, desalted by gel filtration (Nap-5), and purified by reverse-phase HPLC using a gradient of acetonitrile (8%-80%) in 100 mM TEAA (pH 7.0).
For hydrophobic amino acids
To improve the yield of reactions with sparingly soluble amino acids (e.g., cyclohexylstatine), the following protocol was used: 50 μL of 500 mM amino acid in 500 mM NaOH was combined with 50 μL of 50 mg/mL SIA in DMF and agitated for 5 min. To this solution, 50 μL of 1 M sodium phosphate buffer (pH 7.0) was added, followed by the 3′-4-(diphenylphosphino)-benzoic acid-amide linked oligonucleotide solution (100 μL of 1 mM DNA in 10 mM sodium phosphate buffer, pH 7.0). The resulting mixture was agitated for 2 h. Desalting and purification were performed as described above.
The masses of all reagents were confirmed by MALDI-TOF mass spectrometry (see Supporting Information, Figures S5, S6, and S7).
DNA-Templated Reactions and Capping Reactions
DNA-templated reactions were performed at concentrations of 120 nM template, or in the case of combinatorial syntheses, 120 nM per template codon (1.44 μM template total). A concentration of 144 nM of each reagent was used (1.2 equivalents). Small-scale reactions were performed in a non-stick 1.7 mL microcentrifuge tube (VWR). Large-scale reactions (volume > 1.5 mL) were performed in a 30 mL Oak Ridge polypropylene centrifuge tube (VWR).
Steps 1, 2, and 3 amine acylation
Templates and reagents were combined in a solution containing 1 M NaCl, 100 mM pH 6.0 sodium 2-(N-morpholino)ethanesulfonate buffer (MES), 20 mM N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), and 15 mM sulfo-N-hydroxysuccinimide (sNHS). 10% v/v acetonitrile was present during library reactions (templates 24, 26, and 28) to minimize binding to plasticware. The solution was briefly heated to 55 °C and cooled to 25 °C prior to the addition of EDC to facilitate DNA hybridization. After the addition of EDC, the reaction was briefly agitated and left to react for 3 h at 25 °C.
Steps 1 and 2 capping
To the previous reaction mixture (after DNA-templated reaction), 1 μL of acetic anhydride was added per 200 μL of reaction solution (53 mM final concentration). This reaction was briefly agitated and left to react for 2 h at 25 °C.
Steps 1 and 2 cleavage
To the previous reaction mixture (after capping), 25% v/v of 1 M NaOH was added to raise the pH and trigger sulfone linker cleavage.10 The solution was briefly agitated and left to react for 30 min at 25 °C. The pH was adjusted with 0.2 vol of 3 M NaOAc (pH 5.0) with 50 μg glycogen/mL and the material was recovered by precipitation with isopropanol.
Macrocyclization and Purification of Macrocyclic Products
The biotinylated step 3 products were captured from the reaction mixture using PhyTip 1000+ columns loaded with streptavidin-linked resin (Phynexus). The resin was washed once with 100 mM pH 12.0 sodium N-cyclohexyl-3-aminopropanesulfonate buffer (CAPS) at 45 °C, and three times with water (45 °C). The resin was then exposed (∼3 min) to a solution of 50 mM NaIO4, 500 mM NaOAc pH 3.5, washed with 500 mM NaOAc pH 3.5, washed with 25 mM NaOAc pH 3.5, and exposed to 100 mM pH 8.5 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) to induce macrocyclization and elution (1 h). Remaining product was eluted from the resin by washing with 10% acetonitrile in water (v/v). The HEPES solution and the 10% acetonitrile solution were combined and precipitated with ethanol as described above to recover macrocyclic products.
Characterization of DNA-Linked Species by Restriction Digestion and MALDI-TOF for 1a-8a
The 5′ tripeptide scaffold product (0.5-10 pmol) was hybridized with 50 pmol of a 5′- and 3′-biotinylated digestion oligonucleotide (10-mer, 5′-biotin-GTAGACATGC-biotinTEG) in 10-30 μL of NEB 4 buffer (New England Biolabs). The digestion reaction with restriction endonuclease Hpy8I (5 U, Fermentas), which cleaves the sequence 5′-GTNNAC-3′, was added to effect template cleavage after the seventh nucleotide. The digestion reaction was incubated for 1 h at 37 °C. The digestion oligonucleotide byproducts were removed by binding to the streptavidin-linked magnetic beads (80 μL, Roche). The small-molecule products, linked to seven template nucleotides, were collected from the supernatant and desalted using Zip-Tips (Millipore). The sample was eluted directly onto a MALDI analysis plate using 1 μL of 8:1 (50 mg/mL THAP in 1:1 water:acetonitrile):(50 mg/mL ammonium citrate in water). Data was collected on a Voyager DE MALDI-TOF mass spectrometer (PerSeptive Biosystems) operated in reflector mode.
Expected mass (positive ion) for 1a = 2612.6 Da, observed mass = 2615.3.2±6 Da.
Expected mass (positive ion) for 5a =2759.7 Da, observed mass = 2762.4±6 Da.
Expected mass (positive ion) for 6a = 2896.7 Da, observed mass = 2901.2±6 Da.
S1 Nuclease Digestion for High-Resolution LC/MS Analysis
Product was added to 50 μL pH 4.5 NH4OAc buffer in a glass LC/MS vial. S1 nuclease (1 μL, 60 U, Promega) was then added. Digestion was performed at 37 °C for 2 h. The mixture was frozen, lyophilized, and resuspended in 0.1% formic acid for LC/MS analysis.
High resolution LC/MS characterization
Water was obtained from a Milli-Q water purification system (Millipore). All solvents and reagents used were mass spectrometry grade, including methanol and acetonitrile (Fisher Scientific). For the LC/MS analysis of 12-membered library 25 ultra-performance liquid chromatography (UPLC) was carried out with an ACQUITY UPLC system (Waters) using a Waters BEH C18 column (2.1 mm i.d. × 100 mm, 1.7-μm particles) and a linear gradient of 0.1% (v:v) formic acid in acetonitrile (0%-100%) in 0.1% (v:v) aqueous formic acid. The eluent was directly injected into a Q-TOF Premier mass spectrometer (Waters) fitted with an electrospray interface. Data was acquired and processed with MassLynx 4.1 software (Waters). Mass spectra were recorded in the negative mode within a m/z range of 100-1200. The masses for macrocycle libraries 21, 22, and 23 are shown in the Supporting Information, Figures S8, S9, and S10.
NanoLC/MS analysis of 1728-membered library 27
LC/MS analysis was performed on a Q-TOF Premier mass spectrometer equipped with a nano-electrospray interface and the nanoACQUITY UPLC system (Waters). The nano-LC used a flow of a rate of 0.2 μL per min through a BEH C18 nanoLC column (Waters, 75 μm i.d. × 360 μm o.d. 10 cm, 1.7-μm particles) and a gradient of methanol (1% to 99%) in 6 mM aqueous triethylammonium bicarbonate (TEAB). The nanoLC column was connected to a PicoTip Emitter (Waters, tubing i.d. 360 μm, o.d. 20 μm, tip i.d. 10 μm) by means of a ZVD micro-union that allowed for a dead-volume-free butt connection. The column was optimized for maximum sensitivity of a standard raffinose (100 ng/nL) signal in an identical mobile phase. Data was acquired and processed with MassLynx 4.1 software. Mass spectra were recorded in negative ion mode within the m/z range of 750-1300. Masses were identified with a minimum mass accuracy of ±0.05 Da.
Results and Discussion
Development of a Capping-Based DNA-Templated Macrocycle Library Synthesis
Previously we reported a DNA-templated route for the preparation of a library of DNA-linked synthetic macrocycles from a corresponding library of lysine-linked DNA oligonucleotides (1, Figure 1).15 Our original synthesis began with two successive DNA-templated amine acylation reactions, each consisting of carbodiimide-mediated amide bond formation using biotinylated DNA-linked amino acid building blocks (2, 3), capture of the desired reaction intermediate using streptavidin-linked beads, and base-induced linker cleavage to release the coupled reaction product (5, 6). A third DNA-templated amine acylation step was then performed using a biotinylated DNA-linked building block containing a novel Wittig ylide linker (4). After capture of the step 3 reaction product using streptavidin-linked beads, the immobilized product was exposed to aqueous NaIO4 to unmask a side-chain-linked aldehyde (7); elevating the pH of the buffer to 8.5 then induced Wittig olefination and macrocyclization with simultaneous cleavage of the biotinylated linker. The cleavage of this linker caused the desired macrocycle product (but not unreacted macrocycle precursors or step 3 reagents) to self-elute from the beads (8). Using this strategy, we generated a pilot library of 65 unique macrocyclic small molecules in a DNA sequence-programmed manner.15 While effective, this approach required that the products of each DNA-templated step undergo capture with streptavidin-linked beads, washing, elution, and buffer exchange. The removal of these requirements in a manner that does not compromise library quality would significantly enhance the ease and efficiency of DNA-templated library synthesis.
Figure 1.
