

Abstract
The obesity epidemic has generated interest in determining the contribution of various pathways to triglyceride synthesis, including an elucidation of the origin of triglyceride fatty acids and triglyceride glycerol. We hypothesized that a dietary intervention would demonstrate the importance of using glucose versus non-glucose carbon sources to synthesize triglycerides in white adipose tissue. C57BL/6J mice were fed either a low fat, high carbohydrate (HC) diet or a high fat, carbohydrate-free (CF) diet and maintained on 2H2O (to determine total triglyceride dynamics) or infused with [6,6-2H]glucose (to quantify the contribution of glucose to triglyceride glycerol). The 2H2O labeling data demonstrate that although de novo lipogenesis contributed ∼80% versus ∼5% to the pool of triglyceride palmitate in HC- versus CF-fed mice, the epididymal adipose tissue synthesized ∼1.5-fold more triglyceride in CF- versus HC-fed mice, i.e. 37 ± 5 versus 25 ± 3 μmol × day–1. The [6,6-2H]glucose labeling data demonstrate that ∼69 and ∼28% of triglyceride glycerol is synthesized from glucose in HC- versus CF-fed mice, respectively. Although these data are consistent with the notion that non-glucose carbon sources (e.g. glyceroneogenesis) can make substantial contributions to the synthesis of triglyceride glycerol (i.e. the absolute synthesis of triglyceride glycerol from non-glucose substrates increased from ∼8 to ∼26 μmol × day–1 in HC- versus CF-fed mice), these observations suggest (i) the importance of nutritional status in affecting flux rates and (ii) the operation of a glycerol-glucose cycle.
Epidemiological trends in the development of obesity (1–4) and its association with various diseases have generated interest in the study of lipid synthesis and mobilization. In particular, new methods have been developed for quantifying rates of triglyceride turnover in vivo (5–8). For example, using 2H2O, we have demonstrated substantial rates of triglyceride degradation in white adipose tissue during periods of rapid lipid accretion (i.e. growth) and that de novo lipogenesis can make a major contribution to the pool of fatty acids used in the generation of new triglycerides (e.g. >90% of the newly deposited palmitate can be derived from de novo lipogenesis) (6). In addition, we observed a vigorous remodeling of the fatty acid distribution profile (6). Using 2H2O, Hellerstein and colleagues (7, 8) have demonstrated that rates of triglyceride turnover in adipose tissue are depot-specific and sensitive to pharmacological therapy.
Recent investigations have tested hypotheses regarding the contribution of different pathways to the production of α-glycerol-3-phosphate that is used to synthesize triglycerides (7, 9–11). For example, α-glycerol-3-phosphate can be derived from glucose, glycerol, and pyruvate. Although it is clear that the liver can utilize all of these pathways depending on the nutritional/hormonal status, there is an uncertainty regarding the contribution of the various pathways to the formation of α-glycerol-3-phosphate in white adipose tissue in vivo. Although many investigators agree that glucose plays an important role in triglyceride synthesis in adipose tissue and that glycerol makes a minor (if any) contribution to α-glycerol-3-phosphate (because there is low glycerol kinase activity), the potential for converting pyruvate to α-glycerol-3-phosphate (referred to as glyceroneogenesis) is intriguing (7, 9, 12). For example, investigators have suggested that there should be a glyceroneogenic flux to facilitate fatty acid (re)esterification (13), and that hypothesis is supported by experiments in which ∼90% of triglyceride glycerol was apparently derived from glyceroneogenesis (14).
Studies have used various designs to demonstrate the importance of glucose versus glyceroneogenesis to the formation of α-glycerol-3-phosphate (7, 9, 12). For example, Chen et al. (7) have suggested that following the administration of 2H2O, it is possible to quantify the contribution of glucose versus glyceroneogenesis to triglyceride glycerol in vivo; this requires that one measures the proportion of triglyceride glycerol containing one and two 2H and then applies a combinatorial method to determine the number of exchangeable hydrogens (i.e. the “n”). This logic assumes that the production of α-glycerol-3-phosphate from glucose results in an n of 3.5, whereas the production ofα-glycerol-3-phosphate from lactate, pyruvate, etc. results in an n of 5 (7). Investigators have also used a double-labeling approach to sort out the contribution of specific pathways, i.e. measuring total triglyceride synthesis via labeled water and dissecting the contribution of specific pathways via [14C]glucose (14, 15).
We have examined the contribution of different pathways to the synthesis of triglyceride fatty acids and triglyceride glycerol. Mice were fed a high carbohydrate or a carbohydrate-free diet to determine the maximum/minimum potentials of glucose versus non-glucose-derived sources to triglyceride dynamics, 2H2O was used to quantify the rates of de novo lipogenesis and the total rates of triglyceride synthesis and degradation, and [6,6-2H]glucose was used to measure the contribution of glucose to triglyceride glycerol.
EXPERIMENTAL PROCEDURES
Chemicals and Supplies
Unless specified, all chemicals and reagents were purchased from Sigma-Aldrich. 2H2O (99.9 atom % excess) and [6,6-2H]glucose (98 atom % excess) were purchased from Isotec (Miamisburg, OH). Ion exchange resins were purchased from Bio-Rad. Gas chromatography-mass spectrometry (GC-MS)4 supplies were purchased from Agilent Technologies (Wilmington, DE) and Alltech (Deerfield, IL). Enzymes were purchased from Roche Applied Science. Diets were purchased from Research Diets (New Brunswick, NJ). Mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Osmotic pumps (Alzet 2001D) were purchased from Durect Corp. (Cupertino, CA).
