

Abstract
Normal pH sensitivity of the SLC4A2/AE2 anion exchanger requires transmembrane domain (TMD) amino acid (aa) residues not conserved in the homologous but relatively pH-insensitive SLC4A1/AE1 polypeptide. We tested the hypothesis that the nonconserved aa cluster 1075DKPK1078 within the first putative re-entrant loop (RL1) of AE2 TMD contributes to pH sensor function by studying anion exchange function of AE2 mutants in which these and other RL1 aa were systematically substituted with corresponding RL1 aa from AE1. Regulation of Cl–/Cl– and Cl–/HCO–3 exchange by intracellular pH (pHi) or extracellular pH (pHo) was measured as 4,4′-di-isothiocyanatostilbene-2,2′ disulfonic acid-sensitive 36Cl– efflux from Xenopus oocytes. AE2 RL1 mutants 1075AAAQ1078 and 1075AAAQN1079 showed reduced pHi sensitivity and pHo sensitivity was acid-shifted by ∼1 pH unit. Individual mutants D1075A and P1077A exhibited moderately altered pH sensitivity, whereas a range of substitutions at conserved AE2 Ile-1079 substantially altered sensitivity to pHo and/or pHi. Substitution of the complete AE1 RL1 with AE2 RL1 failed to confer AE2-like pH sensitivity onto AE1. Replacement, however, of AE1 RL1 763SGPGAAAQ770 with AE2 1071VAPGDKPK1078 restored pHi sensitivity to the chimera AE2(1–920)/AE1(613–929) without affecting its low sensitivity to pHo. The results show that acute regulation of AE2 by pH requires RL1 of the TMD. We propose that critical segments of RL1 constitute part of an AE2 pH sensor that, together with residues within the N-terminal half of the TMD, constrain the AE2 polypeptide in a conformation required for regulation of anion exchange by pHi.
The mouse SLC4A2/AE2 anion exchanger regulates intracellular pH (pHi),3 chloride concentration, cell volume, and transepithelial ion transport in many tissues and is essential to postweaning life in mice (1). SLC4A2/AE2 and its paralogs SLC4A1/AE1 and SLC4A3/AE3 each mediate Na+-independent electroneutral anion exchange (2). The AE polypeptides are expressed in distinct tissue and cell type-specific patterns and exhibit distinct modes of acute regulation. AE2 and AE3 but not AE1 are acutely regulated by pHi (3). AE2 is also acutely regulated by extracellular pH (pHo), NH+4, hypertonicity, and (in a calmodulin-independent manner) calmidazolium (4, 5). Although the structural basis for these regulatory differences is not completely understood, recent structure-function analysis studies have identified isoform-specific residues in both N-terminal cytoplasmic and C-terminal transmembrane domains of AE2 that are required for acute regulation (4, 6).
The 400–700-aa N-terminal cytoplasmic domains of the SLC4 AE gene products are divergent in sequence, with only 35% aa identity between AE2 and AE1. The better conserved ∼500-aa transmembrane domains (TMDs) of AE1, AE2, and AE3 share ∼65% sequence identity. Even in the absence of their N-terminal cytoplasmic domains, AE TMDs expressed in erythrocytes, Xenopus oocytes, or mammalian cells can mediate electroneutral anion exchange (7–9). However, removal of all or part of the AE2 N-terminal cytoplasmic domain alters its acute regulation by pH, NH+4, hypertonicity, and calmidazolium (5). Mutation of individual aa residues of the N-terminal cytoplasmic domain partially or completely recapitulates these regulatory changes. Moreover, many of these residues are predicted to reside on the surface of the N-terminal cytoplasmic domain (6). Removal of the N-terminal cytoplasmic domain from the AE2 TMD abolishes regulation by pHi (10) and significantly acid-shifts the pHo versus activity profile compared with wild-type AE2 (9). These findings suggest the presence of a pH sensor motif within the AE2 TMD that cooperates with sensor components of the cytoplasmic N-terminal domain to modulate anion exchange activity in response to changing pHo and pHi. Earlier studies of the AE2 TMD pH sensor have identified nonconserved histidines (11), charged residues, and other amino acids of the TMD (12), which contribute to AE2 regulation by pH. We have identified in more recent studies of AE2/AE1 chimeras TMD subdomains of AE2 that contribute to different regulatory phenotypes (4). Together, these studies have indicated that the “pH sensor” of the AE2 TMD probably comprises not a single nonconserved amino acid but rather multiple TMD regions or residues interacting to confer on AE2-mediated anion exchange acute regulation by pH.
Biochemical and cysteine scanning mutagenesis studies of AE1 suggest a structure comprising 12–14 TMDs, including two putative transmembrane re-entrant loops (13) (Fig. 1A). The second putative re-entrant loop (RL2) and the last two transmembrane spans have been proposed to contribute to the AE1 anion selectivity filter (14). Structure-function analysis of the first putative re-entrant loop (RL1) has not been described. Among the ∼38 aa of RL1 (15) are 9 residues that differ between AE1 and AE2 (Fig. 1B). The RL1 of AE2 includes a nonconserved charged stretch of residues 1075DKPK1078 (Fig. 1B) that we hypothesized might be important in regulation of AE2 by pH. Therefore, we tested the consequences to pH sensitivity of AE-mediated 36Cl– transport of systematic introduction of AE1 RL1 residues into their corresponding positions in the RL1 of AE2.
FIGURE 1.

Nonconserved amino acid residues in transmembrane domain re-entrant loop 1 of AE2. A, model of the C-terminal transmembrane domain of AE2 by analogy with that of AE1 proposed by Zhu et al. (13) and based also on data from Refs. 15, 20, and 33. The oval marks putative RL1, and nonconserved amino acid residues, including 1075DKPK1078, are highlighted in gray. B, aligned amino acid sequences of the RL1 regions from mouse anion exchangers AE2 (Slc4a2; J04036), AE1 (Slc4a1; J02756), and AE3 (Slc4a3; AAA40692). Residues in boldface type are nonconserved amino acids. Boxed residues were previously tested for function after mutational substitution (11, 12). Oocytes expressing substitution mutants of boxed italicized residues exhibited loss of anion transport function at pHo 7.4. Schematics below the aligned sequences show alternative transmembrane dispositions of the AE2 RL1 loop as proposed by the models of Refs. 19 (left) and 13 (right). C, 36Cl– efflux rate constants measured at pHo 7.4 in n oocytes previously injected with the indicated ng amounts of cRNA encoding wild-type AE2, wild-type AE1, or the indicated RL1 mutants (mean ± S.E.).
These studies identify conserved and nonconserved RL1 residues of AE2 whose mutation individually or as a group sufficed to alter AE2 sensitivity to pHi, to pHo, or to both. Introduction into the AE1 RL1 of AE2 RL1 aa 1075DKPK1078 or 1072VAPGDKPK1078 failed to confer AE2-like pH sensitivity on AE1. In contrast, introduction of these same AE2 RL1 residues into RL1 of pH-insensitive chimera AE2(1–920)/AE1(613–929) conferred AE2-like sensitivity to pHi but not to pHo. These data identify discrete clusters of amino acid residues within AE2 RL1 as crucial components of a TMD pH sensor that requires functional interaction with region(s) within the first putative 6 TM spans and/or cytoplasmic N-terminal domain for normal regulatory activity.
EXPERIMENTAL PROCEDURES
Reagents—Na36Cl was purchased from GE Healthcare. (2-Aminoethyl) methanethiosulfonate (MTSEA), (2-(trimethylammonium)-ethyl) methanethiosulfonate (MTSET), and sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES) were purchased from Toronto Research Chemicals. Other chemical reagents were of analytical grade and obtained from Sigma, Fluka (St. Louis, MO), or Calbiochem. Restriction enzymes and T4 DNA ligase were from New England BioLabs (Beverly, MA). TaqDNA polymerase was from Roche Biochemicals (Mannheim, Germany), and dNTPs were from Promega (Madison, WI).
Solutions—ND-96 medium consisted of 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, and 2.5 mm sodium pyruvate, pH 7.40. Flux media lacked sodium pyruvate. pH values of 7.0, 8.0, and 8.5 in room air flux media were achieved with 5 mm HEPES. 5 mm MES was used for room air flux media of pH values 5.0 and 6.0. In Cl–-free solutions, NaCl was replaced isosmotically with 96 mm sodium isethionate, and equimolar gluconate salts of potassium, calcium, and magnesium were substituted for the corresponding Cl– salts. CO2/HCO–3-buffered solutions of pH 7.4 containing 24 mm NaHCO–3 and 72 mm sodium isethionate were saturated with 5% CO2, 95% air at room temperature for ∼1 h, and pH was verified prior to the start of each experiment. The addition to flux media of the weak acid salt sodium butyrate was in equimolar substitution for NaCl.
