

Abstract
West Nile virus (WNV) has recently emerged in North America as a significant disease threat to humans and animals. Unfortunately, no approved antiviral drugs exist to combat WNV or other members of the genus Flavivirus in humans. The WNV NS2B-NS3 protease has been one of the primary targets for anti-WNV drug discovery and design since it is required for virus replication. As part of our efforts to develop effective WNV inhibitors, we reexamined the reaction kinetics of the NS2B-NS3 protease and the inhibition mechanisms of newly discovered inhibitors. The WNV protease showed substrate inhibition in assays utilizing fluorophore-linked peptide substrates GRR, GKR, and DFASGKR. Moreover, a substrate inhibition reaction step was required to accurately model kinetic data generated from protease assays with a peptide inhibitor. The substrate inhibition model suggested peptide substrates could bind to two binding sites on the protease. Reaction product analogs also showed inhibition of the protease, demonstrating product inhibition in addition to, and distinct from, substrate inhibition. We propose that small peptide substrates and inhibitors may interact with protease residues that form either the P3-P1 binding surface (i.e., the S3-S1 sites) or the P1′-P3′ interaction surface (i.e., the S1′-S3′ sites). Optimization of substrate analog inhibitors that target these two independent sites may lead to novel anti-WNV drugs.
West Nile virus (WNV), a member of the family Flaviviridae, has become a significant cause of global disease (1). It is transmitted by mosquitoes between numerous species of birds, which are reservoir hosts, and humans and horses which are typically dead-end hosts (2). The virus was documented in 1937 in what was once known as the West Nile district of Uganda (3) and now occurs in Africa (4, 5), Europe (6, 7), the Middle East (8, 9,10), West and Central Asia (11), and recently the Americas. The virus appeared on the east coast of the United States in 1999 (12), and has since spread across the entire country and into Mexico (13), Central and South America (14), and Canada (15). While most infections are either asymptomatic or produce a mild febrile disease, in North America, thousands of clinical cases have progressed to West Nile fever, meningitis, encephalitis, and death. In 2007, the U.S. Centers for Disease Control reported 3,630 clinical cases in the USA, with 2,350 cases of West Nile fever, 1,217 cases of meningitis or encephalitis, and 124 fatalities. There are no vaccines or antiviral therapies approved for use in humans.
The ~11kb genome of WNV is transcribed as a single polyprotein (Figure 1) that is co- and post-translationally cleaved by viral and cellular proteases. The N-terminal domain of the NS3 protein (termed NS3pro) is a serine protease that cleaves after dibasic sites within the viral polyprotein including the NS2A-NS2B, NS2B-NS3, NS3-NS4A, and NS4B-NS5 cleavage sites (16). The central hydrophilic domain of NS2B is a required cofactor for protease activity. Since NS3 protease activity is essential for polyprotein processing and viral replication (17), the NS3 protein is a promising target for the development of flavivirus antiviral drugs (18). Structures of the active form of WNV NS2B-NS3pro (19, 20), or the related dengue 2 virus (DEN2V) NS3pro proteins (19, 21, 22) have guided structure-based studies to discover peptide and small molecule protease inhibitors (23, 24, 25).
Figure 1.

(a) West Nile virus genome including the 5′ and 3′ UTR (untranslated region), the 5′ cap, the structural (C, capsid, PrM, premembrane, and E, envelope) and nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) genes. (b) Schematic representation of the WNV protease expression construct (WNV NS2B-NS3pro) which includes the central hydrophilic domain of NS2B (cofactor) and the N-terminal protease domain of NS3.
Protease inhibition assays utilizing various NS2B-NS3pro constructs of dengue 2 (23, 26) or West Nile (27, 28) viruses have been reported. These assays provided a general platform to characterize activity and specificity of flavivirus proteases (27, 29, 30) and to determine the in vitro efficacy of candidate flavivirus protease inhibitors. Chromophore-linked peptide substrates were used to determine kinetic parameters for wild-type and mutant WNV proteases; these studies revealed important residue interactions between the enzyme and substrate (28, 29, 31). Fluorophore-linked peptide substrates were used to further elucidate the protease cleavage mechanism and to identify enzyme residues important for optimal cleavage (27, 32). Although previous studies of the WNV NS2B-NS3pro utilized a simple Michaelis-Menten model to analyze collected kinetic data, it was not clear if this model provided optimal agreement to the data.
In this paper, extensive kinetic data of WNV NS2B-NS3pro with small peptide substrates was collected to investigate alternative models of WNV protease activity. Surprisingly, a simple Michaelis-Menten model did not fit the highly reproducible experimental data. However, incorporating a substrate inhibition reaction step into the model enabled the data to be reliably fit and produced accurate and reproducible kinetic parameters for the cleavage reaction. Additionally, reaction product peptides inhibited the WNV protease, consistent with previous studies with the DEN2V protease (33). These results suggest new approaches for pursuing protease inhibitor optimization and developing efficacious West Nile antiviral drugs. To our knowledge, this is the first report of substrate inhibition for the WNV protease or flavivirus proteases in general, and the first definitive report of product inhibition for WNV protease.
EXPERIMENTAL PROCEDURES
Expression and Purification of WNV NS2B-NS3 Protease
Plasmid constructs for WNV NS2B-NS3 protease (NS2B-NS3pro) were obtained from Dr. Padmanabhan (Georgetown University) and have been previously described (27). Plasmids were transformed in XL1-blue competent cells. Positive colonies were amplified in 2xYT media using standard protocols, aliquotted, and frozen as glycerol stocks. Two 10 ml aliquots of 2xYT starter culture with 100 μg/ml carbenicillin were inoculated from the glycerol stocks, grown overnight at 37°C, and used to inoculate 2 L of 2xYT media. Bacterial cultures were grown at 250 rpm and 37°C for ~ 3 hrs to OD600 of ~ 0.7 AU, induced with 0.5 mM IPTG and incubated an additional 4 hrs at 37°C and 250 rpm. Cultures were then centrifuged at 3000 rpm for 30 minutes, and bacterial pellets were stored at −80°C.
