

Abstract
Myosin light chain phosphatase plays a critical role in modulating smooth muscle contraction in response to a variety of physiologic stimuli. A downstream target of the RhoA/Rho-kinase and nitric oxide (NO)/cGMP/cyclic GMP-dependent kinase (cGKI) pathways, myosin light chain phosphatase activity reflects the sum of both calcium sensitization and desensitization pathways through phosphorylation and dephosphorylation of the myosin phosphatase targeting subunit (MYPT1). As cerebral blood flow is highly spatio-temporally modulated under normal physiologic conditions, severe perturbations in normal cerebral blood flow, such as in cerebral vasospasm, can induce neurological deficits. In nonpermeabilized cerebral vessels stimulated with U-46619, a stable mimetic of endogenous thromboxane A2 implicated in the etiology of cerebral vasospasm, we observed significant increases in contractile force, RhoA activation, regulatory light chain phosphorylation, as well as phosphorylation of MYPT1 at Thr-696, Thr-853, and surprisingly Ser-695. Inhibition of nitric oxide signaling completely abrogated basal MYPT1 Ser-695 phosphorylation and significantly increased and potentiated U-46619-induced MYPT1 Thr-853 phosphorylation and contractile force, indicating that NO/cGMP/cGKI signaling maintains basal vascular tone through active inhibition of calcium sensitization. Surprisingly, a fall in Ser-695 phosphorylation did not result in an increase in phosphorylation of the Thr-696 site. Although activation of cGKI with exogenous cyclic nucleotides inhibited thromboxane A2-induced MYPT1 membrane association, RhoA activation, contractile force, and regulatory light chain phosphorylation, the anticipated decreases in MYPT1 phosphorylation at Thr-696/Thr-853 were not observed, indicating that the vasorelaxant effects of cGKI are not through dephosphorylation of MYPT1. Thus, thromboxane A2 signaling within the intact cerebral vasculature induces “buffered” vasoconstrictions, in which both the RhoA/Rho-kinase calcium-sensitizing and the NO/cGMP/cGKI calcium-desensitizing pathways are activated.
Physiologic control of cerebral circulation is modulated metabolically via PO2 and PCO2, as well as via eicosanoids, endothelin, and nitric oxide (NO).2 Disruptions in the blood-brain barrier either by traumatic head injury or subarachnoid hemorrhage can cause prolonged and severe perturbations to normal cerebral blood flow. Cerebral vasospasm following subarachnoid hemorrhage is characterized by an extensive prolonged narrowing of cerebral arteries, which may result in neurological deficits (1, 2). Eicosanoids, such as the prostaglandins, leukotrienes, and thromboxanes, have been implicated in the etiology of cerebral vasospasm following subarachnoid hemorrhage (1). The Rho-kinase inhibitor fasudil is used clinically as an effective treatment for cerebral vasospasm (3), suggesting the importance of RhoA signaling in the cerebrovasculature. Thus a better understanding of the signaling pathways regulating normal and pathological cerebral blood flow is warranted and is the basis of this study.
Thromboxane A2 (TXA2), a platelet-secreted, short lived derivative of arachidonic acid, is known to induce vasoconstriction in multiple vascular smooth muscles (4–6), including the cerebral microvessels (7). TXA2 receptors (TXA2R) signal through both Gq, resulting in the activation of phospholipase Cβ catalyzing the generation of inositol 1,4,5-trisphosphate and diacylglycerol, and G12/13, resulting in Ca2+ sensitization through the activation of RhoA (4, 8). Activation of the TXA2R is generally considered to preferentially activate calcium sensitization pathways (9–11). As myosin regulatory light chain (RLC20) phosphorylation reflects the activities of myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP), the extent of RLC20 phosphorylation can be modulated by regulating the activity of MLCK or MLCP. Inhibition of MLCP activity through the phosphorylation of MYPT1, such as it occurs with Ca2+ sensitization, leads to an increase in RLC20 phosphorylation at constant [Ca2+]i. Additionally, in some smooth muscles, activation of the phosphoinhibitory protein CPI-17 by a conventional protein kinase C isoform (12) or Rho-kinase (8) results in reduced MLCP activity thus increasing RLC20 phosphorylation and force. The role of CPI-17 in the cerebral vasculature is not presently known.