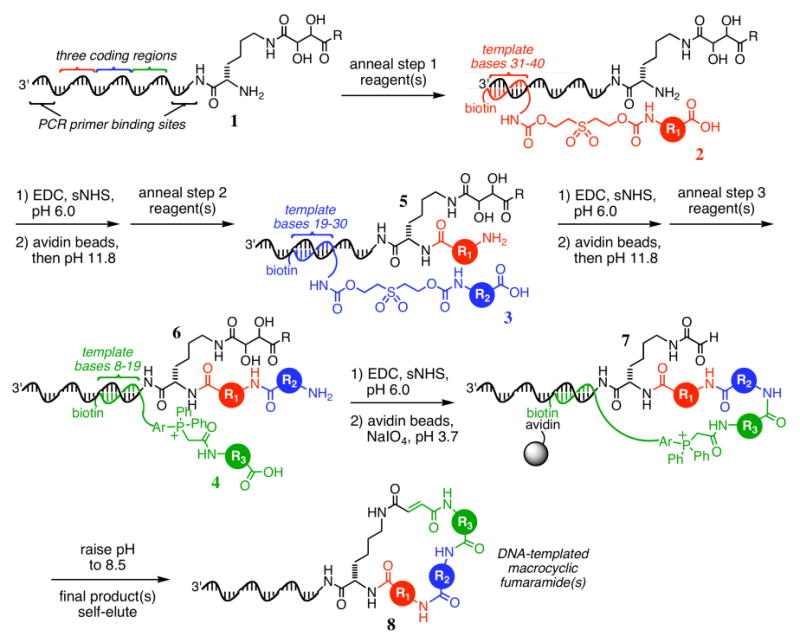
The previously reported15 scheme for the synthesis of a DNA-templated macrocycle library, where R is −NHCH3 or tryptamine, and Ar is −(p-C6H4)–. In contrast with the capping-based method described in the present work, each DNA-templated reaction in this scheme requires a bond formation step, a streptavidin bead capture step, a washing step, and a product elution step.
In order to extend this DNA-templated synthesis to > 100-fold larger libraries, we developed a simpler, more robust route to DNA-templated macrocycles. We hypothesized that the final biotin pulldown, Wittig macrocyclization, and self-elution steps achieve a sufficient purity of final macrocycle product such that the material entering step 3 does not require purification after steps 1 and 2. We further anticipated that hybridized reagent oligonucleotides from previous steps would not impair subsequent reactions, so long as subsequent steps used template bases closer to the site of small-molecule attachment. To avoid truncated macrocycles arising from a failed step 1 or step 2 reaction, we envisioned using acetic anhydride to cap unreacted templates after steps 1 and 2, thereby preventing any unreacted or improperly reacted starting material from participating in step 3 and the macrocyclization reaction (Figure 2). This strategy is reminiscent of the capping procedures used in solid-phase peptide and oligonucleotide synthesis to prevent truncated species from contaminating full-length products.69,70
Figure 2.

A capping-based strategy for DNA-templated macrocycle synthesis. The use of a capping reagent (here, acetic anhydride) prevents unreacted or improperly reacted material from participating in further reactions. The final streptavidin bead capture and macrocyclization steps achieve effective purification of the final product, obviating the need to purify after each DNA-templated reaction as in Figure 1 and reducing the total number of required manipulations.
To test the viability of this approach, we synthesized the single macrocycle 8a from DNA template 1a and the corresponding reagents 2a, 3a, and 4a (Figure 3) using the capping-based method shown in Figure 2. The codons were selected from our previous work15 to ensure template and reactant viability. After each step, we removed an aliquot of the mixture in order to monitor the progress of the synthesis using denaturing polyacrylamide gel electrophoresis (PAGE); the resulting gel reveals efficient product formation during each DNA-templated reaction (Figure 3, lanes b, e, and h). Similarly, each cleavage step (lanes d and g) shows quantitative disappearance of the product band from the preceding lane.
Figure 3.
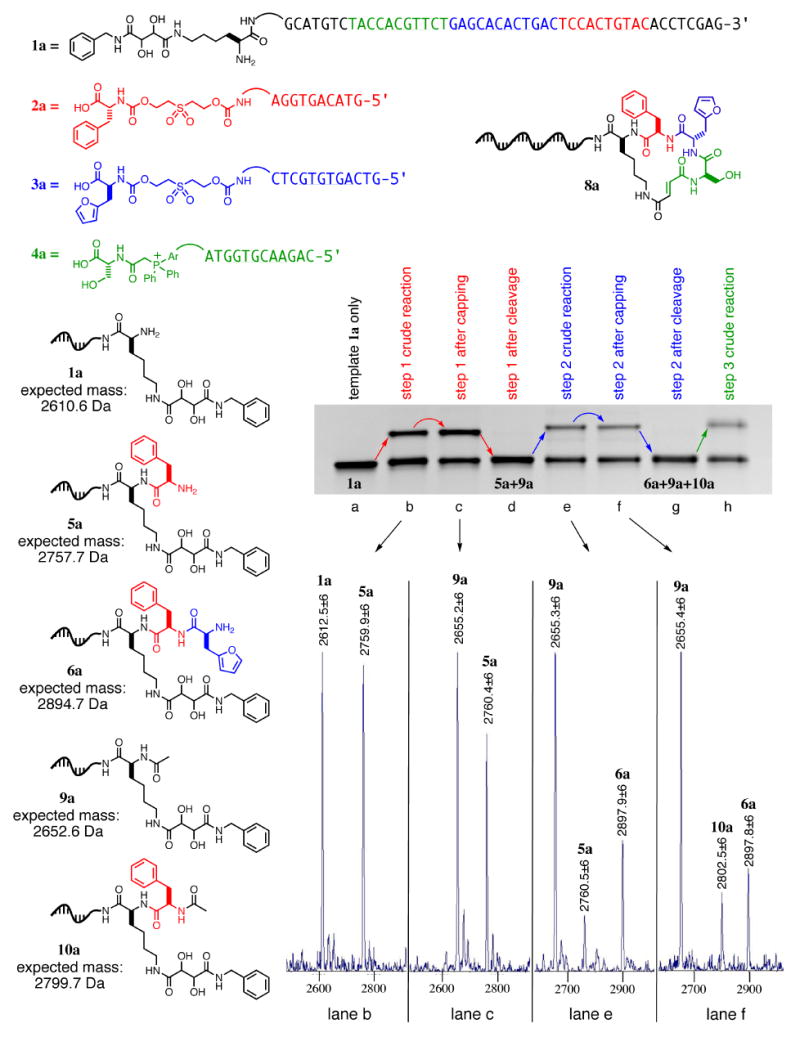
Denaturing PAGE and MALDI-TOF analysis of the capping method applied to DNA-templated macrocycle precursor synthesis.
To gauge the efficiency of the capping step, we removed aliquots from each of the first two rounds of templated synthesis, both before (lanes b and e) and after (lanes c and f) the addition of acetic anhydride; intermediates were then cleaved at high pH, enzymatically digested with Hpy8I to remove all but seven nucleotides of the DNA template, and analyzed by MALDI-TOF mass spectrometry. The resulting spectra show quantitative conversion of unreacted step 1 starting material (1a) to acetylated product (9a) upon addition of acetic anhydride (Figure 3), with no adulteration of the desired product (5a). Similarly, unreacted starting material from the second reaction (5a) was converted to acetylated material (10a) after step 2, with no visible modification to the desired product (6a). The persistence of 9a in the second step confirms that acetylated products are unreactive in subsequent steps (Figure 3).
The product of the third templated reaction (Figure 3, lane h) was purified with streptavidin-linked beads, washed as previously described, and exposed to aqueous sodium periodate to enable cyclization. The final macrocyclic product was eluted from the resin by exposure to pH 8.5 buffer, as confirmed by PAGE analysis (see Supporting Information, Figure S1). Restriction digestion followed by MALDI-TOF analysis (Figure S1) revealed a product mass consistent with the expected macrocycle 8a (expected: 2842.7 Da, found 2846.6±6 Da). The overall yield was calculated by PAGE and densitometry at 1% for all three templated reactions, the streptavidin purification, and macrocyclization. Notably, no significant peaks corresponding to any of the acetylated products (oxidized versions of 9a and 10a) were seen, confirming the effectiveness of the purification step.
This new capping-based strategy offers significant advantages over the previous library-synthesis route. First, it prevents material that has not reacted or that has reacted improperly from continuing to participate in the synthesis, thereby eliminating byproducts including contracted rings. Second, it reduces the total number of manipulations required to generate the final product by eliminating streptavidin capture steps, washing steps, and exposure to plasticware. Indeed, all three DNA-templated reactions are performed within the same vessel using the new strategy. The removal of two sets of streptavidin capture, washing, elution, and buffer exchange steps enabled us to complete the macrocycle synthesis in less than three days starting from reagents and templates, and in less than three weeks starting entirely from commercially available starting materials. Third, the fact that step 1 and step 2 reagents in this new method are no longer biotinylated simplifies their preparation and reduces cost. Finally, we speculate that the reagent DNA strands from completed steps assist in rigidifying template DNA strands by hybridizing to the templates, eliminating template secondary structural motifs that can potentially impede reactivity by blocking proper reagent annealing.68 In summary, this new methodology enhances the ease and efficiency of multistep DNA-templated syntheses, and produces final product in a more robust fashion.
