

Abstract
The development of selective inhibitors for individual PLA2 enzymes is necessary in order to target PLA2-specific signaling pathways; but it is challenging due to the observed promiscuity of known PLA2 inhibitors. In the current work, we present the development and application of a variety of synthetic routes to produce pentafluoro, tetrafluoro and trifluoro derivatives of activated carbonyl groups in order to screen for selective inhibitors and characterize the chemical properties that can lead to selective inhibition. Our results demonstrate that the pentafluoroethyl ketone functionality favors selective inhibition of the GVIA iPLA2, a very important enzyme for which specific, potent reversible inhibitors are needed. We find that 1,1,1,2,2-pentafluoro-7-phenyl-heptan-3-one (FKGK11) is a selective inhibitor of GVIA iPLA2 (XI(50) = 0.0073). Furthermore, we conclude that the introduction of an additional fluorine atom at the α′ position of a trifluoromethyl ketone constitutes an important strategy for the development of new potent GVIA iPLA2 inhibitors.
Keywords: Calcium-independent phospholipase A2, inhibitors, pentafluoroethyl ketones, polyfluoro ketones, phospholipase A2
Introduction
Phospholipase A2 (PLA2) enzymes catalyze the hydrolysis of the sn-2 ester bond of glycerophospholipids producing free fatty acids and lysophospholipids.1,2 Both products are precursor signaling molecules that are involved in a plethora of biological functions. The PLA2 superfamily currently consists of fifteen groups and many subgroups of which a number of enzymes differ in primary sequence, structure and catalytic mechanism.1 Among the various PLA2 enzymes, Group IVA cPLA2 (GIVA cPLA2) is considered the rate-limiting provider of arachidonic acid and lysophospholipids that can be converted into prostaglandins, leukotrienes and PAF, respectively.1–3 Another major intracellular PLA2, the calcium-independent PLA2 (GVIA iPLA2) appears to be the primary phospholipase for basal metabolic functions within the cell.1,2,4,5 Both intracellular enzymes share the same catalytic mechanism of utilizing a serine residue as the nucleophile. The PLA2 superfamily also includes a type of small, secreted phospholipase (sPLA2) that is characterized by a catalytic His/Asp dyad as well as a catalytic Ca2+.1,2,6 A well-studied example of this class is the human Group V secreted phospholipase A2 (GV sPLA2).7 In many cases the activity of sPLA2 has been shown to be dependent on or linked to the activity of GIVA cPLA2.8–10
Various classes of synthetic compounds have been studied as inhibitors of human GIVA cPLA2, GVIA iPLA2 and GV sPLA2; and the results are summarized in recent review articles.11,12 One of the most potent inhibitors of GIVA cPLA2 is pyrrophenone (1, Figure 1).13 Other recently reported inhibitors include 2-propanone derivatives combined with the indole ring (e.g. 2, Figure 1)14–16 and a series of indole derivatives17–19 presented by Wyeth (for example compounds 3a and 3b, Figure 1) of which Efipladib (3b) is currently in phase I clinical trials.19 Our laboratories have reported on the development of 2-oxoamide inhibitors of GIVA cPLA2 (e.g. 4a–d, Figure 1). 20–26
Figure 1.

Some known inhibitors of GIVA cPLA2.
Historically, the first potent inhibitor of GIVA cPLA2 was a trifluoromethyl ketone analogue of arachidonic acid (AACOCF3) in which the carboxyl group was replaced by COCF3 (5, Figure 2).27 This analogue was shown to be a slow- and tight-binding inhibitor of GIVA cPLA2 and its mechanism of inhibition has been characterized via 19F NMR and 13C NMR.28 Trifluoromethyl ketone analogues of H-linolenic and linoleic acid as well as the analogue of palmitic acid (6, Figure 2) also inhibit GIVA cPLA2.29,30 Furthermore, a variety of trifluoromethyl ketones have been analyzed with phospholipid vesicle-, detergent-phospholipid mixed micelle-, and natural membrane-based assays.31
Figure 2.

Trifluoromethyl ketone inhibitors of GIVA cPLA2 and GVIA iPLA2.
AACOCF3 has been used as a tool to study the role of GIVA cPLA2 inhibition in various animal models. Using this inhibitor, it was demonstrated that GIVA cPLA2 plays an important role in the pathogenesis of experimental autoimmune encephalomyelitis (EAE), the animal model of multiple sclerosis.32 AACOCF3 was also used to study possible contributions of central nervous PLA2 enzymes to the development of allodynia after facial carrageenan injection in mice.33 Intrathecal administration of AACOCF3 prevented thermal hyperalgesia induced by intraplantar carrageenan as well as formalin-induced flinching in a dose-dependent manner.34 Intrathecal injection of AACOCF3, at antihyperalgesic doses, decreased the release of prostaglandin PGE-2 into spinal dialysate-evoked N-methyl-D-aspartate (NMDA).35 Similarly, treatment of prion-infected cell lines indicated a pivotal role for PLA2 enzymes in prion diseases.35 Even so, the various in vivo activities of AACOCF3 should be viewed with some caution, since this inhibitor is not selective for GIVA cPLA2 and has been reported to cause cell lysis.36 Additional trifluoromethyl ketone derivatives are also observed to inhibit GIVA cPLA2.37–40 For example, BMS-22972441 (7, Figure 2) was reported to be a tight-binding inhibitor of GIVA cPLA2 possessing anti-inflammatory activity in skin inflammation models.41
Trifluoromethyl ketone analogs of arachidonic and palmitic acids also inhibit GVIA iPLA2.42 Both compounds inhibited macrophage GVIA iPLA2 in a concentration-dependent manner and, in contrast to GIVA cPLA2, GIVA iPLA2 showed a preference for the saturated fatty chain.42 Inhibition studies of a variety of trifluoromethyl ketones as inhibitors of GVIA iPLA2 in mixed-micelle assays found that one trifluoromethyl ketone (8, Figure 2) is a potent inhibitor of GVIA iPLA2 presenting a XI(50) value of 0.0043, which is ten-fold more potent than the corresponding value against GIVA cPLA2.31
Continuing our efforts to synthesize selective inhibitors for the various PLA2 enzyme types, we designed a variety of polyfluoro ketone-based derivatives. In this work, we present routes for the synthesis of polyfluoro ketones and demonstrate their inhibition of the three major human PLA2 enzymes: GIVA cPLA2, GVIA iPLA2 and GV sPLA2, but with vastly different specificities. Of particular note is the development of specific GVIA iPLA2 inhibitors.
Design and Synthesis of Polyfluoro Ketones
We designed a variety of polyfluoro ketones and examples of such activated carbonyl functionalities are depicted in Figure 3. The rationale behind our design of polyfluoro ketones was based on: (a) Increase of the carbonyl reactivity by introduction of additional fluorine atoms at the β- or α′-positions. The inductive effect of additional fluorine atoms may increase carbonyl reactivity against nucleophiles, such as the active-site serine hydroxyl group in GIVA cPLA2 and GVIA iPLA2; and (b) Increase of the inhibitor binding affinity to the target enzymes. Additional fluorine atoms at the β- or α′-position may contribute to the development of additional interactions, further stabilizing the enzyme-inhibitor complex. Recently, it has become clear that fluorine can enhance binding efficacy and selectivity in pharmaceuticals due to a variety of multipolar C-F···H-N, C-F···C=O, and C-F···H-Cα interactions between a fluorinated ligand and protein binding-site.43,44 Since the natural substrates of PLA2 enzymes are long chain phospholipids, we chose to attach the polyfluoro ketone functionality to a long aliphatic chain as well as to short or medium chains carrying a non-substituted or para-alkoxy (or aryloxy) substituted ring.
Figure 3.
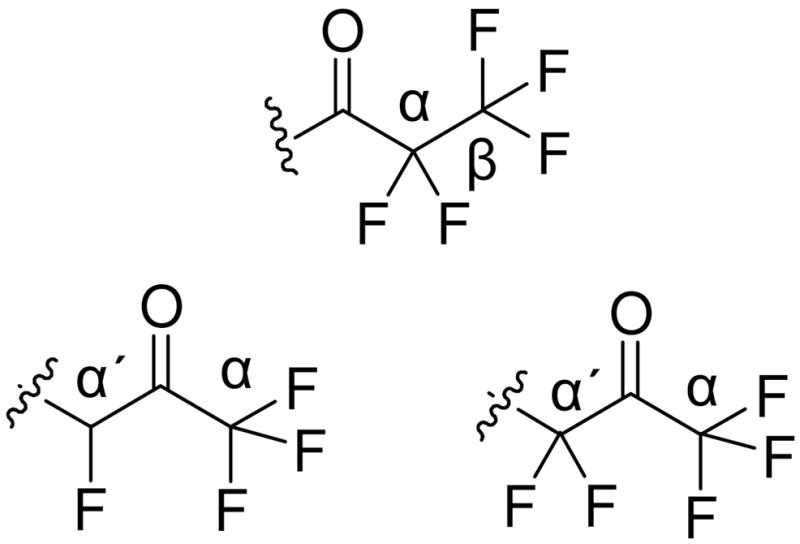
Polyfluoro ketone functionalities.
Among the existing methods, the synthesis of trifluoromethyl ketones through conversion of carboxylic acids into chlorides followed by subsequent treatment with trifluoroacetic anhydride and pyridine45 has found wide application. We observe that simple carboxylic acids, amino acids and peptides,46 and even lipophilic glyceride analogues, as we have demonstrated for the synthesis of potent gastric lipases inhibitors,47 are able to produce trifluoromethyl ketones in satisfactory yields. For the synthesis of pentafluoroethyl ketones, carboxylic acids 9a–c were converted to chlorides by treatment with oxalyl chloride and then to the target compounds 10a–c using pentafluoropropionic anhydride and pyridine (Figure 4). For comparison purposes, we prepared pentafluoroethyl ketone 11 corresponding to palmitic acid as well as trifluoromethyl ketones 12a,b corresponding to pentafluoro derivatives 10b,c.
Figure 4.

Reagents and conditions: (a) i) (COCl2)2, CH2Cl2; ii) (CF3CF2CO)2O, pyridine, CH2Cl2.
The synthesis of various trifluoromethyl and pentafluoroethyl ketones is depicted in Figure 5. The hydroxymethyl group of compounds 13a,b was oxidized to an aldehyde by the NaClO/TEMPO method.48 Wittig olefination of aldehydes 14a,b and Wadworth-Horner-Emmons reaction led to elongation of the chain by two or four carbon atoms, respectively. After hydrogenation and saponification, carboxylic acids 17a,b and 18a,b were converted to fluoroketones 19a,b, 20a,b and 21 as described above. The trifluoromethyl ketone 23 was prepared from he t known carboxylic acid 22 (Figure 6).