Bacterial pellets were resuspended in 10 ml of chilled Buffer A (50 mM Hepes-HCl [pH 7.0], 500 mM NaCl). Lysis buffer (300 μg/ml lysozyme, 30 μg/ml DNAse, and 10 mM MgCl2) was added to the resuspended pellets, and the mixture stirred at 4°C for 30 min. Triton X100 was added to a final concentration of 0.5% (v/v), and the mixture stirred at 4°C for 30 min. The mixture was centrifuged and the soluble fraction applied to a nickel affinity column formed from 1 ml of Nickel Sephadex beads (Amersham Biosciences) pre-equilibrated with buffer A. The beads were washed with Buffer A, and increasing concentrations of imidazole (10 mM, 20 mM, and 30 mM in Buffer A) to remove contaminating proteins. Bound NS2B-NS3pro was eluted from the column with Buffer A and 150 mM imidazole in 1 ml aliquots, dialyzed into a storage buffer (50 mM Tris [pH 7.5], 300 mM NaCl), portioned into 1ml aliquots with 25% glycerol, flash-frozen in liquid nitrogen, and stored at −80°C. Protein concentration was determined by UV spectroscopy.
NS2B-NS3pro Kinetic Assays
Protease activity experiments were performed in vitro using purified NS2B-NS3pro and fluorophore-linked peptide substrates Boc-GRR-AMC, Boc-GKR-AMC, (Bachem, USA), and Ac-DFASGKR-AMC (Genscript). Preliminary activity experiments were performed for 30 min using 100 nM NS2B-NS3pro and 100 μM peptide substrate in cleavage buffer (200 mM Tris [pH 9.0], 20% glycerol) in 100 μl final volume. Reactions were monitored by release of free AMC using a Fluorolog FL3-22 spectrofluorometer (Horiba Jobin Yvon) to record fluorescence emitted at 465 nm following excitation at 380 nm. Assays were performed in duplicate.
Detailed kinetic studies were performed under similar reaction conditions as described above using a broad range of substrate concentrations. Reaction progress was monitored by release of free AMC every five minutes for at least 30 minutes. Assays were performed in triplicate.
To correct for systematic variations in instrument response, an AMC dilution series was measured in conjunction with each protease reaction. These measurements defined the linear range and response of the spectrofluorometer. Relative fluorescence unit data were converted to absolute AMC product concentrations using EXCEL (Microsoft, Redmond, WA), where the data were transformed using the slope from the linear regression of the AMC dilution series. Linear regression analysis was performed using GraphPad Prism (GraphPad Software San Diego, CA) to determine initial velocities for the reaction from AMC product concentrations and reaction times. Errors associated with each initial velocity measurement were consistently < 2%. Analysis of kinetic models for the velocity versus substrate concentration data were performed with the Dynafit program (Biokin, Watertown, MA) (34).
To determine the mechanism of inhibition and inhibition constants for Ac-GRR-NH2 and Ac-DFASGKR (GenScript, Scotch Plains, NJ), 10 μl of four different concentrations of each inhibitor were separately mixed with 70 μl of cleavage buffer and 10 μl of NS2B-NS3pro (100 nM final concentration). The assays were conducted in duplicate in 96-well black plates. Serial dilutions of substrate were added to the wells for final substrate concentrations of 25 μM, 50 μM, 100 μM, 200 μM, 400 μM, and 800 μM. Fluorescence of free AMC fluorophore was monitored at excitation of 380 nm and emission of 465 nm every five minutes for 30 minutes. To convert relative fluorescence units to absolute product concentrations, an AMC dilution series was performed as described above. Errors associated with each initial velocity measurement were < 2%. Model testing and validation was performed with the Dynafit program (34).
Molecular Modeling
Atomic structures of WNV NS2B-NS3pro complexed to either a small peptide inhibitor (PDB identifier 2FP7; 19) or the protein inhibitor aprotinin (PDB identifier 2IJO; 20) were used as reference structures for modeling substrate interactions with the active protease NS2B-NS3pro. Reference structures were uploaded into SWISS PDB, and ligand residues in the crystal structures that interacted with either the protease substrate S3-S1 or S1′-S3′ binding sites were mutated to correspond to the GRR substrate. The aprotinin in the 2IJO structure was used for the majority of the mutagenesis calculations since it was the longer of the two protease ligands and had a larger number of interactions with the protease relative to the ligand in the 2FP7 structure. Protease residues that interacted with a GRR peptide positioned at either the S3-S1 or S1′-S3′ binding sites of the protease were identified. A slight torsion of the P3′ arginine was introduced to remove a single steric clash between the GRR substrate and the protease. The GKR peptide was also modeled into the P1′-P3′ site and interactions delineated. The protease and substrate structures were minimized before analysis.
RESULTS
Protein Expression and Purification
The WNV protease plasmid construct included a 40 residue central hydrophilic domain from WNV NS2B joined to the N-terminal protease domain of the WNV NS3 protein by a glycine linker (Figure 1b) (27). The nucleotide sequence of the plasmid construct was verified by direct sequencing (S. Smith, UTMB). WNV NS2B-NS3pro was expressed and purified to homogeneity as visualized by Comassie blue staining of the protein separated by SDS polyacrylamide gel electrophoresis (data not shown).
NS2B-NS3 Protease Exhibited Substrate Inhibition
A series of detailed kinetic studies was completed to test if the NS2B-NS3pro exhibited Michaelis-Menten kinetics with short peptide substrates. The NS2B-NS3pro was clearly active against Boc-GRR-AMC (Figure 2a) and Boc-GKR-AMC (data not shown). For each substrate concentration, initial reaction velocities were calculated and plotted. Errors associated with initial velocities were consistently < 2%. Interestingly, the NS2B-NS3pro enzyme did not appear to follow simple Michaelis-Menten kinetics with initial reaction velocities reaching a plateau at high substrate concentrations. Instead, the initial reaction velocities reached a peak and then decreased with increasing substrate concentration (Figure 2a, solid curve). Similar kinetic profiles were observed for the GKR substrate although the substrate inhibition appeared more pronounced with the GRR substrate than with the GKR substrate (data not shown).
Figure 2.