Ca2+ desensitization leads to a decrease in RLC20 phosphorylation and relaxation of vascular smooth muscle at constant [Ca2+]i through inhibition of MLCK activity, activation of MLCP, or inhibition of Ca2+-sensitizing pathways (8, 13). Cyclic nucleotide-induced relaxation of vascular smooth muscle occurs through several potential downstream signaling pathways initiating Ca2+ desensitization, which include phospho-inhibition of RhoA·GTP at Ser-188 by cyclic GMP-dependent kinase (cGKI) (14, 15), cGKI phosphorylation of telokin (16–18), and inhibition of RhoA activation through protein kinase A (PKA)-dependent phosphorylation of Gα13 (19). A widely accepted mode of Ca2+ desensitization occurs through the direct interaction of MYPT1 with cGKI through their respective C- and N-terminal leucine zipper motifs (20–22). Although the molecular mechanism whereby cGKI alters MLCP activity remains unknown, recent evidence in permeabilized smooth muscles would suggest that phosphorylation of MYPT1 at Ser-695, resulting in the reduction of phosphorylation at the adjacent inhibitory Thr-696 site (23, 24), may play an important physiologic role.
Cyclic nucleotides, generated in response to endothelium-derived NO, can induce relaxation by decreasing [Ca2+]i through the inhibition of calcium influx (25) or calcium release from the sarcoplasmic reticulum (see Refs. 26, 27 and reviewed in Refs. 28, 29). Activation of cGKI selectively phosphorylates and inhibits the TXA2R α-isoform (30). Furthermore, the nitric oxide donor SNP was shown to decrease CPI-17 phosphorylation coincident in time with the rise in [cGMP] and MLCP activity in porcine carotid arteries (31). It remains uncertain whether activation of cGKI in cerebrovascular smooth muscle similarly inhibits RhoA activation and MYPT1 phosphorylation as in cell culture systems (32), or whether cyclic nucleotide-mediated phosphorylation of telokin, such as in phasic smooth muscles or dephosphorylation of CPI-17, contributes to activation of MLCP in the cerebral vasculature, which is a focus of this study.
Coincident with the Ca2+ sensitization-induced phosphorylation of MYPT1 at Thr-696, MLCP has been observed to translocate from the cytosol to the cell membrane following stimulation with prostaglandin F2α and sphingosine 1-phosphate in freshly isolated ferret portal vein SMCs (33) and in hamster resistance arteries (34), suggesting that removal of MLCP from the actomyosin contractile apparatus may be an additional mechanism of Ca2+ sensitization. Though presently an interesting observation associated with Ca2+ sensitization, the underlying function, magnitude, and mechanism of its translocation to the membrane is presently unknown. In this study we examined the downstream signaling events of TXA2 receptor stimulation in concert with cGKI activation in the intact cerebral vasculature. We demonstrate cGKI-dependent inhibition of MYPT1 membrane association and RhoA activation following TXA2R stimulation, without the anticipated reduction of MYPT1 phosphorylation at Thr-696, recently demonstrated in permeabilized ileum (24) and femoral artery (23). Surprisingly, phosphorylation of the MYPT1 Ser-695 site, generally associated with cyclic nucleotide signaling, is present in the resting state, significantly increased in response to thromboxane A2 signaling, and completely abrogated by l-NAME pretreatment or permeabilization, suggesting a critical role for TXA2 signaling in endothelium-smooth muscle coupling within the cerebral vasculature.
