

Abstract
The mechanisms by which 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) increases the incidence of human cardiovascular disease are not known. We investigated the degree to which cardiovascular disease develops in mice following subchronic TCDD exposure. Adult male C57BL/6 mice were dosed with vehicle or 300 ng TCDD/kg by oral gavage three times per week for 60 days. Blood pressure was recorded by radiotelemetry and aortic endothelial function was assessed by acetylcholine-induced vasorelaxation. Mean arterial pressure of TCDD-exposed mice was increased significantly by day 4 and between days 7–10, 25–35, and 45–60 with two periods of normalization on days 11–24 and days 36–39. Consistent with a prolonged period of systemic hypertension, heart weight was increased and was associated with concentric left ventricular hypertrophy. Significant increases in superoxide production also were observed in the kidney, heart, and aorta of TCDD-exposed mice. Furthermore, increased aortic superoxide resulted in endothelial dysfunction as demonstrated by significant impairment of acetylcholine-induced vasorelaxation in TCDD-exposed mice, which was restored by tempol, a superoxide dismutase (SOD) mimetic. Our model is the first to definitely demonstrate that sustained AhR activation by TCDD increases blood pressure and induces cardiac hypertrophy, which may be mediated, in part, by increased superoxide.
Keywords: Keywords Aryl hydrocarbon receptor, Cardiac hypertrophy, Endothelial dysfunction, Hypertension, Superoxide, TCDD
Introduction
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a ubiquitous toxicant that persists in the environment and is generally resistant to degradation. The public became widely aware of TCDD and its potential toxic effects during the 1960s, when it was identified as a major contaminant in Agent Orange, an herbicide and defoliant abundantly used during the Vietnam War. Today, TCDD and related polychlorinated dibenzo-p-dioxins and dibenzofurans predominantly enter the environment as contaminants in incineration emissions [1].
The toxicity of TCDD is mediated by activation of the aryl hydrocarbon receptor (AhR), an orphan receptor belonging to the basic helix-loop-helix Per-ARNT-Sim family of transcription factors. TCDD and related chemicals bind to the AhR with high affinity, leading to its translocation into the nucleus and dimerization with the AhR nuclear translocator (ARNT). This heterodimeric complex then binds dioxin response elements in the promoters of target genes and regulates their transcriptional expression [2]. Mice in which the AhR has been genetically deleted fail to exhibit any overt toxic responses typically observed after TCDD exposure [3–5], demonstrating that AhR activation is required to mediate TCDD toxicity.
Human TCDD exposure has been associated with cardiovascular diseases. For example, in a study of Korean Vietnam veterans, the incidence of hypertension, stoke, and coronary heart disease was increased in veterans exposed to Agent Orange, compared to unexposed veterans [6]. Moreover, these results are supported by a recent study of Vietnam veterans. This study found that Army Chemical Corps Vietnam veterans who were responsible for spraying Agent Orange around the perimeters of base camps and from helicopters exhibited a significantly higher incidence of heart disease and hypertension compared to Army veterans that did not spray herbicides [7].
Despite the evidence showing an association between human TCDD exposure and cardiovascular diseases, animal models elucidating the mechanisms underlying these effects are limited. One study found that an acute high dose of TCDD resulted in a modest increase in blood pressure in a mouse model [8], while another study reported that chronic exposure of rats to TCDD induced a dose-dependent increase in cardiomyopathy and chronic active arteritis [9]. In a third study of mice with constitutively active AhR (CA-AhR), heart weights were significantly increased in male CA-AhR mice after 58 days of age [10]. These data suggest that continuous AhR activation causes cardiovascular disease. The objective of our study was to investigate the degree to which cardiovascular disease develops in an animal model when exposed subchronically to TCDD with the body burden gradually accumulating over time. Using this model, we tested the hypothesis that subchronic TCDD exposure will induce vascular dysfunction and hypertension. Further, reactive oxygen species (ROS) are commonly elevated in cardiovascular disease, including hypertension [11], and AhR activation by TCDD has been linked to oxidative stress in mice [12]. Thus, we also hypothesized that vascular dysfunction and hypertension induced by subchronic TCDD exposure would be associated with increased ROS.
Materials and Methods
Chemicals
TCDD was a generous gift from Dr. Richard E. Peterson (University of Wisconsin-Madison). Mason’s trichrome stain, bovine serum albumin, phenylephrine (PE), acetylcholine (ACh), tempol, and all components of phosphate buffered saline (PBS), Krebs-Ringer buffer, and physiological saline solution (PSS) were acquired from Sigma–Aldrich (St. Louis, MO).
Animals
Male C57BL6/N mice were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN). Three separate exposure studies were conducted. First, blood pressure was continuously monitored in mice prior to and throughout a 60-day dosing with TCDD. A second study was conducted to assess organ weights on days when TCDD-induced blood pressure peaked, while a third assessed tissue ROS and vascular reactivity after 35 days of dosing since this was the earliest time point that mice were significantly hypertensive and exhibited significant changes in organ weight.
For the first study, mice were dosed from the age of 100 to 160 days with vehicle control (corn oil, 3 ml/kg) or 300 ng TCDD/kg body weight three times per week by oral gavage. This dosing regimen was selected to minimize handling of mice, which could disrupt blood pressure measurements, and to achieve an approximate steady state body burden of 2 μg/kg by day 60.
Blood Pressure Assessment
Fourteen days prior to dosing, a PA-C20 blood pressure telemeter (Data Sciences International, St. Paul, MN) was implanted into each mouse. Thirty minutes prior to surgery, mice were injected with buprenorphine (0.1 mg/kg, sc) for analgesia and then anesthetized with isoflurane (5% for induction, 2.5% for maintenance). The telemeter catheter was inserted into the left carotid artery and fed down to the aortic arch, and the body of the transmitter was placed subcutaneously in the abdomen. Mice were allowed to recover from surgery for 7 days prior to collecting any blood pressure data. Baseline blood pressure was collected for 7 days. Measurement parameters including systolic, diastolic, and mean arterial pressure, and heart rate were collected for 10 s every 15 min. For the duration of the study, telemetry measurements were monitored for quality assurance. At the first signs of a failing battery, when signal strength decreased, or a pinched or occluded catheter, when pulse-wave form was reduced, any further data from that particular mouse were excluded from the study. In addition, since battery life is only guaranteed for 42 days, after collecting baseline BP data some telemeters were turned off until treatment day 35. Sample sizes were vehicle control, n = 4 for the entire treatment period, and TCDD, n = 4 treatment days 1–36 and 54–60, n = 5 treatment days 37–53.
Organ Collection
Following blood pressure assessment, a second study was performed to examine any overt changes in tissue weight at the three time points that blood pressure was highest: 9, 35, and 60 days of treatment. Mice were anesthetized with ketamine (80 mg/kg)/xylazine (4 mg/kg), weighed, and euthanized by exsanguination. Plasma was collected from EDTA-treated blood, divided into aliquots, and frozen at −20°C. The atria were removed from the heart and total ventricle weight measured. Then the left ventricle and ventricular septum were dissected from the right ventricle, weighed, and either frozen at −80°C or fixed in 10% neutral buffered formalin, dehydrated, and embedded in paraffin. Liver and kidneys were also removed, weighed, and frozen at −80°C.