Biological Experiments
Chronic Labeling Study—On arrival, male C57BL/6J mice (5 weeks old, n = 32) were fed a high carbohydrate (HC), low fat diet (D12450B, kcal distribution equal to 10% fat, 70% carbohydrate, and 20% protein). After 5 days, mice were randomized into two groups and either maintained on the HC diet or fed a high fat, carbohydrate-free (CF) diet (D12369B, 90% fat, 0% carbohydrate, and 10% protein). The diet intervention proceeded for 10 days; all mice were then given an intraperitoneal injection of labeled water (20 μl/g of body weight of 9 g of NaCl in 1,000 ml 99% 2H2O). In all cases, to ensure the precision of the injection, mice were sedated by a brief exposure (∼15 s) to isoflurane (Baxter Pharmaceuticals, Deerfield, IL); mice regained consciousness within a minute with no apparent adverse side effects. After injection, mice were returned to their cages (n = four mice per cage) and maintained on 5% 2H-labeled drinking water for the remainder of the study; this design maintained a steady-state 2H labeling of body water at ∼2.75% (6). Mice in each group were fed the respective diets ad libitum, and daily food consumption was measured. Mice were sedated on various days after injection (n = 4 per day per group), and blood and tissue samples were then collected and quick-frozen in liquid nitrogen. Samples were stored at –80 °C until analyses.
Acute Labeling Study—On arrival, male C57BL/6J mice (5 weeks old, n = 30) were fed an HC, low fat diet (D12450B, kcal distribution equal to 10% fat, 70% carbohydrate and 20% protein). After 5 days, mice were randomized into two groups and either maintained on the HC diet or fed a high fat, CF diet (D12369B, 90% fat, 0% carbohydrate, and 10% protein). The diet intervention proceeded for 14 days, and seven mice in each diet group were then randomized to receive an intraperitoneal injection of labeled water (20 μl/g of body weight of 9 g of NaCl in 1,000 ml of 99% 2H2O) under isoflurane anesthesia during which a sham incision was made to the skin. Mice were returned to their cages (n = 4 mice per cage) and maintained on unlabeled drinking water for 24 h, and blood was sampled 2 h after 2H2O injection and at the time of tissue collection. The remaining eight mice in each diet were anesthetized under isoflurane and implanted with Alzet 2001D osmotic pumps loaded with 200 μl of [6,6-2H]glucose (500 mg/ml), which had been primed for 4 h at 37 °C in saline (16). After 24 h, blood and epididymal fat samples were collected from all mice.
Analytical Procedures
2H Labeling of Body Water—The 2H labeling of body water was determined by exchange with acetone (17). Briefly, samples were centrifuged for 1 min at ∼12,000 × g in a microcentrifuge, and 20 μl of sample (or standard) was reacted with 2 μl of 10 n NaOH and 4 μl of a 5% (v/v) solution of acetone in acetonitrile for 24 h at room temperature. Acetone was then extracted by the addition of 600 μl of chloroform followed by the addition of ∼0.5 g of Na2SO4. Samples were vigorously mixed, and a small aliquot of the chloroform was transferred to a GC-MS vial. Acetone was analyzed using an Agilent 5973N-MSD equipped with an Agilent 6890 GC system, and a DB-17MS capillary column (30 m × 0.25 mm × 0.25 μm) was used in all analyses. The temperature program was as follows: 60 °C initial, increase by 20 °C per min to 100 °C, increase by 50 °C per min to 220 °C, and hold for 1 min. The sample was injected at a split ratio of 40:1 with a helium flow of 1 ml/min. Acetone eluted at ∼1.5 min. The mass spectrometer was operated in the electron impact mode (70 eV). Selective ion monitoring of m/z 58 and 59 was performed using a dwell time of 10 ms/ion.
2H Labeling of Triglyceride-bound Glycerol—Total glycerides were extracted from frozen epididymal fat pads by first hydrolyzing both fat pads in 1 n KOH ethanol at 70 °C (6). After 3 h, the hydrolysate was evaporated to dryness, and the dry residue was redissolved in 3 ml of H2O and acidified to ∼pH 1.0 by adding 6 n HCl. Free fatty acids were extracted using diethyl ether (three times with 4 ml). The pH of the aqueous solution was then adjusted to ∼7.0 (using 10 n NaOH). Free glycerol was isolated by passing the sample over a layered ion-exchange bed (AG 50W-X8, hydrogen form and AG 1-X8, formate form) and washing with 20 ml of water, and the eluent containing glycerol was evaporated to dryness. The glycerol labeling was determined following conversion to its triacetate derivative by reacting the dry residue with 100 μl of pyridine:acetic anhydride (1:1, v/v) at 75 °C for 30 min. The 2H labeling of triacetyl glycerol was determined using an Agilent 5973N-MSD equipped with an Agilent 6890 GC system. A DB-17MS capillary column (30 m × 0.25 mm × 0.25 μm) was used in all analyses. The temperature program was 120 °C initial, increased by 10 °C per min to 200 °C, and increased by 50 °C per min to 240 °C, with a 2-min hold time. The split ratio was 40:1 with helium flow of 1 ml/min, and triacetyl glycerol eluted at ∼8.0 min. Ammonia chemical ionization was used, and selective ion monitoring of NH +4 adducts was performed, m/z 236, 237, and 238 (M+0, M+1 and M+2), with a 10-ms dwell time per ion.
Concentration of Triglyceride-bound Glycerol—The concentration of triglyceride-bound glycerol was determined by enzymatic spectrophotometric assay. Briefly, epididymal fat pads were hydrolyzed, and free glycerol was obtained as described above. Glycerol was then redissolved in a known amount of Tris-EDTA buffer (pH 7.4), and its concentration was determined using an enzymatic assay (free glycerol reagent, Sigma).