Figure 5.

Reagents and conditions: (a) NaOCl, TEMPO, NaBr, NaHCO3, toluene/EtOAc, H2O; (b) Ph3P=CHCOOCH3, CH2Cl2; (c) C2H5OOCH=CHCH2P(=O)(OC2H5), LiOH, THF; (d) i) H2, 10% Pd, ii) NaOH, CH3OH; (e) i) (COCl2)2, CH2Cl2, ii) (CF3CO)2O, pyridine, CH2Cl2.
Figure 6.

Reagents and conditions: (a) i) (COCl2)2, CH2Cl2, ii) (CF3CO)2O, pyridine, CH2Cl2.
Tetrafluoro derivative 26 was synthesized as shown in Figure 7. The replacement of the hydroxyl group of methyl 2-hydroxy-hexadecanoate (24) with fluorine was carried out by treatment with diethylaminosulfur trifluoride (DAST), a well-known fluorinating agent.49 Treatment of methyl ester 25 by (trifluoromethyl)trimethylsilane in the presence of a catalytic amount of cesium fluoride, followed by hydrolysis of silyl ether intermediate,50 led directly to tetrafluoro derivative 26. It should be noted that a 2-fluorocarboxylic acid cannot transform into a trifluoromethyl ketone by conversion to chloride and treatment with anhydride and pyridine, probably because the intermediate ketene required for such a transformation45 cannot be formed.
Figure 7.

Reagents and conditions: (a) Deoxofluor, dry CH2Cl2; (b) i) (CH3)3SiCF3, CsF, CH3OCH2CH2OCH3, ii) conc. HCl.
To synthesize pentafluoro derivative 30, we explored two different routes (Figures 8 and 9). Reaction of diethyl oxalate with Grignard reagent51 27 led to 2-oxoester 28 (Figure 8). DAST is an efficient reagent for the conversion of 2-oxoesters to 2,2-difluoroesters;52,53 therefore, 2-oxoester 28 was fluorinated by treatment with DAST and ethyl ester 29 was converted to trifluoromethyl ketone 30 as described above. Alternatively, compound 30 was prepared starting from aldehyde 31 (Figure 9). Formation of cyanohydrin 32 was followed by methanolysis and finally oxidation to produce 2-oxoester 34. By similar procedures to those described above, the pentafluoro derivative 30 was prepared.
Figure 8.

Reagents and conditions: (a) dry Et2O, diethyl oxalate; (b) Et2NSF3; (c) i) (CH3)3SiCF3, CsF, CH3OCH2CH2OCH3, ii) conc. HCl.
Figure 9.

Reagents and conditions: (a) NaHSO3, KCN, CH2Cl2; (b) HCl, MeOH; (c) Dess-Martin periodinate, CH2Cl2; (d) Et2NSF3, CH2Cl2; (e) i) (CH3)3SiCF3, CsF, CH3OCH2CH2OCH3, ii) conc. HCl.
Electrophilic ketones, like fluoroketones, may exist in equilibrium with their corresponding hydrates (gem diols) depending on the environment. Based on the 1H NMR data, the trifluoromethyl ketones and the pentafluoroethyl ketones synthesized in this work were found to exist solely in their ketone forms in chloroform solution. However, tetrafluoro derivative 26 appears to be a mixture of ketone-hydrate form in a ratio 1:2, whereas pentafluoro derivative 30 is completely hydrated (see NMR data in experimental section).19F NMR spectroscopic data confirm the existence of the hydrated form in the cases of compounds 26 and 30.
In Vitro Inhibition of GIVA cPLA2, GVIA iPLA2 and GV sPLA2
All synthesized inhibitors were tested for inhibition of human GIVA cPLA2, GVIA iPLA2 and GV sPLA2 using previously described mixed micelle-based assays.20,21,24,25 The resulting degrees of inhibition are presented in Table 1 as either percent inhibition or XI(50) values. Initially, the percent of inhibition for each PLA2 enzyme at 0.091 mole fraction of each inhibitor was determined; and, XI(50) values were estimated for compounds that displayed greater than 90% inhibition. The XI(50) is the mole fraction of the inhibitor in the total substrate interface required to inhibit the enzyme by 50%.
Table 1. Inhibition of PLA2 by fluoroketones.
Average percent inhibition and standard error (n=3) reported for each compound at 0.091 mole fraction. XI(50) values determined for inhibitors with greater than 90% inhibition. N.D. signifies compounds with less than 25% inhibition (or no detectable inhibition).
| No | Structure | GIVA cPLA2 | GVIA iPLA2 | GV sPLA2 | |||
|---|---|---|---|---|---|---|---|
| % Inhibition | XI(50) | % Inhibition | XI(50) | % Inhibition | XI(50) | ||
| 6 |
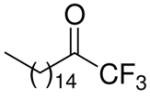
|
96 ± 2 | 0.0223 ± 0.0023 | 92 ± 3 | 0.0195 ± 0.0053 | 79 ± 9 | |
| 11 |
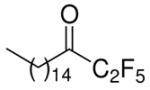
|
N.D. | 50 ± 13 | 43 ± 8 | |||
| 8 |
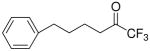
|
38 ± 2 | 96 ± 3 | 0.0096 ±0.0008 | N.D. | ||
| 10a |
|
N.D. | 98 ± 16 | 0.0073 ± 0.0007 | 28 ± 1 | ||
| 12a |
|
62 ± 5 | 96 ± 6 | 0.0025 ± 0.0003 | 48 ± 6 | ||
| 10b |
|
56 ± 4 | 98 ± 5 | 0.0065 ± 0.001 | 46 ± 8 | ||
| 12b |
|
68 ± 6 | 99 ± 10 | 0.0018 ± 0.0005 | 53 ± 14 | ||
| 10c |
|
65 ± 12 | 98 ± 4 | 0.0065 ± 0.0008 | 75 ± 10 | ||
| 19a |
|
91 ± 2 | 0.0199 ± 0.0025 | 85 ± 4 | 0.0328 ±0.0035 | 82 ± 8 | |
| 20a |
|
92 ± 3 | 0.0098 ± 0.0006 | 91 ± 4 | 0.0169 ±0.0021 | 86 ± 2 | |
| 19b |
|
96 ± 2 | 0.0156 ± 0.0019 | 94 ± 8 | 0.0208 ±0.0032 | 80 ± 6 | |
| 20b |
|
95 ± 2 | 0.0116 ± 94 ± 8 | 94 ± 8 | 0.0166 ±0.0022 | 84 ± 7 | |
| 23 |
|
88 ± 1 | 71 ± 14 | 49 ± 12 | |||
| 21 |
|
73 ± 4 | 95 ± 5 | 0.0075 ±0.0011 | 86 ± 4 | ||
| 30 |
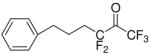
|
27 ± 3 | 49 ± 12 | 59 ± 12 | |||
| 26 |
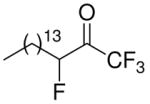
|
94 ± 2 | 0.0167 ± 0.0018 | 93 ± 4 | 0.0011 ± 0.0002 | 86 ± 10 | 0.0236 ± 0.004 |
In accordance with the literature, the long-chain saturated palmitoyl trifluoromethyl ketone 6 inhibits both intracellular enzymes GIVA cPLA2 and GVIA iPLA2 at a similar level. In this work, we show that compound 6 is also a weak inhibitor of GV sPLA2 (79% inhibition at 0.091 mole fraction). However, compound 8 is considered to be a selective inhibitor of GVIA iPLA2 with an observed XI(50) 0.0096, while high mole fraction of the inhibitor causes only 38% inhibition of GIVA cPLA2 and does not affect GV sPLA2.
The introduction of a pentafluoroethyl ketone functionality led to adverse effects depending on the nature of the chain. 1,1,1,2,2-Pentafluoro-7-phenyl-heptan-3-one (10a, FKGK11) presents slightly higher inhibitory activity on GVIA iPLA2 (XI(50) 0.0073) than the corresponding trifluoromethyl derivative 8. The dose-response curve for the inhibition of GVIA iPLA2 by pentafluoroethyl ketone 10a is shown in Figure 10. In addition, it demonstrates selective inhibition for GVIA iPLA2 since high mole fractions (0.091) do not affect GVIA cPLA2 and caused slight inhibition (28%) of GV sPLA2. Interestingly, the long-chain saturated pentafluoroethyl ketone 11 abolished the inhibitory potency and selectivity, demonstrating only 50% inhibition of GVIA iPLA2 and 43% inhibition of GV sPLA2 at 0.091 mole fraction.
Figure 10.

Inhibition curve for pentafluoro ketone 10a in a mixed-micelle assay with human GVIA iPLA2. Non-linear regression (hyperbolic) estimated a XI(50) value of 0.0073 ± 0.0007. Compound 10a inhibited GIVA cPLA2 less than 25% and GV sPLA2 approximately 28% at 0.091 mole fraction.
In pentafluoroethyl derivatives, increasing the chain length (from four to five or six carbon atoms) between the activated carbonyl group and the aromatic ring resulted in decreased selectivity for GVIA iPLA2. Derivatives 10b and 10c (five and six carbon atoms, respectively) inhibit GVIA iPLA2 at a similar level as inhibitor 10a (XI(50) 0.0065). However, both 10b and 10c are weak inhibitors of GIVA cPLA2 (56% and 65%, respectively) and GV sPLA2 (46% and 75%, respectively). For the trifluoromethyl ketone derivatives 12a and 12b, the inhibitory activity increased as the chain length increased between the carbonyl group and the aromatic ring. Both 12a and 12b are more potent inhibitors of GVIA iPLA2 (XI(50) 0.0025 and XI(50) 0.0018, respectively) than compound 8; however, these compounds also weakly inhibit GIVA cPLA2 (62% and 68%, respectively) and GV sPLA2 (48% and 53%, respectively) at 0.091 mole fraction. These results demonstrate that an increase of carbon atoms between the activated carbonyl group and the aromatic ring leads to a loss in selectivity.