Determination of TCDD Tissue Concentration
TCDD was measured in the kidney and liver of control and TCDD-exposed mice after 35 and 60 days of treatment by a method based on EPA Method 1613 (Tetra- through Octa-Chlorinated Dioxins and Furans by Isotope Dilution HRGC/HRMS, 1994) [13]. All chemical standards used for the analysis were purchased from Wellington Laboratories (Guelph, ON). Tissue samples (0.03–0.35 g) were homogenized in saline (200 μl) using disposable pellet pestles and microtubes. An aliquot equivalent to 5 mg (liver) or 50 mg (kidney) was transferred to a Teflon bottle containing methylene chloride:hexane (50:50) (20 ml), spiked with 13C-labeled recovery standards, and vigorously shaken. The extract was filtered through anhydrous sodium sulfate (20 g), solvent exchanged into hexane (10 ml), and applied to an automated dioxin cleanup instrument (Fluid Management Systems, Waltham, MA) for chromatography on tri-phasic silica, basic alumina, and carbon cartridges. 13C-Labeled internal standards were added prior to high resolution gas chromatography/high resolution mass spectrometry (HRGC/HRMS) analysis on an Autospec Ultima mass spectrometer (Waters, Milford, MA) coupled to an Agilent 6890 gas chromatograph. The limit of detection for the 50 and 5 mg samples was 16 and 160 pg/g, respectively.
Left Ventricle Histology
Paraffin-embedded left ventricle tissue from mice treated for 60 days was sliced into 10 μm sections using a Leica RM2135 rotary microtome (Leica Microsystems, Bannockburn, IL) and stained using Mason’s trichrome stain. Images were taken using an Olympus S2X12 dissecting microscope (Olympus America Inc, Center Valley, PA). Left ventricle myocardium area and left ventricle cavity area were determined from the images using the ‘‘Magic Wand tool’’ in Adobe Photoshop (Adobe Systems Incorporated, San Jose, CA), a tool which allows for the discretionary selection of areas within an image that consist of a similar color and reports the area of the selection in pixels.
Determination of Plasma Angiotensin II (Ang II) and Endothelin-1 (ET-1)
The levels of Ang II and ET-1 were determined from 100 μl of plasma from mice after 60 days of treatment, using a radioimmunoassay (RIA) kit (Euro-Diagnostica, Arnhem, The Netherlands) and an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN), respectively. Radioactivity of Ang II RIA samples was quantified for 125I using an automatic gamma counter (Wallac Wizard 3, Perkin Elmer, Waltham, MA), and luminescence of ET-1 ELISA samples were quantified using a multi-label counter (Wallac Victor2, Perkin Elmer).
Gene Expression Associated with AhR and Antioxidant Responses
Total RNA was isolated from left ventricle tissue from mice treated for 60 days with RNeasy Fibrous Tissue Mini Kit (Qiagen, GmbH, Germany). cDNA was synthesized using iScript Select cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA) with the supplied random primers and 250 ng RNA. PCR amplification was performed using an iCycler (Bio-Rad Laboratories) with a reaction mixture comprised of iQ SYBR Green Supermix (Bio-Rad Laboratories) with 500 μM sense and antisense primers (Table 1) and 250 pg cDNA/μl. Cycle threshold data for both the target gene and reference gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), were used to calculate mean normalized expression as previously described [14].
Table 1.
Real-time PCR primer sequences
| Gene | Sense primer | Antisense primer |
|---|---|---|
| Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) | CCAATGTGTCCGTCGTGGATC | TGTAGCCCAAGATGCCCTTCA |
| Cytochrome P4501A1 (CYP1A1) | TGAGTCAGCAGTATGGGGACG | AGGGCCTGCTTGATGGTGTT |
| Cytochrome P4501A1 (CYP1B1) | AATCAATGCGATTCTCCAGCTTTT | CGACCGTATTCTTGGGGATGTAG |
| NF-E2-related factor 2 (Nrf2) | ATCTCCTAGTTCTCCGCTGCT | ACTCCAAGTCCATCATGCTGAG |
| Superoxide dismutase 2 (SOD2) | GCGGCCTACGTGAACAATCT | CTGAAGAGCGACCTGAGTTGTAA |
| Catalase | CAAGATTGCCTTCTCCGGGTG | GCTCCTTCCACTGCTTCATCTG |
| Heme oxygenase-1 (HO-1) | TTCTGGTATGGGCCTCACTGG | ACCTCGTGGAGACGCTTTACA |
| NAD(P)H dehydrogenase, quinone 1 (NQO1) | AACCTCAACTGGTTTACAGCATTG | TCTCCTCCCAGACGGTTTCC |
Reactive Oxygen Species Production in Cardiovascular Organs
A 15 mg cross section of frozen left ventricle, kidney, or aorta was thawed in ice cold PBS and then incubated in ice cold Krebs-Ringer buffer (20 mM HEPES, 10 mM dextrose, 127 mM NaCl, 5.5 mM KCl, 1 mM CaCl2, 2 mM MgSO4, pH 7.4) for 10 min. Each tissue was then transferred to a well of a 96-well white opaque plate containing 200 μl of room temperature Krebs-Ringer buffer with 5 μM lucigenin and 100 μM NADPH. Luminescence was measured three times for 10 s at 2-min intervals using a multi-label counter (Wallac Victor2, Perkin Elmer). The average of the three measures was used to represent the luminescence of each sample. Furthermore, to verify specificity of lucigenin luminescence for representation of superoxide anion production, a second set of TCDD tissue sections was preincubated in ice cold Krebs-Ringer buffer containing 31.6 μM tempol, a superoxide dismutase mimetic, for 30 min prior to measurement of lucigenin luminescence [15, 16].