MATERIALS AND METHODS
Tissue Preparation—Adult male Sprague-Dawley rats (150–175 g) and adult male New Zealand White rabbits (4–5 pounds) were euthanized according to protocols approved by the Animal Care and Use Committee at the University of Virginia. The posterior and middle cerebral as well as the superior cerebellar arteries were removed from the brain, placed in ice-cold HEPES-buffered Krebs solution, and cleaned of their adventitia. Histological sections of the middle cerebral artery (supplemental Fig. 1) show that the vessels consists of 2–3 layers of circular smooth muscle cells with an intact endothelial lining. Morphometric measurements show that endothelial cells contribute ∼16.5 ± 2.9% of the total cross-sectional area of the vessel wall.
Immunofluorescence Microscopy—Rat cerebral arteries were fixed 10 min post-stimulus in 4% paraformaldehyde in phosphate-buffered saline at room temperature for 15 min. Arteries were permeabilized with 0.03% Triton X-100 for 20 min and blocked with 5% normal donkey serum. Primary antibodies were visualized by a rhodamine Red-X conjugated secondary antibody (Jackson ImmunoResearch, West Grove. PA) at 1:500 dilution. Cell nuclei were visualized with TO-PRO-3 Iodide (Invitrogen). All images were acquired under identical laser power and gain settings using an Olympus Fluoview 300 confocal microscope (Olympus).
Protein Phosphorylation—Protein phosphorylation was preserved by freeze substitution in 10% in trichloroacetic acid in acetone at -80 °C. The trichloroacetic acid was removed by successive washes with pure acetone, after which the vessels were allowed to dry, homogenized in SDS buffer, and boiled for 10 min. Following centrifugation at 10,000 × g for 10 min, the total protein content was determined by the RC DC protein assay (Bio-Rad) to ensure equal loading. Following transfer, gels were stained with Coomassie Blue, and the filamin bands were scanned densitometrically as an additional internal loading control.
Membrane Fractionation—Membrane-bound proteins were separated from cytosolic proteins according to the methods described in Gong et al. (35). Briefly, rabbit cerebral vessels were homogenized in ice-cold homogenization buffer (10 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 2 mm EDTA, 250 mm sucrose, 1 mm dithiothreitol) with protease inhibitor mixture (Sigma) at 1:100 dilution and centrifuged at 100,000 × g for 30 min at 4 °C (Optima™ TLX ultracentrifuge, TLA 120.1 rotor, Beckman Instruments). The supernatant was collected as the cytosolic fraction. Pellets were resuspended in ice-cold homogenization buffer supplemented with 1% Triton X-100 and 1% sodium cholate and incubated for 30 min. The detergent-soluble fraction was separated from the detergent-insoluble (pellet) fraction by centrifugation at 1000 × g for 10 min. Fractions were separated by gel electrophoresis and transferred to polyvinylidene difluoride.
Rhotekin Pulldown—RhoA activation was determined by precipitation of active GTP-bound RhoA (RhoA·GTP) with a glutathione S-transferase fusion protein of the Rho-binding domain of the Rho effector rhotekin as described previously (36).
Western Blotting—Proteins were transferred to polyvinylidene difluoride or to nitrocellulose for CPI-17 blotting and visualized using either the Odyssey system (Li-Cor, Lincoln, NE) or ECL. For Odyssey imaging, membranes were blocked with Odyssey Blocking Buffer and probed with primary antibody in Blocking Buffer. Primary antibodies were visualized using either a goat anti-mouse Alexa 680 (Invitrogen) or a goat anti-rabbit IRDye800 (Rockland Immunochemicals, Gilbertsville, PA)-conjugated secondary antibody. Chicken IgY was detected using a rabbit anti-chicken HiLyte 680 (AnaSpec, San Jose CA)-conjugated secondary antibody. Detection and quantification of the infrared signal were performed using the Odyssey system software. ECL blots were processed following previously published methods (37).