Mutant AE2 cDNAs—Murine AE2 (accession number J04036) and AE chimeras (4) encoded in plasmid pΔX (9) or pXT7, mouse erythroid AE1 (accession number X02677) encoded in plasmid pBL, mouse kidney AE1 (16) encoded in plasmid pL2AΔ, and rat cardiac AE3 encoded in plasmid pXT7 (17) were used as templates for PCR. Specific AE chimeras and AE2 and AE3 missense mutants were constructed by a four-primer PCR method (9, 10) using oligonucleotide primers (Biosynthesis, Woodlands, TX); primer sequences are available upon request. Integrity of PCR fragments and ligation junctions was confirmed by DNA sequencing of both strands.
cRNA Expression in Xenopus Oocytes—Ovarian segments were excised from female Xenopus laevis (Department of Systems Biology, Harvard Medical School) anesthetized with 0.17% tricaine according to protocols approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center. Stage V-VI oocytes were manually defolliculated following room temperature incubation of ovarian fragments with 2 mg/ml collagenase Type A (Roche Applied Science) for 60 min in ND-96 solution containing 50 ng/ml gentamycin and 2.5 mm sodium pyruvate. Oocytes were injected on the same day with 50 nl of cRNA or H20. Capped cRNA was transcribed from XbaI-linearized (pXT7), HindIII-linearized (pBL, pL2AΔ), or ClaI-linearized (pΔX) cDNA templates with the T7 MEGAscript kit (Ambion, Austin, TX) and resuspended in diethylpyrocarbonate-treated water. RNA integrity was confirmed by agarose gel electrophoresis in formaldehyde, and RNA concentration was estimated by A260 (Nanodrop, Wilmington, DE). The amount of injected cRNA (0.5–40 ng) was titrated for each AE mutant construct to approximate the 36Cl– efflux rate constant at pHo 7.4 associated with injection of 10 ng of wild-type AE2 cRNA. Injected oocytes were then maintained for 2–6 days at 19 °C in ND-96 culture medium.
Confocal Laser-scanning Immunofluorescence Microscopy—Two days after injection with water or with cRNA encoding wild-type or mutant AE2 bearing the HA epitope YPYDVPDYA inserted between Glu-858 and Ala-859 in the AE2 third extracellular loop, oocytes (n = 10–12/group) were fixed at 4 °C for 30 min in phosphate-buffered saline (PBS) containing 3% paraformaldehyde, washed three times in PBS supplemented with 0.002% sodium azide, and then blocked in PBS with 1% bovine serum albumin and 0.05% saponin (PBS-BSA) for 1 h at 4 °C. Oocytes were then incubated for 1 h at 4 °C with mouse monoclonal anti-HA peptide (dilution 1:100; Sigma), followed by three washes of 10 min at 4 °C. Oocytes were then incubated for 1 h with Cy3-conjugated secondary donkey anti-rabbit Ig (dilution 1:200; Jackson Immunochemicals) and again thoroughly washed in PBS-BSA.
Other oocytes were injected with water or with cRNA encoding wild-type or mutant AE2 fused at the carboxyl termini with green fluorescent protein (GFP). These oocytes were directly fixed for 30 min at room temperature in 1 ml of 3% paraformaldehyde in PBS. Oocytes expressing HA-tagged constructs were aligned in uniform orientation along a Plexiglass groove and sequentially imaged through the 10× objective of a Zeiss LSM510 laser scanning confocal microscope using the 543-nm laser line at 512 × 512 resolution at a uniform setting of 80% intensity, pinhole 160 (2.0 Airy units), detector gain 685, amp gain 1, 0 amp offset. Aligned oocytes expressing GFP fusion constructs were imaged with the 488-nm laser line at the same resolution and with a uniform setting of pinhole 112 (1.4 Airy units), detector gain 918, amp gain 1, 0 amp offset.
Polypeptide abundance at or near each oocyte surface was estimated by quantitation (Image J version 1.38, National Institutes of Health) of specific fluorescence intensity (FI) at the circumference of one quadrant of an equatorial focal plane (supplemental Fig. 1, A and B). Mean background-corrected FI for quadrants of oocytes previously injected with water was subtracted from the background-corrected FI for quadrants of individual cRNA-injected oocytes to yield intensity values for surface-associated specific FI for each oocyte. Surface expression of AE2 mutant polypeptides was normalized to that of the appropriate wild-type AE2 polypeptide. Normalized mutant- and wild type-specific fluorescence intensities were compared with Student's unpaired two-tailed t test (for the AE2-GFP constructs) or Dunnett's two-tailed t test (for AE2-HA tag constructs). p < 0.05 was interpreted as significant.
36Cl– Efflux Measurements—36Cl– efflux was assayed as previously described (6, 12). Oocytes in Cl–-free ND-96 were injected with 50 nl of 130 mm Na36Cl (10,000–12,000 cpm). Following a 5–10-min recovery period, the efflux assay was initiated by transfer of individual oocytes to 6-ml borosilicate glass tubes, each containing 1 ml of efflux solution. At intervals of 3 min, 0.95 ml of this efflux solution was removed for scintillation counting and replaced with an equal volume of fresh efflux solution. Following completion of the assay with a final efflux period in the presence of the anion transport inhibitor 4,4′-diisothiocyanatostilbene-2,2′ disulfonic acid (DIDS; 200 μm), each oocyte was lysed in 100 μl of 2% SDS. Samples were counted for 3–5 min such that the magnitude of 2× S.D. was <5% of the sample mean.
Experimental data were plotted as ln(% cpm remaining in the oocyte) versus time. 36Cl– efflux rate constants were measured from linear fits to data from the last three time points sampled for each experimental condition. Efflux cpm values for water-injected oocytes or for AE2 cRNA-injected oocytes in the presence of DIDS were less than 3-fold higher than machine background (∼20 cpm). Within each experiment, oocytes from the same frog previously injected with water or with cRNA encoding wild-type and mutant AE2 were subjected to parallel measurements. On each experimental day, the activity of tested mutant AE2 polypeptides was compared with WT AE2 activity at pH 7.4. Each AE2 mutant was tested in oocytes from at least three frogs. pHi dependence of 36Cl– efflux was measured as described (6). AE-mediated 36Cl– transport regulation by varying pHi at constant pHo was tested by intracellular acidification (∼0.5 pH units, time constant = 403 ± 55 s, n = 8) with bath addition of 40 mm sodium butyrate and subsequent oocyte alkalinization (∼0.5 pH units, time constant = 315 ± 15 s, n = 6) upon removal of butyrate from the bath (6, 10). Oocytes expressing AE2 RL1 mutants did not differ from oocytes expressing wild-type AE2 either in resting pHi or in magnitude of butyrate-induced acidification (supplemental Fig. 3, B and C). Butyrate is neither an inhibitor nor a substrate of AE2 (6, 10). pHo sensitivity of AE2-mediated 36Cl– efflux at nearly constant pHi was measured as previously validated (6). Individual oocytes were exposed sequentially to ND-96 at pHo 5.0, 6.0, 7.0, 8.0, and 8.5, followed by DIDS (200 μm) at pHo 8.5. Rate constants measured at each pHo value for each oocyte were fit with the following first order logistic sigmoid equation,
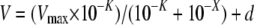 |
(Eq. 1) |
where v is the AE2-mediated Cl– efflux rate constant, Vmax is the maximal AE2-mediated Cl– efflux rate constant, x is the pHo at which the rate constant was measured, K is pHo(50), the pHo at which v is half-maximal, and d ≥ 0. Equation 1 allowed calculation (Microsoft Excel) of Vmax and pHo(50) values for each individual oocyte. Rate constants measured at each pHo value were normalized to that oocyte's Vmax, which was given the value of 100%. Mean normalized data for wild-type AE2 or AE mutants were fit (Sigma Plot version 8.02) to Equation 1 (as in Figs. 2C, 3D, 4C, 5C, 6D, and 8C). The mean normalized data were best fit by Equation 1 when d > 0 (r2 = 0.99).4
FIGURE 2.