2H Labeling and Concentration Profile(s) of Triglyceride-bound Fatty Acids—A known quantity of tissue was hydrolyzed and extracted after adding a known amount of heptadecanoic acid. Fatty acids were analyzed as their trimethylsilyl derivatives using gas chromatography-electron impact ionization mass spectrometry. The 2H enrichment of palmitate was determined by using selective ion monitoring of m/z 313–317 (M+0to M+4), with a 10-ms dwell time per ion (6). The concentration of palmitate (16:0), stearate (18:0), oleate (18:1), and linoleate (18:2) was determined by comparing the corrected abundance of m/z 313–317, 341–345, 339, and 337, respectively, with that of heptadecanoate (17:0, m/z 327). To account for possible differences in the ionization efficiency of each fatty acid, the profile was compared against standards prepared by mixing known quantities of each fatty acid (6).
2H Labeling and Concentration of Blood Glucose—A known amount of [U-13C6]glucose (internal standard) was added to a known amount of blood and then deproteinized by the addition of methanol (10 μl internal standard + 10 μl blood + 200 μl of methanol). The supernatant was evaporated to dryness and reacted with 100 μl of pyridine:acetic anhydride (1:1) at 70 °C for 30 min. The 2H labeling of glucose pentaacetate was determined using an Agilent 5973N-MSD equipped with an Agilent 6890 GC system. A DB-17MS capillary column (30 m × 0.25 mm × 0.25 μm) was used in all analyses. The temperature program was 150 °C initial, increased by 10 °C per min to 250 °C, increased by 50 °C per min to 300 °C, with a 2-min hold time. The split ratio was 40:1 with helium flow of 1 ml/min with glucose pentaacetate eluted at ∼7.5 min. Ammonia chemical ionization was used, and selective ion monitoring of NH +4 adducts was performed, m/z 408–414 (M+0 to M+6), with a 10-ms dwell time per ion.
Calculations
We determined the rates of triglyceride synthesis (μmol × day–1) as described previously (accounting for changes in both the 2H labeling and pool size) (6). In this study, we have accounted for the fact that mice were continuously maintained on 2H2O as compared with previous studies where non-steady-state 2H labeling was used. Triglyceride dynamics were determined using the equation
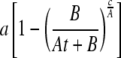 |
(Eq. 1) |
where a is the asymptotic value of the 2H labeling of triglyceride glycerol, B is the pool size of triglyceride on day 0 (μmol), At + B is the rate of increase of adipose tissue triglyceride (assumed to be linear), and c is the rate of triglyceride synthesis (μmol × day–1). Once the rate of synthesis was determined, the rate of degradation was calculated from the net change in triglyceride pool size (which equals synthesis minus degradation). Note that our approach to modeling the reaction rates is conceptually similar to the approach used by Hellerstein and colleagues (8), although the actual flux rates are numerically different (∼10% lower using our approach) due to the manner in which the problem has been defined, i.e. we account for the change in pool size over time (assumed to be linear).
In mice given 2H2O for 3–28 days (chronic labeling study), the percentage of contribution of de novo lipogenesis to the pool of triglyceride palmitate was calculated using the equation
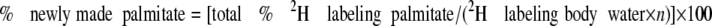 |
(Eq. 2) |
where n is the number of exchangeable hydrogens, assumed to equal 22 (18–20). The percentage of contribution of glyceroneogenesis to triglyceride glycerol was calculated as described by Chen et al. (7) using the equation
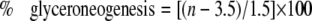 |
(Eq. 3) |
where n is the number of exchangeable carbon-bound hydrogens, determined using the equation
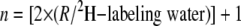 |
(Eq. 4) |
where R is the ratio of excess M2/M1 labeling of triglyceride glycerol (18).
In mice given 2H2O for 24 h (acute labeling study), the “% total newly made triglyceride glycerol” was calculated using the equation
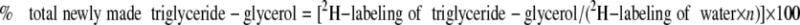 |
(Eq. 5) |
where 2H labeling of triglyceride glycerol is the M1 isotopomer, the 2H labeling of water is the average labeling in a given mouse, and n is the exchange factor (experimentally determined from the M2/M1 ratio of triglyceride glycerol in the chronic labeling study). In mice given [6,6-2H]glucose, the “% newly made triglyceride glycerol from glucose” was calculated using the equation
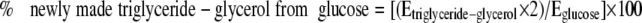 |
(Eq. 6) |
where Etriglyceride glycerol is the M2 labeling of triglyceride glycerol and Eglucose is the M2 labeling of plasma glucose, and the factor of 2 accounts for the dilution of [6,6-2H]glucose during its conversion to triose phosphates. Thus, the contribution of glucose to the total newly made triglyceride glycerol is determined in the acute study by dividing Equation 6 by Equation 5, and the contribution of non-glucose carbon sources is (1 – [Equation 6/Equation 5]). Because we measured the labeling of [6,6-2H]glucose at two points in time, we expect that our estimates of triglyceride synthesis from glucose represent a minimum flux rate, i.e. consumption of food (which occurred primarily during the dark cycle) will transiently dilute the labeling of blood glucose in HC mice. Last, the rate of glucose appearance (Ra) was calculated using the equation
 |
(Eq. 7) |
where I is the infusion rate of [6,6-2H]glucose and Einfusate and Eplasma are the enrichments of the infusate and plasma, respectively (21). The concentration of glucose was determined from the ratio of (M0+M2)/(M6) glucose.
Unless noted, the total 2H labeling of a molecular species is reported, i.e. M1 + 2·M2, etc. where M1, M2, etc. refer to the percentage of excess product molecules with 1, 2, etc. 2H atoms, respectively (18). Data are reported as mean ± S.E. Statistical analyses were done using a two-tailed t test and assuming equal variance.