Isometric Force Measurements—Helical strips (100 μm wide × 3 mm long) were cut from the rat middle cerebral artery (inner diameter ≈75–100 μm) and mounted on a 100-μm stainless steel rod for ease of cutting and to remove the endothelium. In some cases to preserve the endothelium, helical strips were cut from an unmounted MCA. Isometric tension was measured at room temperature with a force transducer (AE 801; SensoNor A.S., Horten, Norway), and a length-adjusting device was mounted on a “bubble plate” (38) or on a newly developed wire myograph system with low bath volumes (900SM, Danish Myo Technology, Aarus, Denmark). Strips were stretched 1.1 × resting length in a bicarbonate Krebs buffer and allowed to equilibrate for 30 min prior to depolarization with 154 mm K+. The bicarbonate Krebs buffer containing (mm) 115.2 NaCl, 22.14 NaHCO3, 7.88 d-glucose, 4.7 KCl, 1.18 KH2PO4, 1.16 MgSO4, 1.80 CaCl2, 0.114 ascorbic acid, and 0.027 Na2EDTA was continuously bubbled with 95% O2, 5% CO2. Strips permeabilized with α-toxin and treated with 10 μm A23187 in G1 solution, as described previously (39, 40), were used to determine the free calcium EC50 (supplemental Fig. 2) and the effects of 8-Br-cGMP on contractile force in the presence of fixed [Ca2+]i. Maximal forces were normalized to that of pCa 5 solution, and curves were fit to the data using Origin-Pro software (Northampton MA).
Cyclic Nucleotide Measurements—Following stimulation, rat cerebral arteries were immediately homogenized in 95% ethanol at pH 3.0, after which the homogenate was left overnight at -20 °C for extraction of cyclic nucleotides (41). Samples were centrifuged at 10,000 × g for 10 min. The supernatant was evaporated to dryness under N2, and cyclic nucleotide content was measured using acetylated radioimmunoassay cAMP and cGMP kits according to the manufacturer's protocol (BTI Inc., Stoughton, MA). The protein pellet was dissolved in 0.5 m NaOH and assayed for protein content as above. Cyclic nucleotide content was normalized to total protein content.
Antibodies and Reagents—The following antibodies were used: mouse monoclonal anti-MYPT1 (Covance) for indirect immunofluorescence localization of MYPT1 and a monoclonal anti-MYPT1 (BD Biosciences, San Jose CA) for Western blotting; phospho-specific rabbit polyclonal antibodies anti-phospho-MYPT1 Thr-696 and anti-phospho-MYPT1 Thr-850 (Upstate, Charlottesville VA); mouse monoclonal anti-MLC20 antibody for total MLC20 (Sigma); total and phospho-vasodilator-stimulated protein (VASP; Ser-157 and Ser-239), as well as the rabbit polyclonal anti-phospho-MLC20 antibodies (Cell Signaling, Danvers MA); mouse monoclonal anti-RhoA (Santa Cruz Biotechnology Inc., Santa Cruz CA); rabbit polyclonal total CPI-17 (42), and a chicken IgY anti-phospho-CPI-17 Thr-38 (43). Rabbit polyclonal anti-phospho-MYPT1 Ser-695 and mouse monoclonal anti-MYPT1 leucine zipper (LZ) antibodies were generous gifts from Dr. Timothy Haystead (Duke University, Durham, NC) and Dr. Frank Brozovich (Mayo Clinic, Rochester, MN), respectively.
Statistical Analysis—Statistical significance with respect to control (*) was determined using the Student's t test (Microsoft Excel). The level of significance was set at p < 0.05.