AE2 RL1 amino acids are essential for regulation of AE2 by pH. A, time course of 36Cl– efflux from representative individual oocytes expressing wild-type AE2 (filled circles) and AE2-AE1(RL1) (open squares) during elevation of pHi by removal of bath butyrate (40 mm), followed by inhibition with DIDS (200 μm). B, normalized rate constants ± S.E. of 36Cl– efflux in the presence of bath butyrate for n oocytes expressing wild-type AE2 or AE2-AE1(RL1). C, regulation by pHo of normalized 36Cl– efflux rate constants exhibited by oocytes expressing wild-type AE2 (filled circles) or AE2-AE1(RL1) (open squares). Values are means ± S.E.; curves were fit to the data as described under “Experimental Procedures.” D, pHo(50) values (mean ± S.E.) of n oocytes expressing wild-type AE2 or AE2-AE1(RL1). B and D, * and gray bar, p < 0.05 compared with wild-type AE2.
FIGURE 3.

Nonconserved subdomains of AE2 RL1 are essential for regulation of AE2 by pH. A, schematic of AE2 RL1 indicating RL1 subdomains (black bars) into which were substituted the corresponding AE1 amino acids. Nonconserved residues are filled. B, time course of 36Cl– efflux from representative individual oocytes expressing AE2 or the indicated mutant polypeptides in the presence and subsequent absence of butyrate, followed by exposure to DIDS (200 μm). C, normalized rate constants in the presence of butyrate in n oocytes expressing wild-type AE2 or the indicated RL1 mutant polypeptides. D, regulation by pHo of normalized 36Cl– efflux from oocytes expressing wild-type AE2 (filled circles) or mutant polypeptides AE2-1075AAAQ1078 (filled triangles) or AE2-1075AAAQN1079 (open squares). Values are mean ± S.E.; curves were fit to the data as described under “Experimental Procedures.” E, pHo(50) values for wild-type AE2 or the indicated RL1 subdomain mutants (mean ± S.E.). C and E, asterisk and gray bars indicate p < 0.05 compared with wild-type AE2.
FIGURE 4.

Individual substitution into AE2 RL1 of corresponding AE1 RL1 amino acids identifies residues involved in regulation of AE2 by pH. A, time course of 36Cl– efflux from representative individual oocytes expressing wild-type AE2 (filled circles) or AE2 mutant polypeptides S1068G (open triangles), A0172G (open circles), or I1079N (filled triangles) during elevation of pHi by the removal of 40 mm butyrate, with subsequent exposure to DIDS (200 μm). B, normalized 36Cl– efflux rate constants ± S.E. in the presence of 40 mm butyrate for n oocytes expressing wild-type AE2 or the indicated AE2 RL1 missense substitution mutants. C, regulation by pHo of normalized 36Cl– efflux from oocytes expressing wild-type AE2 (filled circles), A1072G (open circles), and I1079N (filled triangles). Values are means ± S.E. D, pHo(50) values for n oocytes expressing wild-type AE2 or the indicated AE2 RL1 mutants (mean ± S.E.). For both B and D, asterisk and gray bar indicate p < 0.05 compared with wild-type AE2 (p < 0.05). In B and D, data for AE2 mutants D1075A, K1076A, P1077A, and K1078Q are reproduced from Ref. 12 and included for clarity.
FIGURE 5.

Systematic side chain substitution at Ile-1079 of AE2 RL1 reveals differential regulation by pHi and pHo. A, time course of 36Cl– efflux from representative individual oocytes expressing wild-type AE2 (filled circles), I1079K (open circles), I1079S (open squares), or I1079C (filled squares) during elevation of pHi by removal of 40 mm butyrate, with subsequent exposure to DIDS (200 μm). B, normalized 36Cl– efflux rate constants ± S.E. in the presence of 40 mm butyrate for n oocytes expressing wild-type AE2 or the indicated I1079 substitution mutants. C, time course of 36Cl– efflux from representative individual oocytes expressing WT AE2 (filled circles), I1079C (gray squares), I1079E (gray circles), and I1079K (filled triangles) during sequential increases in pHo (top bar). D, regulation by pHo of normalized 36Cl– efflux from oocytes expressing WT AE2 (filled circles), I1079C (filled squares), I1079N (open squares), I1079E (gray circles), and I1079K (filled triangles). Values are means ± S.E.; curves were fit to the data as described under “Experimental Procedures.” E, pHo(50) values for n oocytes expressing WT AE2 or the indicated Ile-1079 substitution mutants (mean ± S.E.). For both B and E, asterisk and gray bars indicate significant difference in values for Ile-1079 single point mutants when compared with WT AE2 (p < 0.05). F, normalized 36Cl– efflux rate constants ± S.E. in the presence of 40 mm butyrate for n oocytes expressing AE2 mutant I1079C preincubated 10 min without or with the indicated methanethiosulfonate reagent (see “Experimental Procedures” for details).
FIGURE 6.

Mutation of AE2 RL1 residues alters Cl–/HCO–3 exchange and its regulation by pHi. A, time course of 36Cl– efflux from representative individual oocytes expressing wild-type AE2 (filled circles), I1079N (filled triangles), 1075AAAQN1079 (filled squares), and H2O (open circles) sequentially exposed to 96 mm Cl– (a), to 24 mm HCO–3 in the presence of 72 mm Cl– (b), and then to Cl–-free bath in continued presence of HCO–3 (c), terminated by the addition of 200 μm DIDS (d). B, 36Cl– efflux rate constants of Cl–i/HCO–3o exchange normalized to those of Cl–i/Cl–o exchange in the same oocytes ((rate constant of period c (Kc)/rate constant of period a (Ka)) × 100%). Values are means ± S.E. of n oocytes. C, time course of 36Cl– efflux from representative individual oocytes expressing wild-type AE2 (filled circles) or AE2 RL1 mutants 1075AAAQ1078 (open circles) and 1075AAAQN1079 (filled squares) during elevation of pHi by removal of bath butyrate (40 mm) in the continued presence of HCO–3, with subsequent inhibition by DIDS (200 μm). D, normalized 36Cl– efflux rate constants ± S.E. ((rate constant of period a (ka)/rate constant of period b (kb)) × 100%). for n oocytes expressing wild-type AE2 or the indicated AE2 mutants in the presence of 40 mm butyrate at 5% CO2/24 mm HCO–3, pHo 7.4. B and D, * and gray bars, p < 0.05 compared with wild-type AE2.
FIGURE 8.

pH insensitivity of chimeric polypeptide AE2(1–920)/AE1(613–929) is rescued by introduction of AE2 RL1 residues. A, time course of 36Cl– efflux from representative individual oocytes expressing wild-type AE2 or the indicated chimeric AE polypeptides (defined as in B) during elevation of pHi by the removal of 40 mm butyrate, with subsequent exposure to DIDS (200 μm). B, normalized 36Cl– efflux rate constants ± S.E. in the presence of 40 mm butyrate for n oocytes expressing wild-type AE2 or the indicated chimeric AE polypeptides. Inset, RL1 schematic. Gray RL1 residues are those mutated in the constructs studied. C, regulation by pHo of normalized 36Cl– efflux from oocytes expressing wild-type AE2 (filled circles), wild-type AE1 (open circles), or the indicated chimeric AE polypeptides. Values are means ± S.E. The curves were fit to the data as described under “Experimental Procedures.” D, pHo(50) values (means ± S.E.) for n oocytes expressing wild-type AE2, wild-type AE1, or the indicated chimeric polypeptides. Data for construct 1 (AE2(1–920)/AE1(613–929)) are from Ref. 4 and are shown here for clarity. For both B and D, asterisk and gray bars indicate p < 0.05 compared with wild-type AE2.
Methanethiosulfonate (MTS) reagents were dissolved immediately before the start of the experiment in Cl–-free ND-56 containing 40 mm butyrate at final concentrations of 10 mm MTSET, 10 mm MTSES, or 2.5 mm MTSEA. Oocytes were preincubated with MTS reagents for 10 min prior to the start of measurement of pHi dependence of AE2-mediated 36Cl– efflux. MTS reagents did not alter wild-type AE2-mediated 36Cl– efflux at pHo 7.4 (data not shown).
Statistics—Data are reported as mean ± S.E. Analysis of variance was performed among groups of mutants compared with wild-type AE2 by Dunnett's two-way t test. Statistical significance was also assessed by Student's paired and unpaired t tests, for which the level of significance was p < 0.05.