RESULTS
Fig. 1 demonstrates a net increase in triglyceride-bound fatty acids with time and is consistent with our earlier study (6) where we observed a disproportionate increase in the amount of unsaturated versus saturated fatty acids. Also, the fatty acid profile of the tissue did not reflect that of the respective diets. Consistent with an increase in total fatty acids, we observed an increase in triglyceride glycerol between 3 and 28 days, i.e. in HC-fed mice epididymal glycerol increased from 326 ± 36 to 393 ± 49 μmol (p < 0.01) and in CF-fed mice from 329 ± 27 to 512 ± 66 μmol (p < 0.01).
FIGURE 1.

Distribution of triglyceride-bound fatty acids. Total lipids were extracted from epididymal fat pads, and the absolute quantity of various fatty acids was determined following hydrolysis (A, high carbohydrate diet; B, carbohydrate-free diet). Data were compared on day 3 and day 28 after administration of 2H2O, n = 4 per day per group, *, p < 0.05 within a group.
Fig. 2 demonstrates the 2H labeling of triglyceride-bound palmitate in mice chronically exposed to 2H2O. As expected, we observed a continuous increase in the proportion of heavily labeled palmitate over the course of the study in mice fed the HC diet (panel A). However, in mice fed the CF diet, very little of the palmitate was derived from de novo lipogenesis (panel B), i.e. 5 ± 2% of the palmitate is newly made in mice fed the CF diet versus 80 ± 4% newly made palmitate in mice fed the HC diet.
FIGURE 2.

2H labeling of triglyceride palmitate. A demonstrates the incorporation of 2H-labeled palmitate in mice fed a high carbohydrate diet, n = four mice per day. B compares the 2H labeling profile of palmitate from mice fed a high carbohydrate versus a carbohydrate-free diet for 28 days, n = 4 mice per diet group.
Fig. 3 demonstrates the incorporation of 2H over 28 days into triglyceride glycerol in epididymal fat pads of mice fed the HC versus the CF diet. The fractional synthetic rates were determined by fitting the increase in 2H labeling to a single exponential, i.e. 7.7 ± 0.2% versus 11.8 ± 1.2% newly made triglyceride per day in HC- versus CF-fed mice, respectively. The calculated rates of triglyceride synthesis were 25 ± 3 and 37 ± 5 μmol made per day, and the calculated rates of degradation were 22 ± 3 and 30 ± 5 μmol per day in HC- and CF-fed mice, respectively.
FIGURE 3.

2H labeling of triglyceride glycerol. The total 2H labeling of triglyceride glycerol was determined (n = 4 per day per group), and the fractional rates of turnover were calculated by fitting the respective data to single exponential curves, i.e. 7.7 ± 0.2% versus 11.8 ± 1.2% newly made triglyceride per day (mean ± S.E.) in HC- versus CF-fed mice, respectively. The inset compares the absolute rates of triglyceride synthesis and breakdown, determined as described under “Experimental Procedures.”
We used two approaches to dissect the contribution of glucose versus non-glucose carbon sources to triglyceride glycerol. First, in the “chronic labeling study,” we measured the labeling pattern of triglyceride glycerol and applied the equations proposed by Chen et al. (7); we determined that glucose contributes 48 ± 8% versus 10 ± 7% of the triglyceride glycerol in mice fed the HC (n = 4.28 ± 0.11) versus the CF (n = 4.85 ± 0.12) diet, respectively. Second, in the “acute labeling study,” we compared the M2 labeling of triglyceride glycerol with that of plasma glucose and then compared (i.e. normalized) those data against the respective groups that were given labeled water; we determined that glucose contributes 69 ± 4 and 28 ± 3% of the triglyceride glycerol in mice fed the HC and in mice fed the CF, respectively (Table 1). In regard to the source(s) of triglyceride glycerol, in both diet groups, the “acute labeling data” (double tracer approach) are different from the “chronic labeling data” (mass isotopomer distribution approach) (p < 0.01).
TABLE 1.
2H labeling in mice undergoing the “acute double-tracer protocol”
Mice in each diet group were randomized and either given a bolus of 2H2O or continuously infused with [6,6-2H]glucose from 0 to 24 h. The “theoretical maximum” plasma glucose labeling was assumed to be “water labeling × 7”. To determine the “% newly made triglyceride-glycerol” the triglyceride-glycerol labeling was divided by 4.28 and 4.85 in mice fed the high carbohydrate and carbohydrate-free diets, respectively. Those factors account for the number of exchangeable hydrogens that are incorporated into triglyceride-glycerol and are derived from the mass isotopomer distribution analyses of the M2/M1 labeling ratios from the “chronic labeling study” (the theoretical maximum is 5). The “contribution of glucose to triglyceride-glycerol synthesis” was determined by dividing the individual values obtained from mice given [6,6-2H]glucose by the mean value obtained from mice given 2H2O for a given diet group. Data are presented as mean ± S.E. (n = 6 to 8 per group), “mpe” represents mole percent excess. ND, not detected.