Trifluoromethyl ketones 19a, 19b, 20a and 20b containing a medium (hexyloxy) or a long (decyloxy) chain substituent at the para position of the aromatic ring inhibit both GIVA cPLA2 and GVIA iPLA2. The dose-response curves for the inhibition of GVIA iPLA2 and GIVA cPLA2 by 1,1,1-trifluoro-6-(4-hexyloxy-phenyl)-hexan-2-one (20a, FKGK2) are shown in Figure 11. Comparison of 19a with 20a and 19b with 20b shows that the increase of the chain length between the carbonyl group and the aromatic ring from two to four carbon atoms results in increased inhibitory potency for both GIVA cPLA2 and GVIA iPLA2. All of these compounds (19a, 19b, 20a and 20b) also inhibit GV sPLA2. Thus, trifluoromethyl ketones containing an alkoxy group at the para position of the aromatic group can be considered to be pan inhibitors of the all three enzymes: GIVA cPLA2, GVIA iPLA2 and GV sPLA2. In particular, compound 20a is an inhibitor suitable for applications involving the inhibition of both intracellular and extracellular PLA2 enzymes. The replacement of the hexyloxy by a benzyloxy group led to derivative 23, which weakly inhibited all the three PLA2 enzymes. Comparison of inhibitors 8, 20a, 20b and 23 demonstrates that the introduction of an alkoxy or a benzyloxy group in the aromatic ring destroys the selectivity for GVIA iPLA2.
Figure 11.

Inhibition curves for trifluoromethyl ketone 20a in a mixed-micelle assay with human GIVA cPLA2 and GVIA iPLA2. Non-linear regressions (hyperbolic) estimated XI(50) values of 0.0169 ± 0.0021 and 0.0098 ± 0.0006 for GIVA cPLA2 and GVIA iPLA2, respectively. Compound 20a inhibited GV sPLA2 approximately 86% at 0.091 mole fraction.
Comparison of pentafluoroethyl ketone 21 with the corresponding trifluoromethyl ketone 19a reinforces our observation that pentafluoroethyl ketone functionality favors the inhibition of GVIA iPLA2 (XI(50) 0.0075). However, the presence of a hexyloxy substituent leads to loss of selectivity for GVIA iPLA2, since compound 21 weakly inhibits GIVA cPLA2 (73%) and GV sPLA2 (86%) at 0.091 inhibitor mole fraction.
Comparison of compound 26 with 6 shows that the introduction of an additional fluorine atom at the α′ position in a long chain saturated derivative results in a derivative with slightly better activity for GIVA cPLA2 (XI(50) 0.0167) than the parent trifluoromethyl ketone 6 (XI(50) 0.0223). More importantly, tetrafluoro derivative 26 is approximately twenty-fold more potent inhibitor of GVIA iPLA2 (XI(50) 0.0011) than the trifluoro derivative 6 (XI(50) 0.0195). To our knowledge, compound 26 is the most potent inhibitor of GVIA iPLA2 reported, indicating that introduction of an additional fluorine atom at the α′ position constitutes an important strategy for the development of new potent GVIA iPLA2 inhibitors. However, the tetrafluoro derivative 26 also inhibits GIVA and GVA PLA2. Interestingly, the introduction of two fluorine atoms at the α′ position in an aromatic ring containing derivative destroyed the inhibitory potency and the selectivity for GVIA iPLA2. For example, at 0.091 mole fraction, derivative 30 is a weak inhibitor of GVIA iPLA2 (49%), GV sPLA2 (59%), and presents no significant inhibition of GIVA cPLA2 (27%).
Our data indicates the importance of screening selective inhibitors against multiple enzyme classes within the PLA2 superfamily. As mentioned above, our work shows that the known inhibitor palmitoyl trifluoromethyl ketone 6, reported to strictly inhibit intracellular GVIA iPLA2 and GIVA cPLA2, also weakly inhibits GV sPLA2. Similarly, some of our synthesized trifluoromethyl, pentafluoroethyl and tetrafluoro derivatives (for example, compounds 20a, 21, 26) were found to inhibit GV sPLA2. Futhermore, Gelb et al. demonstrated that difluoro ketones similar to 36 (Figure 12) inhibit cobra venom PLA2.54 Therefore activated ketones, such as polyfluoro ketones, are likely to inhibit serine enzymes, GIVA cPLA2 and GVIA iPLA2, as well as histidine enzymes like secreted PLA2.
Figure 12.

Structures of difluoro ketone inhibitor 36 of cobra venom PLA2 and BEL inhibitor 37.
Bromoenol lactone (BEL) 37 (Figure 12) is considered to be a selective and irreversible GVIA iPLA2 and has been widely applied to study potential biological roles for GVIA iPLA2.55,56 However, Turk et al. have recently reported that BEL inactivates GVIA iPLA2 by generating a diffusible bromomethyl keto acid that alkylates cysteine thiols, rather than creating an acyl-enzyme intermediate with the active-site serine.57 Therefore, it is likely that BEL affects multiple enzymes and should be used with appropriate caution when studying potential roles of GVIA iPLA2.57 These observations lead us to design selective inhibitors of GVIA iPLA2 such as the pentafluoroethyl ketone 10a.
In conclusion, we developed and applied a variety of synthetic routes to produce various pentafluoro, tetrafluoro and trifluoro derivatives containing activated carbonyl groups. We studied their in-vitro activity on the three major human PLA2 enzyme classes and demonstrated that the pentafluoroethyl ketone functionality favors GVIA iPLA2 inhibition. Furthermore, 1,1,1,2,2-pentafluoro-7-phenyl-heptan-3-one (10a) was shown to be a selective inhibitor of GVIA iPLA2. Additionally, introduction of an additional fluorine atom at the α′ position of a trifluoromethyl ketone constitutes an important strategy for the development of new potent GVIA iPLA2 inhibitors. The tetrafluoro derivative of palmitic acid 26 is observed to be the most potent inhibitor of GVIA iPLA2 to date; however, it also inhibits GIVA cPLA2 and GV sPLA2. Polyfluoro ketones displaying an array of selectivities for the major PLA2 enzyme classes will prove to be valuable tools for the in-vivo characterization of the roles of PLA2 enzymes. Furthermore, we found that these compounds do not show cytotoxcity toward cells in culture and we are currently utilizing these polyfluoro ketone derivatives for the comparison of intracellular versus extracellular PLA2 enzyme roles in animal models of neurological disorders such as multiple sclerosis, spinal cord injury and peripheral nerve injury.58
Experimental Section
Synthesis of fluoroketone inhibitors
Melting points were determined on a Buchi 530 apparatus and are uncorrected. Nuclear magnetic resonance spectra were obtained on a Varian Mercury spectrometer (1H NMR recorded at 200 MHz, 13C NMR recorded at 50 MHz, 19F NMR recorded at 188 MHz) and are referenced in ppm relative to TMS for 1NMR and 13C NMR, and relative to TFA as an internal standard for 19F NMR. Thin layer chromatography (TLC) plates (silica gel 60 F254) and silica gel 60 (230–400 mesh) for flash column chromatography were purchased from Merck. Visualization of spots was effected with UV light and/or phosphomolybdic acid, in EtOH stain. Tetrahydrofuran (THF), toluene, and Et2O were dried by standard procedures and stored over molecular sieves or Na. All other solvents and chemicals were reagent grade and used without further purification. All the products gave satisfactory elemental analysis results.
General Procedure for the Synthesis of Pentafluoroethyl Ketones
Oxalyl chloride (0.38 g, 3 mmol) and N,N-dimethylformamide (40 μL) were added to a solution of carboxylic acid (1 mmol) in dry dichloromethane (40 mL). After 3 h stirring at room temperature, the solvent and excess reagent were evaporated under reduced pressure and the residue was dissolved in dry dichloromethane (10 mL). Pyridine (0.64 mL, 8 mmol) and pentafluoropropionic anhydride (0.85 mL, 6 mmol) were added dropwise to this solution at 0 °C consecutively. After stirring at 0 °C for 30 min and at room temperature for 1.5 h, the reaction mixture was cooled again at 0 °C and water (2 mL) was added dropwise. After stirring for 30 min at 0 °C and another 30 min at room temperature, the reaction mixture was diluted with dichloromethane (10 mL). The organic phase was then washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residual oil was purified by flash column chromatography [EtOAc-petroleum ether (bp 40–60 °C) 1/9].
1,1,1,2,2-Pentafluoro-7-phenyl-heptan-3-one (10a)
Yield 53%; yellowish oil; 1H NMR (CDCl3): δ 7.31-7.17 (5H, m, Ph), 2.80 (2H, t, J=6.2 Hz, CH2), 2.66 (2H, t, J=6.6 Hz, CH2), 1.73-1.67 (4H, m, 2×CH2); 13C NMR: δ 194.2 (t, JC-C-F=26 Hz, CO), 141.6 (Ph), 128.4 (Ph), 128.3 (Ph), 125.9 (Ph), 117.8 (qt, JC-F3=287 Hz, JC-CF2=34 Hz, CF3), 106.8 (tq, JC-F2=267 Hz, JC-CF3=38 Hz, CF2), 37.1 (CH2), 35.5 (CH2), 30.3 (CH2), 21.9 (CH2); 19F NMR: δ -4.1 (CF3), -45.5 (CF2); MS (ESI) m/z (%): 279 (M−, 100). Anal. (C13H13F5O) C, H.
1,1,1,2,2-Pentafluoro-8-phenyl-octan-3-one (10b)
Yield 75%; yellowish oil; 1H NMR (CDCl3): δ 7.35-7.21 (5H, m, Ph), 2.79 (2H, t, J=6.8 Hz, CH2), 2.68 (2H, t, J=7.4 Hz, CH2), 1.80-1.68 (4H, m, 2×CH2), 1.48-1.40 (2H, m, CH2); 13C NMR: δ 194.3 (t, JC-C-F=26 Hz, CO), 142.2 (Ph), 128.3 (Ph), 128.2 (Ph), 125.7 (Ph), 117.8 (qt, JC-F3=285 Hz, JC-CF2=34 Hz, CF3), 106.9 (tq, JC-F2=265 Hz, JC-CF3=37 Hz, CF2), 37.2 (CH2), 35.6 (CH2), 31.0 (CH2), 28.2 (CH2), 22.1 (CH2); 19F NMR: δ -4.2 (CF3), -45.6 (CF2); MS (ESI) m/z (%): 293 (M−, 100). Anal. (C14H15F5O) C, H.
1,1,1,2,2-Pentafluoro-9-phenyl-nonan-3-one (10c)
Yield 60%; yellowish oil; 1H NMR (CDCl3): δ 7.31-7.18 (5H, m, Ph), 2.76 (2H, t, J=6.8 Hz, CH2), 2.64 (2H, t, J=8.0 Hz, CH2), 1.72-1.58 (4H, m, 2×CH2), 1.44-1.34 (4H, m, 2×CH2); 13C NMR: δ 194.4 (t, JC-C-F=26 Hz, CO), 142.5 (Ph), 128.4(Ph), 128.3 (Ph), 125.7 (Ph), 117.8 (qt, JC-F3=285 Hz, JC-CF2=34 Hz, CF3), 106.9 (tq, JC-F2=265 Hz, JC-CF3 = 37 Hz, CF2), 37.3 (CH2), 35.8 (CH2), 31.1 (CH2), 28.8 (CH2), 28.5 (CH2), 22.2 (CH2); 19F NMR: δ -4.2 (CF3), -45.6 (CF2); MS (ESI) m/z (%): 307 (M−, 100). Anal. (C15H17F5O) C, H.