RESULTS
Putative RL1 of mouse AE2 encompasses aa 1050–1083 within the AE2 transmembrane domain (Fig. 1A) and shares with the corresponding regions of AE1 and cAE3 respective sequence identities of ∼75 and ∼85% (Fig. 1B). To test the hypothesis that all or part of RL1 is important for AE2 regulation by pH, we tested the pH-sensitive anion exchange activity of AE2 mutant polypeptides in which all or part of RL1 was replaced with corresponding aa residues from RL1 of the relatively pH-insensitive mouse AE1 polypeptide and vice versa. As described under “Experimental Procedures” and shown in Fig. 1C, injected cRNA quantities were chosen for each mutant construct to result in 36Cl– efflux rate constants as close as possible to that of wild-type AE2. Thus, paralogous substitution of neither the entire RL1 nor of its most divergent subdomain resulted in significantly decreased rates of 36Cl–/Cl– exchange at pHo 7.4. Nominal surface expression of C-terminally GFP-tagged AE2 mutant 1075AAAQN1079 did not differ from that of WT AE2-GFP (supplemental Fig. 1A). This finding was consistent with our previous demonstration that anion exchange rates measured at pHo 7.4 for intact and GFP-tagged wild-type and (other) mutant AE2 polypeptides correlate well with nominal surface expression of the GFP fusion proteins in Xenopus oocytes (4). Additional experiments with AE2 polypeptides bearing an HA epitope tag introduced into the third extracellular loop of the TMD and immunodetected at 4 °C in unfixed oocytes confirmed that wild-type AE2 and AE2 RL1 mutant polypeptides do not differ detectably in their expression at the oocyte surface (supplemental Fig. 1B).
Replacement of the AE2 RL1 Sequence with That of AE1 Alters AE2 Regulation by pHi and pHo—We assessed the contribution of RL1 to regulation of AE2 by pHi and by pHo through study of a chimeric polypeptide in which the RL1 sequence from mouse AE1 was inserted into mouse AE2. As shown in Fig. 2, A and B, chimera AE2-AE1RL1-mediated 36Cl– efflux exhibited a significant decrease in sensitivity to pHi. Fig. 2C compares the pHo dependence of normalized 36Cl– efflux activity of AE2-AE1RL1 with that of wild-type AE2. As summarized in Fig. 2D, the pHo(50) value for AE2-AE1RL1 (6.44 ± 0.06, n = 11) was significantly acid-shifted compared with AE2 (6.90 ± 0.06, n = 12; p < 0.05). These data demonstrate the importance of AE2-specific aa residues of RL1 in the acute regulation of AE2 by pH.
We have previously shown that individual mutagenesis of AE2 RL1 residue Asp-1075 and Pro-1077 modestly alters AE2 regulation by pHo and pHi, respectively (12), suggesting the involvement of multiple TMD aa residues in regulation by pH. We therefore mutated contiguous groups of nonconserved amino acids within AE2 RL1 to their corresponding AE1 residues in order to identify subdomains of RL1 potentially important to regulation by pH. Thus, AE2 RL1 sequences 1068SKAVA1072 and 1075DKPK1078 were changed (Fig. 3A) to the corresponding AE1 sequences 1068GKASG1072 and 1075AAAQ1078 (in which AE2 numbering is retained). We also tested a mutant produced during the mutagenesis reactions in which AE2 Ile-1079 was additionally changed to Asn, yielding AE2-1075AAAQN1079. Fig. 3B shows 36Cl– efflux time courses from oocytes expressing wild-type or mutant AE2 polypeptides in the presence (acid pHi) or absence (more alkaline pHi) of 40 mm butyrate. The normalized pHi sensitivity data in Fig. 3C show that AE2 mutants 1068GKASG1072 and 1075AAAQN1079 both exhibited significant loss of sensitivity to pHi. AE2 mutant 1075AAAQ1078 also exhibited a moderate decrease in pHi sensitivity. This region is also important in function of the homologous, pHi-sensitive anion exchanger, cardiac AE3 (cAE3), as evidenced by the reduction in its pHi sensitivity resulting from replacement of AE3 residues 869DKPQI873 with residues AAAQN (supplemental Fig. 2). These data demonstrate the importance of contiguous amino acid subdomains within the AE2 RL1 to regulation by pHi.
Since pHo regulates AE2 activity (3), we also assessed the response of AE2 RL1 subdomain mutants to varying pHo. Fig. 3, D and E, shows that replacement of AE2 RL1 residues with the corresponding AE1 residues acid-shifts the pHo activity profile of those mutants compared with wild-type AE2 by ∼1 pH unit. In contrast to AE2 regulation by pHi (Fig. 3, B and C), the acid-shifted pHo(50) values of AE2 mutants 1075AAAQ1078 and 1075AAAQN1079 did not differ. This latter finding suggested that AE2 Ile-1079, although important to pHi sensitivity, appeared to be dispensable for regulation by pHo.
Nonconserved Residues of the AE2 RL1 Are Involved in pHi and pHo Regulation—Since the proposed structure of AE2 RL1 predicts accessibility to both intra- and extracellular compartments, we tested the roles in AE2 regulation by both pHi and pHo of individual nonconserved aa residues of AE2 RL1, systematically changing each to the corresponding AE1 residue. As shown in Fig. 4A and summarized in Fig. 4B, AE2 mutant polypeptides in which nonconserved RL1 residues Ser-1068, Val-1071, and Ala-1072 were substituted individually with their AE1 counterparts lacked sensitivity to pHi. This contrasted with the lack of effect of similar substitutions of nonconserved RL1 residues Asp-1075, Lys-1076, and Lys-1078 and the modest decrease in pHi sensitivity produced by substitution of Pro-1077 (reproduced in Fig. 4B from Ref. 12 for the purpose of comparison). Interestingly, mutation of the conserved Ile-1079 to Asn greatly reduced pHi sensitivity, probably explaining the different pHi sensitivities of the AE2 mutants 1075AAAQ1078 and 1075AAAQN1079 (Fig. 3B).
Fig. 4C compares pHo dependence of normalized 36Cl– efflux activity for AE2 RL1 mutants A1072G and I1079N with that of wild-type AE2. As summarized in Fig. 4D, the pHo(50) value for A1072G (6.04 ± 0.13, n = 17) was significantly acid-shifted from that of wild-type AE2 (6.81 ± 0.04, n = 27; p < 0.05), whereas the pHo(50) of I1079N did not differ from the wild-type value (p > 0.05). AE2 mutants S1068G, V1071S, and D1075A were less dramatically acid-shifted in their pHo versus activity profiles. Thus, nonconserved AE2 RL1 residues Ser-1068, Val-1071, and Ala-1072 are important for AE2 sensitivity to both pHi and pHo, whereas Pro-1077 and Ile-1079 are important only for pHi sensitivity and Asp-1075 is important only for pHo sensitivity. These effects on regulation by pH are not likely to be due to variations in mutant polypeptide surface abundance, since the injected cRNA quantity chosen for each AE2 mutant yielded 36Cl–/Cl– exchange rates at pHo 7.4 indistinguishable from that of wild-type AE2 (supplemental Fig. 3), with one exception. That exception was AE2 K1078Q, which exhibited a wild-type phenotype for pH sensitivity (Fig. 4, B and D) despite a 36Cl– efflux rate constant at pH0 7.4 slightly lower than that of wild-type AE2 (p < 0.05; supplemental Fig. 3).
The N-terminal end of AE2 RL1 contains two alanine residues in close proximity (Fig. 1B). Although their dual mutation to the corresponding AE1 residues did not change basal activity at pHo 7.4 (data not shown) or AE2 regulation by pHi (Fig. 4B), the pHo(50) value of the mutant A1051S/A1053T (7.21 ± 0.09 (n = 13) was significantly alkaline-shifted from the wild-type value 6.81 ± 0.04 (n = 27; p < 0.05).