Development of an Optimized Codon Set for Library Synthesis
The codons (variable regions) within a library of DNA templates play crucial roles during and after library translation. They mediate changes in effective molarity that promote the efficient reactivity of otherwise highly dilute reactants, determine the sequence specificity of DNA hybridization (and therefore the specificity of building-block reactivity), and reveal the structures of active library members after in vitro selection and PCR amplification. We recently characterized the relationship between template secondary structure and DNA-templated reactivity, and discovered that optimal reactivity arises from templates that have an intermediate degree of secondary structure that is sufficient to promote template compaction but insufficient to block reagent hybridization.68 We found that the window for maximum reactivity is in the range of -7 to -3 kcal/mol of predicted template folding energy.
Using the codon-generating algorithm shown in Figure 4, we produced three new sets of 40 codons each such that a minimum of three mismatches are present between any mismatched (noncognate) template and reagent pair. We screened in silico templates containing these codons using the Oligonucleotide Modeling Platform (OMP),71,72 and eliminated those codons that frequently resulted in templates with insufficient or excessive folding energies.68 This strategy generated codon sets that result in templates closely fitting the ideal energetic profile.
Figure 4.

Method for the computational generation, modeling, and screening of a codon set suitable for DNA-templated library synthesis.
To encode 1,728 building-block combinations in a 12×12×12 arrangement, we used 36 codons (3 sets of 12 each) from the refined codon set. We synthesized 12 representative templates (1A-2A-3A, 1B-2B-3B, etc. through 1L-2L-3L) such that all 36 codons were represented within the 12-template library. The corresponding 36 complementary reagent oligonucleotides (step 1 reagents complementary to codons 1A to 1L, step 2 reagents complementary to codons 2A to 2L, and step 3 reagents complementary to codons 3A to 3L) were also synthesized. Each reagent contained an 11-base coding region and a 4-base Ω constant region, as previously described.73 The templates were synthesized with a 5′ amine to enable amine acylation; reagents were prepared with a 3′ (D)-Phe group ending in a carboxylic acid68
To rigorously test the sequence fidelity of templates containing the new codon set, we exposed each of the 12 representative templates (and by extension, each of the 36 codons) to each of the 36 DNA-linked reagents under DNA-templated amine acylation conditions, and measured the amount of resulting product by PAGE. Product yields arising from sequence-matched reactivity are shown along the diagonals of the matrices in Figure 5; mismatched reactivity is represented by all off-diagonal yields. The matched reagents regularly resulted in reaction yields in the 60-70% range, with only five matched combinations dropping below the 50% yield mark. In contrast, none of the 396 mismatched combinations exhibited reactivity resulting in more than 8% yield, and only five combinations exceeded 5% yield. Notably, this study tested codon sequence fidelity under more stringent conditions than would be present during normal library synthesis. In the above experiments, one reagent was mixed with one template (matched or mismatched) and allowed to react; in an actual library synthesis, the matched reagent is always present in solution and its hybridization to the template precludes the hybridization of mismatched reagents, reducing undesirable reactivity.
Figure 5.
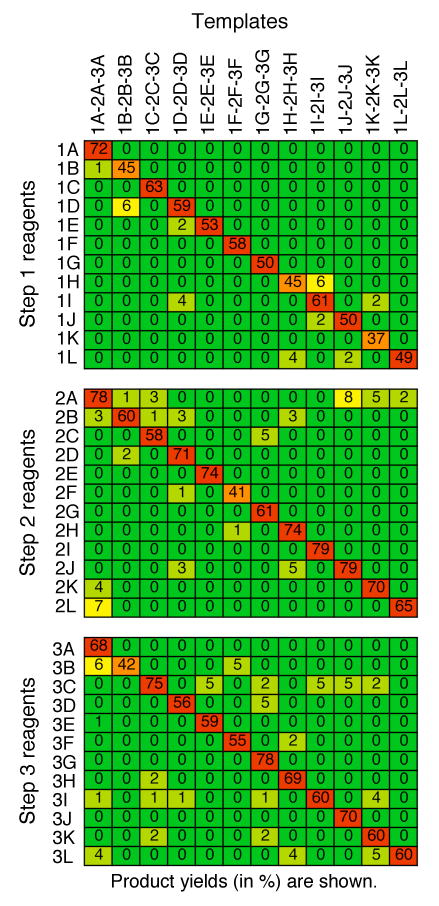
DNA-templated reaction yields for all possible matched and mismatched combinations of templates and reagents used in this work. Product yields arising from sequence-matched reactivity are shown on the diagonals of the matrices; mismatched reactivity yields are shown in off-diagonal cells. Cells are colored by percent yield from highest (red) to lowest (green) in spectral order.
To test the codon set for use in multistep DNA-templated macrocycle synthesis, we repeated the synthesis of macrocycle 8a using a representative template (1b, Figure 6) from the new codon set side-by-side with a template from the original, smaller codon set.15 The template using the original codons produced 8a in 1% final overall yield of pure macrocycle. The new codon set provided identical macrocycle 8b in 3% overall yield of pure product. (The improvement in yield arises largely from the use of the Ω architecture,73 as the end-of-helix reagents using the same template 1b resulted in a 0.8% final overall yield.) To further characterize the structure of the macrocycle 8b, we digested the product with S1 nuclease to liberate the macrocycle from the DNA template. S1 nuclease digestion leaves a single nucleotide 5′-monophosphate at the 3′ end of its digestion products,74 producing chemical species that are sufficiently small to be analyzed by high-resolution electrospray LC/MS. The crude digested sample yielded a mass spectrum consistent with the expected structure (expected: 972.35 Da, observed: 972.34±0.05 Da), as shown in Figure 6. Collectively, the above results indicate that the new codon set exhibits an excellent combination of efficient reactivity towards matched reagents and low reactivity towards mismatched reagents, while maintaining compatibility with multistep DNA-templated macrocycle synthesis.
Figure 6.
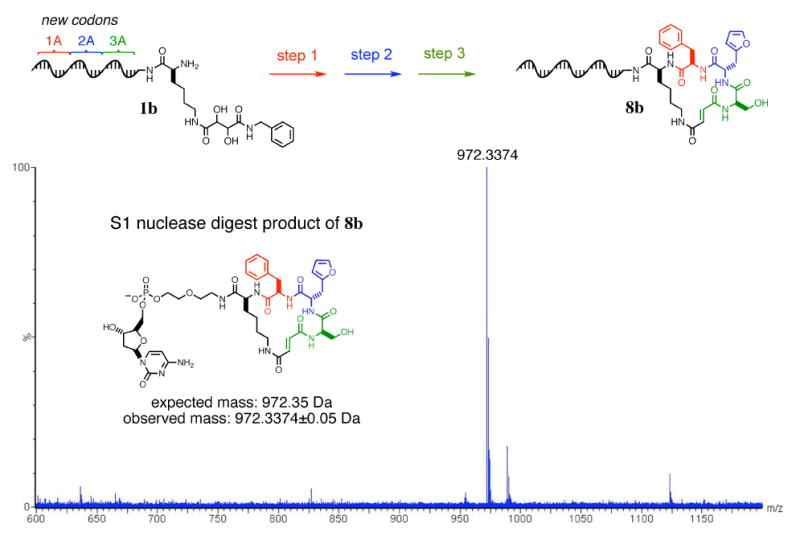
High-resolution LC/MS analysis of macrocycle 8b after S1 nuclease digestion. Macrocycle 8b was synthesized using the capping-based approach from template 1b using the new codons 1A, 2A, and 3A.
These results also validate the template design principles we recently described68 that relate DNA-templated reaction efficiency with predicted template folding energy. High-quality codon sets are critical to the success of any nucleic acid-templated synthesis. The capability developed here to accurately and rapidly design and test the reactivities of many library codons and templates is essential to current and future multiplexed DNA-templated syntheses.
The Effect of Building-Block and Scaffold Variability on DNA-Templated Macrocycle Synthesis
Next we performed a series of experiments to determine the range of building blocks compatible with an expanded macrocycle library synthesis. Based on our previous in vitro selection studies,75 we estimated that maintaining final product abundances within a range of ∼100-fold in the final library would facilitate in vitro selection by minimizing problems associated with more extreme stoichiometry differences between products, such as the need for more library material entering selection to enable active but underrepresented members to survive selection and PCR amplification.