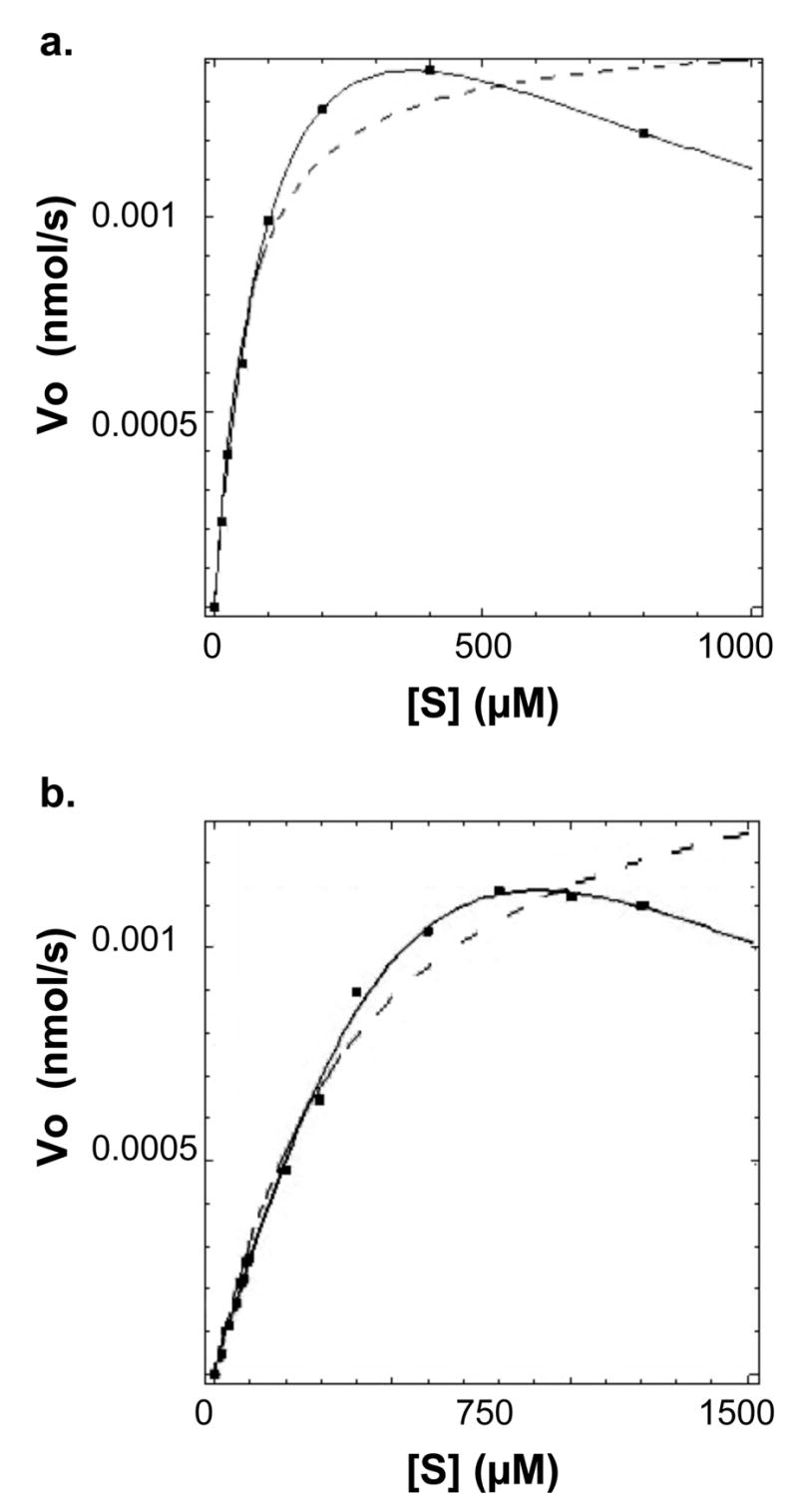
(a) WNV protease demonstrated substrate inhibition of the small peptide substrate Boc-Gly-Arg-Arg-AMC. (b) WNV protease demonstrated substrate inhibition of the longer peptide substrate Ac-Asp-Phe-Ala-Ser-Gly-Lys-Arg-AMC. Data was analyzed with the program Dynafit according to Scheme 1. Curves representing the substrate inhibition and the classical Michaelis-Menten models are indicated by solid and dashed lines, respectively. Initial velocity data points showed < 2% error.
Graphpad Prism was initially used to examine how accurately a simple Michaelis-Menten (35) model could fit the NS2B-NS3pro reaction kinetic data. Since a simple Michaelis-Menten model could not fit the NS2B-NS3pro kinetic data (Figure 2a, dashed curve), Graphpad Prism was used to test if an analytical closed–form solution (Equation 1) for Michaelis-Menten kinetics with substrate inhibition could fit the experimental data.
| Equation 1 |
In equation 1, [S] refers to substrate concentration, Vo to initial reaction velocity, and Km and Vmax were the classical Michaelis-Menten parameters. In Scheme 1, E, S, P, ES, and ESS refer to enzyme, substrate, product, enzyme-substrate intermediate complex, and the substrate-inhibited enzyme-substrate complex, respectively. Clearly, the Michaelis-Menten model with substrate inhibition (Figure 2a, solid curve) was fit to the kinetic data with a much higher R2 value and lower parameter errors than those calculated with a classical Michaelis-Menten model (Figure 2a, dashed curve). The kinetic parameters and data fitting were confirmed using the program Dynafit (34) which solved systems of differential rate equations to determine the best-fit of the model to the kinetic data (36). Identical kinetic parameters and curve fits were obtained with the GraphPad and Dynafit programs (Table 1). A product inhibition model was also examined and did not fit the data. This is not surprising since the concentration of accumulated product was not high enough to contribute to the total inhibition.
Scheme 1.
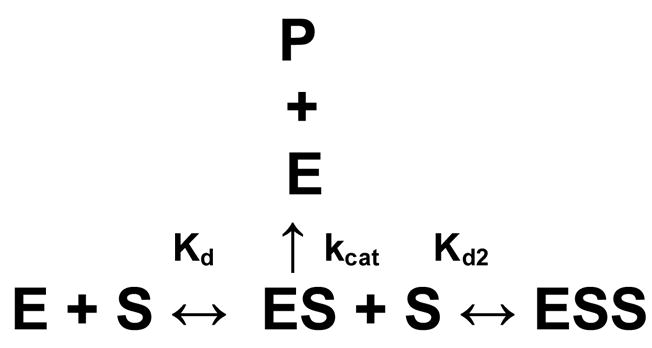
Table 1.