Vascular Reactivity
A third study was performed to determine functional capacity of the vasculature. This study was conducted after 35 days of TCDD treatment since mice were hypertensive at this time point and exhibited significant increases in superoxide production in cardiovascular tissues, including aorta. After 35 days of treatment, mice were anesthetized with ketamine (80 mg/kg)/xylazine (4 mg/kg) and euthanized by exsanguination. Aortae were carefully removed, immediately placed in ice cold PSS (130 mM NaCl, 4.7 mM KCl, 1.18 mM KH2PO4, 1.17 mM MgSO4, 14.9 mM NaHCO3, 5.5 mM glucose, 26 μM CaNa2EDTA, 1.8 mM CaCl2, pH 7.4), trimmed of connective tissue, and cut into 3 mm segments. Two metal hangers were placed inside the lumen of each aorta and then quickly placed in the tissue bath (Radnoti Glass Technology Inc., Monrovia, CA) with the lower hanger attached to a fixed stage and the upper hanger attached to a force transducer (Grass Technologies, West Warwick, RI). Aortic rings were run in duplicate. PSS and the tissue baths were constantly bubbled with 21% O2, 6% CO2 balance N2 and refreshed every 10 min. Initial resting tension was brought up to 250 mg for the first 10 min, followed by resting tensions of 500 mg for 10 min, 1000 mg for 10 min, and 1500 mg for 30 min. Rings were then constricted with 100 mM KCl and 10 μM PE for 10 min followed by washout for 30 min, which was repeated for a total of three cycles. Endothelium viability was confirmed by relaxation to 10 μM ACh in aortae preconstricted with 10 μM PE. Vessels failing to vasorelax at least 60% to Ach (10 μM) were removed from the study. Following a 40-min washout period, vessels were preconstricted with 3.16 μM PE for 10 min. An ACh dose response (1 nM, 3.16 nM, 10 nM, 31.6 nM, 100 nM, 316 nM, 1 μM, 3.16 μM, 10 μM) was then performed, increasing the concentration of ACh every 5 min. Following another 40-min washout period, aortae were incubated with tempol (31.6 μM), a superoxide dismutase mimetic, for 20 min prior to repeating the ACh dose response in preconstricted vessels.
Statistical Analysis
Each 24 h of systolic, diastolic, and mean arterial pressure (MAP) data was averaged for 12-h periods, daytime and nighttime. Systolic, diastolic, MAP, and ACh dose response of aortic rings were compared using a two-way repeated measures ANOVA with post-hoc Holm-Sidak pairwise comparisons for a given time point. Body weight, organ weights, morphometric measures of left ventricle, plasma Ang II, and plasma ET-1 were compared between treatment groups by Student’s t-test. Lucigenin luminescence was compared between treatment groups by one-way ANOVA with post-hoc Holm-Sidak pairwise comparisons. A P-value <0.05 was considered significant.
Results
Tissue TCDD Concentrations
To determine the degree to which TCDD accumulated in the liver and kidney when dosing 300 ng TCDD/kg three times per week, the hepatic and renal TCDD concentrations were determined on days 35 and 60. Tissue concentrations of TCDD in all control mice, except the liver of one on day 60 (249 pg/g), were below the limit of detection of 160 pg/g for liver and 16 pg/g for kidney (Table 2). In contrast, TCDD was detected in the liver and kidney of TCDD-treated mice on both days 35 and 60. While the mean liver concentration increased between day 35 and 60 and the mean kidney concentration decreased, the respective tissue concentrations were not significantly different between days. The degree of accumulation of TCDD in the liver and kidney using this dosing regimen was very similar to that reported when dosing 150 ng TCDD/kg 5 days per week [17]. We chose to dose three times per week to minimize handling of mice to limit disruption of the blood pressure measurements.
Table 2.
Effect of exposure to TCDD on tissue TCDD concentrations
| Day 35 |
Day 60 |
|||
|---|---|---|---|---|
| Control | TCDD | Control | TCDD | |
| Kidney (pg/g)a | ND | 187 ± 15 | ND | 96 ± 38 |
| Liver (pg/g)b | ND | 6,814 ± 1,663 | ND—249c | 9,212 ± 512 |
n = 4
ND not detectable
Limit of detection = 16 pg/g
Limit of detection = 160 pg/g
ND in all mice except one, which had a reading of 249 pg/g
Expression of AhR Response Genes
To verify AhR stimulation, mRNA expression of the AhR-inducible genes CYP1A1 and CYP1B1 was examined in the aorta, kidney, and left ventricle after 35 days of exposure. CYP1A1 mRNA expression was significantly upregulated in all tissues of TCDD-exposed mice, most dramatically in the left ventricle (Fig. 1a); while CYP1B1 mRNA expression was only significantly upregulated in the heart of TCDD-exposed mice (Fig. 1b).
Fig. 1.

Effect of exposure to TCDD for 35 days on CYP1A1 (a) and CYP1B1 (b) mRNA expression in aorta, kidney, and left ventricle. Target mRNA was normalized to GAPDH mRNA and expressed relative to control. n = 5. * P < 0.05, compared to respective control
To confirm sustained induction of CYP1A1 and CYP1B1, mRNA expression was also examined in the left ventricle after 60 days of exposure. Induction of CYP1A1 mRNA expression of TCDD-exposed mice was similar to values seen after 35 days of exposure (122.6 ± 15.9 fold). However, the induction of CYP1B1 mRNA expression in TCDD-exposed mice was significantly greater after 60 days of exposure (7.6 ± 2.8 fold, P = 0.05).
Systemic Arterial Blood Pressure
We assessed blood pressure 24 h/day during subchronic TCDD exposure. We found that TCDD exposure increased both daytime and nighttime MAP (Figs. 2b and 3b). MAP was significantly increased in TCDD-exposed mice within the first week of treatment. This increase in MAP peaked at day 10 and was followed by a normalization. MAP remained normal for approximately 12 days until a second increase began, which peaked on day 35 of treatment. After another short normalization period of about 8 days, MAP was significantly increased for the remainder of the 60-day study. These changes in MAP were paralleled by similar changes in systolic and diastolic blood pressure and at the end of the study both were also significantly elevated in TCDD-exposed mice (Figs. 2a, c and 3a, c). No significant alterations in daytime and nighttime heart rate, activity, or circadian rhythms were observed (data not shown).
Fig. 2.

Effect of exposure to TCDD on daytime (sleep period) systolic (a), mean (b), and diastolic (c) arterial pressure. Vehicle control, n = 4; TCDD, n = 4 days 1–36 and 54–60, n = 5 days 37–53. * P < 0.05, compared to control on the same day
Fig. 3.

Effect of exposure to TCDD on nighttime (active period) systolic (a), mean (b), and diastolic (c) arterial pressure. Vehicle control, n = 4; TCDD, n = 4 days 1–36 and 54–60, n = 5 days 37–53. * P < 0.05, compared to control on the same day
Body and Organ Weights
We next conducted a study to assess organ weights on the approximate days when TCDD-induced blood pressure peaked (i.e., days 9, 35, and 60; Table 3). Neither body weight nor body weight gain was altered in TCDD-exposed mice, demonstrating the lack of overt toxicity. Increased liver weight, a well-documented response to TCDD exposure, was observed on days 35 and 60, and at all three time points in TCDD-exposed mice, when expressed as a percentage of total body weight. Raw heart weight was increased in the TCDD treatment group only on day 35.
Table 3.