|
High carbohydrate diet
|
Carbohydrate-free diet
|
|||
|---|---|---|---|---|
| 2 h | 24 h | 2 h | 24 h | |
| 2H2O group | ||||
| Water labeling (mpe) | 2.73 ± 0.08 | 2.22 ± 0.05 | 2.85 ± 0.05 | 2.47 ± 0.09 |
| Plasma glucose labeling (M1, mpe) | 7.57 ± 0.53 | 8.98 ± 0.94 | 15.36 ± 0.82 | 13.99 ± 0.74 |
| Plasma glucose labeling (% of theoretical maximum) | 40 ± 3 | 58 ± 6 | 87 ± 5 | 92 ± 4 |
| Triglyceride-glycerol labeling (M1, mpe) | ND | 1.01 ± 0.09 | ND | 1.82 ± 0.11 |
| % of newly made triglyceride-glycerol (total) | ND | 9.6 ± 0.9 | ND | 16.9 ± 1.4 |
| [6,6-2H]glucose group | ||||
| Plasma glucose labeling (M2, mpe) | 8.29 ± 0.97 | 9.39 ± 0.85 | 17.41 ± 1.03 | 19.08 ± 1.42 |
| Triglyceride-glycerol labeling (M2, mpe) | ND | 0.32 ± 0.04 | ND | 0.46 ± 0.04 |
| % of newly made triglyceride-glycerol (from glucose) | ND | 6.8 ± 0.4 | ND | 5.1 ± 0.6 |
| Relative triglyceride-glycerol flux | ||||
| Contribution of glucose (%) | ND | 69 ± 4 | ND | 28 ± 3 |
| Contribution of non-glucose carbon sources (%) | ND | 31 ± 4 | ND | 72 ± 3 |
DISCUSSION
We aimed to determine the contribution of glucose versus non-glucose carbon sources to triglyceride synthesis in vivo. For example, although glucose is generally regarded as a good substrate for de novo lipogenesis, less is known about the source(s) of triglyceride glycerol. The ability of adipose tissue to generate triglyceride glycerol from non-glucose substrates was demonstrated by several groups nearly 40 years ago (22–24), which Hanson and colleagues (24) defined as glyceroneogenesis. The study by Hanson's group (24) is particularly elegant in that attention was directed toward examining the synthesis of triglyceride glycerol in the presence/absence of different substrates and hormones. For example, although pyruvate was incorporated into triglyceride glycerol, the addition of glucose and insulin led to a ∼5-fold reduction in the conversion of pyruvate to triglyceride glycerol, suggesting a dynamic control of triglyceride glycerol production (24). Since its discovery, the glyceroneogenic pathway was mostly overlooked; however, recent interest in the role of peroxisome proliferator-activated receptor-γ agonists has demonstrated that glyceroneogenic flux is sensitive to pharmacological manipulation (10, 12, 25, 26). Although most investigators probably agree that modulation of plasma free fatty acid flux (via esterification in white adipose tissue) offers therapeutic benefits, it is difficult to determine whether manipulation of glyceroneogenesis is solely responsible and/or whether altered glucose flux in adipose tissue plays a role. Namely, contrary to the seminal work of Hanson and colleagues (24), recent in vitro studies typically quantify pyruvate flux to triglyceride glycerol in the absence of glucose (10, 12, 25, 26).
We became intrigued by the notion that glyceroneogenesis could play an important, if not predominant, role in affecting triglyceride glycerol production in white adipose tissue in vivo (7, 9–12); however, the experimental methods used in those studies deserve consideration because artifacts in the tracer techniques can affect the interpretation of the data (discussed below). We reasoned that a somewhat extreme dietary manipulation would highlight the potential contribution of different pathways to triglyceride synthesis; therefore, C57BL/6J mice were fed either an HC or a CF diet (15), and rates of flux were estimated using stable isotope tracer methods.
Consistent with our previous investigation (6), we found a net increase in epididymal fat and a substantial remodeling of triglyceride-bound fatty acids (Fig. 1). The latter was independent of dietary fat composition and further supports observations regarding bias when using one labeled fatty acid to trace the fate of other fatty acids (27, 28). The 2H labeling of triglyceride palmitate was consistent with the generally accepted hypothesis that dietary carbohydrate provides an excellent substrate for de novo lipogenesis (Fig. 2). These data agree with our earlier report (6) and with the pioneering work of Schoenheimer and Rittenberg (29), i.e. because mice are capable of storing a limited amount of glycogen, de novo lipogenesis constitutes an important pathway for temporarily storing energy. Note that it is not possible to draw conclusions regarding the site(s) of de novo lipogenesis using our experimental design.
Turning our attention toward triglyceride glycerol dynamics in adipose tissue, we observed reasonable rates of triglyceride turnover in mice fed the HC diet as compared with data in our previous studies (5, 6). However, we were surprised by the substantial rates of triglyceride turnover in mice fed the CF diet. They synthesized ∼1.5 times as much triglyceride as compared with mice fed the HC diet (Fig. 3); note that the caloric intake was comparable in the different groups. To determine the source(s) of the triglyceride glycerol, we initially applied the logic developed by Chen et al. (7). Those calculations suggested that in mice fed the HC diet, ∼48% of the triglyceride glycerol came from glucose, whereas in mice fed the CF diet, ∼10% of the triglyceride glycerol came from glucose. Although our data seemed reasonable considering the macronutrient intake, the approach outlined by Chen et al. (7) assumes that glucose enters the adipocyte unlabeled. Contrary to their data (7), we observed substantial and rapid labeling of glucose (Fig. 4 and Table 1); thus, the method proposed by Chen et al. is not valid in our experimental setting.
FIGURE 4.

2H labeling of water, triglyceride glycerol and blood glucose. The total 2H labeling of water was determined and expressed as a multiple of the maximum possible n for each end product, i.e. 5 and 7 for triglyceride glycerol and blood glucose, respectively. Data are shown for samples collected on day 3 following the administration of 2H2O. Note: A steady-state 2H labeling of water and blood glucose was observed between days 3 and 28 (data not shown).
The discrepancy between our data and those reported by Chen et al. (7) raises a critical issue that deserves consideration. As shown, we found that plasma glucose was heavily labeled (∼54–86% of its theoretical maximum in HC- and CF-fed mice, Fig. 4), whereas Chen et al. (7) report that blood glucose reached ∼7–10% of its theoretical maximum, i.e. ∼2.3–3.2% 2H labeling of glucose versus 7 ×∼4.5% (or ∼31.5%) 2H labeling of body water. Note that to compare the data, the labeling of water should be multiplied by ∼7 because each carbon-bound hydrogen of glucose can be derived from water, i.e. at relatively low labeling of body water (precursor labeling), the total 2H labeling of a product is approximately equal to the number of copies of the precursor (n) multiplied by the labeling of the precursor (18). Thus, the data reported by Chen et al. (7) suggest that less than one of the seven carbon-bound hydrogens was labeled.