Kinetic Parameters for the WNV NS2B-NS3 Protease a
| Substrate | Kd (μM) | Kd2 (μM) | kcat (s−1) | kcat/Kd (M−1 s−1) |
|---|---|---|---|---|
| Boc-GRR-AMC | 117 ± 19 | 1366 ± 386 | 0.026 ± 0.002 | 222 ± 35 |
| Ac-DFASGKR-AMC | 800 ± 210 | 1204 ± 483 | 0.196 ± 0.067 | 245 ± 84 |
GRR experiments were repeated three times in triplicate. DFASGKR experiments were repeated twice in triplicate.
After observing that substrate inhibition occurred with short peptide substrates, the kinetic behavior of longer peptide substrates was examined since these substrates could potentially have greater relevance to the mechanism of virus replication in infected cells. NS2B-NS3pro activity was analyzed using the substrate Ac-DFASGKR-AMC, which mimics the P7-P1 residues of the NS3-NS4A natural cleavage site residues in the WNV polyprotein. Significantly, substrate inhibition was also observed with the longer substrate (Figure 2b), though it was observed at higher substrate concentrations compared to the tripeptide substrates.
It was clear that the kinetic data for the peptide substrates were fit better using a substrate inhibition model (Figure 2, solid curves) as opposed to a simple Michaelis-Menten model (Figure 2, dashed curves). The modeled curves in these figures were the same using either the Graphpad or Dynafit programs. Thus the reaction mechanism was a substrate inhibition model for both the short (Boc-GRR-AMC, Boc-GKR-AMC) and longer (Ac-DFASGKR-AMC) peptide substrates (Scheme 1). While all 3 substrates demonstrated inhibition at high concentrations, inhibition was more pronounced with Boc-GRR-AMC than with Boc-GKR-AMC (data not shown) or Ac-DFASGKR-AMC. Kinetic parameters for the protease and tested substrates are shown in Table 1. The enzyme demonstrated tighter binding (lower Kd) to the Boc-GRR-AMC substrate relative to the Ac-DFASGKR-AMC substrate. However, the higher kcat for the longer substrate relative to the tripeptide substrate implied that the protease had similar specificity (kcat/Kd) for both the Ac-DFASGKR-AMC and Boc-GRR-AMC substrates.
To ensure that inner-filter effects were not responsible for the observed decrease in initial velocity at high substrate concentrations, the fluorescence intensity of AMC was measured in the presence of substrate concentrations similar to those used in the protease assay. For these control experiments, AMC concentrations were similar to those measured in the protease assay. No change in AMC fluorescence signal was observed as a function of peptide substrate concentration (data not shown).
Kinetic Models for NS2B-NS3pro Inhibitors
Analogs of protease substrates or cleavage products have been used as strategies for the development of protease inhibitor molecules (37, 38). The tripeptide cleavage product Ac-GRR-NH2 was synthesized and tested to determine if it would inhibit NS2B-NS3pro activity. Analysis of kinetic reaction data clearly showed that this peptide behaved as an inhibitor (Figure 3). Several inhibition mechanisms, including competitive, uncompetitive, simple noncompetitive, and mixed noncompetitive, were tested independently. Each was constructed using the Dynafit program and model parameters were optimized to best fit each model to the experimental data points (data not shown). Comparison of models based on parameter errors, fitting accuracy, and model discrimination analysis within the Dynafit program (34) consistently highlighted the mixed noncompetitive inhibition model (Scheme 2) with substrate inhibition (hereafter referred to as a mixed inhibitor-substrate inhibition model) as the best model to replicate the kinetic data (Figure 3a). Substrate inhibition reaction steps were required to accurately fit the data since simple mixed noncompetitive models did not reproduce the kinetic data (Figure 3b).
Figure 3.

(a) The peptide Ac-GRR-NH2 inhibits WNV NS2B-NS3 protease according to a substrate inhibition and mixed inhibition model. Concentrations of inhibitor tested were 0 (triangles), 350 (circles), 700 (squares) μM, and 1 mM (diamonds). Data was analyzed with the program Dynafit according to Scheme 2. (b) Curves reflect an analysis of Ac-GRR-NH2 inhibition utilizing a mixed inhibition model without substrate inhibition. Initial velocity data points showed < 2% error.
Scheme 2.
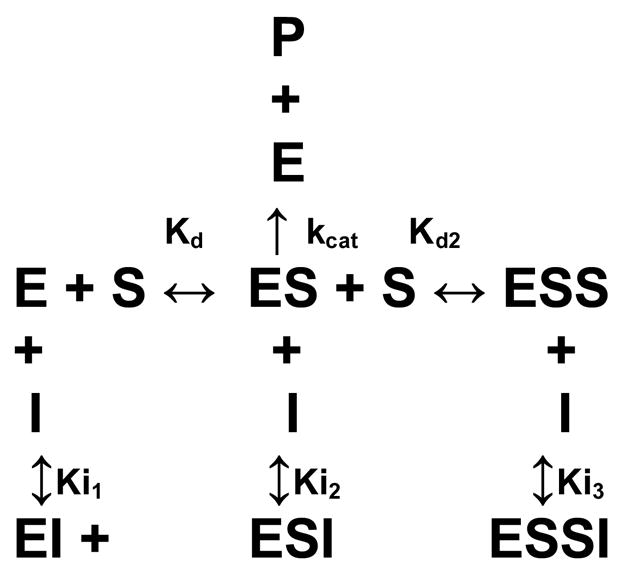
The kinetic model suggested that the inhibitor interacted primarily with the apo-enzyme (Ki1), and the enzyme-substrate complex (Ki2). An additional possibility is interaction with the substrate-inhibited enzyme-substrate complex (Ki3), and analyses of the model with and without this interaction were similar reflecting a large Ki3 which did not play a substantial role in the overall inhibition. Further studies will be necessary to fully determine the role, if any, of Ki3.