Systematic Side Chain Substitution of AE2 I1079 Reveals Differential Effects on Regulation by pHi and by pHo—Mutation of conserved AE2 RL1 residue Ile-1079 abolished pHi sensitivity while retaining regulation by pHo. We hypothesized that Ile-1079 was important for the structural integrity of AE2 RL1 or adjacent regions and thus investigated the consequences of systematic side chain substitution at this position to acute AE2 regulation by pH. Fig. 5, A and B, show that change of AE2 Ile-1079 to Lys but not to Ser reduced pHi sensitivity compared with wild-type AE2, whereas change to Cys increased pHi sensitivity. Substitution of Ile-1079 with the hydrophobic Leu or polar residue Ser had no effect on AE2 pHi sensitivity, but substitution with the larger hydrophobic residue Met, with aromatic residues Phe or Tyr, with larger polar residues Thr or Asn, or with charged residues Lys or Glu each substantially reduced the sensitivity of AE2 to pHi. Fig. 5, C–E, reveals reduced pHo sensitivity of AE2 mutants I1079E and I1079K, with pHo(50) values acid-shifted ∼0.8 pH unit compared with wild-type AE2 and to mutant I1079N. In contrast, mutant I1079C exhibited increased pHo sensitivity, with the pHo(50) value alkaline-shifted by 0.7 units (Fig. 5E). Taken together, these data show that various side chain substitutions at the functionally important residue AE2 Ile-1079 can produce mutant polypeptides with a range of pH sensitivity phenotypes. We tested further the impact of side chain variation in the context of the AE2 mutant I1079C by modification with MTS reagents. Since I1079C is predicted to be accessible to extracellular reagents (Figs. 1 and 3), we tested the consequences of its modification with membrane-permeant MTSEA as well as with membrane-impermeant MTSES and MTSET. In contrast to isoleucine modification to Lys or Glu, none of the MTS reagents altered the enhanced pHi dependence of 36Cl– efflux mediated by the AE2 mutant I1079C (Fig. 5F).
Regulation of Cl–/HCO–3 Exchange Is Altered by Mutation of Residues of the AE2 RL1—AE2 pH sensitivity structure-function relationships derived from Cl–/Cl– exchange measurements in our previously published work (6, 10) have applied equally to pH sensitivity of AE2-mediated Cl–/HCO–3 exchange. We therefore tested the consequences of mutation of functionally important RL1 residues on AE2-mediated Cl–/HCO–3 exchange (Fig. 6A). 36Cl– efflux was measured in the presence of bath Cl– (period a representing 36Cl–/Cl– exchange) and then during exposure to CO2/HCO–3 in the continued presence (period b) and subsequent absence of bath Cl– (period c representing 36Cl–/HCO–3 exchange), followed by confirmation of efflux specificity by inhibition with 200 μm DIDS (period d). Wild-type AE2-mediated 36Cl–/HCO–3 exchange (period c) was only ∼20% of the rate of 36Cl–/Cl– exchange (period a), in contrast to the higher relative rates of Cl–/HCO–3 exchange exhibited by AE2 mutants I1079N, 1075AAAQ1078, and 1075AAAQN1079 (Fig. 6B; p < 0.05). Cl–/HCO–3 exchange mediated by each of these three mutants was pHi-insensitive (Fig. 6, C and D), as previously noted for Cl–/Cl– exchange. The higher relative HCO–3 transport rates of these mutants evident in Fig. 6, A and B, may reflect altered substrate selectivity.
The AE2 RL1 Is Not Sufficient to Confer AE2-like pHi Sensitivity on AE1—Since the AE2 RL1 was necessary for normal pH sensitivity of AE2, we tested the hypothesis that AE2 RL1 or a subdomain might suffice to confer AE2-like pH sensitivity on the relatively pH-insensitive AE1. Fig. 7, A and B, shows that neither AE1 mutant polypeptide containing the AE2 RL1 subdomain 1075DKPK1078 nor that containing subdomain 1071VAPGDKPK1078 conferred regulation by pHi. Replacement of the complete AE1 RL1 with that from AE2 RL1 (AE1-AE2(RL1)) also failed to confer regulation by pHi (Fig. 7B). Moreover, regulation by pHo of AE1-AE2(RL1) was indistinguishable from that of WT AE1 (Fig. 7C). The pHo(50) value for AE1-AE2(RL1) was acid-shifted >1.3 units (5.57 ± 0.05, n = 8) with respect to that of wild-type AE2 (6.90 ± 0.06, n = 14; p < 0.05) (Fig. 7D).
FIGURE 7.

pH insensitivity of AE1 is not modified by introduction into AE1 of AE2 RL1. A, time course of 36Cl– efflux from representative individual oocytes expressing wild-type AE2 (filled circles), wild-type AE1 (open squares), AE1-767DKPK770 (filled triangles), or AE1-760SKAVAPGDKPK770 (filled squares) during elevation of pHi by removal of 40 mm butyrate and subsequent exposure to DIDS (200 μm). B, normalized 36Cl– efflux rate constants in the presence of 40 mm butyrate for n oocytes ± S.E. expressing wild-type AE2, wild-type AE1, or the indicated AE1 RL1 mutants. C, regulation by pHo of normalized 36Cl– efflux from oocytes expressing wild-type AE2 (filled circles), wild-type AE1 (open squares), or AE1-AE2(RL1) (open circles). Values are means ± S.E.; curves were fit to the data as described under “Experimental Procedures.” D, pHo(50) values (mean ± S.E.) for n oocytes expressing the indicated wild-type or mutant polypeptides. For both B and D, asterisk and gray bar indicate p < 0.05 compared with wild-type AE2.
Replacement of the AE1 RL1 Sequence with That of AE2 Restores pHi Regulation to the pHi-insensitive Chimera AE2(1–920)/AE1(613–929)—We recently proposed that multiple regions of the AE2 TMD are required for physiological regulation of AE2 by pH (4). Replacement of AE2 aa 921–1237 with the corresponding C-terminal section of the AE1 TMD severely attenuated both sensitivities to pHi and pHo. Since the AE1-encoded C-terminal TMD region of chimera AE2(1–920)/AE1(613–929) includes RL1, we tested the hypothesis that reintroduction into this chimeric polypeptide of all or part of the AE2 RL1 might restore to it AE2-like regulation by pH.
Fig. 8, A and B, shows in the pHi-insensitive AE chimera, AE2(1–920)/AE1(613–929), that replacement of AE1 RL1 residues 767AAAQ770 with the corresponding AE2 residues 1075DKPK1078 was insufficient to restore AE2-like pHi sensitivity. In contrast, replacing the larger group of contiguous nonconserved AE2 aa 1072VAPGDPKP1078 rescued pHi sensitivity similar to that shown for wild-type AE2. Complete replacement of the AE1 RL1 with that from AE2 also restored pHi sensitivity to chimera AE2(1–920)/AE1(613–929). Thus, these data suggest that nonconserved AE2 RL1 amino acid residues are able, together with previously described portions of the AE2 N-terminal cytoplasmic domain, to determine pHi regulation of AE2 activity.
Since chimeric polypeptide AE2(1–920)/AE1(613–929) lacks regulation by pHo (4), we assessed its pHo sensitivity following substitution of its AE1-derived RL1 with functionally important residues from the RL1 of AE2. Substitution of AE1 RL1 residues with AE2 residues, 1075DKPK1078 or 1072VAPGDPKP1078, failed to rescue AE2-like regulation by pHo (Fig. 8, C and D). The pHo versus activity profile for AE chimera, AE2(1–920)/AE1(613–929) with complete replacement of its AE1 RL1 by that of AE2, could not be determined due to low transport activity of oocytes expressing this mutant polypeptide. Thus, it remains possible that replacement in this chimera of AE1 aa Ser-743 and Thr-745 with AE2 aa Ala-1051 and Ala-1053 alone or together with the other nonconserved aa of RL1 may have restored AE2-like pHo regulation. Together, the data show clearly distinct differences in regulation by pHi and by pHo resulting from mutagenesis of individual or multiple residues of RL1. Thus, unlike restoration of the chimera's pHi sensitivity, introduction into the AE1-derived RL1 of groups of nonconserved AE2 RL1 residues does not rescue AE2-like regulation by pHo.
DISCUSSION
The anion exchangers SLC4A2/AE2 and SLC4A3/AE3 differ from their close homolog SLC4A1/AE1 in their pH-sensitive regulation across the physiological range. AE2 is regulated independently by pHi and by pHo, with a pHo(50) value of ∼6.8. The AE2 N-terminal cytoplasmic domain is critical for sensitivity to pHi and for modulating regulation of transport activity by pHo (6, 18). The molecular contributors to the transmembrane domain “pH sensor” of AE2 remain incompletely understood (4, 11, 12). In the present work, we have identified putative RL1 as essential for regulation of AE2-mediated Cl–/Cl– exchange by both pHi and pHo and further defined individual amino acid residues within RL1 that are likely to be responsible for this regulatory function. We have also demonstrated the importance of RL1 C-terminal residues for regulation by pHi of AE2-mediated Cl–/HCO–3 exchange and of Cl–/Cl– exchange mediated by the closely related AE3 anion exchanger.