We chose six different amino acids to represent a diverse range of chemical functionalities (Figure 7): glycine (small and flexible), 1-amino-1-cyclohexyl-carboxylic acid (α,α-disubstituted), proline (N-substituted and constrained), statine (extended backbone), N′,N′,N′-trimethyllysine (charged), and 4-benzoyl-(D)-phenylalanine (D-stereochemistry and hydrophobic). Using macrocycle 8a as the centerpiece of our study, we synthesized three separate series of macrocycles (11, 12, and 13), each containing amino acid substitutions at one of the three variable macrocycle positions (macrocycle series 11 for substitutions at R1, series 12 for R2, and series 13 for R3). Each synthesis was started from the last common template starting material (1a, 5a, and 6a, for 11, 12, and 13, respectively) and the codons used throughout the study were identical between templates to enable the direct comparison of reaction yields. The analogs of reagents 2a, 3a, and 4a containing each of the six building blocks in Figure 7 were also prepared. Macrocycle 8a was synthesized side-by-side with each sublibrary to provide a benchmark of reaction efficiency.
Figure 7.
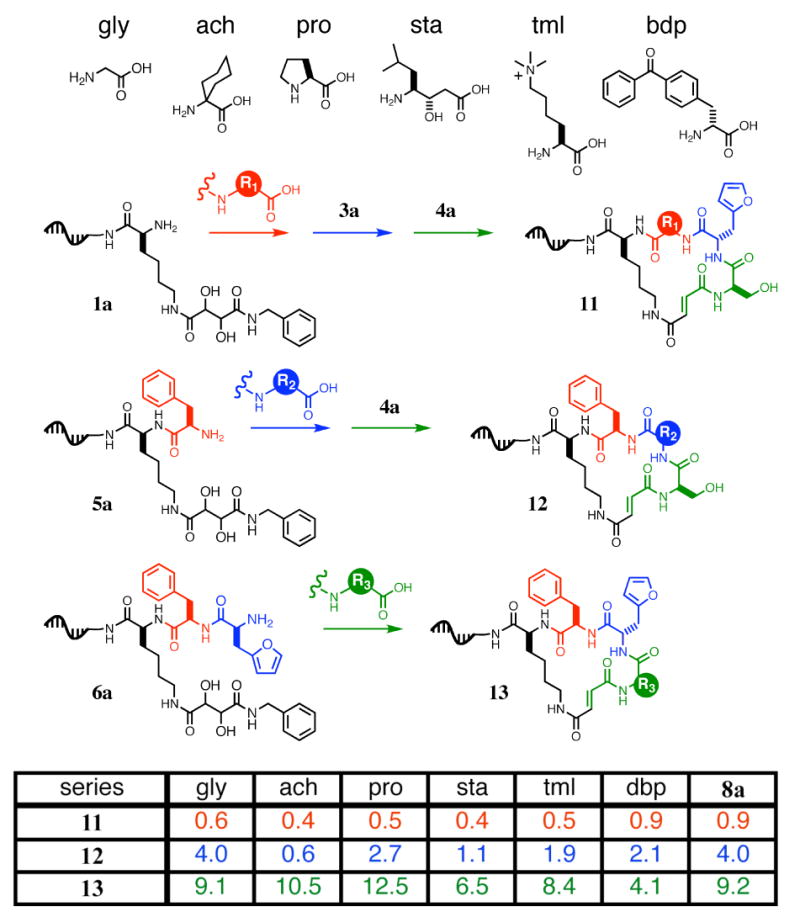
A systematic study of building-block compatibility with DNA-templated macrocycle synthesis. Using macrocycle 8a as a benchmark of “normal” reactivity, we selected a set of six distinct amino acids for substitution, one at a time, into the macrocycle at each of the three building block positions (R1, R2, and R3). Macrocycle series 11 (substitution at R1), 12 (substitution at R2), and 13 (substitution at R3) were synthesized using the corresponding reagent sets and starting materials 1a, 5a, and 6a, respectively. Percent yields of each macrocycle as analyzed by PAGE using internal standards are shown in the table.
The relative yields for all three series of macrocycles are shown in Figure 7, as calculated using densitometry with internal standards. With the exception of 2- to 3-fold lower yield from the use of the α,α-disubstituted building block in step 1 and step 2, reactivity was quite uniform (Figure 7). In all 18 cases, the products containing these six building blocks were viable substrates for the remaining steps of the library synthesis, including macrocyclization. These findings suggest that a wide variety of building-block parameters including size, flexibility, charge, hydrophobicity, and stereochemistry can be accommodated by our DNA-templated macrocycle synthesis. Given the range of building-block reactivities observed over the three sites of substitution, we estimate the ratio between the lowest potential macrocycle yield and the highest, assuming independent reactivity, to be ∼1:50; this ratio is within our target range of 100-fold. To further decrease this range, we avoided α,α-disubstituted amino acids in the first two steps in subsequent macrocycle library syntheses.
To further expand the size and diversity of the final library, the starting scaffold of template 1b was varied among lysine, ornithine (14), diamino-butyric acid (15), or diamino-propionic acid (16, Figure 8). Each of these diamino acids incrementally contracts the macrocycle ring by reducing the number of side-chain methylenes and enables access to significantly different preferred macrocycle conformations. In addition, the tartaramide group that gives rise to the aldehyde during macrocyclization, originally located on the side chain ε-amine of lysine in 1b, can alternatively be placed on either the α-amine or side-chain amine (“s-amine”) of the diamino acid in question (scaffolds 17, 18, 19 and 20, for the four respective diamino acids). These two choices generate macrocycles of opposite building-block orientation (Figure 8). Between the two different orientations and four different amino acids, a total of eight different scaffolds are possible. The effect of changing scaffolds on macrocycle shape and functional-group presentation is demonstrated in Figure 9, which shows that energy-minimized macrocycles with the same three building blocks but using two different scaffolds can have dramatically different predicted conformations.
Figure 8.
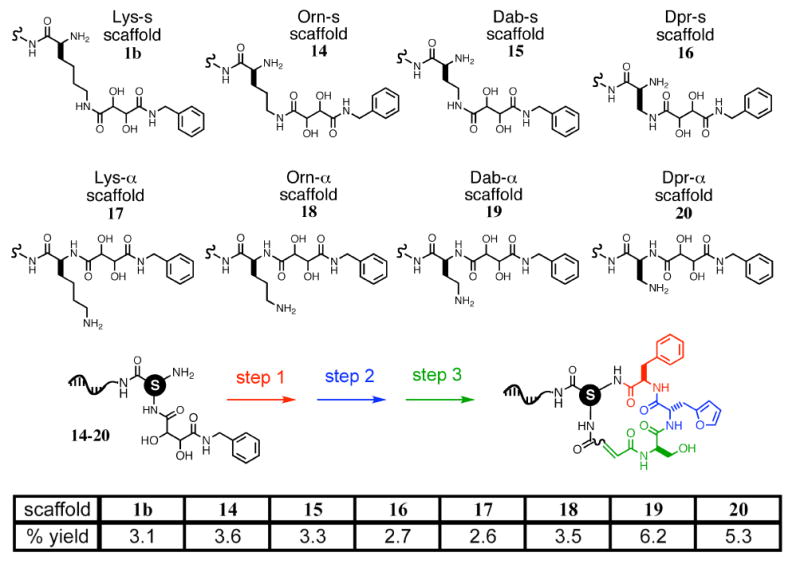
Eight scaffolds for DNA-templated macrocycle syntheses. The tartaramide group (aldehyde precursor) can be placed on either the α-amine or side-chain (“s”) amine, resulting in two different orientations of building blocks within the resulting macrocycles. The eight scaffolds (denoted by the circled S) were transformed into their corresponding macrocycles using the same reagents used in the synthesis of macrocycle 8b from scaffold 1b; the respective overall percent yields for final purified macrocycles are shown in the table above.
Figure 9.
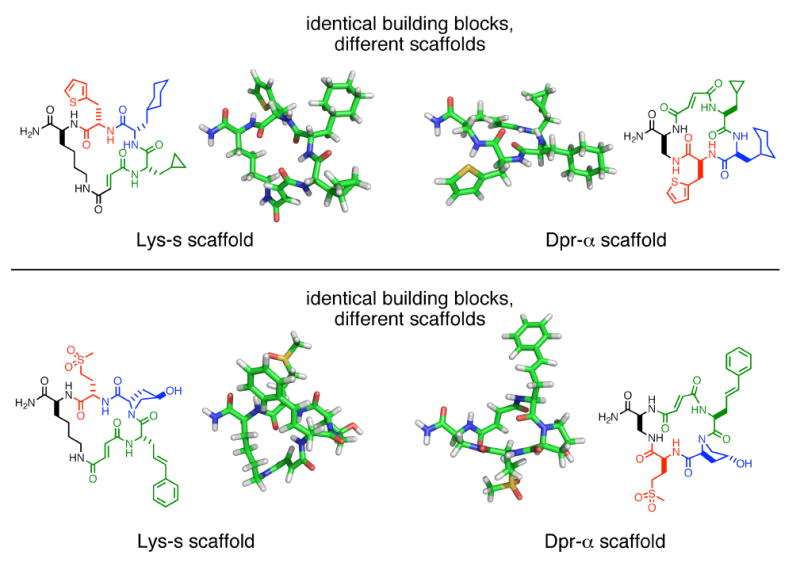
Three-dimensional models for two sets of MM2 energy-minimized macrocycles. The macrocycles within each set incorporate identical building blocks, but use a different scaffold (either the Lys-s or Dpr-α scaffold). The different scaffolds can result in very different preferred conformations.