The kinetic parameters for the inhibitor are shown in Table 2. While the inhibitor behaved primarily as a competitive inhibitor, it also interacted with the ES complex as an uncompetitive inhibitor. Although Ki2 was a weaker interaction than Ki1, its inclusion in the kinetic model was necessary to accurately determine the kinetic parameters and inhibition constants and may elucidate new strategies for protease inhibition. Furthermore, it was crucial that the substrate inhibition step be included to model the data. Without its inclusion, the Kd and kcat were not accurate (Table 2). Interestingly, Kd and Ki1 were similar (Table 2), and there was only an ~ 2-fold difference between Kd2 and Ki2. This was not surprising since the inhibitor had the same peptide structure as the substrate. This might also imply that the Boc and AMC moieties either did not contribute to the interaction with the protease, or their interactions were compensatory since the peptide did not include the Boc or AMC groups.
Table 2.
Ac-GRR-NH2 Inhibition of WNV Protease
| Michaelis-Menten models | Kd (μM) | Kd2 (μM) | kcat (s−1) | Ki1 (μM) | Ki2 (μM) |
|---|---|---|---|---|---|
| Substrate & mixed inhibition | 122 ± 10 | 1265 ± 174 | 0.027 ± 0.001 | 180 ± 13 | 549 ± 44 |
| Simple mixed inhibition | 60 ± 6 | - | 0.018 ± 0.0005 | 146 ± 25 | 726 ± 105 |
| Simple competitive inhibition | 51 ± 10 | - | 0.017 ± 0.001 | 59 ± 11 |
Although the primary interaction was competitive (Ki1), if the kinetic data was analyzed according to simple Michaelis-Menten kinetics with competitive inhibition, then Ki1 was ~ 2-fold lower than when analyzed using substrate inhibition and mixed inhibition (Table 2). The error estimates for Kd and Ki1 significantly increased for the simple Michaelis-Menten competitive inhibition model relative to a model that included substrate inhibition and mixed inhibition (Table 2). Inaccurate estimations of Ki1 could lead to inefficient utilization of time and resources during drug development.
To determine whether peptide inhibition was observed for additional peptides, a second peptide Ac-DFASGKR (similar to the 7-mer peptide substrate used in the above kinetic studies) was tested to see if it inhibited the protease. Peptide inhibition was observed with the 7-mer peptide, and the data fit best to the mixed inhibitor-substrate inhibition model. In this analysis, inhibition constants were Ki1 = 1184 ± 151 and Ki2 = 2050 ± 415. Similar trends were seen for the longer inhibitor as with the tri-peptide with the primary interaction being with the apoenzyme and a weaker interaction with the enzyme-substrate complex. In addition, Kd and Ki1 were again very similar, and there was an ~2-fold difference between Kd2 and Ki2. As with the tri-peptide, model discrimination analysis with Dynafit revealed that the mixed inhibitor-substrate inhibition model was the most accurate model to describe the reaction kinetics.
Molecular Modeling
The aprotinin-WNV protease structure (PDB 2IJO; 20) was used to develop models for the binding of the peptide substrate Gly-Arg-Arg to WNV NS2B-NS3pro (Figure 4). The initial site examined was the conventional substrate binding pocket with interactions with substrate P3, P2, and P1 residues. The residues in aprotinin were Pro-Cys-Lys, and these were changed to Gly-Arg-Arg to mimic the peptide substrate. These substitutions resulted in no steric clashes between the substrate and protease, and increased favorable interactions relative to the aprotinin protease structure. This was expected as the assay substrate was designed to mimic a natural WNV polyprotein substrate.
Figure 4.

Peptide GRR interacting at two binding sites of WNV NS2B-NS3 protease. SwissPDB was used to mutate residues of aprotinin (PDB reference code 2IJO) from PCK to GRR in the P3-P1 positions (green), and from ARL to GRR in the P1′-P3′ positions (blue). Substrate positions for binding to polyprotein are designated according to standard nomenclature. The protease is colored grey, and active site is colored magenta. PYMOL (41) was used to generate the figure.
A second potential binding site was analyzed by examining interactions of the peptide substrate with the protease S1, ′ S2, ′ and S3′ residues. The second peptide binding site for substrate inhibition was modeled by mutating the P1′-P3′ residues of aprotinin in the 2IJO structure to Gly-Arg-Arg. The P1′ alanine of aprotinin made one contact with Thr132. A glycine substituted at this position made a similar contact with Thr132 and following minimization, an additional contact with Ser135 of the active site. The P2′ arginine was mutated to Arg; this control was performed to test that the computational substitutions and minimization largely reconstituted the observed crystallographic interactions. The P3′ isoleucine in aprotinin did not make any significant contacts with the protease. However, replacing this residue with arginine led to four additional contacts between the peptide substrate and the protease, including interaction between the guanidinium group of the P3′ arginine and the backbone carbonyl of the active site His51. In addition, the P3′ arginine made contacts with other residues near the active site (Thr52 at 2.29 Å, Thr53 at 2.89 Å, and Lys54 at 2.74 Å). The binding of the Gly-Lys-Arg substrate was also examined at the P1′-P3′ site, and observed to fit into the protease binding site.
The 2IJO structure showed a second loop of aprotinin containing Gly36-Gly37-Cys38-Arg39 that made significant contacts with the WNV protease. The tripeptide substrate was tested for binding at this site, and the modeled GRR substrate could also fit at this location with no steric clashes to the protease. This position allowed interactions with the NS2B cofactor as opposed to the NS3pro. This position could be examined at a later date to determine if this potential third binding site might account for the Ki3 inhibitory interaction.
The aprotinin-WNV protease structure was used to identify residues within 6 Å and 3.5 Å of each of the P3-P3′ substrate residues. Protease residues that made contacts with P3-P1 substrate residues were more highly conserved across the flaviviruses than were protease residues that contacted the P1′-P3′ substrate residues (Table 3). While there was sequence conservation for residues that contacted the P1′-P3′ substrate among the flavivirues, the conservation was much higher between the encephalitic flaviviruses (WNV and Japanese encephalitis virus) than the hemorrhagic flaviviruses. This implies that substrate inhibition may likely occur for the NS2B-NS3pro of Japanese encephalitis virus.
Table 3.