RESULTS
Effects of Thromboxane A2 and cGMP Signaling on Middle Cerebral Artery Contractility—To assess the general effects of TXA2R stimulation and cGMP signaling in the intact cerebral vasculature, we examined the effects of the stable thromboxane A2 mimetic U-46619 and 8-Br-cGMP on the contractility of the intact middle cerebral artery (MCA, Fig. 1). The maximal contractile force induced by 300 nm U-46619 (10 min) is reversible upon addition of the Rho-kinase inhibitor Y-27632 (Fig. 1A, inset), although not completely, suggesting a component of TXA2R-mediated contractile force is independent of Rho-kinase. Additionally, small changes in [Ca2+]i above resting values may contribute to the Rho-kinase-independent component of the TXA2-signaled contractile force, as in the rat caudal artery (6). Pretreatment with 8-Br-cGMP (10 min) significantly inhibited the U-46619-induced contraction by ∼40% (Fig. 1A, n = 4, p < 0.05), similar to the nearly 40% reduction in U-46619-induced contractile force upon addition of (post-treatment) 8-Br-cGMP (Fig. 1B). The efficacy of 8-Br-cGMP to inhibit (pretreatment) or reverse (post-treatment) U-46619-induced contractile force was not significantly different (Fig. 1C). MCA preparations were permeabilized with α-toxin and exhibited a normal pCa-tension relationship (supplemental Fig. 2). Addition of 8-Br-cGMP reduced pCa 6.3 force by ∼70% (n = 4, p < 0.05; data not shown) under conditions where Ca2+ was clamped with EGTA buffer. This finding plus the ability of the Rho-kinase inhibitor to relax the U-46619-contracted nonpermeabilized MCA demonstrate that 8-Br-cGMP-induced relaxation is not solely because of decreases in cytoplasmic [Ca2+].
FIGURE 1.

Effects of thromboxane A2 and cGMP on rat MCA contractility. A, representative force trace from endothelium-denuded, nonpermeabilized MCA stimulated with 154 mm K+ and 300 nm U-46619 with and without 25 μm 8-Br-cGMP pretreatment. Contractile force induced by 300 nm U-46619 is reversed by treatment with 10 μm Y-27632 (inset). B, representative force trace from endothelium-denuded, nonpermeabilized MCA stimulated with 154 mm K+ and 25 μm 8-Br-cGMP following maximal sustained 300 nm U-46619 contraction. C, summary of the effects of 8-Br-cGMP added pre- or post-stimulation with U-46619. The effects of 8-Br-cGMP on contractile force are normalized to that of the average maximal U-46619 contraction (dashed line). Error bars represent mean ± S.D.; *, p < 0.05 with respect to control, n = 4 independent experiments.
8-Br-cGMP Pretreatment Inhibits TXA2 Receptor-coupled RhoA Activation—To elucidate the convergence of signaling pathways activated by both 8-Br-cGMP and U-46619 as it relates to TXA2R-mediated contractile force, we performed rhotekin pulldowns on homogenates of intact cerebral vessels to measure changes in RhoA activation (Fig. 2A). We observe a greater than 2.5-fold significant increase (2.72 ± 0.57, n = 3, p < 0.05) in active RhoA following stimulation with U-46619 as compared with control (Fig. 2B). Pretreatment with 8-Br-cGMP significantly inhibits U-46619-induced RhoA activation by ∼38% (n = 3, p < 0.05), resulting in a net 1.6-fold increase (1.66 ± 0.21, n = 3, p < 0.05) in active RhoA as compared with control.
FIGURE 2.

U-46619-induced RhoA activation is inhibited with 8-Br-cGMP pretreatment. A, representative RhoA Western blots following Rhotekin pulldown of stimulated intact cerebral vasculature. Increased RhoA in pulldown fraction with respect to whole homogenate (WH) are observed following U-46619 treatment. B, quantification of changes in RhoA activation following U-46619 alone, and in the presence of (following pretreatment) 8-Br-cGMP. Rabbit cerebral vessels were used for the rhotekin pulldown assay because of protein requirements. Error bars represent mean ± S.D; *, p < 0.05 with respect to control, n = 3 independent experiments.