1,1,1,2,2-Pentafluoro-octadecan-3-one (11)
Yield 24%; colorless oil; 1H NMR (CDCl3): δ 2.75 (2H, t, J=7.4 Hz, CH2), 1.67 (2H, t, J=7.0 Hz, CH2), 1.38-1.20 (24H, m, 12×CH2), 0.88 (3H, t, J=7.0 Hz, CH3); 13C NMR: δ 194.5 (t, JC-CF2=26 Hz, CO), 117.8 ppm (qt, JC-F3=285 Hz, JC-CF2=34 Hz, CF3), 106.9 ppm (tq, JC-F2=265 Hz, JC-CF3 = 38 Hz, CF2), 37.4 (CH2), 31.9 (CH2), 30.3 (CH2), 29.7 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4 (CH2), 29.2 (CH2), 28.7 (CH2), 22.7 (CH2), 22.3 (CH2), 14.1 (CH3); 19F NMR: δ -4.2 (CF3), -45.6 (CF2); MS (ESI) m/z (%): 357 (M−, 93). Anal. (C18H31F5O) C, H.
1,1,1,2,2-Pentafluoro-5-(4-hexyloxy-phenyl)-pentan-3-one (21)
Yield 76%; yellowish oil; 1H NMR (CDCl3): δ 7.10 (2H, d, J=8.6 Hz, Ph), 6.85 (2H, d, J=8.6 Hz, Ph), 3.94 (2H, t, J=6.6 Hz, CH2O), 3.02 (2H, t, J=7.0 Hz, CH2), 2.96 (2H, t, J=7.0 Hz, CH2), 1.86-1.73 (2H, m, CH2), 1.60-1.25 (6H, m, 3×CH2), 0.93 (3H, t, J=6.4 Hz, CH3). 13C NMR (CDCl3): δ 193.5 (t, JC-C-F=26 Hz, CO), 157.9 (Ph), 130.9 (Ph), 129.2 (Ph), 117.8 (qt, JC-F3 =286 Hz, JC-CF2=34 Hz, CF3), 114.7 (Ph), 106.8 (tq, JC-F2=265 Hz, JC-CF3=38 Hz, CF2), 68.0 (CH2O), 39.4 (CH2), 31.6 (CH2), 29.3 (CH2), 27.5 (CH2), 25.7 (CH2), 22.6 (CH2), 13.9 (CH3). 19F NMR: δ -4.2 (CF3), -45.6 (CF2). MS (ESI) m/z (%): 351 (M−, 100). Anal. (C17H21F5O2) C, H.
Synthesis of Trifluoromethyl Ketones
The synthesis of trifluoromethyl ketones was carried out following the procedure described above for pentafluoromethyl ketones, except that trifluoroacetic anhydride was used instead of pentafluoropropionic anhydride. The products were purified by flash column chromatography [EtOAc-petroleum ether (bp 40–60 °C) 3/7].
1,1,1-Trifluoro-7-phenylheptan-2-one (12a).59
Yield 45%; yellowish oil; 1H NMR (CDCl3): δ 7.34-7.19 (5H, m, Ph), 2.76-2.62 (4H, m, 2×CH2), 1.77-1.66 (4H, m, 2×CH2), 1.46-1.39 (2H, m, CH2); 13C NMR: δ 191.8 (q, JC-C-F=35 Hz, COCF3), 142.2 (Ph), 128.3 (Ph), 128.2 (Ph), 125.7 (Ph), 115.5 (q, JC-F=290 Hz, CF3), 36.2 (CH2), 35.7 (CH2), 30.9 (CH2), 28.2 (CH2), 22.2 (CH2); 19F NMR: δ -1.5 (CF3); MS (ESI) m/z (%): 243 (M−, 100).
1,1,1-Trifluoro-8-phenyloctan-2-one (12b).59
Yield 42%; yellowish oil; 1H NMR (CDCl3): δ 7.28-7.17 (5H, m, Ph), 2.72-2.60 (4H, m, 2×CH2), 1.70-1.61 (4H, m, 2×CH2), 1.42-1.24 (4H, m, 2×CH2); 13C NMR: δ 191.4 (q, JC-C-F=35 Hz, COCF3), 142.5 (Ph), 128.3 (Ph), 128.2 (Ph), 125.7 (Ph), 115.6 (q, JC-F=291 Hz, CF3), 36.3 (CH2), 35.8 (CH2), 31.1 (CH2), 28.7 (CH2), 28.6 (CH2), 22.3 (CH2); 19F NMR: δ -1.5 (CF3); MS (ESI) m/z (%): 257 (M−, 100).
1,1,1-Trifluoro-4-(4-hexyloxy-phenyl)-butan-2-one (19a)
Yield 53%; yellowish oil; 1H NMR (CDCl3): δ 7.10 (2H, d, J=8.6 Hz, Ph), 6.84 (2H, d, J=8.6 Hz, Ph), 3.93 (2H, t, J=6.2 Hz, CH2O), 3.10-2.92 (4H, m, 2×CH2), 1.82-1.62 (2H, m, CH2), 1.55-1.22 ( 6H, m, 3×CH2), 0.91 (3H, t, J=6.6 Hz, CH3); 13C NMR: δ 190.7 (q, JC-C-F = 35 Hz, COCF3), 157.9 (Ph), 131.0 (Ph), 129.2 (Ph), 115.5 (q, JC-F=292 Hz, CF3), 114.7 (Ph), 68.0 (OCH2), 38.3 (CH2), 31.6 (CH2), 29.2 (CH2), 27.5 (CH2), 25.7 (CH2), 22.6 (CH2), 13.9 (CH3); 19F NMR: δ -1.5 (CF3); MS (ESI) m/z (%): 301 (M−, 100). Anal. (C16H21F3O2) C, H.
4-(4-Decyloxy-phenyl)-1,1,1-trifluoro-butan-2-one (19b)
Yield 46%; yellowish oil; 1H NMR (CDCl3): δ 7.12 (2H, d, J=8.6 Hz, Ph), 6.85 (2H, d, J=8.6 Hz, Ph), 3.95 (2H, t, J=6.6 Hz, CH2O), 3.05-2.85 (4H, m, 2×CH2), 1.81-1.62 (2H, m, CH2), 1.56-1.22 (14H, m, 7×CH2), 0.92 (3H, t, J=6.8 Hz, CH3); 13C NMR: δ 190.5 (q, JC-C-F=35 Hz, COCF3), 157.9 (Ph), 131.0 (Ph), 129.2 (Ph), 115.5 (q, JC-F=292 Hz, CF3), 114.6 (Ph), 68.0 (CH2O), 38.3 (CH2), 31.8 (CH2), 29.6 (CH2), 29.3 (CH2), 29.2 (CH2), 27.4 (CH2), 26.0 (CH2), 22.7 (CH2), 14.0 (CH3); 19F NMR: δ -1.5 (CF3); MS (FAB) m/z (%): 358 (M+, 85). Anal. (C20H29F3O2) C, H.
1,1,1-Trifluoro-6-(4-hexyloxy-phenyl)-hexan-2-one (20a)
Yield 45%; yellowish oil; 1H NMR (CDCl3): δ 7.09 (2H, d, J=8.0 Hz, Ph), 6.85 (2H, d, J=8.0 Hz, Ph), 3.95 (2H, t, J=6.6 Hz, CH2), 2.74 (2H, t, J=6.6 Hz, CH2), 2.60 (2H, t, J=6.2 Hz, CH2), 1.82-1.62 (6H, m, 3×CH2), 1.46-1.25 (6H, m, 3×CH2), 0.94 (3H, t, J=6.8 Hz, CH3); 13C NMR: δ 191.4 (q, JC-C-F=34 Hz, COCF3), 157.9 (Ph), 133.4 (Ph), 129.1 (Ph), 115.4 (q, JC-F=290 Hz, CF3), 114.4 (Ph), 67.9 (CH2O), 36.1 (CH2), 34.5 (CH2), 31.6 (CH2), 30.6 (CH2), 29.3 (CH2), 25.7 (CH2), 22.6 (CH2), 21.8 (CH2), 13.9 (CH3); 19F NMR: δ -1.6 (CF3); MS (FAB) m/z (%): 330 (M+, 23). Anal. (C18H25F3O2) C, H.
6-(4-Decyloxy-phenyl)-1,1,1-trifluoro-hexan-2-one (20b)
Yield 46%; yellowish oil; 1H NMR (CDCl3): δ 7.08 (2H, d, J=8.6 Hz, Ph), 6.84 (2H, d, J=8.6 Hz, Ph), 3.94 (2H, t, J=6.6 Hz, CH2O), 2.73 (2H, t, J=6.6 Hz, CH2), 2.59 (2H, t, J=7.0 Hz, CH2), 1.82-1.62 (6H, m, 3×CH2), 1.45-1.22 (14H, m, 7×CH2), 0.90 (3H, t, J=6.8 Hz, CH3); 13C NMR: δ 191.6 (q, JC-C-F=35 Hz, COCF3), 157.7 (Ph), 133.7 (Ph), 129.4 (Ph), 115.8 (q, JC-F = 292 Hz, CF3), 114.6 (Ph), 68.2 (CH2O), 36.4 (CH2), 34.8 (CH2), 32.1 (CH2), 30.9 (CH2), 29.8 (CH2), 29.7 (CH2), 29.6 (CH2), 29.5 (CH2), 26.6 (CH2), 26.3 (CH2), 22.9 (CH2), 22.1 (CH2), 14.32 (CH3); 19F NMR: δ -1.5 (CF3); MS (FAB) m/z (%): 386 (M+, 100). Anal. (C22H33F3O2) C, H.
4-(4-Benzyloxy-phenyl)-1,1,1-trifluoro-butan-2-one (23)
Yield 43%; yellowish solid; mp 71–72 °C; 1H NMR (CDCl3): δ 7.46-7.35 (5H, m, Ph), 7.15 (2H, d, J=8.4 Hz, Ph), 6.95 (2H, d, J=8.4 Hz, Ph), 5.07 (2H, s, PhCH2), 3.12-2.85 (4H, m, 2×CH2); 13C NMR: δ 190.7 (q, JC-C-F = 35 Hz, COCF3), 157.4 (Ph), 136.9 (Ph), 131.5 (Ph), 129.2 (Ph), 128.5 (Ph), 127.9 (Ph), 127.4 (Ph), 115.4 (q, JC-F=290 Hz, CF3), 114.9 (Ph), 70.0 (CH2O), 38.3 (CH2), 27.4 (CH2); 19F NMR: δ -1.4 (CF3); MS (ESI) m/z (%): 307 (M−, 100). Anal. (C17H15F3O2) C, H.