We believe that the glucose labeling data reported by Chen et al. (7) are impossibly low. For example, Rognstad et al. (30) demonstrated that ∼85% of the possible seven carbon-bound hydrogens are labeled when glucose is made from lactate/pyruvate; they also found substantial labeling from other physiologically relevant gluconeogenic substrates, e.g. the conversion of glycerol to glucose led to the labeling of ∼50% of the possible seven carbon-bound hydrogens. Recent studies using NMR further support the notion that glucose will be substantially and rapidly labeled when made in the presence of 2H2O (31, 32); 2H is incorporated into all of the carbon-bound hydrogens of glucose isolated from overnight fasted subjects, and the total labeling is equal to ∼47% of the theoretical maximum (32). Last, the recent report by Chacko et al. (33) is predicated on the fact that glucose becomes readily labeled in the presence of 2H2O (34). Thus, our data are in agreement with reports in the literature, i.e. one expects some dilution of blood glucose labeling in HC-fed mice because ∼70% of the dietary energy is derived from carbohydrate, whereas in the mice fed the CF diet, virtually all blood glucose must be derived from gluconeogenesis; therefore, the labeling should approach that of water. Certainly one factor that impacts the glucose labeling data of Chen et al. (7) has to do with how they measured the 2H labeling of glucose, i.e. by using the “aldonitrile derivative,” they removed 2H bound to carbon 1 of glucose. We believe that 2H2O cannot be used to unequivocally determine the sources of carbon that are used in the generation of α-glycerol-3-phosphate (7), since the generation of 2H-labeled glucose (i.e. a secondary tracer) confounds the interpretation of the data.
Because our “chronic labeling studies” demonstrated that 2H2O alone cannot answer questions regarding the source(s) of triglyceride glycerol, we turned our attention toward the use of a “double-labeling strategy,” i.e. using 2H2O to determine the total triglyceride glycerol synthesis and using labeled glucose to quantify glucose flux, the difference being equal to the contribution of glyceroneogenesis (assuming minimal contribution of glycerol in the adipocyte) to triglyceride formation. This strategy was used by Botion et al. (15) and Nye et al. (14) and has led to the conclusion(s) that glyceroneogenesis plays a major, if not predominant, role in affecting triglyceride glycerol synthesis. Certain points require consideration as we believe that those tracer data also require a reinterpretation.
Botion et al. (15) determined total triglyceride glycerol synthesis by dividing the total 3H labeling of triglyceride glycerol by 3.3 (the presumed number of exchangeable hydrogens). The factor of 3.3 is an underestimation of the true exchange factor because substrates that are converted to glycerol-phosphate via the glyceroneogenic pathway, e.g. conversion of pyruvate to triglyceride glycerol, yield approximately five 3H atoms (7). Underestimating the exchange factor leads to an overestimation of total triglyceride glycerol synthesis. Botion et al. (15) appear to have also underestimated the contribution of glucose because the 14C labeling of triglyceride glycerol should be multiplied by 2, i.e. the labeling of the product (triglyceride glycerol) is expected to be half that of the precursor (glucose), noted by Nye et al. (14). Also, the manner in which glucose flux to triglyceride glycerol is calculated is not an ideal (or true) precursor: product labeling ratio; rather, it is a relative glucose flux index (15, 35). Therefore, we believe that it is not possible to determine the flux rates using the approach outlined by Botion et al. (15).
Nye et al. (14) also used 3H2O and [U-14C]glucose to quantify flux rates in vivo. Although they include the factor of 2 when estimating the contribution of glucose, the (re)cycling of 14C via lactate (and pyruvate) is problematic. Namely, uncertainty regarding dilution factors that account for relative rates of citric acid cycle and glyceroneogenic flux in adipose tissue makes it difficult to interpret the data (as the authors acknowledge). To quantify the glyceroneogenic flux, Nye et al. (14) also compared the total 3H labeling in triglyceride glycerol (multiplied by 2/5) against the total 3H labeling in pyruvate (multiplied by 2/3); they assume that the conversion of glucose to triglyceride glycerol does not label the hydrogen bound to carbon 3 of α-glycerol-3-phosphate. This logic neglects known pathways that affect the positional labeling of triglyceride glycerol (7). For example, the entry of cold glucose into the adipocyte does label the hydrogen bound to carbon 3 of glycerol-phosphate. Also, consistent with our data, Nye et al. (14) demonstrated substantial 3H labeling of blood glucose, which, on entering the adipocytes, further complicates the interpretation of the data (i.e. because some glucose is already labeled before entering the adipocyte, more 3H will end up bound to carbon 3 of triglyceride glycerol). Last, because their analysis does not consider the positional location of 3H in triglyceride glycerol, their precursor:product labeling ratio more closely yields a measure of the total newly made triglyceride glycerol and not glyceroneogenesis, i.e. to estimate glyceroneogenesis, one would need to assume that 3H on C3 of triglyceride glycerol can only arise from pyruvate flux (which is incorrect) and then compare the labeling of 3H bound to C3 of triglyceride glycerol with that of pyruvate (or water). Therefore, we believe that it is not possible to determine the flux rates using the approach outlined by Nye et al. (14).