Effect of exposure to TCDD on body and organ weights
| Weight (g) (% body weight) | Day 9 |
Day 35 |
Day 60 |
|||
|---|---|---|---|---|---|---|
| Control n = 10 | TCDD n = 10 | Control n = 5 | TCDD n = 5 | Control n = 9 | TCDD n = 8 | |
| Body | 26.94 ± 0.31 | 26.97 ± 0.35 | 28.14 ± 0.71 | 30.01 ± 0.61 | 34.15 ± 1.36 | 33.90 ± 1.36 |
| Liver | 1.492 ± 0.038 (5.54 ± 0.13) | 1.587 ± 0.052 (5.92 ± 0.20)* | 1.490 ± 0.075 (5.29 ± 0.18) | 1.922 ± 0.062* (6.40 ± 0.09)* | 1.728 ± 0.071 (5.08 ± 0.16) | 1.979 ± 0.088* (5.86 ± 0.21)* |
| Heart | 0.123 ± 0.001 (0.46 ± 0.01) | 0.124 ± 0.003 (0.46 ± 0.01) | 0.114 ± 0.005 (0.41 ± 0.01) | 0.134 ± 0.004* (0.45 ± 0.02) | 0.129 ± 0.004 (0.38 ± 0.01) | 0.134 ± 0.005 (0.40 ± 0.01) |
| LV + S | 0.085 ± 0.002 (0.32 ± 0.01) | 0.087 ± 0.002 (0.32 ± 0.01) | 0.083 ± 0.004 (0.29 ± 0.01) | 0.099 ± 0.003* (0.33 ± 0.01)* | 0.089 ± 0.003 (0.26 ± 0.01) | 0.095 ± 0.004 (0.28 ± 0.01)* |
| RV | 0.39 ± 0.002 (0.14 ± 0.01) | 0.037 ± 0.002 0.14 ± 0.01 | 0.031 ± 0.003 0.11 ± 0.01 | 0.034 ± 0.003 0.11 ± 0.01 | 0.040 ± 0.003 (0.12 ± 0.01) | 0.039 ± 0.004 (0.12 ± 0.01) |
P < 0.05, compared to control on the same day
However, when the left ventricle and septum (LV + S) were dissected from the right ventricle (RV), a significant increase in LV + S weight was observed in TCDD-exposed mice on day 35, and on both day 35 and 60 when expressed as a percentage of total body weight. There was no difference in RV weight in TCDD-exposed mice, compared to vehicle controls. No changes in kidney weight were observed on days 9, 35, or 60 of treatment (data not shown).
Cardiac Histology
Due to the observed increase in left ventricle weight and that MAP had been significantly elevated for 13 of the previous 16 days, the histology of the left ventricle from mice after 60 days of treatment was examined from paraffin-embedded tissue sections. Concentric hypertrophy, where the wall of ventricle increases and radius of the ventricle cavity decreases, was observed in the left ventricle of TCDD-exposed mice (Fig. 4a), which is consistent with a chronic increase in systemic arterial blood pressure [18].
Fig. 4.

Effect of exposure to TCDD for 60 days on left ventricle histology (a, b) and morphometrics (c). n = 4. * P < 0.05, compared to respective control
To quantify the changes observed in the left ventricle histology, morphometric measures of ventricular myocardium and cavity were taken. Left ventricular myocardium area was significantly increased in TCDD-exposed mice and left ventricular cavity area was significantly decreased in TCDD-exposed mice (Fig. 4b), consistent with the observed concentric hypertrophy.
Plasma Vasoactive Agents
Systemic hypertension is frequently associated with increases in the vasoconstricting peptides, Ang II and ET-1. Thus, plasma samples from mice on day 60 of treatment were analyzed for Ang II and ET-1. No significant alterations were observed in plasma Ang II or ET-1 (data not shown).
Indices of Oxidative Stress
To assess the potential TCDD-induced oxidative stress, mRNA expression of antioxidant genes was examined in left ventricle. Expression of Nrf2, a transcription factor governing antioxidant response elements [19, 20], was significantly upregulated by 1.5-fold. There was a trend for an increase in the expression of SOD2 and catalase, enzymes which reduce superoxide and hydrogen peroxide, respectively. Expression of HO-1, an inducible isoform of heme oxygenase to oxidative stress, was significantly increased by 1.4-fold. Moreover, expression of NQO1, an enzyme which reduces vitamin E and coenzyme Q10, was significantly increased by 1.3-fold (Table 4).
Table 4.
Effect of exposure to TCDD for 60 days on AhR and oxidative stress response gene expression
| Genea | Controlb | TCDDb | P-value |
|---|---|---|---|
| Nrf2 | 1.0 ± 0.1 | 1.5 ± 0.2 | 0.033 |
| SOD2 | 1.0 ± 0.1 | 1.2 ± 0.1 | 0.062 |
| Catalase | 1.0 ± 0.1 | 1.2 ± 0.1 | 0.094 |
| HO-1 | 1.0 ± 0.1 | 1.4 ± 0.1 | 0.050 |
| NQO1 | 1.0 ± 0.1 | 1.3 ± 0.1 | 0.047 |
n = 5
Normalized to GAPDH mRNA expression
Normalized to control expression
Due to modest increases in expression of antioxidant response genes, we measured ROS directly by assessing superoxide production in cardiovascular organs of interest, the left ventricle, kidney, and aorta using lucigenin luminescence. We focused on animals exposed for 35 days since these mice were significantly hypertensive and this was the first time point when we detected significant changes in organ weights. Increased lucigenin luminescence was observed in all three cardiovascular organs from TCDD-exposed mice. This increase in lucigenin luminescence was verified to be attributed to an increase in superoxide production by a normalization of lucigenin luminescence to control levels in tissue from TCDD-exposed mice with preincubation with tempol, an SOD mimetic and antioxidant [21] (Fig. 5a–c).
Fig. 5.