Intermediate compounds 14a,b and 22 were prepared by known methods and their spectroscopic data were in accordance with those in the literature.60,61
Horner-Wadsworth-Emmons Olefination
A suspension of aldehyde 14a or 14b (1 mmol), triethyl 4-phosphonocrotonate (0.37 g, 1.5 mmol), lithium hydroxide (0.036 g, 1.5 mmol) and molecular sieves (beads, 4–8 mesh, 1.5 g/mmol aldehyde) in dry tetrahydrofuran (10 mL) was refluxed under argon for 24 h. The reaction mixture was then cooled to room temperature, filtered through a thin pad of celite and the solvent evaporated under reduced pressure. The residual oil was purified by chromatography on silica gel eluting with ether-petroleum ether (bp 40–60 °C) 1/9.
Ethyl (2E,4E)-5-(4-Hexyloxy-phenyl)-penta-2,4-dienoate (16a)
Yield 71%; white solid; mp 68–69 °C; 1H NMR (CDCl3): δ 7.48-7.20 (3H, m, CH, Ph), 6.90-6.75 (3H, m, CH, Ph), 6.71 (1H, d, J=15.4 Hz, CH), 5.94 (1H, d, J=15.4 Hz, CHCOO), 4.23 (2H, q, J=7.4 Hz, OCH2CH3), 3.97 (2H, t, J=6.2 Hz, CH2O), 1.85-1.62 (2H, m, CH2CH2O), 1.45-1.02 (9H, m, 3×CH2, CH3), 0.92 (3H, t, J=6.8 Hz, CH3); 13C NMR: δ 167.2 (COO), 160.0 (Ph), 145.0 (CH), 140.2 (CH), 131.9 (Ph), 128.6 (Ph), 123.9 (CH), 119.9 (CH), 114.7 (Ph), 68.0 (CH2O), 60.2 (OCH2CH3), 31.6 (CH2), 29.1 (CH2), 25.7 (CH2), 22.6 (CH2), 14.3 (CH3), 14.0 (CH3). Anal. (C19H26O3) C, H.
Ethyl (2E,4E)-5-(4-Decyloxy-phenyl)-penta-2,4-dienoate (16b)
Yield 65%; white solid; mp 80–81 °C; 1H NMR (CDCl3): δ 7.45-7.38 (3H, m, CH, Ph), 6.88-6.80 (3H, m, CH, Ph), 6.78 (1H, d, J=12 Hz, CH), 5.94 (1H, d, J=15.4 Hz, CHCOO), 4.23 (2H, q, J=7.4 Hz, OCH2CH3), 3.97 (2H, t, J=6.6 Hz, CH2O), 1.81-1.75 (2H, m, CH2CH2O), 1.50-1.14 (17H, m, 7×CH2, CH3), 0.89 (3H, t, J=6.8 Hz, CH3); 13C NMR: d167.3 (COO), 160.0 (Ph), 145.1 (CH), 140.2 (CH), 131.9 (Ph), 128.6 (Ph), 124.0 (CH), 119.9 (CH), 114.8 (Ph), 68.1 (CH2O), 60.2 (CH2), 31.9 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 26.0 (CH2), 22.7 (CH2), 14.3 (CH3), 14.1 (CH3). Anal. (C23H34O3) C, H.
Wittig Olefination
A solution of aldehyde 14a or 14b (1 mmol) and methyl (triphenylphosphanylidene)acetate (0.334 g, 1 mmol) in dry dichloromethane (3 mL) was refluxed under argon for 24 h. The reaction mixture was then cooled to room temperature and the solvent evaporated under reduced pressure. The residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 1/9.
Methyl (E)-3-(4-Hexyloxy-phenyl)-acrylate (15a)
Yield 93%; white solid; mp 84–85 °C; 1H NMR (CDCl3): δ 7.63 (1H, d, J=15.8 Hz, CH=CHCO), 7.43 (2H, d, J=8.8 Hz, Ph), 6.87 (2H, d, J=8.8 Hz, Ph), 6.28 (1H, d, J=15.8 Hz, CHCOO), 3.95 (2H, t, J=6.4 Hz, CH2O), 3.77 (3H, s, OCH3), 1.76 (2H, m, CH2CH2O), 1.46-1.21 (6H, m, 3×CH2), 0.89 (3H, t, J=6.8 Hz, CH3); 13C NMR: δ 167.7 (COO), 161.0 (Ph), 144.6 (CH), 129.6 (Ph), 126.8 (Ph), 115.0 (CH), 114.7 (Ph), 68.1 (CH2O), 51.5 (OCH3), 31.5 (CH2), 29.0 (CH2), 25.6 (CH2), 22.5 (CH2), 13.9 (CH3). Anal. (C16H22O3) C, H.
Methyl (E)-3-(4-Decyloxy-phenyl)-acrylate (15b)
Yield 92%; white solid; mp 75–76 °C; 1H NMR (CDCl3): δ 7.63 (1H, d, J=15.8 Hz, CH=CHCOO), 7.37 (2H, d, J=8.8 Hz, Ph), 6.85 (2H, d, J=8.8 Hz, Ph), 6.23 (1H, d, J=15.8 Hz, CHCOO), 3.87 (2H, t, J=6.6 Hz, CH2O), 3.71 (3H, s, OCH3), 1.78-1.62 (2H, m, CH2CH2O), 1.40-1.22 (14H, m, 7×CH2), 0.84 (3H, t, J=7 Hz, CH3); 13C NMR: δ 167.4 (COO), 160.8 (Ph), 144.3 (CH), 129.4 (Ph), 126.6 (Ph), 114.8 (CH), 114.5 (Ph), 67.8 (CH2O), 51.2 (OCH3), 31.7 (CH2), 29.4 (CH2), 29.2 (CH2), 29.1 (CH2), 29.0 (CH2), 25.8 (CH2), 22.5 (CH2), 13.9 (CH3). Anal. (C20H30O3) C, H.
Hydrogenation and Saponification of Unsaturated Esters
A mixture of the unsaturated ester (0.7 mmol) in dry 1,4-dioxane (7 mL) and 10% palladium on activated carbon (0.07 g) was hydrogenated for 12 h under atmospheric conditions. After filtration through a pad of celite, the solvent was removed in vacuo to give the saturated compound.
The solution of the saturated ester in methanol (1.4 mL) was treated with sodium hydroxide 1N (1 mL, 1 mmol). The mixture was stirred at room temperature for 12 h, acidified with 1N HCl and extracted with EtOAc (3 × 10 mL). The solvent was removed in vacum to afford the saturated acid as a white solid.
3-(4-Hexyloxy-phenyl)-propanoic acid (17a)
Yield 90%; white solid; mp 70–72 °C; 1H NMR (CDCl3): δ 7.14 (2H, d, J=8.2 Hz, Ph), 6.86 (2H, d, J=8.2 Hz, Ph), 3.96 (2H, t, J=6.6 Hz, CH2O), 2.93 (2H, t, J=7.6 Hz, CH2), 2.67 (2H, t, J=7.6 Hz, CH2), 1.76-1.60 (2H, m, CH2), 1.41-1.30 (6H, m, 3×CH2), 0.92 (3H, t, J=6.7 Hz, CH3); 13C NMR: δ 179.0 (COO), 157.6 (Ph), 132.0 (Ph), 129.1 (Ph), 114.5(Ph), 67.9 (CH2O), 35.9 (CH2), 31.5 (CH2), 29.7 (CH2), 29.2 (CH2), 25.7 (CH2), 22.5 (CH2), 14.0 (CH3). Anal. (C15H22O3) C, H.
3-(4-Decyloxy-phenyl)-propanoic acid (17b)
Yield 96%; white solid; mp 74–76 °C; 1H NMR (CDCl3): δ 7.14 (2H, d, J=8.2 Hz, Ph), 6.86 (2H, d, J=8.2 Hz, Ph), 3.95 (2H, t, J=6.5 Hz, CH2O), 2.93 (2H, t, J=7.7 Hz, CH2CH2COO), 2.67 (2H, t, J=7.7 Hz, CH2COO), 1.85-1.68 (2H, m, CH2CH2O), 1.50-1.21 (14H, br s, 7×CH2), 0.92 (3H, t, J=6.2 Hz, CH3); 13C NMR: δ 179.3 (COO), 157.6 (Ph), 132.0 (Ph), 129.1 (Ph), 114.5 (Ph), 67.9 (CH2O), 35.9 (CH2), 31.9 (CH2), 29.7 (CH2), 29.5 (CH2), 29.3 (CH2), 26.0 (CH2), 22.6 (CH2), 14.1 (CH3). Anal. (C19H30O3) C, H.
5-(4-Hexyloxy-phenyl)-pentanoic acid (18a)
Yield 96%; white solid; mp 90–91 °C; 1H NMR (CDCl3): δ 7.03 (2H, d, J=8.4 Hz, Ph), 6.77 (2H, d, J=8.4 Hz, Ph), 3.88 (2H, t, J=6.2 Hz, CH2O), 2.52 (2H, t, J=6.8 Hz, CH2), 2.32 (2H, t, J=6.7 Hz, CH2COO), 1.80-1.60 (6H, m, 3×CH2), 1.60-1.21 (6H, m, 3×CH2), 0.89 (3H, t, J=6.7 Hz, CH3); 13C NMR: δ 180.1 (COO), 157.3 (Ph), 133.9 (Ph), 129.2 (Ph), 114.4 (Ph), 68.0 (CH2O), 34.6 (CH2), 33.9 (CH2), 31.6 (CH2), 31.0 (CH2), 29.3 (CH2), 25.7 (CH2), 24.2 (CH2), 22.6 (CH2), 14.0 (CH3). Anal. (C17H26O3) C, H.
5-(4-Decyloxy-phenyl)-pentanoic acid (18b)
Yield 94%; white solid; mp 101–102 °C; 1H NMR (CDCl3): δ 7.08 (2H, d, J=8.4 Hz, Ph), 6.82 (2H, d, J=8.4 Hz, Ph), 3.93 (2H, t, J=6.2 Hz, CH2O), 2.57 (2H, t, J=6.8 Hz, PhCH2), 2.37 (2H, t, J=7 Hz, CH2COOH), 1.80-1.60 (6H, m, 3×CH2), 1.51-1.22 (14H, m, 7×CH2), 0.89 (3H, t, J=6.6 Hz, CH3); 13C NMR: δ 179.5 (COO), 157.2 (Ph), 133.8 (Ph), 129.1 (Ph), 114.3 (Ph), 67.9 (CH2O), 34.5 (CH2), 33.8 (CH2), 31.8 (CH2), 30.9 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 26.0 (CH2), 24.2 (CH2), 22.6 (CH2), 14.0 (CH3). Anal. (C21H34O3) C, H.