To avoid problems related to tracer recycling, we measured the incorporation of 2H from [6,6-2H]glucose into triglyceride glycerol (21). We believe that there are several striking observations regarding triglyceride flux; some are consistent with the literature, and others bring new knowledge regarding triglyceride dynamics. First, in HC-fed mice, glucose appears to be the predominant carbon source for making triglyceride, i.e. most of the palmitate is deposited from de novo lipogenesis and most of the triglyceride glycerol is derived from glucose. Second, there appears to be an increased triglyceride synthesis in the absence of dietary carbohydrate, which is consistent with the relative observations reported by Botion et al. (15) (i.e. regardless of whether they applied the correct factor for the 3H labeling data, they observed an increased triglyceride synthesis in CF-fed animals). Third, although studies in HC-fed mice suggest a role for glyceroneogenesis (generating ∼31% of the triglyceride glycerol), our experiments have considered a highly integrative measurement. Because mice experienced a fed-fasted transition in 24 h, it is not possible to immediately draw conclusions regarding the influence of specific factors on the pathways, e.g. does glyceroneogenic flux increase when glucose availability becomes limiting? However, data from CF-fed mice suggest that glyceroneogenesis is sensitive to nutrient availability and that flux is up-regulated when glucose is limiting in vivo. For example, absolute rates of glyceroneogenesis increased from ∼8 to ∼26 μmol per day when plasma glucose concentration decreased from 9.6 ± 0.3 to 7.2 ± 0.6 mm (p < 0.01), and the apparent glucose turnover decreased from 126 ± 12 to 53 ± 8 μmol × kg–1 × min–1 (p < 0.01) in HC- versus CF-fed mice, respectively (absolute glucose flux to triglyceride glycerol changed to a lesser degree, ∼17 versus ∼11 μmol per day in HC-versus CF-fed mice, respectively).
In summary, we have demonstrated that triglyceride synthesis in adipose tissue is not down-regulated when carbohydrate appears to be limiting; there is an extensive triglyceride turnover during conditions where ∼0% of the calories are derived from dietary carbohydrate. Our data suggest that the contribution of glucose and non-glucose carbon sources shifts with changes in nutritional status. The observation that glucose makes a substantial contribution to triglyceride glycerol when carbohydrate appears to be limiting may seem counterintuitive (13). However, because glycerol flux is ∼10–20% of glucose flux (36, 37) and because one molecule of glucose can give rise to one or two molecules of α-glycerol-3-phosphate, the absolute requirement for glucose to (re)esterify fatty acids is relatively small and could be self-sustaining based on the proposed shuttling mechanism(s) (Fig. 5). Our observations suggest (i) a dynamic control of glucose and non-glucose sources of triglyceride glycerol in adipocytes in vivo and (ii) the operation of a glycerol-glucose cycle between adipose tissue and presumably liver and kidney (analogous to Cori cycling, glucose → lactate → glucose).
FIGURE 5.

Glycerol-glucose cycling between adipose tissue and liver. Two modes of operation of a glucose-glycerol cycle can be envisioned; each is self-sustaining based on carbon balance. First, one molecule of glucose can contribute two molecules of α-glycerol-3-phosphate; this would require a source of ATP and NADH in adipose tissue. Second, one molecule of glucose can provide one molecule of α-glycerol-3-phosphate and one molecule of pyruvate; this would generate the necessary ATP and NADH that are required to run the cycle in adipose tissue.
Acknowledgments
We thank Frederick Allen, Paul Miller, and the CWRU-Mouse Metabolic Phenotyping Center for support and for allowing access to their equipment. S. F. Previs thanks Drs. Kalhan, Hanson, and Nye for exchanging views during the writing of this manuscript and dedicates this work to Dr. Bernard R. Landau for mentoring and for enthusiastic commitment to tracer studies in metabolic research.
This work was supported, in whole or in part, by National Institutes of Health Metabolomics Roadmap Initiative Grant R33DK070291. This work was also supported by supported by grants from the American Diabetes Association (Grant 1-04-JF-40) and the Mt. Sinai Health Care Foundation (Cleveland, OH). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: GC-MS, gas chromatography-mass spectrometry; HC, high carbohydrate; CF, carbohydrate-free.
References
- 1.Behn, A., and Ur, E. (2006) Curr. Opin. Cardiol. 21 353–360 [DOI] [PubMed] [Google Scholar]
- 2.Bray, G. A., and Bellanger, T. (2006) Endocrine 29 109–117 [DOI] [PubMed] [Google Scholar]
- 3.Daniels, S. R. (2006) Future Child 16 47–67 [DOI] [PubMed] [Google Scholar]
- 4.Dietz, W. H. (2004) N. Engl. J. Med. 350 855–857 [DOI] [PubMed] [Google Scholar]
- 5.Bederman, I. R., Dufner, D. A., Alexander, J. C., and Previs, S. F. (2006) Am. J. Physiol. 290 E1048–E1056 [DOI] [PubMed] [Google Scholar]