8-Br-cGMP Pretreatment Uncouples MLCP Inhibitory Phosphorylation of MYPT1 from RhoA·GTP in the Intact Cerebral Vasculature—To determine whether the observed decreases in RhoA activation (Fig. 2) following U-46619 stimulation in the presence of 8-Br-cGMP results in decreased MLCP inhibitory phosphorylation of MYPT1, we performed quantitative Western blot analysis on MYPT1 phosphorylation at Thr-696 and Thr-853. Representative Western blots are shown depicting the effects of U-46619 and 8-Br-cGMP treatment on the phosphorylation of MYPT1 at Thr-696 and Thr-853 by both LiCor (Fig. 3A) and ECL (supplemental Fig. 4A) detection methodologies. Treatment with U-46619 results in significant increases in RLC20 phosphorylation, as well as MYPT1 phosphorylation at both Thr-696 and Thr-853 (Fig. 3B), suggesting inhibition of MLCP activity, and consistent with prior studies demonstrating large contractile force with minimal increases in [Ca2+]i (9–11) on other smooth muscles (8). Surprisingly, pretreatment with 8-Br-cGMP has no significant effect on U-46619-induced phosphorylation of MYPT1 at either Thr-696 or Thr-853 (Fig. 3B), despite a significant reduction in RhoA·GTP levels (Fig. 2B), suggesting that the activity of the kinase(s) that phosphorylate these sites remain(s) active in the presence of increased cGKI activity. 8-Br-cGMP treatment alone does not significantly alter basal phosphorylation of MYPT1 at either Thr-696 or Thr-853 (supplemental Fig. 4B). However, pretreatment with 8-Br-cGMP significantly inhibits U-46619-induced RLC20 phosphorylation by nearly 40% (n = 5, p < 0.05), and treatment with 8-Br-cGMP alone reduces RLC20 phosphorylation by ∼20% (n = 5, p < 0.05) with respect to control (Fig. 3B), consistent with a decrease in MLCK activity, rather than disinhibition of MLCP. Furthermore, the phospho-inhibitory protein CPI-17 does not appear to play a role in TXA2 signaling at the time points examined, as we observed no change in CPI-17 phosphorylation at Thr-38 following U-46619 stimulation, unlike our observed increase in Thr-38 phosphorylation following phorbol 12,13-dibutyrate stimulation as positive control (Fig. 3A). Similarly, telokin, previously shown to potentiate 8-Br-cGMP-mediated relaxation in smooth muscles by activation of MLCP (18, 44), likely does not play a role in the observed effects of 8-Br-cGMP signaling as its expression is below the Western blot detection threshold in the cerebral vasculature (data not shown).
FIGURE 3.

8-Br-cGMP pretreatment inhibits maximal TXA2R-mediated myosin RLC20 phosphorylation without reducing MLCP inhibitory phosphorylation of MYPT1. A, representative Western blots of U-46619- and 8-Br-cGMP-induced changes in the phosphorylation states of MYPT1, CPI-17, and RLC20 in the intact cerebral vasculature. PDBu, phorbol 12,13-dibutyrate. B, pretreatment with 8-Br-cGMP has no significant effect on U-46619-induced phosphorylation of MYPT1 at both Thr-696 and Thr-853, while significantly reducing maximal U-46619-induced myosin RLC20 phosphorylation with respect to U-46619 alone. Error bars represent mean ± S.E.; *, p < 0.05 with respect to control, n = 5 independent experiments.