Percent Identity of Contact Residues a
| Site (6Å/3.5Å) | S3 (6/1) b | S2 (14/2) b | S1 (22/7) b | S1′ (10/2) b | S2′ (10/2) b | S3′ (10/4) b |
|---|---|---|---|---|---|---|
| WNV | 100/100 c | 100/100 | 100/100 | 100/100 | 100/100 | 100/100 |
| JEV | 83/100 | 82/100 | 77/100 | 90//50 | 80/50 | 78/100 |
| DEN1V | 75/100 | 82/100 | 64/100 | 70/50 | 50/0 | 44/50 |
| DEN2V | 75/100 | 82/100 | 68/100 | 70/50 | 50/0 | 44/50 |
| DEN3V | 75/100 | 82/100 | 64/100 | 70/50 | 50/0 | 44/50 |
| DEN4V | 75/100 | 82/100 | 73/100 | 70/50 | 50/0 | 44/50 |
| LGTV | 63/100 | 82/100 | 55/71 | 70/50 | 60/0 | 44/50 |
| YFV | 50/100 | 73/100 | 64/100 | 70/50 | 60/0 | 44/50 |
Sequences analyzed included WNV, Japanese encephalitis virus (JEV), the four dengue serotypes (DEN1V-DEN4V), Langat virus (LGTV), and yellow fever virus (YFV).
Number of contact residues within 6 Å and 3.5 Å, respectively, of the indicated residue site.
First and second numbers represent the percent identity of residues within 6 Å and 3.5 Å, respectively, of the indicated residue site.
To model interactions between a longer peptide substrate and the WNV NS2B-NS3pro, the P7-P1 residues of aprotinin were mutated to the DFASGKR sequence. The GKR residues interacted as indicated above, and the P7-P4 DFAS residues showed no steric clashes or additional contacts with the protease. The longer peptide substrate could also be accommodated at the potential P1′-P3′ substrate inhibition site with no steric clashes.
DISCUSSION
Since the NS2B-NS3 protease is required for flavivirus replication, it is a principle target for the development of small molecule and peptide-based antiviral drugs (25, 27, 33, 39). In vitro flavivirus protease inhibition assays used in the development of flavivirus antivirals are almost exclusively based on the cleavage of chromophore or fluorophore-linked small peptide substrates. Our research clearly showed that the WNV protease underwent substrate inhibition with three tested AMC-linked peptide substrates. Although the kinetics of the WNV protease have been previously described (27, 28), to our knowledge, substrate inhibition has not been previously reported for the WNV NS2B-NS3 protease. Substrate concentrations used in previous WNV protease assays may not have been sufficiently high to observe the substrate inhibition reaction mechanism. Additionally, optimization of the WNV protease purification protocol (27), followed by rapid flash-freezing, produced tighter binding protease preparations that enabled substrate inhibition to be observed at lower substrate concentrations relative to previous studies.
The observed substrate inhibition was not dependent upon short peptide substrates, since the longer peptide substrate (Ac-DFASGKR-AMC) also showed substrate inhibition. It is possible that substrate inhibition plays a biological role in WNV replication, perhaps serving as a feedback mechanism to tightly regulate WNV translation. However, the size of natural WNV polyprotein substrates may preclude the multiple substrate-protease states suggested by our substrate inhibition model. The occurrence and biological significance of protease substrate inhibition in WNV replication will be investigated in future studies.
Regardless of the biological role of the observed WNV substrate inhibition, it is important to account for this phenomenon to accurately determine WNV protease kinetic parameters and inhibition constants. An incorrect kinetic model can produce inaccurate and imprecise parameter values for a protease inhibition experiment. Since protease assays are routinely used to discover and improve WNV protease inhibitors, accurate assay interpretations are critical to effectively characterize and optimize WNV antivirals. In addition, the observed substrate inhibition mechanism may partly account for efficacy differences between in vitro and ex vivo experiments.
Our substrate inhibition model suggested that peptide substrates may simultaneously be bound at two sites on the protease. The catalytic substrate binding site would be the S3-S1 binding pocket of the protease which positions the substrate for cleavage. Molecular modeling indicated that dibasic peptide substrates will favorably fit into the S3-S1 binding pocket. In addition, molecular modeling showed that the tri- and heptapeptide substrates used in the WNV protease assays could also fit into at least two other sites on NS2B-NS3pro. One potential substrate inhibitory binding site was at the protease S1′-S3′ pocket, where the peptide substrate was predicted to make several favorable contacts with the protease including contacts with the protease catalytic site residues. These interactions may altar the conformation of the catalytic site so that it is unable to cleave substrate bound in the catalytic substrate binding pocket. While it would be reasonable to hypothesize that the Boc and AMC groups might contribute to the substrate inhibition, the similarities between Kd and Kd2 to Ki1 and Ki2, respectively, make this unlikely.
Competitive product inhibition has been demonstrated for the related hepatitis C (40) and dengue viruses (33), and suggested for WNV (24, 30). Our results clearly showed the product peptides Ac-GRR-NH2 and Ac-DFASGKR inhibited the WNV protease through a mixed non-competitive mechanism (with both competitive and uncompetitive interactions). This inhibition was in addition to substrate inhibition, and was only observed at higher product concentrations than would have been present in the substrate cleavage assays. These results suggest that it might be possible to develop substrate and/or product analogs as uncompetitive inhibitors targeting the substrate inhibitory binding site(s). An uncompetitive inhibitor is attractive as an antiviral since its inhibition would not be compromised at high substrate concentrations. Moreover, complementary inhibitors might be optimized for the separate competitive and uncompetitive binding sites on the protease. Since molecular modeling suggested these two binding sites may be adjacent to each other, the two separate inhibitors might be linked and developed as a single bidentate inhibitor. Such an inhibitor may reduce the likelihood of selecting for resistant WNV escape mutants, and lead to the development of a highly effective antiviral drug whose efficacy is not imperiled by the threat of escape mutations.
While further investigation will be needed to elucidate if a biological role exists for substrate and product inhibition in the cell, this study provides novel insights into WNV protease activity with small peptide substrates and offers new avenues to explore for the optimization of protease inhibitors that can be developed into effective antiviral drugs.
Acknowledgments
We thank N. Mueller and Dr. R. Padmanabhan for providing the WNV protease plasmids, and R. Malmstrom and Drs. A. Barrett, S. Gilbertson, J. Halpert, J. Lee, and J. Stevens for helpful discussions.