- 6.Brunengraber, D. Z., McCabe, B. J., Kasumov, T., Alexander, J. C., Chandramouli, V., and Previs, S. F. (2003) Am. J. Physiol. 285 E917–E925 [DOI] [PubMed] [Google Scholar]
- 7.Chen, J. L., Peacock, E., Samady, W., Turner, S. M., Neese, R. A., Hellerstein, M. K., and Murphy, E. J. (2005) J. Biol. Chem. 280 25396–25402 [DOI] [PubMed] [Google Scholar]
- 8.Turner, S. M., Murphy, E. J., Neese, R. A., Antelo, F., Thomas, T., Agarwal, A., Go, C., and Hellerstein, M. K. (2003) Am. J. Physiol. 285 E790–E803 [DOI] [PubMed] [Google Scholar]
- 9.Brito, S. C., Festuccia, W. L., Kawashita, N. H., Moura, M. F., Xavier, A. R., Garofalo, M. A., Kettelhut, I. C., and Migliorini, R. H. (2006) Metabolism 55 84–89 [DOI] [PubMed] [Google Scholar]
- 10.Forest, C., Tordjman, J., Glorian, M., Duplus, E., Chauvet, G., Quette, J., Beale, E. G., and Antoine, B. (2003) Biochem. Soc. Trans. 31 1125–1129 [DOI] [PubMed] [Google Scholar]
- 11.Hanson, R. W., and Reshef, L. (2003) Biochimie (Paris) 85 1199–1205 [DOI] [PubMed] [Google Scholar]
- 12.Leroyer, S. N., Tordjman, J., Chauvet, G., Quette, J., Chapron, C., Forest, C., and Antoine, B. (2006) J. Biol. Chem. 281 13141–13149 [DOI] [PubMed] [Google Scholar]
- 13.Reshef, L., Olswang, Y., Cassuto, H., Blum, B., Croniger, C. M., Kalhan, S. C., Tilghman, S. M., and Hanson, R. W. (2003) J. Biol. Chem. 278 30413–30416 [DOI] [PubMed] [Google Scholar]
- 14.Nye, C. K., Hanson, R. W., and Kalhan, S. C. (2008) J. Biol. Chem. 283 27565–27574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Botion, L. M., Brito, M. N., Brito, N. A., Brito, S. R., Kettelhut, I. C., and Migliorini, R. H. (1998) Metabolism 47 1217–1221 [DOI] [PubMed] [Google Scholar]
- 16.Xu, J., Xiao, G., Trujillo, C., Chang, V., Blanco, L., Joseph, S. B., Bassilian, S., Saad, M. F., Tontonoz, P., Lee, W. N., and Kurland, I. J. (2002) J. Biol. Chem. 277 50237–50244 [DOI] [PubMed] [Google Scholar]
- 17.Yang, D., Diraison, F., Beylot, M., Brunengraber, D. Z., Samols, M. A., Anderson, V. E., and Brunengraber, H. (1998) Anal. Biochem. 258 315–321 [DOI] [PubMed] [Google Scholar]
- 18.Lee, W. N., Bassilian, S., Ajie, H. O., Schoeller, D. A., Edmond, J., Bergner, E. A., and Byerley, L. O. (1994) Am. J. Physiol. 266 E699–E708 [DOI] [PubMed] [Google Scholar]
- 19.Lee, W. N., Bassilian, S., Guo, Z., Schoeller, D., Edmond, J., Bergner, E. A., and Byerley, L. O. (1994) Am. J. Physiol. 266 E372–E383 [DOI] [PubMed] [Google Scholar]
- 20.Diraison, F., Pachiaudi, C., and Beylot, M. (1997) J. Mass Spectrom. 32 81–86 [DOI] [PubMed] [Google Scholar]
- 21.Wolfe, R. R., and Chinkes, D. L. (2004) Isotope Tracers in Metabolic Research: Principles and Practice of Kinetic Analysis, Wiley-Liss, New York
- 22.White, L. W., Williams, H. R., and Landau, B. R. (1968) Arch. Biochem. Biophys. 126 552–557 [DOI] [PubMed] [Google Scholar]
- 23.Kneer, P., and Ball, E. G. (1968) J. Biol. Chem. 243 2863–2870 [PubMed] [Google Scholar]
- 24.Ballard, F. J., Hanson, R. W., and Leveille, G. A. (1967) J. Biol. Chem. 242 2746–2750 [PubMed] [Google Scholar]
- 25.Cadoudal, T., Distel, E., Durant, S., Fouque, F., Blouin, J. M., Collinet, M., Bortoli, S., Forest, C., and Benelli, C. (2008) Diabetes 57 2272–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tordjman, J., Chauvet, G., Quette, J., Beale, E. G., Forest, C., and Antoine, B. (2003) J. Biol. Chem. 278 18785–18790 [DOI] [PubMed] [Google Scholar]
- 27.Bessesen, D. H., Vensor, S. H., and Jackman, M. R. (2000) Am. J. Physiol. 278 E1124–E1132 [DOI] [PubMed] [Google Scholar]
- 28.Summers, L. K., Barnes, S. C., Fielding, B. A., Beysen, C., Ilic, V., Humphreys, S. M., and Frayn, K. N. (2000) Am. J. Clin. Nutr. 71 1470–1477 [DOI] [PubMed] [Google Scholar]
- 29.Schoenheimer, R., and Rittenberg, D. (1936) J. Biol. Chem. 114 381–396 [Google Scholar]
- 30.Rognstad, R., Clark, G., and Katz, J. (1974) Eur. J. Biochem. 47 383–388 [DOI] [PubMed] [Google Scholar]
- 31.Burgess, S. C., Nuss, M., Chandramouli, V., Hardin, D. S., Rice, M., Landau, B. R., Malloy, C. R., and Sherry, A. D. (2003) Anal. Biochem. 318 321–324 [DOI] [PubMed] [Google Scholar]
- 32.Jones, J. G., Perdigoto, R., Rodrigues, T. B., and Geraldes, C. F. (2002) Magn. Reson. Med. 48 535–539 [DOI] [PubMed] [Google Scholar]
- 33.Chacko, S. K., Sunehag, A. L., Sharma, S., Sauer, P. J., and Haymond, M. W. (2008) J Appl. Physiol 104 944–951 [DOI] [PubMed] [Google Scholar]
- 34.Burgess, S. C., Chandramouli, V., Browning, J. D., Schumann, W. C., and Previs, S. F. (2008) J Appl. Physiol. 104 1852–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baker, N., and Huebotter, R. J. (1972) J. Lipid Res. 13 716–724 [PubMed] [Google Scholar]
- 36.Nurjhan, N., Campbell, P. J., Kennedy, F. P., Miles, J. M., and Gerich, J. E. (1986) Diabetes 35 1326–1331 [DOI] [PubMed] [Google Scholar]
- 37.Siler, S. Q., Neese, R. A., Christiansen, M. P., and Hellerstein, M. K. (1998) Am. J. Physiol. 275 E897–E907 [DOI] [PubMed] [Google Scholar]