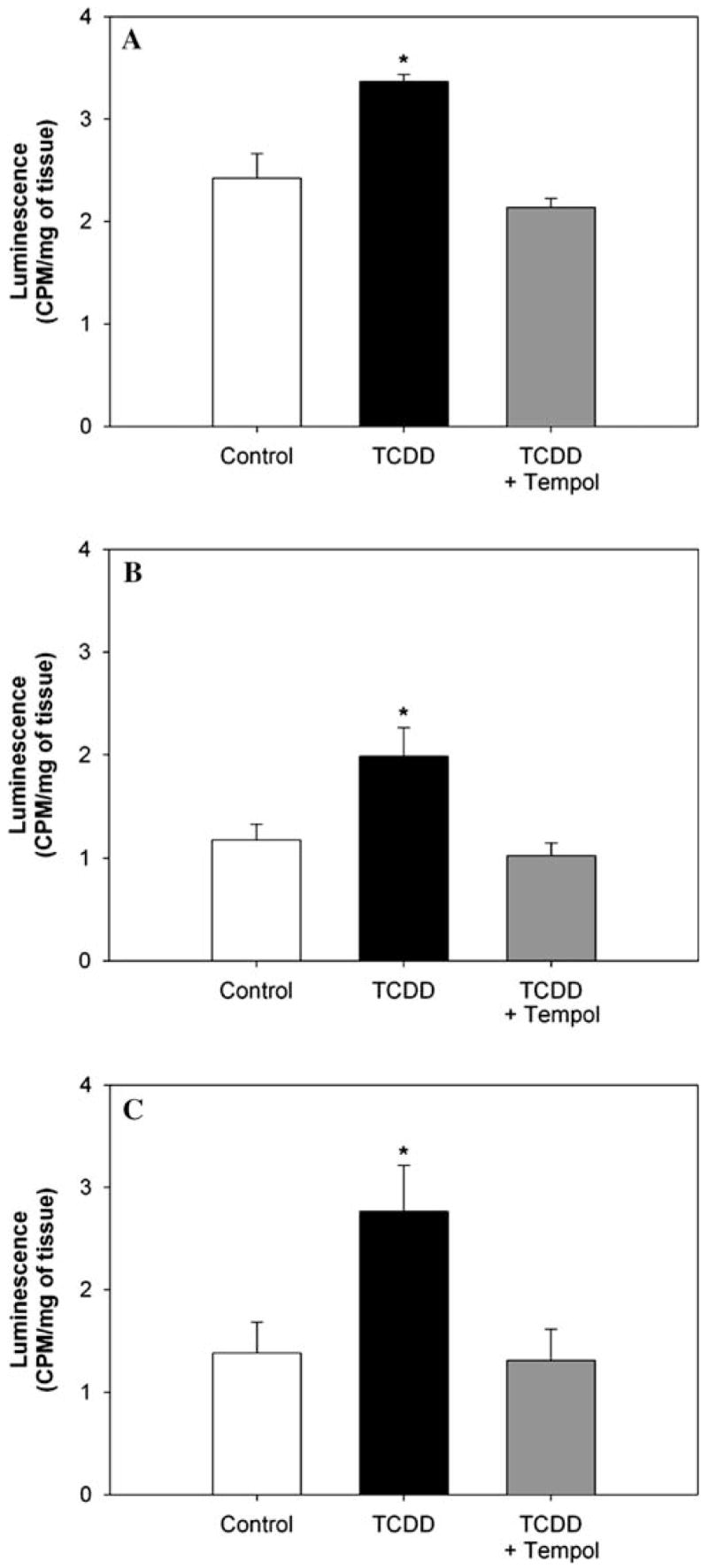
Effect of exposure to TCDD for 35 days on lucigenin luminescence in aorta (a), left ventricle (b), and kidney (c). n = 5. * P < 0.05, compared to control
Vascular Reactivity
With increased superoxide and the presence of hypertension, we examined the vasorelaxation response to ACh, which is mediated in the mouse aorta by endothelial production of nitric oxide (NO). Since superoxide can react with NO and reduce its ability to stimulate vasorelaxation, we further examined the ACh-response in the presence of tempol, an SOD mimetic and antioxidant. While aortic rings from control mice responded normally to ACh in a dose-dependent manner, the response to ACh in aortic rings from TCDD-exposed mice was significantly attenuated (Fig. 6a). However, when the vessels were preincubated with tempol, this attenuated response was normalized (Fig. 6b).
Fig. 6.

Effect of exposure to TCDD for 35 days on basal acetylcholine-induced vasorelaxation (a) (Control, n = 7; TCDD, n = 6) and acetylcholine-induced vasorelaxation in the presence of tempol (b) (n = 4). * P < 0.05, compared to respective control
Discussion
Our results demonstrate that subchronic TCDD exposure increases systemic arterial blood pressure, left ventricle weight and wall thickness, cardiovascular superoxide, and induces endothelial dysfunction characterized by a reduction in NO-dependent vasorelaxation. This study represents the first animal model that definitively demonstrates that sustained AhR activation by TCDD induces hypertension and cardiac hypertrophy, associated with impaired vascular reactivity.
Systolic blood pressure above 130 mmHg is considered hypertensive in mice [22]. Therefore, the blood pressure of 131/106 mmHg observed in TCDD-exposed mice at the termination of the study would classify them as hypertensive. This increase in blood pressure is consistent with previously reported increases in blood pressure from a mouse model of acute TCDD exposure as measured by a tail cuff system [8]. However, we observe an initial increase in blood pressure as early as day 2–3, while Dalton et al. [8] did not see significant increases in blood pressure until day 23–29. This may be due in part to the differences of dose and exposure, as well as the increased sensitivity that radiotelemetry provides for detecting smaller changes in blood pressure [23]. Moreover, the telemetry recordings allowed us to observe that the TCDD-exposed mice developed a nondipping pattern of diurnal blood pressure, characterized by less than 10% differences in daytime and nighttime blood pressure. Hypertensive target organ damage is more prevalent in individuals with a nondipper blood pressure pattern [24, 25], suggesting that TCDD-induced hypertension may lead to significant organ damage as observed with the left ventricle hypertrophy. Furthermore, the increase in LV + S weight, but not in RV weight, in TCDD-treated mice is indicative of an increase in systemic arterial pressure in the absence of an increase in pulmonary arterial pressure.
The TCDD-induced hypertension is also associated with a significant impairment of NO-dependent vasodilation that is normalized by a superoxide dismutase mimetic tempol. Superoxide is frequently elevated in human and experimental hypertension [11] and an important consequence of superoxide production is the loss of endothelium-derived NO. NO is as a potent vasodilator and genetic deletion of the enzyme that produces it, endothelial nitric oxide synthase (eNOS), results in loss of endothelial-dependent vasodilation (i.e., endothelial dysfunction) and hypertension [26]. Increased vascular superoxide reacts with NO to produce peroxynitrite and thus represents a key mechanism by which NO bioavailability is reduced, leading to diminished endothelial-dependent vasorelaxation. Furthermore, treatment with antioxidants, including tempol, improves vascular function and attenuates hypertension in many animal models [27], similar to our results in isolated artery studies. Moreover, chronic TCDD exposure increases the severity of atherosclerotic plaques in apolipoprotein E null mice [8], which may be attributed to a decrease in bioavailable NO and its anti-atherosclerotic activity [18]. Nonetheless, we cannot rule out the possibility that TCDD exposure reduces NO-dependent signaling in the vascular smooth muscle. Future studies of the aortic vasorelaxation responses to NO donors would establish whether downstream NO signaling is also altered by TCDD.
It has been well established that TCDD and other AhR ligands induce oxidative stress in humans and experimental animal models. TCDD exposure has been shown to induce oxidative stress in brain [28, 29], liver [28]), and kidney tissue [12] of mice. In rats, oxidative stress is increased in brain [30], liver [30], and reproductive tissue [30] by exposure to TCDD, polychlorinated dibenzofurans, or polychlorinated biphenyls. In humans, serum polychlorinated dibenzo-p-dioxin and polychlorinated dibenzofuran levels and plasma lipid peroxidation were positively correlated in metal recovery workers [31]. Moreover, concomitant antioxidant supplementation has been shown be protective in TCDD-induced toxicity in mice [32].
In the cardiovascular system, 3,3′,4,4′-tetrachlorobi-phenyl (PCB 77), a TCDD-like AhR agonist, has been shown to increase ROS in endothelial cells [33]. Moreover, exposure of zebrafish embryos to TCDD results in decreased blood flow in the mesencephalic vein, which is abolished by simultaneous exposure to an antioxidant [34]. Additionally, occupational exposure of pesticide production workers to TCDD is associated with impaired microvascular reactivity, which was negatively correlated with superoxide dismutase activity [35], indicative of vascular superoxide and endothelial dysfunction. Thus, activation of AhR appears to increase ROS in multiple organ systems.