Although the RL1 region is crucial for wild-type regulation by pH of AE2 and AE3, introduction of the AE2 RL1 into the relatively pH-insensitive AE1 polypeptide does not suffice to confer AE2-like pH sensitivity. However, introduction of the AE2 RL1 into the pH-insensitive chimera AE2(1–921)/AE1(613–929) rescues AE2-like pHi-sensitivity selectively, without effect on the chimera's acid-shifted, AE1-like pHo sensitivity. Thus, the AE2 RL1 is sufficient, in the presence of the AE2 N-terminal cytoplasmic domain and the first six TM spans of the AE2 transmembrane domain, to confer AE2-like pHi sensitivity. Individual residues from RL1, defined here, jointly contribute with undefined residues from within the first six TM spans of AE2 to form a structure (Fig. 9) that, together with residues of the N-terminal cytoplasmic domain (6), can sense changes in pHi and in response adjust the rate of AE2-mediated anion exchange (Fig. 9). The ability of RL1 to contribute effectively to physiological regulation of AE2 by pHo requires the presence of additional regions of AE2 yet to be defined.
FIGURE 9.

RL1 of the AE2 TMD plays a critical role in the acute regulation of anion exchange by pH. A, summary of TMD subdomains (shaded boxes) and individual amino acid residues identified from mutagenesis studies as contributing importantly to regulation of AE2 activity by pH, NH+4, and calmidazolium. Residues of RL1 interact with as yet unidentified amino acids within TM1–6 and together contribute to “pH sensor” functions in the AE2 TMD. Residues involved in regulation by pHo are gray, those involved in regulation by pHi are white, and those involved in regulation by both pHi and pHo are black. White boxes marked × represent residues that when mutated yielded oocytes with functional activity too low to study. B, aligned RL1 amino acid sequences from AE1, AE2, and AE3. Boldface residues in gray blocks are the AE2 residues, mutation of which alters regulation by pH. Also Boldface in gray blocks are AE3 and AE1 residues identical to these AE2 residues. Right, terminal amino acid residue numbers of these sequences.
Topographical Disposition of the RL1 Region of the AE2 TMD—The amino acid sequence differences between the TMD portions of AE2 and AE1 proposed to span the membrane bilayer are at their greatest in RL1 and RL2. Nine among the ∼36 aa of RL1 differ between AE2 and AE1, suggesting RL1 as a candidate component of the AE2 pH-sensing mechanism. Early hydropathy profiles of the AE1 sequence (19) envisaged the RL1 region as a helical hairpin, resulting in placement of several charged and polar residues within the plane of the lipid bilayer (Fig. 1B). This model placed AE1 Lys-743 (corresponding to mouse AE1 Lys-761 and AE2 Lys-1069) at the TMD cytoplasmic face consistent with the sensitivity of this site to tryptic cleavage in low ionic strength conditions in resealed human red cell ghosts, in inside-out red cell membrane vesicles, and in microsomes from HEK-293 cells expressing recombinant human eAE1 (20). In contrast, scanning cysteine accessibility mutagenesis studies of the recombinant Cys-less human AE1 expressed in HEK-293 cells (21) demonstrated accessibility to extracellular biotin maleimide and Lucifer Yellow iodoacetamide of individual Cys residues reintroduced across the entire RL1 region, and an extracellular facing RL1 was proposed (22). N-Glycosylation scan mutagenesis of recombinant human AE1 N642D also suggested extracellular disposition of RL1 when tested by microsomal insertion during in vitro translation in reticulocyte lysate (15). However, lack of N-glycosylation of the same mutant expressed in HEK-293 and COS-7 cells was consistent with intracellular disposition of Lys-743 of RL1 (corresponding to mouse AE2 Lys-1069). Engineered glycosylation sites at neighboring RL1 residues of Ser-731 (mouse AE2 S1057) and Gln-754 (mouse AE2 Q1080) were N-glycosylated when expressed in the same mammalian cells, suggesting extracellular location of both residues, each only 11–12 residues to either side of the intracellular tryptic site Lys-743 (15). Therefore, RL1 was proposed to exhibit post-translational conformational flexibility manifest as a transition from an extracellular disposition in the endoplasmic reticulum membrane to a refolded intrabilayer/intracellular conformation subsequent to departure from the endoplasmic reticulum and prior to or upon arrival at the cell surface. The schematic of Fig. 1A attempts to reconcile these data. The absence of helical structure need not be invoked in RL1 if the low ionic strength required for tryptic susceptibility of Lys-743 (mouse AE2 Lys-1069) allows protease access to the backbone amide substrate. The absence of Cys residues in AE1 might stabilize the extracellular disposition of RL1 in the construct subjected to scanning cysteine accessibility mutagenesis and prevent its post-ER insertional refolding (20, 21). The 40% reduced surface expression and ∼60% reduced specific Cl–/HCO–3 exchange activity exhibited by Cys-less human AE1 (23) may reflect such an altered conformation. Thus, the structure and orientation of the AE1 RL1 remains controversial.
The schematic in Fig. 9 incorporates our current and previous functional data with previously proposed transmembrane structures. Thus, individual mutation of the proposed exofacial residues Glu-888, Lys-889, Glu-981, Lys-982, Ala-1051, and Ala-1053 selectively alters pHo sensitivity, whereas mutation of proposed endofacial residues Arg-921, Phe-922, and Arg-1107 selectively alters regulation by pHi. Among the 54 AE2 TMD residues individually mutated to date, mutation of only four of these (Ser-1068, Val-1071, Ala-1072, and Ile-1079; Fig. 9, black circles) alters both regulation by pHo and by pHi, and all four are located within the putative RL1. Although RL1 remains a region of topographical uncertainty within the AE2 polypeptide, the presence of these four residues in RL1 strongly supports accessibility of RL1, or parts of RL1, to both extracellular and intracellular solvent.
AE2 RL1 Nonconserved Residues Important for AE2 Regulation by pH—The nine amino acid residues of AE2 RL1 not conserved in AE1 (in boldface type) comprise three subdomains: 1051AAA1053, 1068SKAVA1072, and 1075DKPK1078. The AE1-based RL1 model of Fig. 1 predicts location of AE2 1075DKPK1078 close to the extracellular surface, whereas cytoplasmic exposure is predicted for 1068SKAVA1072, in which Lys-1069 corresponds to intracellular tryptic cleavage site Lys-743 of human AE1. Group substitution of either cluster with corresponding AE1 residues alters AE2 regulation by both pHi and pHo (Fig. 3), suggesting accessibility of both subdomains to cytoplasmic and extracellular protons or an important role for RL1 in maintaining the conformations required to sense and/or to respond to changing pH in either compartment.
Systematic introduction of individual AE1 RL1 amino acid residues in place of each of the nine nonconserved AE2 RL1 residues (Fig. 4) resulted in only two substitutions that left regulation of AE2 by pH unaltered, K1076A and K1078Q. The seven other RL1 residues not conserved in AE1 are present in all mammalian AE2 polypeptides. The selective alteration of pHo sensitivity by the mutation at Ala-1051/Ala-1053 is consistent with the proposed extracellular location of this initial part of RL1 but suggests a conformational effect on titratable residues not contiguous in the linear sequence. Selective alteration of pHo sensitivity by mutant D1075A and of pHi sensitivity by mutant P1077A does not conform easily with the RL1 model of Fig. 1A. These data suggest a more complex structure in which individual mutations can project pH-sensitive conformational change to more remote titration sites. Alternatively, RL1 and other portions of this model may need revision.
Regulation by pH of Other SLC4 Anion Transporters—Among the seven nonconserved AE2 RL1 residues defined by individual mutation as of regulatory importance, only four are identical in the RL1 of pHi-sensitive AE3: Ala-1053, Ala-1072, Asp-1075, and Pro-1077. Substitution of the latter two in cAE3 with the corresponding AE1 residues sufficed to reduce greatly the pHi sensitivity of AE3 (supplemental Fig. 2). Among the AE2 RL1 residues defined through mutation as functionally important in AE2 regulation by pH, Ala-1053 is conserved in all SLC4 polypeptides except AE1. AE2 residues Ala-1072 and Pro-1077 are identical in all SLC4 polypeptides except AE1 and the putative borate transporter SLC4A11. The residue corresponding to AE2 Asp-1075 (identical in AE3) is conservatively replaced by Glu in all other SLC4 polypeptides.
Correlation of RL1 sequence conservation with pH-sensitive anion transport function of other SLC4 polypeptides is limited by available data. Rat AE3 is sensitively regulated by pHi, but its regulation by pHo is less sensitive than that of AE2.5 Guinea pig cardiomyocyte Cl–/OH– exchange is regulated independently by pHi and by pHo (24), but its molecular identity remains unknown. Studies of acute regulation by pH have not yet been reported for recombinant Na+-dependent SLC4 polypeptides.