To test the viability of each of the eight scaffolds, we synthesized a series of templates using codons identical to those of 5′-lysine(tartaramide) template 1b (codons 1A, 2A, and 3A), but bearing the different scaffolds on their respective 5′ ends (14-20). Using conditions and reagents identical to those used to translate template 1b into macrocycle 8b, we translated templates 14-20 into their corresponding macrocycles. Final yields of macrocycles, ranging from 6.2% (for 19) to 2.6% (for 17), are shown in Figure 8 and indicate efficient synthesis with all eight scaffolds. These results indicate the viability of using variable macrocycle scaffolds as a source of library diversity. The use of these eight scaffolds significantly augments the variety of macrocyclic structures that can be accessed within the framework of the library synthesis scheme.
Validation of 36 Building Blocks By Macrocycle Sublibrary Synthesis
Guided by the above findings, we selected three sets of 12 amino acid building blocks for step 1, step 2, and step 3 of the macrocycle library synthesis (Figure 10). The building blocks were chosen on the basis of their diverse chemical functionalities, compatibility with the synthetic route (no side-chain free amines or carboxylic acids), and variable backbone lengths. To assess their suitability for simultaneous reaction in a single pot during each DNA-templated library synthesis step and to establish their viability as macrocycle building blocks, we synthesized three 12-membered DNA-templated sublibraries by installing all 12 library building blocks for a given step into the macrocycle while keeping the other two amino acids of the macrocycle constant (libraries 21, 22, and 23, for variation at R1, R2, and R3, respectively). These three test libraries collectively tested the ability of all 36 library building blocks to react in their designated positions and to generate corresponding macrocyclic products.
Figure 10.

Amino acid building blocks for macrocycle libraries 21, 22, and 23.
We prepared all 36 of the necessary DNA-linked library reagents using the new codon set shown in Figure 11 by standard DNA synthesis, followed by small-molecule conjugation and HPLC purification. Through a convergent, modular ligation-based strategy (described below), we also generated three corresponding 12-membered template libraries containing the (L)-lysine s-amine scaffold linked to each of 12 oligonucleotides, representing a 12×1×1 library (testing all 12 step 1 reagents, for 21), a 1×12×1 library (testing all 12 step 2 reagents, for 22), and a 1×1×12 library (testing all 12 step 3 reagents, for 23).
Figure 11.
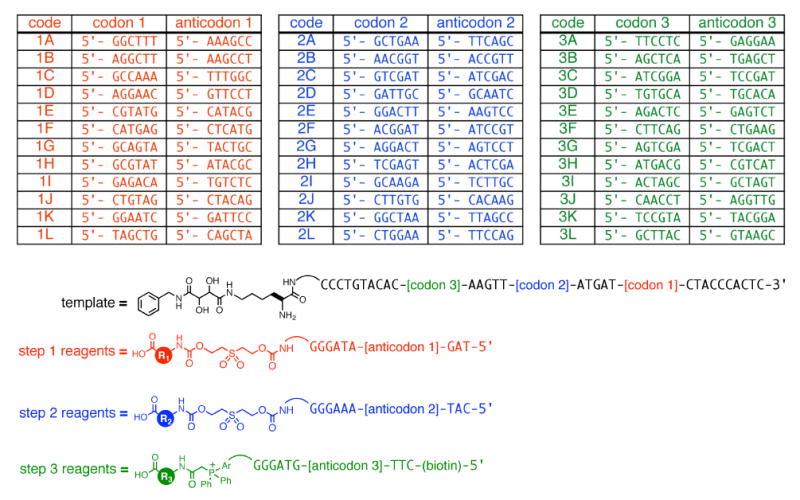
Template and reagent sequences that mediate the synthesis of macrocycle libraries 21, 22, and 23. The optimized template codons and corresponding reagent anticodons resulting from the method shown in Figure 4 are shown.
Each template library was then translated using the new capping-based macrocycle synthesis method (Figure 2) to generate its corresponding macrocycle library. Macrocycle libraries 21, 22, and 23 were synthesized in overall yields of 1.9%, 2.4%, and 1.1%, respectively. While some of our earlier macrocycle pilot library syntheses were sufficiently simple to be analyzed by low-sensitivity, low-resolution MALDI mass spectrometry of heptanucleotide-macrocycle conjugates,15 the characterization of these 12-membered test libraries (and larger subsequent libraries) requires an improved analysis method. As we previously did with macrocycle 8b, we therefore digested libraries 21, 22, and 23 with S1 nuclease and analyzed the resulting products by high-resolution LC/MS analysis. The removal of all but a single nucleotide 5′-monophosphate by S1 nuclease digestion enables the mass characterization of these DNA-templated products to an accuracy of ±0.05 Da or better.
The resulting spectra (Figure 12) confirm the presence of 34/36 (94%) of the total possible macrocyclic products in these three test libraries (explicit masses are listed in the Supporting Information, Figures S8, S9, and S10). In addition, the number of unexpected masses observed in significant abundance represented only a small fraction of the total observed masses, suggesting the effectiveness of the macrocycle selection step as a means of simultaneously effecting the macrocyclization and purification of final products. Taken together, these results validate the codons, starting scaffolds, and building blocks that make up a diverse, densely functionalized 8×12×12×12 (13,824-membered) DNA-templated macrocycle library.
Figure 12.
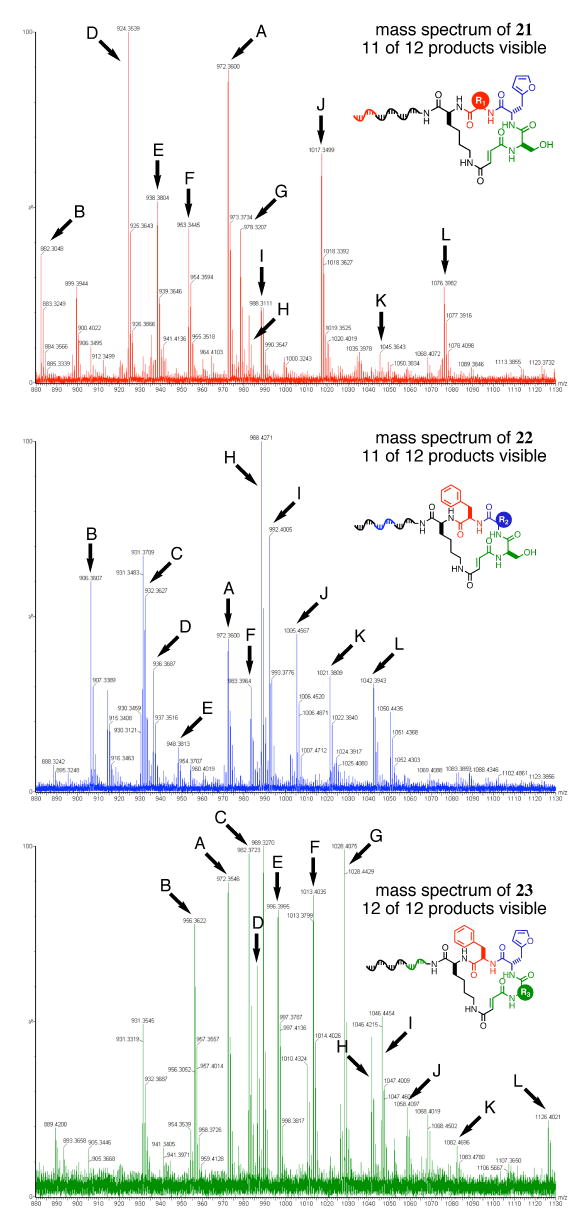
LC/MS analysis of 12-membered macrocycle libraries 21, 22, and 23 following S1 nuclease digestion. In total, 34 of the 36 expected macrocyclic species (94%) are visible.
Evaluating Sequence Fidelity During Library Translation
While the three 12-membered sublibraries assess the ability of the 36 building blocks to participate in DNA-templated macrocycle synthesis, they do not confirm the sequence fidelity of each DNA-templated library synthesis reaction in which 12 reagents and thousands of templates are all present in one solution. The codon sequence fidelity tests (Figure 5) indicate that each DNA-templated coupling reaction can proceed sequence specifically, but they were not conducted in the context of a multistep macrocycle synthesis. To rigorously evaluate the sequence specificity of macrocycle translation, we synthesized a sublibrary of exactly 12 templates using codon combinations 1A-2A-3A through 1L-2L-3L, such that each codon was represented within the template set once; all other features (5′ scaffold, primer binding sites, and constant regions) of the templates were identical. This template library (24, Figure 13) therefore encodes the synthesis of 12 distinct macrocycles, each comprising three unique building blocks, that collectively use all of the 36 DNA-linked reagents.
Figure 13.