Methyl 2-Fluoro-hexadecanoate (25)
Compound 24 (1 mmol) was added to a solution of bis(2-methoxyethyl)amino-sulfur-trifluoride, Deoxofluor (0.2 mL, 1 mmol) in dry dichloromethane (0.2 mL) at −78 °C. After stirring for 2 h at −78 °C and another 3 h at room temperature, the reaction mixture quenched with saturated aqueous NaHCO3 (2.5 mL). The organic phase was then washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 3/7. Yield 64%; yellowish oil; 1H-NMR (CDCl3): δ 4.86 (1H, dt, JH-F=49.2 Hz, JH-H=6.6 Hz, CH), 3.74 (3H, s, OCH3), 2.00-1.72 (2H, m, CH2), 1.45-1.10 (24H, br, 12×CH2), 0.83 (3H, t, J=6.2 Hz, CH3); 13C NMR: δ 170.4 (d, JC-C-F=24 Hz, COO), 88.9 (d, JC-F=183 Hz, CF), 52.0 (OCH3), 32.3 (d, JC-C-F=21, CH2), 31.9 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 29.0 (CH2), 24.6 (CH2), 24.4 (CH2), 24.3 (CH2), 22.7 (CH2), 14.0 (CH3); 19F NMR: δ -120.8 (m, CF). Anal. (C17H33FO2) C, H.
1,1,1,3-Tetrafluoro-heptadecan-2-one (in equilibrium with 1,1,1,3-Tetrafluoro-heptadecane-2,2-diol) (26)
A solution of compound 25 (173 mg, 0.6 mmol) and trifluoromethyltrimethylsilane (170 μL, 1.15 mmol) in ethylene glycol dimethyl ether (0.55 mL) at 0 °C was treated with cesium fluoride (3 mg). After stirring for 30 min at 0 °C and another 18 h at 25 °C the reaction mixture was treated with concentrated HCl (1 mL). After stirring for another 18 h at 25 °C, the reaction mixture was diluted with EtOAc (10 mL). The organic phase was then washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 3/7. Yield 58%; white solid; mp 34–35 °C; 1H-NMR (CDCl3): δ 5.23 (1/3H, dt, JH-F=48.2 Hz, JH-H=6.2 Hz, CH), 4.65 (2/3H, dt, JH-F=49.4 Hz, JH-H=6.6 Hz, CH), 3.74 (2/3H, s, OH), 3.49 (2/3H, s, OH), 2.08-1.27 (26H, m, 13×CH2), 0.89 (3H, t, J=7 Hz, CH3); 13C NMR: δ 122.6 (q, JC-F3=286 Hz, CF3), 115.4 (q, JC-F3=290 Hz, CF3) 92.9 [C(OH)2], 92.4 (d, JC-F=186 Hz, CF), 32.1 (CH2), 31.6 (d, JC-C-F=20 Hz, CH2), 29.9 (CH2), 29.8 (CH2), 29.7 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4 (CH2), 29.1 (CH2), 28.5 (CH2), 28.1 (CH2), 22.7 (CH2), 22.3 (CH2), 14.3 (CH3); 19F NMR: δ 1.6 (CF3), -5.3 (CF3), -121.7 (CF); MS (ESI) m/z (%): 343 (M−, 100).
Ethyl 2,2-Difluoro-5-phenyl-pentanoate (29)
To a stirring mixture of magnesium (350 mg, 14.6 mmol) and iodine in dry THF (10 ml), (3-bromo-propyl)-benzene (2.87 g, 14.4 mmol) was added dropwise under N2 atmosphere. Once the Grignard reagent was formed, the resulting mixture was added dropwise to a cooled (−78 °C) solution of diethyl oxalate (1.6 mL, 11.8 mmol) in dry ether (17.3 mL). The reaction mixture was stirred at −78 °C for 45 min and then was quenched with 1N HCl. The aqueous layer was extracted with ether (3 × 25 mL) and the combined organic layers were washed with brine, dried (Na2SO4) and the solvent was evaporated in vacuo. After flash column chromatography, a mixture of methyl 2-oxo-5-phenyl-pentanoate (28) with diethyl oxalate was obtained and treated with DAST (1 eq) at room temperature. After stirring for 4 h at 45 °C, the reaction mixture was quenched with ice water. The reaction mixture was diluted with dichlolomethane and the organic phase was then washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 1/9. Yield 69%; yellowish oil; 1H NMR (CDCl3): δ 7.38-7.12 (5H, m, Ph), 4.30 (2H, q, J=6.8 Hz, OCH2), 2.68 (2H, t, J=7.4 Hz, PhCH2), 2.21-1.93 (2H, m, CH2CF2), 1.90-1.75 (2H, m, CH2), 1.34 (3H, t, J=6.8 Hz, CH3); 13C NMR: δ 164.2 (t, JC-C-F=24 Hz, COO), 140.9 (Ph), 128.4 (Ph), 128.3 (Ph), 126.1 (Ph), 116.2 (t, JC-F=248 Hz, CF2), 62.7 (OCH2), 34.9 (CH2), 33.8 (t, JC-C-F=23 Hz, CH2CF2), 23.0 (t, JC-C-C-F=4 Hz, CH2CH2CF2), 13.8 (CH3); 19F NMR: δ - 28.0 (t, J=17 Hz, CF2). Anal. (C13H16F2O2) C, H.
1,1,1,3,3-Pentafluoro-6-phenyl-hexane-2,2-diol (30)
It was prepared following the method used for the synthesis of compound 26. Yield 35%; yellowish oil; 1H NMR (CDCl3): δ 7.41-7.18 (5H, m, Ph), 3.93 (2H, br, 2×OH), 2.69 (2H, t, J=7.6 Hz, PhCH2), 2.22-1.88 (4H, m, 2×CH2); 13C NMR: δ 141.3 (Ph), 128.4 (Ph), 126.3 (Ph), 126.1 (Ph), 121.5 (q, J=286 Hz, CF3), 120.7 (t, J=249 Hz, CF2), 92.3 [C(OH)2], 35.2 (CH2), 30.8 (t, JC-C-F2=23 Hz, CH2CF2), 22.5 (t, JC-C-C-F2=2.4 Hz, CH2CH2CF2); 19F NMR: δ –3.2 (CF3), –36.4 (CF2); Ms (ESI) m/z (%): 283 (M−, 65), 213 (100). Anal. (C12H13F5O2) C, H.
2-Hydroxy-5-phenyl-pentanenitrile (32).62
A solution of 4-phenylbutanal 31 (0.56 g, 3.78 mmol) and NaHSO3 (0.59 g in 1 ml H2O) in dichloromethane was stirred for 30 min at room temperature. After the formation of the white salt, the organic solvent was evaporated and water (3.8 mL) was added. The mixture cooled to 0 °C and an aqueous solution of KCN (0.368 g, 567 mmol in 1 mL H2O) was added dropwise. The reaction mixture was stirred for another 18 h at room temperature and then CH2Cl2 (10 mL) and water (10 mL) were added. The organic phase was washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 2/8 to give 0.653 g (99%) of the title compound as a clear oil; 1H-NMR (CDCl3): δ 7.32-7.15 (5H, m, Ph), 4.40 (1H, t, J=8.8 Hz, CH), 2.63 (2H, t, J=6.6 Hz, CH2), 1.90-1.70 (4H, m, 2×CH2); 13C NMR: 141.5 (Ph), 128.7 (Ph), 128.6 (Ph), 126.3 (Ph), 120.4 (CN), 61.2 (CH), 35.3 (CH2), 34.7 (CH2), 26.4 (CH2). Anal. (C11H13NO) C, H.
Methyl 2-Hydroxy-5-phenyl-pentanoate (33).63
Compound 32 (0.63 g, 3.59 mmol) was treated with HCl (0.6 mL. 6N) in MeOH for 18 h at room temperature. The organic solvent was evaporated and an aqueous solution of K2CO3 was added to neutralize the pH of the mixture. After extraction with EtOAc (3 × 15 mL), the combined organic phases were washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 3/7 to give 0.56 g (79%) of the title compound as a clear oil; 1H-NMR (CDCl3): δ 7.30-7.12 (5H, m, Ph), 4.22 (1H, t, J=4.0 Hz, CH), 3.74 (3H, s, OCH3), 3.18 (1H, s, OH), 2.66 (2H, t, J=6.6 Hz, CH2), 1.85-1.62 (4H, m, 2×CH2); 13C NMR: 175.4 (COO), 141.6 (Ph), 128.1 (Ph), 128.0 (Ph), 125.5 (Ph), 70.1 (CHOH), 52.1 (OCH3), 35.2 (CH2), 33.6 (CH2), 26.3 (CH2). Anal. (C12H16O3) C, H.
Methyl 2-Oxo-5-phenyl-pentanoate (34)
Compound 33 (0.20 g, 0.96mmol) was dissolved in CH2Cl2 (20 mL) and treated with Dess-Martin periodinane (0. 43 g) under stirring for 40 min. The organic phase was washed with brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residual oil was purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 3/7 to give 0.195 g (99%) of the title compound as an yellowish oil; 1H-NMR (CDCl3): δ 7.31-7.15 (5H, m, Ph), 3.85 (3H, s, OCH3), 2.86 (2H, t, J=6.6 Hz, CH2), 2.63 (t, J=6.6 Hz, 2H, CH2), 1.71-1.62 (2H, m, CH2); 13C NMR: 194.0 (CO), 161.2 (COO), 141.8 (Ph), 128.3 (Ph), 128.0 (Ph), 125.8 (Ph), 52.8 (OCH3), 35.5 (CH2), 30.5 (CH2), 22.5 (CH2). Anal. (C12H14O3) C, H.