Side Chain Scan of Conserved RL1 Residue Ile-1079 of AE2 Reveals Steric and Charge Constraints for AE2 Regulation by pH—In addition to the four nonconserved RL1 residues whose mutation alters AE2 regulation by pH, the AE2 residue Ile-1079, conserved in mammalian AE1 and AE3, also proved important in governing pH sensitivity (Fig. 5). In mouse AE2, Ile-1079 substitution with Asn greatly attenuated pHi sensitivity without effect on regulation by pHo. Ile-1079 substitution with Phe, Tyr, or Thr also greatly reduced sensitivity to pHi. Phe replaces Ile-1079 in the Na+-dependent SLC4 HCO–3 transporters, and Thr does so in the putative borate transporter SLC4A11. Ile-1079 replacement with Lys or with Glu reduced AE2 sensitivity to both pHi and pHo, whereas replacement with Cys increased sensitivity to both pHi and pHo. However, treatment of AE2 I1079C with cysteine modifier MTSES, MTSEA, or MTSET did not alter the enhanced pHi sensitivity of the mutant. This lack of effect contrasts with the accessibility of human AE1 A751 (corresponding to mouse AE2 A1077) to extracellular biotin maleimide (21), further suggesting that RL1 conformation in the human AE1 constructs subjected to cysteine scan studies was indeed altered, as suggested by trypsin susceptibility experiments (20). Thus, the topographical model of Fig. 1B (right) reflecting I1079 accessibility to the extracellular environment may not reflect the conformation of native, nonrecombinant polypeptide.
Alternatively, Cys at position 1079 could be accessible to and reactive with the MTS reagents, but the resultant adducts may not phenocopy substitutions with Lys or Glu for steric reasons. Application of MTS reagents has had limited success in other SLC4 studies. MTSEA and MTSET only slightly inhibited Cl–/ HCO–3 exchange mediated by Cys-less AE1 with single Cys residues reintroduced into RL1 in positions of accessibility to extracellular biotin maleimide (14). Similarly, MTSEA inhibited most NBCe1 constructs with individual Cys substitutions in TM8 by ≤20% (25). Side chain charge, size, and hydrophobicity may each contribute to maintenance of one or more conformation(s) competent to respond to changing pHi and/or pHo with normal regulation of AE2-mediated anion exchange. pHo(50) values of the Ile-1079 substitution series correlated moderately well (supplemental Fig. 4) with interfacial free energies for phosphatidylcholine/water and for octanol/water (26), consistent with the extracellular interfacial location proposed for AE2 I1079 (Fig. 1B, right). In contrast, no correlation with pHi sensitivity was apparent (not shown). Neither pHi sensitivity nor pHo(50) values of the Ile-1079 substitution series correlated with side chain molecular weight, partial molar volume (27), van der Waals radius, solvent-accessible surface area (28), or charge separation index (29). The aa 1079 side chain may either block access to or modify the pK of a nearby titratable residue, as proposed for pH-sensitive K+ channels (30).
Potential Mechanisms by Which RL1 aa Residues Mediate Regulation of AE2 by pH—Mutation of individual AE2 RL1 residues to their AE1 counterparts produced three classes of pH phenotype: altered regulation by pHo only (at Asp-1075 and Ala-1051/Ala-1053), by pHi only (at Pro-1077), and by both pHo and pHi (at Ser-1068, Val-1071, and Ala-1072). Mutation of conserved AE2 residue Ile-1079 in one case selectively altered pHi sensitivity (I1079N), but other substitutions (I1079E, I1079K, and I1079C) altered regulation by both pHo and pHi. Whereas AE2 I1079C exhibited enhanced sensitivity to inhibition by both intra- and extracellular protons, both sensitivities were decreased for all five other mutants of this class. In none of these mutants did the direction of changes in pHi sensitivity and pHo sensitivity diverge. These findings, along with the selective rescue of pHi sensitivity (but not pHo sensitivity) of a pH-insensitive chimeric AE polypeptide by reintroduction of part of the AE2 RL1, demonstrate independent regulation of AE2 by pH change on either side of the membrane. In AE2 mutants, such altered sensitivity to both pHo and pHi might reflect changes in pH dependence of anion binding affinity or of post-translocation dissociation rate at a side chain component of the anion translocation site alternately accessible to both intracellular and extracellular compartments. Similarly concordant changes of pH sensitivity on both sides of the plasma membrane characterize wild-type activities of the acid-extruding Na+/H+ exchanger (probably NHE1) of synaptosomes (31) and Purkinje fiber cardiomyocytes (32).
The pattern of pH sensitivity exhibited by AE1 replacement mutants in the nonconserved positions of the AE2 RL1 might also explain the wide variation noted in their “basal” 36Cl– efflux rates (supplemental Fig. 3). Thus, to achieve a comparable pHo 7.4 “basal” activity level expressed by oocytes previously injected with 10 ng of wild-type AE2 cRNA, only 0.5–2.0 ng of cRNA was required for the three mutants altered in both pHi and pHo sensitivities: S1068G, V1071S, and A1072G. Thus, the variation in “basal” transport rates among these RL1 mutants reflects in large part the presence or absence of the normal inhibition exerted by protons on both sides of the membrane. Inhibition by protons may be exerted through Cl– binding affinity (more likely for Cl–i than Cl–o) or through elevation of the free energy barrier for the conformational change associated with anion translocation.
Interaction between RL1 and TM1–6 of AE2 in Regulation by pH—It is unlikely that Ile-1079 is directly protonated or acting as a pH sensor. Ile-1079 more likely controls proton access to TMD protonation sites and/or regulates conformation of and possibly interacts directly with a specific nonconserved AE2 residue to mediate regulation by pH. This site may be Asp-1075, shown to be important in regulation by pHo, or another RL1 residue or among the additional components of the TMD proton sensing apparatus located within TM1–6 (Figs. 8 and 9). Although Lys-1069 within the second RL1 subdomain, 1068SKAVA1072, could be titratable, substitution of Lys-1069 with the corresponding AE3 residue Thr does not alter regulation by pH (12). This subdomain might hydrogen-bond with protonatable residues of the N-terminal portion of the AE2 TMD.
Thus, the minimum required complement of AE2 RL1 residues found necessary to rescue pHi sensitivity of the pHi-insensitive AE chimera, AE2(1–922)/AE1(623–929), included conserved AE2 Ile-1079 and nonconserved AE2 residues 1075DKPK1078 and AE2 1071VA1072 (Fig. 8B). In contrast, regulation of AE2(1–922)/AE1(623–929) by pHo was not restored by replacing most of the AE1 RL1 with AE2 RL1 (Fig. 8D). Note that the pHi -rescued chimera AE2(1–922)/AE1(623–929)/AE2(1068S-VA-DKPK1078) lacked the two nonconserved AE2 residues Ale-1051 and Ala-1053 necessary for normal regulation by pHo but not required for pHi sensitivity (Fig. 4).
The observation of Ref. 33 that deletion of TM1–3 from the human AE1 TMD expressed in COS cells enhances N-glycosylation of appropriately engineered RL1 suggests an immature RL1 conformation in the absence of TM1–3. Also consistent with the postulated interaction between RL1 and TM1–6 are several studies with coexpression of split in vitro translated AE1 constructs. Thus, human AE1 construct TM9-Cterm (including RL1) accumulated in Xenopus oocytes only in the presence of co-expressed TM1–8 (34). Human AE1 fragment TM1–6 was co-immunoprecipitated with AE1 fragments TM8–14 or fragment 9–14, but TM1–4 interacted only weakly with the distal domains (35). Moreover, coexpression in Xenopus oocytes of AE1 fragments TM1–5 or TM1–7 with their distal complements reconstituted Cl– transport function, whereas TM1–3 and its coexpressed complement were nonfunctional (36). These studies suggest a dominant role for the C-terminal half of AE2 TM1–6 in its regulatory interaction with the RL1 of AE2.
Conclusion—We have identified a requirement for the putative RL1 of AE2 in normal acute regulation of AE2-mediated Cl–/anion exchange by pHi and pHo. We have further identified within the AE2 RL1 several nonconserved and one conserved residue playing critical roles in that regulation and shown that RL1 harbors all known individual missense mutations of the AE2 TMD that alter both regulation by pHo and by pHi. RL1 and the first six transmembrane spans of the AE2 TMD together are sufficient to confer wild-type regulation by pHi, but regulation by pHo requires the presence of other AE2 regions. These results strengthen the evidence favoring independent sensors of pHi and pHo in AE2. The structure of these sensors is likely to be complex. Further details on their mechanism and structure will require additional investigation of intramolecular contacts and, ultimately, electron micrographic and crystal structure of the AE2 TMD.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DK43495 (to S. L. A.) and RR017927 (Shared Instrument Grant) to Beth Israel Deaconess Medical Center. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4.