Abbreviations
- WNV
West Nile virus
- DEN2V
dengue 2 virus
- YFV
yellow fever virus
- JEV
Japanese encephalitis virus
- NS
nonstructural
- C
capsid
- prM
premembrane
- E
envelope
- GRR
glycine-arginine-arginine
- GKR
glycine-lysine-arginine
- DFASGKR
aspartate-phenylalanine-alanine-serine-glycine-lysine-arginine
- AMC
7-amino-4-methylcoumarin
Footnotes
This research was supported in part by NIH/NIAID AI066160 (SJW), The Welch Foundation H-1642 (SJW), and a training fellowship to SMT from the Computational and Structural Biology in Biodefense Training Program of the W. M. Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia (NIAID Grant No. 1 T32 AI065396-01).
References
- 1.Campbell G, Marfin A, Lanciotto R, Gubler D. West Nile virus. Lancet Infect Dis. 2002;2:519–529. doi: 10.1016/s1473-3099(02)00368-7. [DOI] [PubMed] [Google Scholar]
- 2.Fields B, Knipe D, Howley P, Chanock R, Melnick J, Monath T, Roizman B, Strauss S. Field’s Virology. 3. Lippincott Williams & Wilkins; Philadelphia: 1996. Flaviviruses; pp. 961–1034. [Google Scholar]
- 3.Brinton MA. The molecular biology of West Nile virus: a new invader of the Western hemisphere. Annu Rev Microbiol. 2002;56:371–402. doi: 10.1146/annurev.micro.56.012302.160654. [DOI] [PubMed] [Google Scholar]
- 4.Hurlbut H, Rizk F, Taylor R, Work T. A study of the ecology of West Nile virus in Egypt. Am J Trop Med Hyg. 1956;5:579–620. doi: 10.4269/ajtmh.1956.5.579. [DOI] [PubMed] [Google Scholar]
- 5.McIntosh B, Madsen W, Dickinson D. Ecological studies on Sindbis and West Nile viruses in South Africa. VI. The antibody response of wild birds. S Afr J Med Sci. 1969;34:83–91. [PubMed] [Google Scholar]
- 6.Pfleiderer C, Blumel J, Schmidt M, Roth W, Houfar M, Eckert J, Chudy M, Menichetti E, Lechner S, Nubling C. West Nile virus and blood product safety in Germany. J Med Virol. 2008;80:557–563. doi: 10.1002/jmv.21110. [DOI] [PubMed] [Google Scholar]
- 7.Del Giudice P, Schuffenecker I, Vandenbos F, Counillon E, Zellet H. Human West Nile virus, France. Emerg Infect Dis. 2004;10:1885–1886. doi: 10.3201/eid1010.031021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samina I, Margalit J, Peleg J. Isolation of viruses from mosquitoes of the Negev, Israel. Trans R Soc Trop Med Hyg. 1986;80:471–472. doi: 10.1016/0035-9203(86)90348-2. [DOI] [PubMed] [Google Scholar]
- 9.Ozer N, Ergunay K, Simsek F, Kaynas S, Alten B, Caglar S, Ustacelebi S. West Nile virus studies in the Sanliurfa province of Turkey. J Vector Ecol. 2007;32:202–206. doi: 10.3376/1081-1710(2007)32[202:wnvsit]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 10.Weinberger M, Pitlik S, Gandacu D, Lang R, Nassar F, Ben D, Rubinstein E, Izthaki A, Mishal J, Kitzes R, Siegman-Ingra Y, Giladi M, Pick N, Mendelson E, Bin H, Shohat T. West Nile fever outbreak, Israel, 2000: epidemiologic aspects. Emerg Infect Dis. 2001;7:686–691. doi: 10.3201/eid0704.010416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bondre V, Jadi R, Mishra A, Yergolkar P, Arankalle V. West Nile virus isolates from India: evidence for a distinct genetic lineage. J Gen Virol. 2007;88:875–884. doi: 10.1099/vir.0.82403-0. [DOI] [PubMed] [Google Scholar]
- 12.Lanciotti R, Roehrig J, Deubel V, Smith J, Parker M, Steele K, Crise B, Volpe K, Crabtree M, Scherret J, Hall R, Mackenzie J, Cropp C, Panigrahy B, Ostlund E, Schmitt B, Malkinson M, Banet C, Weissman J, Komar N, Savage H, Stone W, McNamara T, Gubler D. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286:2333–2337. doi: 10.1126/science.286.5448.2333. [DOI] [PubMed] [Google Scholar]
- 13.Deardorff E, Estrada-Franco J, Brault A, Navarro-Lopez R, Campomanes-Cortes A, Paz-Ramirez P, Solis-Hernandez M, Ramey W, Davis C, Beasley D, Tesh R, Barrett A, Weaver S. Introductions of West Nile virus strains to Mexico. Emerg Infect Dis. 2006;12:314–318. doi: 10.3201/eid1202.050871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berrocal L, Pena J, Gonzalez M, Mattar S. West Nile virus; ecology and epidemiology of an emerging pathogen in Colombia. Rev Salud Publica (Bogata) 2006;8:218–228. doi: 10.1590/s0124-00642006000200010. [DOI] [PubMed] [Google Scholar]
- 15.Schellenberg T, Anderson M, Drebot M, Vooght M, Findlater A, Curry P, Campbell C, Osei W. Seroprevalence of West Nile virus in Saskatchewan’s Five Hills Health Region, 2003. Can J Public Health. 2006;97:369–373. doi: 10.1007/BF03405344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers T, Nestorowics A, Amberg S, Rice C. Mutagenesis of the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J Virol. 1993;67:6797–6807. doi: 10.1128/jvi.67.11.6797-6807.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falgout B, Pethel M, Zhang Y, Lai C. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol. 1991;65:2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leyssen P, de Clercq E, Neyts J. Perspectives for the treatment of infections with Flaviviridae. Clin Microbiol Rev. 2000;13:67–82. doi: 10.1128/cmr.13.1.67-82.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Arcy A, Chaillet M, Schiering N, Villard F, Pheng Lim S, Lefeuvre P, Erbel P. Purification and crystallization of dengue and West Nile virus NS2B-NS3 complexes. Acta Cryst. 2006;62:157–162. doi: 10.1107/S1744309106001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aleshin AE, Shiryaev SA, Strongin AY, Liddington RC. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 2007;16:795–806. doi: 10.1110/ps.072753207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murthy H, Clum S, Padmanabhan R. Dengue virus NS3 serine protease. Crystal structure and insights into interaction of the active site with substrates by molecular modeling and structural analysis of mutational effects. J Biol Chem. 1999;274:5573–5580. doi: 10.1074/jbc.274.9.5573. [DOI] [PubMed] [Google Scholar]