There are a number of potential mechanisms for these observed increases in ROS. First, antioxidant enzyme activity could be suppressed, increasing susceptibility to endogenous ROS. TCDD exposure has been shown to decrease catalase and glutathione peroxidase activity in rat brain tissue [36] and decrease glutathione peroxidase, glutathione reductase, and SOD activity in chicken liver [37]. Since we see modest increases, rather than decreases, in mRNA expression of antioxidant response genes, these data suggest that suppressed antioxidant responses are not the primary cause of the increased ROS.
A second potential mechanism is increased mitochondrial production of ROS. Mitochondria are responsible for the majority of ROS production in most cells. However, ROS production and elimination by antioxidant systems is balanced under normal conditions [38]. Mitochondrial dysfunction that elevates ROS release can lead to the development of cardiovascular disease, particularly atherosclerosis and hypertension [39], and AhR-dependent mitochondrial production of ROS has been shown to be increased following TCDD exposure in mouse liver [40]. Interestingly, we saw an increase in mRNA expression of the mitochondrial isoform of SOD (SOD2 or MnSOD) in the left ventricle of TCDD-exposed mice. These data suggest that the source of the ROS could be mitochondrial, potentially linked to the mitochondrial accumulation of CYP450s that follow TCDD exposure [41].
Uncoupling of eNOS is a third potential source of ROS, where the enzyme transfers an electron from NADPH to oxygen, rather than the substrate, L-arginine, resulting in the formation of superoxide instead of NO [42]. The primary mechanism by which eNOS uncoupling occurs is from a decrease in the availability of the co-factor tetra-hydrobiopterin (BH4) [43]. BH4 levels can be reduced as a result of impaired synthesis and/or increased oxidation, both of which can lead to eNOS uncoupling, endothelial dysfunction, and hypertension [44–46]. Microarray studies have shown that TCDD exposure decreases the mRNA expression of cyclohydrolase 1 (GTPCH), the rate limiting step in BH4 synthesis, and increases the mRNA expression of GTP cyclohydrolase negative feedback protein, which inhibits BH4 synthesis [47, 48]. Thus, sustained AhR activation by TCDD may lead to BH4 depletion, eNOS uncoupling, and increased vascular superoxide.
Lastly, uncoupling of the induced cytochrome P450s is a fourth potential source of ROS. It has been demonstrated that P450-enriched human microsomes are capable of producing ROS [49]. Moreover, TCDD-induced mouse liver microsomes have increased production of superoxide [50]. CYP1A1 is of particular interest as a potential source of ROS. It is dramatically induced in the tissues of TCDD-exposed mice in our study, including the vasculature, and CYP1A and CYP1A1 are inducible in endothelial cells by AhR activation [51]. Furthermore, CYP1A and CYP1A1 have been shown to contribute to TCDD-induced toxicity in zebrafish and mice, respectively [52]. Future studies will investigate the role of CYP1A1 in ROS production and cardiovascular toxicity.
Hypertension in both humans and animal models is often associated with activation of the sympathetic nervous system and/or elevation of circulating Ang II and ET-1 [53]. Heart rate was not significantly altered in TCDD-exposed mice, suggesting that sympathetic nervous system activity was not increased. Similarly, neither plasma Ang II nor ET-1 was significantly altered in TCDD-exposed mice. While these data do not provide evidence of the involvement of these pathways, our results also do not rule them out. For example, increased sympathetic tone of peripheral resistance vessels can increase blood pressure without increasing heart rate [53], while plasma ET-1 levels do not necessarily reflect the amount of vasoactive ET-1 [54]. Therefore, future studies using pharmacological inhibitors of these pathways will help to further define their contribution to TCDD-induced hypertension.
While the TCDD exposure resulted in hypertension, it also induced periodic normalizations of the blood pressure. The mechanism underlying these normalizations in blood pressure is unknown, but may include a compensatory decrease in autonomic nervous system activity, changes in the tissue distribution of TCDD over time, or pressure natriuresis, an increase in sodium and water renal excretion in response to an increase in renal arterial pressure. A decrease in autonomic activity commonly is an acute compensatory response occurring over seconds to minutes rather than days, while significant changes in TCDD tissue distribution would not be expected to occur during these periods of blood pressure normalizations, based on previous reports of the toxicokinetics of TCDD in the mouse [17, 55]. Pressure natriuresis is a possible explanation, which could be verified by determining if urine volume and sodium excretion increase during the normalization periods. Moreover, if pressure natriuresis is responsible for the normalizations, the observed hypertension would be salt sensitive. Future studies of the mechanisms responsible for TCDD-induced increases in blood pressure will likely help to elucidate the reasons for the periodic normalizations.
In conclusion, our model is the first to definitively demonstrate that sustained AhR activation by subchronic TCDD exposure induces hypertension, vascular dysfunction, and cardiac hypertrophy. These data are consistent with the epidemiology studies of Vietnam veterans exposed to Agent Orange and validate the need to continue epidemiology studies of such cohorts as they reach the age when cardiovascular morbidity and mortality increase further. This model should provide valuable insight into the mechanisms underlying TCDD-induced cardiovascular pathogenesis, including the role of vascular ROS as potential mediators of TCDD-induced hypertension.
Acknowledgments
Funding National Institute of Health (ES12335 and HL078914 to MKW, ES12072 to UNM, 5T32HL007736 to UNM). Phillip Kopf is recognized as an American Foundation for Pharmaceutical Education Pre-Doctoral Fellow. The authors thank Drs. Nancy L. Kanagy and Matthew J. Campen for their valuable assistance.
Footnotes
The use of trade, firm, or corporation names in this publication is for the information and convenience of the reader. Such use does not constitute an official endorsement or approval by the United States Department of Agriculture or the Agricultural Research Service of any product or service to the exclusion of others that may be suitable.
Contributor Information
Phillip G. Kopf, Email: pkopf@salud.unm.edu.
Janice K. Huwe, Email: Janice.Huwe@ars.usda.gov.
Mary K. Walker, Email: mwalker@salud.unm.edu.