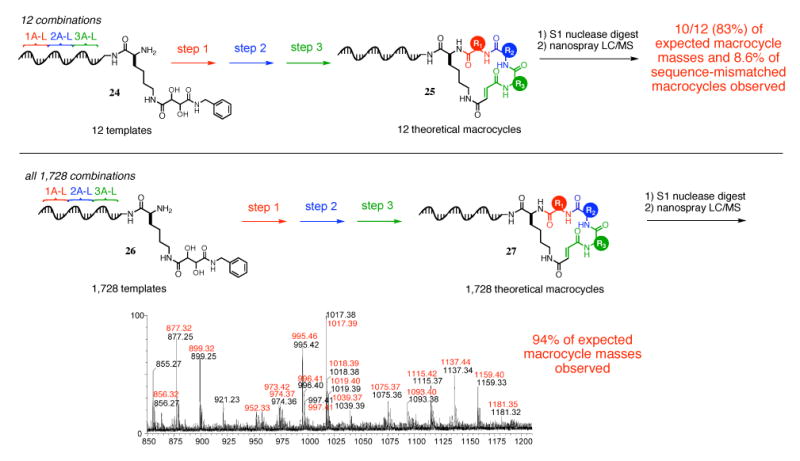
High-resolution LC/MS analysis of the 12-membered (25) and 1,728-membered (27) DNA-templated macrocycle sublibraries. From library 25, a stringent test of sequence specificity, 10 of 12 expected macrocycle masses were detected, and only 8.6% of masses corresponding to sequence-mismatched products were observed. For library 27, 94% of expected masses were observed. The macrocycles eluted over a total of ∼2,400 seconds; the representative mass spectrum shown here is of a 35-second elution window. Expected masses are in red and observed masses are in black.
If translation occurs with perfect sequence dependence, only the 12 expected macrocyclic products would result from DNA-templated library synthesis starting with 24. Cross-reactivity between mismatched templates and reagents would appear as unintended macrocycle products, of which 1,716 ([12×12×12]-12) are possible. For the translation of this library, we used three complete sets of reagents linked to anticodons complementary to 1A-1L, 2A-2L, and 3A-3L. Compared with the reagent sets used for the 1,728-membered sublibrary, the reagent sets for this 12-membered sequence-specificity test contain two amino acid substitutions to facilitate analysis by replacing the lowest yielding building blocks, as indicated by the spectra of sublibraries 21 and 22 (Figure 12).
Execution of the library synthesis on template library 24 with the step 1, 2 and 3 reagents resulted in the formation of macrocycle library 25 (Figure 13) in 1.4% overall yield. High-resolution LC/MS analysis of this library after S1 nuclease digestion produced a spectrum in which 10 of the 12 expected macrocycle masses were present with a signal above a threshold of 3,000 ion counts. Of the 1,716 possible macrocycles corresponding to unanticipated cross reactivity, masses consistent with only 148 (8.6%) were seen at or above the same signal threshold. The low fraction of non-sequence-programmed products formed during this stringent test of sequence specificity, consistent with the explicit tests of mismatched codon-mediated reactivity described above (Figure 5), suggests that non-sequence-programmed macrocycle formation should represent only a small fraction of total macrocycles in a full 1,728-membered macrocycle sublibrary.
DNA-Templated Synthesis and Characterization of a 1,728-Membered Macrocycle Sublibrary
Next we synthesized and characterized the complete 1,728-membered sublibrary representing one scaffold and all possible combinations of the 36 macrocycle building blocks. Template library 26 was generated by a combination of split-pool solid-phase oligonucleotide synthesis and enzymatic ligation; this template library contains the codons for all 36 unique reagents (1A-1L, corresponding to the step 1 reagent set, 2A-2L for step 2, and 3A-3L for step 3) as did template library 24, but in a combinatorial 12×12×12 format (Figure 13). Whereas library 24 encoded 12 distinct macrocycles, library 26 therefore encodes all 1,728 possible macrocycles.
Template library 26 was then reacted as described above with 12-membered step 1, step 2 and step 3 reagent sets, followed by streptavidin capture and macrocyclization, to yield final macrocycle library 27 (Figure 13). The final yield, across all three amide-bond formations, purification, and cyclization was 1.0%. In total, 48 picomoles of purified product were generated.
This library of up to 1,728 macrocycles was then digested with S1 nuclease and subjected to high-resolution nano-LC/MS analysis (Figure 13). The resulting LC/MS spectrum of 27 was computationally compared to the expected macrocycle masses of all 1,728 products by searching for the presence of all expected product masses based on a minimum distinguishable signal threshold empirically determined immediately prior to injection of the library sample. Of the 1,394 discrete expected masses, 1,317 (94%) were detected within the spectrum at a signal level exceeding the threshold of 1,000 ion counts. In addition, an examination of representative portions of the LC/MS spectrum revealed relatively few unanticipated masses (see Figure 13 for an example). These results are consistent with the presence of up to 1,633 (95%) of the 1,728 theoretical macrocyclic species in the library. Taken together, these findings suggest the robustness of the DNA-templated macrocycle library synthesis scheme and the viability of synthesizing a large scale DNA-templated library suitable for in vitro selection.
DNA-Templated Synthesis of a 13,824-Membered Macrocycle Library
Integrating the above developments, we synthesized the complete library of 13,824 DNA-templated macrocycles using all eight validated scaffolds (Figure 8) and all 36 building blocks (Figure 10). The starting template pool (28) was generated by split-pool DNA synthesis and enzymatic ligation as described above, but using eight different scaffolds (14-20) instead of one scaffold. Since the structural elucidation of any library member surviving selection requires the identification of the starting scaffold as well as the three building blocks, it is necessary to encode the identity of the scaffold in each template sequence. As the scaffold-encoding sequence does not direct any chemical reactions through DNA hybridization, it can be of minimal length, and placed anywhere within the template between the two PCR primer binding sites. We chose to encode the scaffold by using a three-base sequence near the 3′ (non-reacting) end of the templates (Figure 14; scaffold codons are shown in Supporting Information, Figure S2), which were otherwise prepared as described above.
Figure 14.
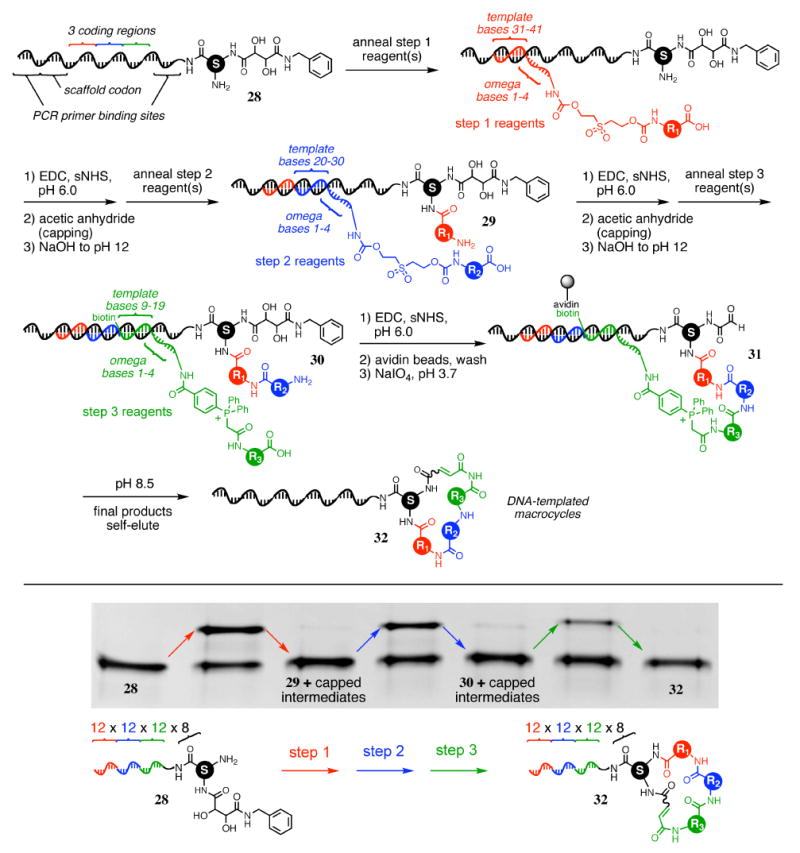
Synthesis of a 13,824-membered macrocycle library (32) from DNA template library 28. Eight template library-linked scaffolds were processed in a single solution through three DNA-templated reactions, each using 12 building blocks each, to produce a final macrocycle library with 13,824 members. The synthesis was performed in triplicate, with an average overall purified yield of 1.6% as determined by PAGE (one example is shown in the gel above). In total, > 375 pmol of library was produced, sufficient material for hundreds of in vitro selections.
The synthesis scheme for the 13,824-membered library is shown in Figure 14. Using the capping strategy, the template library (28) was reacted with the step 1 reagent set using EDC/sNHS to effect amine acylation; products were capped with acetic anhydride, and cleaved by pH elevation with hydroxide to generate intermediate library 29. After recovery by precipitation, 29 was reacted with the step 2 reagent set under standard amine acylation conditions; products were cleaved, capped, and precipitated as before to produce intermediate 30. This mixture was reacted with the step 3 reagent set under amine acylation conditions. The crude product was captured with streptavidin-linked beads, washed extensively, and treated with aqueous periodate to unmask the scaffold aldehyde group (intermediate 31). Upon exposure to pH 8.5 solution, the macrocyclized material eluted from the streptavidin-linked beads to afford the final macrocycle library product (32).