Methyl 2,2-Difluoro-5-phenyl-pentanoate (35)
A solution of compound 34 (0.404 g, 1.67 mmol) in CH2Cl2 (3.3 mL) was treated dropwise with DAST (0.489 mL, 3.6 mmol) at room temperature. After heating at 55 °C for 5 h, it was poured into H2O, cautiously neutralized by the addition of solid K2CO3, and extracted with CHCl3 (2 × 15 mL). The organic solvent was dried over Na2SO4, filtered and evaporated, and the crude product purified by flash column chromatography on silica gel eluting with EtOAc-petroleum ether (bp 40–60 °C) 1/9 to give 0.202 g (50%) of the title compound as an yellowish oil; 1H NMR (CDCl3): δ 7.32-7.12 (5H, m, Ph), 4.10 (3H, s, OCH3), 2.69 (2H, t, J=7.4 Hz, PhCH2), 2.21-1.90 (2H, m, CH2CF2), 1.80-1.72 (2H, m, CH2); 13C NMR: δ 164.2 (t, JC-C-F=33 Hz, COO), 140.9 (Ph), 128.4 (Ph), 128.3 (Ph), 126.1 (Ph), 116.2 (t, JC-F=248 Hz, CF2), 52.7 (OCH3), 34.9 (CH2), 33.8 (t, JC-C-F=23 Hz, CH2CF2), 23.0 (t, JC-C-C-F=2.4 Hz, CH2CH2CF2); 19F NMR: δ -28.0 (2F, t, J=17 Hz, CF2). MS (ESI) m/z (%): 229 (M++1, 100). Anal. (C12H14F2O2) C, H.
In-vitro PLA2 Assays
Phospholipase A2 activity was determined using the previously described modified Dole assay20 with buffer and substrate conditions optimized for each enzyme as described previously20,21,24,25: (i) GIVA cPLA2 substrate mixed-micelles were composed of 400 μM Triton X-100, 97 μM PAPC, 1.8 μM 14C-labeled PAPC, and 3 μM PIP2 in buffer containing 100 mM HEPES pH 7.5, 90 μM CaCl2, 2 mM DTT and 0.1 mg/ml BSA; (ii) GVI iPLA2 substrate mixed-micelles were composed of 400 μM Triton X-100, 99 μM DPPC, and 1.5 μM 14C-labeled DPPC in buffer containing 200 mM HEPES pH 7.0, 1 mM ATP, 2 mM DTT and 0.1 mg/ml BSA; and (iii) GV sPLA2 substrate mixed-micelles were composed of 400 μM Triton X-100, 99 μM DPPC, and 1.5 μM 14C-labeled DPPC in buffer containing 50 mM Tris pH 8.0 and 5 mM CaCl2.
In-vitro PLA2 Inhibition Studies
Initial screening of compounds at 0.091 mole fraction inhibitor in mixed-micelles was carried out. We considered compounds displaying 25% or less inhibition to have no inhibitory affect (designated N.D.). We report average percent inhibition (and standard error, n=3) for compounds displaying more than 25% and less than 90% enzyme inhibition. If percent inhibition was greater than 90%, we determined its XI(50) by plotting percent inhibition vs. inhibitor molar fraction (7 points; typically 0.005 to 0.091 mole fraction). Inhibition curves were modeled in Graphpad Prism using either a linear (x, y intercept = 0) or non-linear regression (one-site binding model - hyperbola, BMAX = 100) to calculate the reported XI(50) and associated error values.
Supplementary Material
Elemental analysis results for the compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH GM 20,501 and GM64611 (E.A.D.). The project is co-funded by the European Social Fund and National Resources – (EPEAEK II) (G.K.)
Abbreviations
- AACOCF3
arachidonyl trifluoromethyl ketone
- ATP
adenosine triphosphate
- BEL
bromoenol lactone, DAST, diethylaminosulfur trifluoride
- Deoxofluor
bis(2-methoxyethyl)amino-sulfur-trifluoride
- DIBALH
diisobutylaluminium hydride
- DPPC
1,2-dipalmitoylphosphotidylcholine
- DTT
dithiothreitol
- EAE
experimental autoimmune encephalomyelitis
- EtOAc
ethyl acetate
- GIVA cPLA2
Group VIA cytosolic phospholipase A2
- GV sPLA2
Group V secreted phospholipase A2
- GVIA iPLA2
Group VIA calcium-independent phospholipase A2
- NMDA
N-methyl-D-aspartate
- PAF
platelet activating factor
- PAPC
1-palmitoyl, 2-arachidonal phosphatidylcholine
- PIP2
phosphatidyl inositol (4,5)-bisphosphate
- TBAF
tetra-n-butylammonium fluoride
- TEMPO
2,2,6,6-tetramethylpiperidine-1-yloxy free radical
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- TMS
tetramethylsilane
References
- 1.Schaloske RH, Dennis EA. The phospholipase A(2) superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 2.Kudo I, Murakami M. Phospholipase A(2) enzymes. Prostag Oth Lipid M. 2002:68–69. 3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 3.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Winstead MV, Balsinde J, Dennis EA. Calcium-independent phospholipase A(2): structure and function. Biochim Biophys Acta. 2000;1488:28–39. doi: 10.1016/s1388-1981(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 5.Balsinde J, Balboa MA. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A(2) in activated cells. Cellular Signalling. 2005;17:1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Berg OG, Gelb MH, Tsai M-D, Jain MK. Interfacial enzymology: The secreted phospholipase A2-paradigm. Chem Rev. 2001;101:2613–2654. doi: 10.1021/cr990139w. [DOI] [PubMed] [Google Scholar]
- 7.Balestrieri B, Arm JP. Group V sPLA2: Classical and novel functions. Biochim Biophys Acta. 2006;1761:1280–1288. doi: 10.1016/j.bbalip.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Mounier CM, Ghomashchi F, Lindsay MR, James S, Singer AG, Parton RG, Gelb MH. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-α. J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 9.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirai Y, Balsinde J, Dennis EA. Localization and functional interrelationships among cytosolic Group IV, secreted Group V, and Ca2+-independent Group VI phospholipase A2s in P388D1 macrophages using GFP/RFP constructs. Biochim Biophys Acta. 2005;1735:119–129. doi: 10.1016/j.bbalip.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Magrioti V, Kokotos G. Synthetic inhibitors of Group IVA and Group VIA phospholipase A2. Anti-Inflammatory Anti-Allergy Agents Med Chem. 2006;5:189–203. [Google Scholar]
- 12.Reid RC. Inhibitors of secretory phospholipase A2 Group IIA. Curr Med Chem. 2005;12:3011–3026. doi: 10.2174/092986705774462860. [DOI] [PubMed] [Google Scholar]
- 13.Seno K, Okuno T, Nishi K, Murakami Y, Yamada K, Nakamoto S, Ono T. Pyrrolidine inhibitors of human cytosolic phospholipase A2. Part 2: synthesis of potent and crystallized 4-triphenylmethylthio derivative ‘Pyrrophenone’. Bioorg Med Chem Lett. 2001;11:587–590. doi: 10.1016/s0960-894x(01)00003-8. [DOI] [PubMed] [Google Scholar]
- 14.Ludwig J, Bovens S, Brauch C, Elfringhoff AS, Lehr M. Design and synthesis of 1-indol-1-yl-propan-2-ones as inhibitors of human cytosolic phospholipase A(2)alpha. J Med Chem. 2006;49:2611–2620. doi: 10.1021/jm051243a. [DOI] [PubMed] [Google Scholar]
- 15.Hess M, Elfringhoff AS, Lehr M. 1-(5-Carboxy- and 5-carbamoylindol-1-yl)propan-2-ones as inhibitors of human cytosolic phospholipase A2α: Bioisosteric replacement of the carboxylic acid and carboxamide moiety. Bioorg Med Chem. 2007;15:2883–2891. doi: 10.1016/j.bmc.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Fritsche A, Elfringhoff AS, Fabian J, Lehr M. 1-(2-Carboxyindol-5-yloxy)propan-2-ones as inhibitors of human cytosolic phospholipase A2α: Synthesis, biological activity, metabolic stability, and solubility. Bioorg Med Chem. 2008;16:3489–3500. doi: 10.1016/j.bmc.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 17.Lee KL, Foley MA, Chen LR, Behnke ML, Lovering FE, Kirincich SJ, Wang WH, Shim J, Tam S, Shen MWH, Khor SP, Xu X, Goodwin DG, Ramarao MK, Nickerson-Nutter C, Donahue F, Ku MS, Clark JD, Mckew JC. Discovery of ecopladib, an indole inhibitor of cytosolic phospholipase A(2)alpha. J Med Chem. 2007;50:1380–1400. doi: 10.1021/jm061131z. [DOI] [PubMed] [Google Scholar]
- 18.Lee KL, Behnke ML, Foley MA, Chen L, Wang W, Vargas R, Nunez J, Tam S, Mollova N, Xu X, Shen MWH, Ramarao MK, Goodwin DG, Nickerson-Nutter CL, Abraham WM, Williams C, Clark JD, McKew JC. Benzenesulfonamide indole inhibitors of cytosolic phospholipase A2α: Optimization of in vitro potency and rat pharmacokinetics for oral efficacy. Bioorg Med Chem. 2008;16:1345–1358. doi: 10.1016/j.bmc.2007.10.060. [DOI] [PubMed] [Google Scholar]
- 19.McKew JC, Lee KL, Shen MWH, Thakker P, Foley MA, Behnke ML, Hu B, Sum FW, Tam S, Hu Y, Chen L, Kirincich SJ, Michalak R, Thomason J, Ipek M, Wu K, Wooder L, Ramarao MK, Murphy EA, Goodwin DG, Albert L, Xu X, Donahue F, Ku MS, Keith J, Nickerson-Nutter CL, Abraham WM, Williams C, Hegen M, Clark JD. Indole cytosolic phospholipase A2 α inhibitors: Discovery and in vitro and in vivo characterization of 4-{3-[5-Chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(diphenylmethyl)-1H-indol-3-yl]propyl}benzoic acid, efipladib. J Med Chem. 2008;51:3388–3413. doi: 10.1021/jm701467e. [DOI] [PubMed] [Google Scholar]
- 20.Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. Novel 2-oxoamide inhibitors of human Group IVA phospholipase A2. J Med Chem. 2002;45:2891–2893. doi: 10.1021/jm025538p. [DOI] [PubMed] [Google Scholar]
- 21.Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, Kotsovolou S, Chiou A, Beltzner CC, Dennis EA. Inhibition of Group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells and in vivo. J Med Chem. 2004;47:3615–3628. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- 22.Constantinou-Kokotou V, Peristeraki A, Kokotos CG, Six DA, Dennis EA. Synthesis and activity of 2-oxoamides containing long chain beta-amino acids. J Pept Sci. 2005;11:431–435. doi: 10.1002/psc.628. [DOI] [PubMed] [Google Scholar]
- 23.Yaksh TL, Kokotos G, Svensson CI, Stephens D, Kokotos CG, Fitzsimmons B, Hadjipavlou-Litina D, Hua XY, Dennis EA. Systemic and intrathecal effects of a novel series of phospholipase A(2) inhibitors on hyperalgesia and spinal prostaglandin E-2 release. J Pharmacol Exp Ther. 2006;316:466–475. doi: 10.1124/jpet.105.091686. [DOI] [PubMed] [Google Scholar]