References
- 1.ATSDR. Toxicological profile for chlorinated dibenzo-p-dioxins. Atlanta: Agency for Toxic Substance and Disease Registry; 1998. [PubMed] [Google Scholar]
- 2.Swanson HI, Bradfield CA. The AH-receptor: Genetics, structure and function. Pharmacogenetics. 1993;3:213–230. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicology and Applied Pharmacology. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- 4.Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes to Cells. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- 5.Peters JM, Narotsky MG, Elizondo G, Fernandez-Salguero PM, Gonzalez FJ, Abbott BD. Amelioration of TCDD-induced teratogenesis in aryl hydrocarbon receptor (AhR)-null mice. Toxicological Sciences. 1999;47:86–92. doi: 10.1093/toxsci/47.1.86. [DOI] [PubMed] [Google Scholar]
- 6.Kim JS, Lim HS, Cho SI, Cheong HK, Lim MK. Impact of Agent Orange exposure among Korean Vietnam veterans. Industrial Health. 2003;41:149–157. doi: 10.2486/indhealth.41.149. [DOI] [PubMed] [Google Scholar]
- 7.Kang HK, Dalager NA, Needham LL, Patterson DG, Jr, Lees PS, Yates K, et al. Health status of Army Chemical Corps Vietnam veterans who sprayed defoliant in Vietnam. American Journal of Industrial Medicine. 2006;49:875–884. doi: 10.1002/ajim.20385. [DOI] [PubMed] [Google Scholar]
- 8.Dalton TP, Kerzee JK, Wang B, Miller M, Dieter MZ, Lorenz JN, et al. Dioxin exposure is an environmental risk factor for ischemic heart disease. Cardiovascular Toxicology. 2001;1:285–298. doi: 10.1385/CT:1:4:285. [DOI] [PubMed] [Google Scholar]
- 9.Jokinen MP, Walker NJ, Brix AE, Sells DM, Haseman JK, Nyska A. Increase in cardiovascular pathology in female Sprague-Dawley rats following chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin and 3,3′,4,4′,5-pentachloro-biphenyl. Cardiovascular Toxicology. 2003;3:299–310. doi: 10.1385/CT:3:4:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunnberg S, Andersson P, Lindstam M, Paulson I, Poellinger L, Hanberg A. The constitutively active Ah receptor (CA-Ahr) mouse as a potential model for dioxin exposure-effects in vital organs. Toxicology. 2006;224:191–201. doi: 10.1016/j.tox.2006.04.045. [DOI] [PubMed] [Google Scholar]
- 11.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension. 2004;44:248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 12.Slezak BP, Hatch GE, DeVito MJ, Diliberto JJ, Slade R, Crissman K, et al. Oxidative stress in female B6C3F1 mice following acute and subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicological Sciences. 2000;54:390–398. doi: 10.1093/toxsci/54.2.390. [DOI] [PubMed] [Google Scholar]
- 13.Huwe JK, Smith DJ. Laboratory and on-farm studies on the bioaccumulation and elimination of dioxins from a contaminated mineral supplement fed to dairy cows. Journal of Agricultural and Food Chemistry. 2005;53:2362–2370. doi: 10.1021/jf0480997. [DOI] [PubMed] [Google Scholar]
- 14.Simon P. Q-Gene: Processing quantitative real-time RT-PCR data. Bioinformatics (Oxford, England) 2003;19:1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 15.Lund AK, Peterson SL, Timmins GS, Walker MK. Endothelin-1-mediated increase in reactive oxygen species and NADPH Oxidase activity in hearts of aryl hydrocarbon receptor (AhR) null mice. Toxicological Sciences. 2005;88:265–273. doi: 10.1093/toxsci/kfi284. [DOI] [PubMed] [Google Scholar]
- 16.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594. 87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diliberto JJ, DeVito MJ, Ross DG, Birnbaum LS. Subchronic exposure of [3H]-2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in female B6C3F1 mice: Relationship of steady-state levels to disposition and metabolism. Toxicological Sciences. 2001;61:241–255. doi: 10.1093/toxsci/61.2.241. [DOI] [PubMed] [Google Scholar]
- 18.Levick JR. Introduction to cardiovascular physiology. London: Hodder Arnold; 2003. [Google Scholar]
- 19.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and Biophysical Research Communications. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 20.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muscoli C, Cuzzocrea S, Riley DP, Zweier JL, Thiemermann C, Wang ZQ, et al. On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. British Journal of Pharmacology. 2003;140:445–460. doi: 10.1038/sj.bjp.0705430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsukahara C, Sugiyama F, Paigen B, Kunita S, Yagami K. Blood pressure in 15 inbred mouse strains and its lack of relation with obesity and insulin resistance in the progeny of an NZO/HILtJ x C3H/HeJ intercross. Mammalian Genome. 2004;15:943–950. doi: 10.1007/s00335-004-2411-3. [DOI] [PubMed] [Google Scholar]
- 23.Gross V, Luft FC. Exercising restraint in measuring blood pressure in conscious mice. Hypertension. 2003;41:879–881. doi: 10.1161/01.HYP.0000060866.69947.D1. [DOI] [PubMed] [Google Scholar]
- 24.Kario K, Matsuo T, Kobayashi H, Imiya M, Matsuo M, Shimada K. Nocturnal fall of blood pressure and silent cerebrovascular damage in elderly hypertensive patients. Advanced silent cerebrovascular damage in extreme dippers. Hypertension. 1996;27:130–135. doi: 10.1161/01.hyp.27.1.130. [DOI] [PubMed] [Google Scholar]
- 25.Shimada K, Kawamoto A, Matsubayashi K, Nishinaga M, Kimura S, Ozawa T. Diurnal blood pressure variations and silent cerebrovascular damage in elderly patients with hypertension. Journal of Hypertension. 1992;10:875–878. [PubMed] [Google Scholar]
- 26.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 27.Pollock DM. Endothelin, angiotensin, and oxidative stress in hypertension. Hypertension. 2005;45:477–480. doi: 10.1161/01.HYP.0000158262.11935.d0. [DOI] [PubMed] [Google Scholar]
- 28.Bagchi D, Balmoori J, Bagchi M, Ye X, Williams CB, Stohs SJ. Comparative effects of TCDD, endrin, naphthalene and chromium (VI) on oxidative stress and tissue damage in the liver and brain tissues of mice. Toxicology. 2002;175:73–82. doi: 10.1016/S0300-483X(02)00062-8. [DOI] [PubMed] [Google Scholar]
- 29.Hassoun EA, Wilt SC, DeVito MJ, Van BA, Alsharif NZ, Birnbaum LS, et al. Induction of oxidative stress in brain tissues of mice after subchronic exposure to 2,3,7, 8-tetrachlorodibenzo-p-dioxin. Toxicological Sciences. 1998;42:23–27. doi: 10.1006/toxs.1997.2411. [DOI] [PubMed] [Google Scholar]
- 30.Hassoun EA, Li F, Abushaban A, Stohs SJ. The relative abilities of TCDD and its congeners to induce oxidative stress in the hepatic and brain tissues of rats after subchronic exposure. Toxicology. 2000;145:103–113. doi: 10.1016/S0300-483X(99) 00221-8. [DOI] [PubMed] [Google Scholar]