The synthesis was repeated three times, with an average yield of 1.6% as indicated by PAGE and densitometry analysis (Figure 14). In total, sufficient material (> 375 pmols) was synthesized and isolated for hundreds of in vitro selections.15,75 The sublibrary characterization experiments described above (Figures 12 and 13) suggest that this library contains more than 13,000 correctly translated macrocycle members; as such, the library represents one of the largest collections of synthetic macrocycles of this size (17- to 25-membered rings) reported to date.76-82
This DNA-templated macrocycle library provides a rich starting point for the discovery of functional synthetic macrocycles through in vitro selections against biological targets. We previously described in vitro selections of small molecule-DNA conjugates that exhibited enrichment factors of ∼100- to 1,000-fold per round.75 When such selections are applied to the 13,824-membered macrocycle library described above, two rounds of selection in principle could result in a single active library member predominating the molecules surviving selection. As these in vitro selections are quite general, require only very simple equipment, and can be performed in less than one day regardless of library size, efforts are currently underway to select this unique library of DNA-templated macrocycles against a wide variety of protein and RNA targets of biological and biomedical interest.
Conclusion
Biology-inspired approaches to the discovery of functional synthetic molecules bring the efficiency of in vitro selection, the sensitivity of DNA amplification, and the ease of DNA sequence analysis to bear on structures that can only be accessed through synthetic organic chemistry. Previously, we used multistep DNA-templated synthesis to translate a modest library of 65 DNA templates into a corresponding library of synthetic macrocycles15.
In the present work, we expanded the scope of DNA-templated macrocycle library synthesis considerably through a series of studies that each addressed a specific limitation of our original approach. We increased the robustness and efficiency of the synthetic methodology by adopting a streamlined capping-based synthesis strategy that reduces the total number of manipulations, eliminates impurities, and reduces cost. In addition, we designed and computationally screened a new set of library codons in accordance with the principles revealed in our recent study on the relationship between template sequence and DNA-templated reactivity.68 We experimentally validated the ability of this new codon set to mediate DNA-templated reactions efficiently and in a sequence-specific manner. We also examined the range of building blocks that are compatible with macrocycle synthesis by incorporating a representative set of different types of amino acids into corresponding macrocycles. We expanded the structural diversity of the library through the incorporation of a diverse set of 36 building blocks and eight different scaffolds that alter the size and orientation of the macrocyclic ring. Finally, we evaluated the efficacy and sequence fidelity of our library syntheses by generating a series of relevant macrocycle sublibraries and developing high-resolution LC/MS-based methods for characterizing each possible product.
Integrating these developments, we synthesized a final library of > 13,000 DNA-linked synthetic macrocycles. To our knowledge this library represents one of the largest collections of medium-sized synthetic macrocycles reported to date. Sufficient material of the completed macrocycle library was generated for many in vitro selections against biological targets of interest. In addition, the principles and strategies described here facilitate future DNA-templated synthesis efforts.
Supplementary Material
Additional DNA sequences, procedures, experimental results, and the complete author list for reference 60. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by the NIH/NIGMS (R01GM065865) and the Howard Hughes Medical Institute. T.M.S. and B.N.T. gratefully acknowledge NSF Graduate Research Fellowships. T.M.S. also acknowledges the support of an ACS Division of Organic Chemistry Fellowship sponsored by Organic Reactions, Inc. We thank Zev J. Gartner, Matthew W. Kanan, and Christopher T. Calderone for many helpful discussions.
References
- 1.Ellington AD, Szostak JW. Nature. 1990;346:818–22. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C, Gold L. Science. 1990;249:505–10. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 3.Singer BS, Shtatland T, Brown D, Gold L. Nucleic Acids Res. 1997;25:781–6. doi: 10.1093/nar/25.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shamah SM, Healy JM, Cload ST. Acc Chem Res. 2008;41:130–8. doi: 10.1021/ar700142z. [DOI] [PubMed] [Google Scholar]
- 5.Roberts RW, Szostak JW. Proc Natl Acad Sci U S A. 1997;94:12297–302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi TT, Austin RJ, Roberts RW. Trends Biochem Sci. 2003;28:159–65. doi: 10.1016/S0968-0004(03)00036-7. [DOI] [PubMed] [Google Scholar]
- 7.Hanes J, Pluckthun A. Proc Natl Acad Sci U S A. 1997;94:4937–42. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson DR, Finlay BB. Can J Microbiol. 1998;44:313–29. [PubMed] [Google Scholar]
- 9.Gartner ZJ, Liu DR. J Am Chem Soc. 2001;123:6961–3. doi: 10.1021/ja015873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gartner ZJ, Kanan MW, Liu DR. J Am Chem Soc. 2002;124:10304–6. doi: 10.1021/ja027307d. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Gartner ZJ, Tse BN, Liu DR. J Am Chem Soc. 2004;126:5090–2. doi: 10.1021/ja049666+. [DOI] [PubMed] [Google Scholar]
- 12.Calderone CT, Liu DR. Angew Chem Int Ed Engl. 2005;44:7383–6. doi: 10.1002/anie.200502899. [DOI] [PubMed] [Google Scholar]
- 13.Sakurai K, Snyder TM, Liu DR. J Am Chem Soc. 2004;127:1660–1661. doi: 10.1021/ja0432315. [DOI] [PubMed] [Google Scholar]
- 14.Rosenbaum DM, Liu DR. J Am Chem Soc. 2003;125:13924–5. doi: 10.1021/ja038058b. [DOI] [PubMed] [Google Scholar]
- 15.Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305:1601–5. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wrenn SJ, Weisinger RM, Halpin DR, Harbury PB. J Am Chem Soc. 2007;129:13137–43. doi: 10.1021/ja073993a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halpin DR, Harbury PB. PLoS Biol. 2004;2:E173. doi: 10.1371/journal.pbio.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halpin DR, Harbury PB. PLoS Biol. 2004;2:E174. doi: 10.1371/journal.pbio.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halpin DR, Lee JA, Wrenn SJ, Harbury PB. PLoS Biol. 2004;2:E175. doi: 10.1371/journal.pbio.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melkko S, Scheuermann J, Dumelin CE, Neri D. Nat Biotechnol. 2004;22:568–74. doi: 10.1038/nbt961. [DOI] [PubMed] [Google Scholar]
- 21.Dumelin CE, Scheuermann J, Melkko S, Neri D. Bioconjug Chem. 2006;17:366–70. doi: 10.1021/bc050282y. [DOI] [PubMed] [Google Scholar]
- 22.Melkko S, Zhang Y, Dumelin CE, Scheuermann J, Neri D. Angew Chem Int Ed Engl. 2007;46:4671–4. doi: 10.1002/anie.200700654. [DOI] [PubMed] [Google Scholar]
- 23.Scheuermann J, Dumelin CE, Melkko S, Zhang Y, Mannocci L, Jaggi M, Sobek J, Neri D. Bioconjug Chem. 2008 doi: 10.1021/bc7004347. [DOI] [PubMed] [Google Scholar]
- 24.Schreiber SL. Bioorg Med Chem. 1998;6:1127–52. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 25.Stockwell BR. Nat Rev Genet. 2000;1:116–25. doi: 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobson CM. Nature. 2004;432:824–8. doi: 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- 27.Stockwell BR. Nature. 2004;432:846–54. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spring DR. Chem Soc Rev. 2005;34:472–82. doi: 10.1039/b312875j. [DOI] [PubMed] [Google Scholar]