- 22.Murthy H, Judge K, Delucas L, Padmanabhan R. Crystal structure of dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: implications for flaviviral polyprotein processing and drug design. J Mol Biol. 2000;301:759–767. doi: 10.1006/jmbi.2000.3924. [DOI] [PubMed] [Google Scholar]
- 23.Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J Biol Chem. 2001;276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- 24.Knox J, Ling Ma N, Yin Z, Patel S, Wang W, Chan W, Rao K, Wang G, Ngew X, Patel V, Beer D, Pheng Lim S, Vasudevan S, Keller T. Peptide inhibitors of West Nile NS3 protease: SAR study of tetrapeptide aldehyde inhibitors. J Med Chem. 2006;49:6585–6590. doi: 10.1021/jm0607606. [DOI] [PubMed] [Google Scholar]
- 25.Ganesh VK, Muller N, Judge K, Luan CH, Padmanabhan R, Murthy KH. Identification and characterization of nonsubstrate-based inhibitors of the essential dengue and West Nile virus proteases. Bioor Med Chem. 2005;13:257–264. doi: 10.1016/j.bmc.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 26.Yusof R, Clum S, Wetzel M, Murthy H, Padmanabhan R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J Biol Chem. 2000;275:9963–9969. doi: 10.1074/jbc.275.14.9963. [DOI] [PubMed] [Google Scholar]
- 27.Mueller NH, Yon C, Ganest VK, Padmanabhan R. Characterization of the West Nile virus protease substrate specificity and inhibitors. Int J Biochem Cell Biol. 2007;39:606–614. doi: 10.1016/j.biocel.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 28.Chappell K, Nall T, Stoermer M, Fang N, Tyndall J, Fairlie D, Young P. Site-directed mutagenesis and kinetic studies of the West Nile virus NS3 protease identify key enzyme-substrate interactions. J Biol Chem. 2005;28:2896–2903. doi: 10.1074/jbc.M409931200. [DOI] [PubMed] [Google Scholar]
- 29.Chapell K, Stoermer M, Fairlie D, Young P. Generation and characterization of proteolytically active and highly stable truncated and full-length recombinant West Nile virus NS3. Protein Expr Purif. 2007;53:87–96. doi: 10.1016/j.pep.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 30.Bera AK, Juhn RJ, Smith JL. Functional characterization of cis and trans activity of the Flavivirus NS2B-NS3 protease. J Biol Chem. 2007;282:12883–12892. doi: 10.1074/jbc.M611318200. [DOI] [PubMed] [Google Scholar]
- 31.Nall T, Chappell K, Stoermer M, Fang N, Tyndall J, Young P, Fairlie D. Enzymatic characterization of a homology model of a catalytically active recombinant West Nile virus NS3 protease. J Biol Chem. 2004;279:48535–48542. doi: 10.1074/jbc.M406810200. [DOI] [PubMed] [Google Scholar]
- 32.Shiryaev S, Ratnikov B, Aleshin A, Kozlov I, Nelson N, Lebl M, Smith J, Liddington R, Strongin A. Switching the substrate specificity of the two-component NS2B-NS3 flavivirus proteinase by structure based mutagenesis. J Virol. 2007;81:4501–4509. doi: 10.1128/JVI.02719-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chanprapaph S, Saparpakorn P, Sangma C, Niyomrattanakit P, Hannongbua S, Angsuthanasombat C, Katzenmeier G. Competitive inhibition of the dengue virus NS3 serine protease by synthetic peptides representing polyprotein cleavage sites. Biochem Biophys Res Commun. 2005;330:1237–1246. doi: 10.1016/j.bbrc.2005.03.107. [DOI] [PubMed] [Google Scholar]
- 34.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 35.Segel I Wiley Classics Library. Enzyme Kinetics Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley & Sons, Inc; New York: 1993. Kinetics of Unireactant Enzymes; pp. 18–89. [Google Scholar]
- 36.Kuzmic P, Cregar L, Millis S, Gopldman M. Mixed-type noncompetitive inhibition of lethal factor protease by aminoglycosides. FEBS J. 2006;273:3054–3062. doi: 10.1111/j.1742-4658.2006.05316.x. [DOI] [PubMed] [Google Scholar]
- 37.Turk B. Discovery and development of anthrax lethal factor metalloproteinase inhibitors. Curr Pharm Biotechnol. 2008;9:24–33. doi: 10.2174/138920108783497604. [DOI] [PubMed] [Google Scholar]
- 38.Lai L, Han X, Chen H, Wei P, Huang C, Liu S, Fan K, Zhou L, Liu Z, Pei J, Liu Y. Quaternary structure, substrate selectivity and inhibitor design for SARS 3C-like proteinase. Curr Pharm Des. 2006;12:4555–4564. doi: 10.2174/138161206779010396. [DOI] [PubMed] [Google Scholar]
- 39.Yin Z, Patel S, Wang W, Wang G, Chan W, Rao K, Alan J, Jeyaraj D, Ngew X, Patel V, Beer D, Lim S, Vasudevan S, Keller T. Peptide inhibitors of dengue virus NS3 protease. Part 1: Warhead. Bioorg Med Chem Lett. 2006;16:36–39. doi: 10.1016/j.bmcl.2005.09.062. [DOI] [PubMed] [Google Scholar]
- 40.Steinhuhler C, Biasiol G, Brunetti M, Urbani A, Koch U, Cortese R, Pessi A, De Francesco R. Product inhibition of the hepatitis C virus NS3 protease. Biochemistry. 1998;37:8899–8905. doi: 10.1021/bi980313v. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. DeLano Scientific LLC; Palo Alto, CA, USA: 2007. The PyMOL Molecular Graphics System. http://www.pymol.org. [Google Scholar]