- 31.Chen HL, Hsu CY, Hung DZ, Hu ML. Lipid peroxidation and antioxidant status in workers exposed to PCDD/Fs of metal recovery plants. The Science of the Total Environment. 2006;372:12–19. doi: 10.1016/j.scitotenv.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Alsharif NZ, Hassoun EA. Protective effects of vitamin A and vitamin E succinate against 2,3,7,8-tetrachlo-rodibenzo-p-dioxin (TCDD)-induced body wasting, hepatomegaly, thymic atrophy, production of reactive oxygen species and DNA damage in C57BL/6J mice. Basic & Clinical Pharmacology & Toxicology. 2004;95:131–138. doi: 10.1111/j.1742-7843.2004.950305.x. [DOI] [PubMed] [Google Scholar]
- 33.Slim R, Toborek M, Robertson LW, Lehmler HJ, Hennig B. Cellular glutathione status modulates poly-chlorinated biphenyl-induced stress response and apoptosis in vascular endothelial cells. Toxicology and Applied Pharmacology. 2000;166:36–42. doi: 10.1006/taap.2000.8944. [DOI] [PubMed] [Google Scholar]
- 34.Dong W, Teraoka H, Yamazaki K, Tsukiyama S, Imani S, Imagawa T, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin toxicity in the zebrafish embryo: Local circulation failure in the dorsal midbrain is associated with increased apoptosis. Toxicological Sciences. 2002;69:191–201. doi: 10.1093/toxsci/69.1.191. [DOI] [PubMed] [Google Scholar]
- 35.Pelclova D, Prazny M, Skrha J, Fenclova Z, Kalousova M, Urban P, et al. 2,3,7,8-TCDD exposure, endothelial dysfunction and impaired microvascular reactivity. Human and Experimental Toxicology. 2007;26:705–713. doi: 10.1177/0960327107 083971. [DOI] [PubMed] [Google Scholar]
- 36.Hassoun EA, Al-Ghafri M, Abushaban A. The role of antioxidant enzymes in TCDD-induced oxidative stress in various brain regions of rats after subchronic exposure. Free Radical Biology and Medicine. 2003;35:1028–1036. doi: 10.1016/S0891-5849(03)00458-1. [DOI] [PubMed] [Google Scholar]
- 37.Lim J, DeWitt JC, Sanders RA, Watkins JB, III, Henshel DS. Suppression of endogenous antioxidant enzymes by 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced oxidative stress in chicken liver during development. Archives of Environmental Contamination and Toxicology. 2007;52:590–595. doi: 10.1007/s00244-006-0168-2. [DOI] [PubMed] [Google Scholar]
- 38.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry Biokhimiia. 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 39.Puddu P, Puddu GM, Cravero E, De PS, Muscari A. The putative role of mitochondrial dysfunction in hypertension. Clinical and Experimental Hypertension. 2007;29:427–434. doi: 10.1080/10641960701613852. [DOI] [PubMed] [Google Scholar]
- 40.Senft AP, Dalton TP, Nebert DW, Genter MB, Hutchinson RJ, Shertzer HG. Dioxin increases reactive oxygen production in mouse liver mitochondria. Toxicology and Applied Pharmacology. 2002;178:15–21. doi: 10.1006/taap.2001.9314. [DOI] [PubMed] [Google Scholar]
- 41.Genter MB, Clay CD, Dalton TP, Dong H, Nebert DW, Shertzer HG. Comparison of mouse hepatic mitochondrial versus microsomal cytochromes P450 following TCDD treatment. Biochemical and Biophysical Research Communications. 2006;342:1375–1381. doi: 10.1016/j.bbrc.2006.02.121. [DOI] [PubMed] [Google Scholar]
- 42.Verhaar MC, Westerweel PE, van Zonneveld AJ, Rabelink TJ. Free radical production by dysfunctional eNOS. Heart (British Cardiac Society) 2004;90:494–495. doi: 10.1136/hrt.2003.029405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:2439–2444. doi: 10.1161/01.ATV.00002439 24.00970.cb. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell BM, Dorrance AM, Webb RC. GTP cyclohydrolase 1 inhibition attenuates vasodilation and increases blood pressure in rats. American Journal of Physiology Heart and Circulatory Physiology. 2003;285:H2165–H2170. doi: 10.1152/ajpheart.00253.2003. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell BM, Dorrance AM, Webb RC. GTP cyclohydrolase 1 downregulation contributes to glucocorticoid hypertension in rats. Hypertension. 2003;41:669–674. doi: 10.1161/01.HYP.0000051889.62249.5D. [DOI] [PubMed] [Google Scholar]
- 46.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. The Journal of Clinical Investigation. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andreasen EA, Mathew LK, Tanguay RL. Regenerative growth is impacted by TCDD: Gene expression analysis reveals extracellular matrix modulation. Toxicological Sciences. 2006;92:254–269. doi: 10.1093/toxsci/kfj118. [DOI] [PubMed] [Google Scholar]
- 48.Carney SA, Chen J, Burns CG, Xiong KM, Peterson RE, Heideman W. Aryl hydrocarbon receptor activation produces heart-specific transcriptional and toxic responses in developing zebrafish. Molecular Pharmacology. 2006;70:549–561. doi: 10.1124/mol.106.025304. [DOI] [PubMed] [Google Scholar]
- 49.Puntarulo S, Cederbaum AI. Production of reactive oxygen species by microsomes enriched in specific human cytochrome P450 enzymes. Free Radical Biology and Medicine. 1998;24:1324–1330. doi: 10.1016/S0891-5849(97)00463-2. [DOI] [PubMed] [Google Scholar]
- 50.Shertzer HG, Clay CD, Genter MB, Chames MC, Schneider SN, Oakley GG, et al. Uncoupling-mediated generation of reactive oxygen by halogenated aromatic hydrocarbons in mouse liver microsomes. Free Radical Biology and Medicine. 2004;36:618–631. doi: 10.1016/j.freeradbiomed.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 51.Annas A, Brittebo EB. Localization of cytochrome P4501A1 and covalent binding of a mutagenic heterocyclic amine in blood vessel endothelia of rodents. Toxicology. 1998;129:145–156. doi: 10.1016/S0300-483X(98)00087-0. [DOI] [PubMed] [Google Scholar]
- 52.Teraoka H, Dong W, Tsujimoto Y, Iwasa H, Endoh D, Ueno N, et al. Induction of cytochrome P450 1A is required for circulation failure and edema by 2,3,7,8-tetrachlo-rodibenzo-p-dioxin in zebrafish. Biochemical and Biophysical Research Communications. 2003;304:223–228. doi: 10.1016/S0006-291X(03)00576-X. [DOI] [PubMed] [Google Scholar]
- 53.Kaplan NM. In: Kaplan’s clinical hypertension. Kaplan NM, editor. Philadelphia: Lippincott Williams & Wilkins; 2002. pp. 56–135. [Google Scholar]
- 54.Schiffrin EL. Vascular endothelin in hypertension. Vascular Pharmacology. 2005;43:19–29. doi: 10.1016/j.vph.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Diliberto JJ, Burgin D, Birnbaum LS. Effects of CYP1A2 on disposition of 2,3,7,8-tetrachlorodibenzo-p-dioxin, 2,3,4,7,8-pentachlorodibenzofuran, and 2,2′,4,4′,5,5′-hexachloro-biphenyl in CYP1A2 knockout and parental (C57BL6N and 129/Sv) strains of mice. Toxicology and Applied Pharmacology. 1999;159:52–64. doi: 10.1006/taap.1999.8720. [DOI] [PubMed] [Google Scholar]