- 29.Eggert US, Mitchison TJ. Curr Opin Chem Biol. 2006;10:232–7. doi: 10.1016/j.cbpa.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Walsh DP, Chang YT. Chem Rev. 2006;106:2476–530. doi: 10.1021/cr0404141. [DOI] [PubMed] [Google Scholar]
- 31.Schreiber SL. Science. 2000;287:1964–9. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 32.Ding S, Schultz PG. Curr Top Med Chem. 2005;5:383–95. doi: 10.2174/1568026053828402. [DOI] [PubMed] [Google Scholar]
- 33.Marcaurelle LA, Johannes CW. Prog Drug Res. 2008;66:187, 189–216. doi: 10.1007/978-3-7643-8595-8_3. [DOI] [PubMed] [Google Scholar]
- 34.Driggers EM, Hale SP, Lee J, Terrett NK. Nat Rev Drug Discov. 2008;7:608–24. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 35.Deber CM, Madison V, Blout ER. Acc Chem Res. 1976;9:106–113. [Google Scholar]
- 36.Fairlie DP, Abbenante G, March DR. Curr Med Chem. 1995;2:654–686. [Google Scholar]
- 37.Tyndall JD, Fairlie DP. Curr Med Chem. 2001;8:893–907. doi: 10.2174/0929867013372715. [DOI] [PubMed] [Google Scholar]
- 38.Clardy J, Walsh C. Nature. 2004;432:829–37. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 39.Wessjohann LA, Ruijter E, Garcia-Rivera D, Brandt W. Mol Divers. 2005;9:171–86. doi: 10.1007/s11030-005-1314-x. [DOI] [PubMed] [Google Scholar]
- 40.Patton GC, van der Donk WA. Curr Opin Microbiol. 2005;8:543–51. doi: 10.1016/j.mib.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 41.Fung S, Hruby VJ. Curr Opin Chem Biol. 2005;9:352–8. doi: 10.1016/j.cbpa.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craik DJ, Cemazar M, Daly NL. Curr Opin Drug Discov Devel. 2006;9:251–60. [PubMed] [Google Scholar]
- 43.Kopp F, Marahiel MA. Nat Prod Rep. 2007;24:735–49. doi: 10.1039/b613652b. [DOI] [PubMed] [Google Scholar]
- 44.Turner RA, Oliver AG, Lokey RS. Org Lett. 2007;9:5011–4. doi: 10.1021/ol702228u. [DOI] [PubMed] [Google Scholar]
- 45.Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Proc Natl Acad Sci U S A. 1999;96:13638–43. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tavassoli A, Benkovic SJ. Nat Protoc. 2007;2:1126–33. doi: 10.1038/nprot.2007.152. [DOI] [PubMed] [Google Scholar]
- 47.Gilon C, Halle D, Chorev M, Selinger Z, Byk G. Biopolymers. 1991;31:745–50. doi: 10.1002/bip.360310619. [DOI] [PubMed] [Google Scholar]
- 48.Cherney RJ, Wang L, Meyer DT, Xue CB, Wasserman ZR, Hardman KD, Welch PK, Covington MB, Copeland RA, Arner EC, DeGrado WF, Decicco CP. J Med Chem. 1998;41:1749–51. doi: 10.1021/jm970850y. [DOI] [PubMed] [Google Scholar]
- 49.Gudmundsson OS, Vander Velde DG, Jois SD, Bak A, Siahaan TJ, Borchardt RT. J Pept Res. 1999;53:403–13. doi: 10.1034/j.1399-3011.1999.00077.x. [DOI] [PubMed] [Google Scholar]
- 50.Gudmundsson OS, Jois SD, Vander Velde DG, Siahaan TJ, Wang B, Borchardt RT. J Pept Res. 1999;53:383–92. doi: 10.1034/j.1399-3011.1999.00076.x. [DOI] [PubMed] [Google Scholar]
- 51.Dinsmore CJ, Bogusky MJ, Culberson JC, Bergman JM, Homnick CF, Zartman CB, Mosser SD, Schaber MD, Robinson RG, Koblan KS, Huber HE, Graham SL, Hartman GD, Huff JR, Williams TM. J Am Chem Soc. 2001;123:2107–8. doi: 10.1021/ja003673q. [DOI] [PubMed] [Google Scholar]
- 52.Dathe M, Nikolenko H, Klose J, Bienert M. Biochemistry. 2004;43:9140–50. doi: 10.1021/bi035948v. [DOI] [PubMed] [Google Scholar]
- 53.Udugamasooriya G, Saro D, Spaller MR. Org Lett. 2005;7:1203–6. doi: 10.1021/ol0475966. [DOI] [PubMed] [Google Scholar]
- 54.Charpentier B, Dor A, Roy P, England P, Pham H, Durieux C, Roques BP. J Med Chem. 1989;32:1184–90. doi: 10.1021/jm00126a007. [DOI] [PubMed] [Google Scholar]
- 55.Rizo J, Gierasch LM. Annu Rev Biochem. 1992;61:387–418. doi: 10.1146/annurev.bi.61.070192.002131. [DOI] [PubMed] [Google Scholar]
- 56.Craik DJ, Simonsen S, Daly NL. Curr Opin Drug Discov Devel. 2002;5:251–60. [PubMed] [Google Scholar]
- 57.Shibata K, Suzawa T, Soga S, Mizukami T, Yamada K, Hanai N, Yamasaki M. Bioorg Med Chem Lett. 2003;13:2583–6. doi: 10.1016/s0960-894x(03)00476-1. [DOI] [PubMed] [Google Scholar]
- 58.Tugyi R, Mezo G, Fellinger E, Andreu D, Hudecz F. J Pept Sci. 2005;11:642–9. doi: 10.1002/psc.669. [DOI] [PubMed] [Google Scholar]
- 59.Illuminati G, Mandolini L. Acc Chem Res. 1981;14:95–102. [Google Scholar]
- 60.Woodward RB, et al. J Am Chem Soc. 1981;103:3213–3215. [Google Scholar]
- 61.Davies JS. J Pept Sci. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 62.Blankenstein J, Zhu J. Eur J Org Chem. 2005;2005:16. [Google Scholar]
- 63.Liau BB, Gnanadesikan V, Corey EJ. Org Lett. 2008;10:1055–7. doi: 10.1021/ol702868z. [DOI] [PubMed] [Google Scholar]
- 64.Besser D, Olender R, Rosenfeld R, Arad O, Reissmann S. J Pept Res. 2000;56:337–45. doi: 10.1034/j.1399-3011.2000.00735.x. [DOI] [PubMed] [Google Scholar]
- 65.Haddadi ME, Cavelier F, Vives E, Azmani A, Verducci J, Martinez J. J Pept Sci. 2000;6:560–70. doi: 10.1002/1099-1387(200011)6:11<560::AID-PSC275>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 66.Taunton J, Collins JL, Schreiber SL. J Am Chem Soc. 1996;118:10412–10422. [Google Scholar]
- 67.Li CJ, Chan TH. Organic reactions in aqueous media. Wiley and Sons; New York: 1997. [Google Scholar]
- 68.Snyder TM, Tse BN, Liu DR. J Am Chem Soc. 2008;130:1392–401. doi: 10.1021/ja076780u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Merrifield B. Science. 1986;232:341–7. doi: 10.1126/science.3961484. [DOI] [PubMed] [Google Scholar]
- 70.Caruthers MH, Beaton G, Wu JV, Wiesler W. Methods Enzymol. 1992;211:3–20. doi: 10.1016/0076-6879(92)11003-2. [DOI] [PubMed] [Google Scholar]
- 71.SantaLucia J, Jr, Hicks D. Annu Rev Biophys Biomol Struct. 2004;33:415–40. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 72.SantaLucia J., Jr Methods Mol Biol. 2007;402:3–34. doi: 10.1007/978-1-59745-528-2_1. [DOI] [PubMed] [Google Scholar]
- 73.Gartner ZJ, Grubina R, Calderone CT, Liu DR. Angew Chem Int Ed Engl. 2003;42:1370–5. doi: 10.1002/anie.200390351. [DOI] [PubMed] [Google Scholar]
- 74.Vogt VM. Methods Enzymol. 1980;65:248–55. doi: 10.1016/s0076-6879(80)65034-4. [DOI] [PubMed] [Google Scholar]
- 75.Doyon JB, Snyder TM, Liu DR. J Am Chem Soc. 2003;125:12372–3. doi: 10.1021/ja036065u. [DOI] [PubMed] [Google Scholar]
- 76.Spatola AF, Crozet Y. J Med Chem. 1996;39:3842–6. doi: 10.1021/jm9604078. [DOI] [PubMed] [Google Scholar]
- 77.McBride JD, Freeman HN, Leatherbarrow RJ. Eur J Biochem. 1999;266:403–12. doi: 10.1046/j.1432-1327.1999.00867.x. [DOI] [PubMed] [Google Scholar]
- 78.Jefferson EA, Arakawa S, Blyn LB, Miyaji A, Osgood SA, Ranken R, Risen LM, Swayze EE. J Med Chem. 2002;45:3430–9. doi: 10.1021/jm010437x. [DOI] [PubMed] [Google Scholar]
- 79.Kohli RM, Walsh CT, Burkart MD. Nature. 2002;418:658–61. doi: 10.1038/nature00907. [DOI] [PubMed] [Google Scholar]
- 80.Sedrani R, Kallen J, Martin Cabrejas LM, Papageorgiou CD, Senia F, Rohrbach S, Wagner D, Thai B, Jutzi Eme AM, France J, Oberer L, Rihs G, Zenke G, Wagner J. J Am Chem Soc. 2003;125:3849–59. doi: 10.1021/ja021327y. [DOI] [PubMed] [Google Scholar]
- 81.Schmidt DR, Kwon O, Schreiber SL. J Comb Chem. 2004;6:286–292. doi: 10.1021/cc020076m. [DOI] [PubMed] [Google Scholar]
- 82.Goto Y, Ohta A, Sako Y, Yamagishi Y, Murakami H, Suga H. ACS Chem Biol. 2008;3:120–9. doi: 10.1021/cb700233t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional DNA sequences, procedures, experimental results, and the complete author list for reference 60. This material is available free of charge via the Internet at http://pubs.acs.org.