- 24.Stephens D, Barbayianni E, Constantinou-Kokotou V, Peristeraki A, Six DA, Cooper J, Harkewicz R, Deems RA, Dennis EA, Kokotos G. Differential inhibition of Group IVA and Group VIA phospholipases A(2) by 2-oxoamides. J Med Chem. 2006;49:2821–2828. doi: 10.1021/jm050993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Six DA, Barbayianni E, Loukas V, Constantinou-Kokotou V, Hadjipavlou-Litina D, Stephens D, Wong AC, Magrioti V, Moutevelis-Minakakis P, Baker S, Dennis EA, Kokotos G. Structure-activity relationship of 2-oxoamide inhibition of group IVA cytosolic phospholipase A2 and group V secreted phopholipase A2. J Med Chem. 2007;50:4222–4235. doi: 10.1021/jm0613673. [DOI] [PubMed] [Google Scholar]
- 26.Moutevelis-Minakakis P, Neokosmidi A, Filippakou M, Stephens D, Dennis EA, Kokotos G. Synthesis of lipophilic 2-oxoamides based on H-aminobutyric and δ-aminovaleric analogues and their activity against phospholipase A2. J Pept Sci. 2007;13:634–641. doi: 10.1002/psc.889. [DOI] [PubMed] [Google Scholar]
- 27.Street IP, Lin HK, Laliberte F, Ghomashchi F, Wang Z, Perrier H, Tremblay NM, Huang Z, Weech PK, Gelb MH. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 28.Trimble LA, Street IP, Perrier H, Tremblay NM, Weech PK, Bernstein MA. NMR structural studies of the tight complex between a trifluoromethyl ketone inhibitor and the 85-kDa human phospholipase A2. Biochemistry. 1993;32:12560–12565. doi: 10.1021/bi00210a002. [DOI] [PubMed] [Google Scholar]
- 29.Amandi-Burgermeister E, Tibes U, Kaiser BM, Friebe WG, Scheuer WV. Suppression of cytokine synthesis, integrin expression and chronic inflammation by inhibitors of cytosolic phospholipase A2. Eur J Pharmacol. 1997;326:237–250. doi: 10.1016/s0014-2999(97)85419-2. [DOI] [PubMed] [Google Scholar]
- 30.Conde-Frieboes K, Reynolds LJ, Lio YC, Hale MR, Wasserman HH, Dennis EA. Activated ketones as inhibitors of intracellular Ca2+-dependent and Ca2+-independent phospholipase A2. J Am Chem Soc. 1996;118:5519–5525. [Google Scholar]
- 31.Ghomashchi F, Loo R, Balsinde J, Bartoli F, Apitz-Castro R, Clark JD, Dennis EA, Gelb MH. Trifluoromethyl ketones and methyl fluorophosphonates as inhibitors of group IV and VI phospholipases A2: structure-function studies with vesicle, micelle, and membrane assays. Biochim Biophys Acta. 1999;1420:45–56. doi: 10.1016/s0005-2736(99)00056-5. [DOI] [PubMed] [Google Scholar]
- 32.Kalyvas A, David S. Cytosolic phospholipase A2 plays a key role in the pathogenesis of multiple sclerosis-like disease. Neuron. 2004;41:323–335. doi: 10.1016/s0896-6273(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 33.Yeo JF, Ong WY, Ling SF, Farooqui AA. Intracerebroventricular injection of phospholipases A2 inhibitors modulates allodynia after facial carrageenan injection in mice. Pain. 2004;112:148–155. doi: 10.1016/j.pain.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Svensson CI, Lucas KK, Hua XY, Powell HC, Dennis EA, Yaksh TL. Spinal phospholipase A2 in inflammatory hyperalgesia: Role of the small, secretory phospholipase A2. Neuroscience. 2005;133:543–553. doi: 10.1016/j.neuroscience.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 35.Bate C, Reid S, Williams A. Phospholipase A2 inhibitors or platelet-activating factor antagonists prevent prion replication. J Biol Chem. 2004;279:36405–36411. doi: 10.1074/jbc.M404086200. [DOI] [PubMed] [Google Scholar]
- 36.Risse D, Elfringhoff AS, Lehr M. Determination of the cell lytic properties of amphiphilic inhibitors of the cytosolic phospholipase A2 against human platelets by measuring the liberation of serotonin with high-performance liquid chromatography and fluorescence detection. J Chromatogr B. 2002;769:185–190. doi: 10.1016/s1570-0232(02)00013-2. [DOI] [PubMed] [Google Scholar]
- 37.Kazuyoshi, Y., Kenjiro, O., Mayumi, K., Sei K. Patent JP09268153A, 1997.
- 38.Banville, J., Marinier, A., Gai, Y., Plamondon, S., Roy, S., Balasubramanian, N. Patent US 6,414,179 B1, 2002.
- 39.Banville, J., Plamondon, S., Gai, Y., Balasubramanian, N. Patent US 6,492,550 B2, 2001.
- 40.Banville, J., Remillard, R., Balasubramanian, N., Bouthillier, G., Martel, A. Patent US 6,924,391 B2, 2005.
- 41.Burke JR, Davern LB, Stanley PL, Gregor KR, Banville J, Remillard R, Russell JW, Brassil PJ, Witmer MR, Johnson G, Tredup JA, Tramposch KM. BMS-229724 is a tight-binding inhibitor of cytosolic phospholipase A2 that acts at the lipid/water interface and possesses anti-inflammatory activity in skin inflammation models. J Pharmacol Exp Ther. 2001;298:376–385. [PubMed] [Google Scholar]
- 42.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 43.Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 44.Bohm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Muller K, Obst-Sander U, Stahl M. Fluorine in medicinal chemistry. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 45.Boivin J, El Kaim L, Zard SZ. A new and efficient synthesis of trifluoromethyl ketones from carboxylic acids. Part I Tetrahedron. 1995;51:2573–2584. [Google Scholar]
- 46.Gelb MH, Svaren JP, Abeles RH. Fluoro ketone inhibitors of hydrolytic enzymes. Biochemistry. 1985;24:1813–1821. doi: 10.1021/bi00329a001. [DOI] [PubMed] [Google Scholar]
- 47.Kokotos G, Kotsovolou S, Verger R. Novel trifluoromethyl ketones as potent gastric lipase inhibitors. ChemBioChem. 2003;4:90–95. doi: 10.1002/cbic.200390019. [DOI] [PubMed] [Google Scholar]
- 48.Leanna MR, Sowin TJ, Morton HE. Synthesis of α-amino and α-alkoxy aldehydes via oxoammonium oxidation. Tetrahedron Lett. 1992;33:5029–5032. [Google Scholar]
- 49.Middleton WJ. New fluorinating reagents. Dialkylaminosulfur fluorides. J Org Chem. 1975;5:574–578. [Google Scholar]
- 50.Singh RP, Cao G, Kirchmeier RL, Shreeve JM. Cesium fluoride catalysed trifluoromethylation of esters, aldehydes, and ketones with (trifluoromethyl)trimethylsilane. J Org Chem. 1999;64:2873–2876. doi: 10.1021/jo982494c. [DOI] [PubMed] [Google Scholar]
- 51.Creary X. Reaction of organometallic reagents with ethyl trifluoroacetate and diethyl oxalate. Formation of trifluoromethyl ketones and α-keto esters via stable tetrahedral adducts. J Org Chem. 1987;52:5026–5030. [Google Scholar]
- 52.Erni B, Khorana HG. Fatty acids containing photoactivable carbene precursors. Synthesis and photochemical properties of 3,3-bis(1,1-difluorohexyl)diazirine and 3-(1,1-difluorooctyl)-3H-diazirine. J Am Chem Soc. 1980;102:3888–3896. [Google Scholar]
- 53.Middleton WJ, Bingham EM. α,α-Difluoroarylacetic acids: preparation from (diethylamino)sulphur trifluoride and α-oxoarylacetates. J Org Chem. 1980;45:2883–2887. [Google Scholar]
- 54.Yuan W, Berman RJ, Gelb MH. Synthesis and evaluation of phospholipid analogues as inhibitors of cobra venom phospholipase A2. J Am Chem Soc. 1987;109:8071–8081. [Google Scholar]
- 55.Balsinde J, Dennis EA. Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J Biol Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- 56.Balsinde J, Dennis EA. Function and inhibition of intracellular calcium-independent phospholipase A2. J Biol Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- 57.Song H, Ramanadham S, Bao S, Hsu FF, Turk JA. Bromoenol lactone suicide substrate inactivates group VIA phospholipase A2 by generating a diffiusible bromomethyl keto acid that alkylates cysteine thiols. Biochemistry. 2006;45:1061–1073. doi: 10.1021/bi052065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.López-Vales R, Navarro X, Shimizu T, Baskakis C, Kokotos G, Constantinou-Kokotou V, Stephens D, Dennis EA, David S. Intracellular phospholipase A2 group IVA and group VIA play important roles in Wallerian degeneration and axon regeneration after peripheral nerve injury. Brain. 2008 doi: 10.1093/brain/awn188. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leung D, Du W, Hardouin C, Cheng H, Hwang I, Cravatt BF, Boger DL. Discovery of an exceptionally potent and selective class of fatty acid amide hydrolase inhibitors enlisting proteome-wide selectivity screening: concurrent optimization of enzyme inhibitor potency and selectivity. Bioorg Med Chem Lett. 2005;15:1423–1428. doi: 10.1016/j.bmcl.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 60.Pez D, Leal I, Zuccotto F, Boussard C, Brun R, Croft SL, Yardley V, Ruiz Perez LM, Gonzalez Pacanowska D, Gilbert IH. 2,4-Diaminopyrimidines as inhibitors of leishmanial and trypanosomal dihydrofolate reductase. Bioorg Med Chem. 2003;11:4693–4711. doi: 10.1016/j.bmc.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 61.Lwein AH, Szewczyk J, Wilson JW, Ivy Carroll F. Galanthamine analogs: 6H-benzofuro[3a,3,2,-e,f][1]benzazepine and 6H-benzofuro[3a,3,2,-e,f][3]benzazepine. Tetrahedron. 2005;61:7144–7152. [Google Scholar]
- 62.Roda G, Riva S, Danieli B. Almond oxynitrilase-catalyzed transformation of aldehydes is strongly influenced by napthyl and alkoxy substituents. Tetrahedron: Asymmetry. 1999;10:3939–3949. [Google Scholar]
- 63.Rho HS, Ko BS. Regioselective deoxygenation of the cyclic thionocarbonates of 2,3-dihydroxy esters with magnesium in methanol. Synth Commun. 1999;29:2875–2880. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis results for the compounds. This material is available free of charge via the Internet at http://pubs.acs.org.