Footnotes
The abbreviations used are: pHi, intracellular pH; pHo, extracellular pH; TMD, transmembrane domain; aa, amino acid(s); MES, 4-morpholineethanesulfonic acid; MTSEA, (2-aminoethyl) methanethiosulfonate; MTSET, (2-(trimethylammonium)-ethyl) methanethiosulfonate; MTSES, (2-(trimethylammonium)-ethyl) methanethiosulfonate; PBS, phosphate-buffered saline; BSA, bovine serum albumin; HA, hemagglutinin; GFP, green fluorescent protein; FI, fluorescence intensity; DIDS, 4,4′-di-isothiocyanatostilbene-2,2′ disulfonic acid; WT, wild type; MTS, methanethiosulfonate.
An alternative method normalized each rate constant to that individual oocyte's rate constant measured at pHo 8.5, which was given a value of 100%. The normalized rate constants at every pHo value were fit with Equation 1. Data calculated by this approach yielded pHo(50) values not significantly different in most cases from those presented. However, application of this alternative method to Equation 1 yielded poor fits (r2 as low as 0.6) for some AE2 mutants.
A. K. Stewart, P. Papageorgiou, and S. L. Alper, unpublished observations.
References
- 1.Gawenis, L. R., Ledoussal, C., Judd, L. M., Prasad, V., Alper, S. L., Stuart-Tilley, A., Woo, A. L., Grisham, C., Sanford, L. P., Doetschman, T., Miller, M. L., and Shull, G. E. (2004) J. Biol. Chem. 279 30531–30539 [DOI] [PubMed] [Google Scholar]
- 2.Stewart, A. K., Kurschat, C. E., and Alper, S. L. (2007) The Kidney (Alpern, R. I., and Hebert, S. C., eds) Vol. 2, 4th Ed., pp. 1499–1537, Elsevier Academic Press, New York [Google Scholar]
- 3.Stewart, A. K., Chernova, M. N., Shmukler, B. E., Wilhelm, S., and Alper, S. L. (2002) J. Gen. Physiol. 120 707–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart, A. K., Kurschat, C. E., Vaughan-Jones, R. D., Shmukler, B. E., and Alper, S. L. (2007) J. Physiol. (Lond.) 584 59–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chernova, M. N., Stewart, A. K., Jiang, L., Friedman, D. J., Kunes, Y. Z., and Alper, S. L. (2003) Am. J. Physiol. 284 C1235–C1246 [DOI] [PubMed] [Google Scholar]
- 6.Stewart, A. K., Kerr, N., Chernova, M. N., Alper, S. L., and Vaughan-Jones, R. D. (2004) J. Biol. Chem. 279 52664–52676 [DOI] [PubMed] [Google Scholar]
- 7.Grinstein, S., Ship, S., and Rothstein, A. (1978) Biochim. Biophys. Acta 507 294–304 [DOI] [PubMed] [Google Scholar]
- 8.Kopito, R. R., Lee, B. S., Simmons, D. M., Lindsey, A. E., Morgans, C. W., and Schneider, K. (1989) Cell 59 927–937 [DOI] [PubMed] [Google Scholar]
- 9.Zhang, Y., Chernova, M. N., Stuart-Tilley, A. K., Jiang, L., and Alper, S. L. (1996) J. Biol. Chem. 271 5741–5749 [DOI] [PubMed] [Google Scholar]
- 10.Stewart, A. K., Chernova, M. N., Kunes, Y. Z., and Alper, S. L. (2001) Am. J. Physiol. 281 C1344–C1354 [DOI] [PubMed] [Google Scholar]
- 11.Stewart, A. K., Kurschat, C. E., Burns, D., Banger, N., Vaughan-Jones, R. D., and Alper, S. L. (2007) Am. J. Physiol. 292 C909–C918 [DOI] [PubMed] [Google Scholar]
- 12.Stewart, A. K., Kurschat, C. E., and Alper, S. L. (2007) Pflugers Arch. 454 373–384 [DOI] [PubMed] [Google Scholar]
- 13.Zhu, Q., Lee, D. W., and Casey, J. R. (2003) J. Biol. Chem. 278 3112–3120 [DOI] [PubMed] [Google Scholar]
- 14.Zhu, Q., and Casey, J. R. (2004) J. Biol. Chem. 279 23565–23573 [DOI] [PubMed] [Google Scholar]
- 15.Popov, M., Li, J., and Reithmeier, R. A. (1999) Biochem. J. 339 269–279 [PMC free article] [PubMed] [Google Scholar]
- 16.Brosius, F. C., III, Alper, S. L., Garcia, A. M., and Lodish, H. F. (1989) J. Biol. Chem. 264 7784–7787 [PubMed] [Google Scholar]
- 17.Linn, S. C., Kudrycki, K. E., and Shull, G. E. (1992) J. Biol. Chem. 267 7927–7935 [PubMed] [Google Scholar]
- 18.Kurschat, C. E., Shmukler, B. E., Jiang, L., Wilhelm, S., Kim, E. H., Chernova, M. N., Kinne, R. K., Stewart, A. K., and Alper, S. L. (2006) J. Biol. Chem. 281 1885–1896 [DOI] [PubMed] [Google Scholar]
- 19.Wood, P. (1992) Prog. Cell Res. 2 325–352 [Google Scholar]
- 20.Kuma, H., Shinde, A. A., Howren, T. R., and Jennings, M. L. (2002) Biochemistry 41 3380–3388 [DOI] [PubMed] [Google Scholar]
- 21.Fujinaga, J., Tang, X. B., and Casey, J. R. (1999) J. Biol. Chem. 274 6626–6633 [DOI] [PubMed] [Google Scholar]
- 22.Tang, X. B., Kovacs, M., Sterling, D., and Casey, J. R. (1999) J. Biol. Chem. 274 3557–3564 [DOI] [PubMed] [Google Scholar]
- 23.Tang, X. B., Fujinaga, J., Kopito, R., and Casey, J. R. (1998) J. Biol. Chem. 273 22545–22553 [DOI] [PubMed] [Google Scholar]
- 24.Niederer, S. A., Swietach, P., Wilson, D. A., Smith, N. P., and Vaughan-Jones, R. D. (2007) Biophys. J. 94 2385–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McAlear, S. D., and Bevensee, M. O. (2006) J. Biol. Chem. 281 32417–32427 [DOI] [PubMed] [Google Scholar]
- 26.White, S. H., and Wimley, W. C. (1999) Annu. Rev. Biophys. Biomol. Struct. 28 319–365 [DOI] [PubMed] [Google Scholar]
- 27.Hackel, M., Hinz, H. J., and Hedwig, G. R. (1999) Biophys. Chem. 82 35–50 [DOI] [PubMed] [Google Scholar]
- 28.Miller, S., Janin, J., Lesk, A. M., and Chothia, C. (1987) J. Mol. Biol. 196 641–656 [DOI] [PubMed] [Google Scholar]
- 29.Matta, C. F., and Bader, R. F. (2003) Proteins 52 360–399 [DOI] [PubMed] [Google Scholar]
- 30.Sackin, H., Nanazashvili, M., Palmer, L. G., and Li, H. (2006) Biophys. J. 90 3582–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jean, T., Frelin, C., Vigne, P., Barbry, P., and Lazdunski, M. (1985) J. Biol. Chem. 260 9678–9684 [PubMed] [Google Scholar]
- 32.Vaughan-Jones, R. D., and Wu, M. L. (1990) J. Physiol. (Lond.) 428 441–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanki, T., Sakaguchi, M., Kitamura, A., Sato, T., Mihara, K., and Hamasaki, N. (2002) Biochemistry 41 13973–13981 [DOI] [PubMed] [Google Scholar]
- 34.Groves, J. D., Wang, L., and Tanner, M. J. (1998) Biochem. J. 332 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groves, J. D., and Tanner, M. J. (1999) Biochem. J. 344 687–697 [PMC free article] [PubMed] [Google Scholar]
- 36.Wang, L., Groves, J. D., Mawby, W. J., and Tanner, M. J. (1997) J. Biol. Chem. 272 10631–10638 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.