TXA2 Signaling Results in Increased MYPT1 Phosphorylation at Ser-695—To fully elucidate the signaling events occurring in response to treatment by U-46619 and 8-Br-cGMP downstream of RhoA·GTP, we examined MYPT1 phosphorylation at Ser-695, a site recently identified as a target of both cGKI and PKA, that when phosphorylated results in decreased Thr-696 phosphorylation of MYPT1 and enhanced MLCP activity (23, 24). Unexpectedly, U-46619 stimulation alone results in a significant increase in Ser-695 phosphorylation in the intact cerebral vasculature, an effect not observed in either intact pulmonary artery, both with (Ser-695 (OD)/filamin (OD), p = 0.08, n = 4) and without an intact endothelium (Fig. 4A), or permeabilized cerebral vessels (Fig. 4, B and C). Analysis of the respective ECL time-dependent monoexponential (Ser-659 (OD)/filamin or MYPT1 (OD)) indicates that the pulmonary artery (control and U-46619) Ser-695 OD signal is smaller than cerebral vessel (control) at all time points (supplemental Fig. 5). Together these results suggest that the TXA2-induced increase in MYPT1 Ser-695 phosphorylation is specific to the intact cerebral vasculature. In addition, although not statistically significant, we observe a general trend in which basal Ser-695 phosphorylation levels are greater in the cerebral vasculature than in the intact pulmonary artery, an observation most clearly shown when all samples are run under identical conditions on the same gel (supplemental Fig. 5). Interestingly, in intact preparations, neither 6-Bnz-cAMP nor 8-Br-cGMP treatment results in an increase in Ser-695 phosphorylation (Fig. 4, A and C), despite our observed 8-Br-cGMP-mediated reductions in maximal U-46619-induced contractile force (Fig. 1A), RhoA activation (Fig. 2), and RLC20 phosphorylation (Fig. 3). Together these results indicate that the 25 μm 8-Br-cGMP, applied extracellularly (unknown intracellular concentration), is unable to activate cGKI to the magnitude necessary to elicit changes in MYPT1 Ser-695 phosphorylation, suggesting that the 8-Br-cGMP-induced reduction in contractile force (Fig. 1A) is not preferentially through disinhibition of MYPT1 in vivo, but rather reflects cGKI-mediated inhibition of RhoA activation (Fig. 2) and likely decreases in [Ca2+]i. Furthermore, neither 6-Bnz-cAMP nor 8-Br-cGMP treatment results in increased VASP phosphorylation at either Ser-157 or Ser-239 (Fig. 4A), sites generally associated with PKA and cGKI activities (45). However, upon membrane permeabilization, treatment with both 6-Bnz-cAMP and 8-Br-cGMP (Fig. 4, B and C) results in large increases in MYPT1 phosphorylation at Ser-695, consistent with the prior studies of Nakamura et al. (23) and Wooldridge et al. (24) following cyclic nucleotide treatment of permeabilized rabbit femoral artery and ileum, respectively. Changes in MYPT1 Ser-695 phosphorylation in response to 6-Bnz-cAMP in permeabilized samples (Fig. 4B) were not quantified because of the control (pCa 6.2) phosphorylation levels being at or below detection threshold. In permeabilized cerebral vessels, we similarly observed significant increases in VASP phosphorylation at Ser-239 in response to 8-Br-cGMP (6.53 ± 2.13-fold, p < 0.05) and 6-Bnz-cAMP (7.00 ± 0.22-fold, p < 0.05) stimulation, suggesting that in vivo, activation of PKA and/or cGKI activities alone are insufficient to induce phosphorylation of both MYPT1 and VASP at the serine sites examined. Together, these results strongly suggest that membrane integrity and/or endothelium-smooth muscle coupling plays a significant role in modulating cyclic nucleotide signaling pathways within the cerebral vasculature.
FIGURE 4.

TXA2R activation, not 8-Br-cGMP stimulation, leads to increased MYPT1 phosphorylation at Ser-695 in the intact cerebral vasculature. A, representative Western blots depicting increased MYPT1 phosphorylation at Ser-695 in response to U-46619 stimulation with and without 8-Br-cGMP pretreatment in the intact cerebral vasculature and pulmonary artery. Stimulation with either 25 μm 8-Br-cGMP or 10 μm 6-Bnz-cAMP alone does not result in increased Ser-695 phosphorylation. The absence of changes in basal MYPT1 Ser-695 phosphorylation following U-46619 stimulation in intact rat pulmonary artery (Pulm. A.) indicate that the TXA2R activation-induced increase in MYPT1 Ser-695 phosphorylation is specific to the cerebral vasculature. Endothelium-denuded pulmonary artery, both control and U-46619-stimulated, has lower MYPT1 Ser-695 phosphorylation levels than their intact endothelial counterparts. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. B, membrane permeabilization abrogates basal and U-46619-induced Ser-695 phosphorylation in the cerebral vasculature. Stimulation with either 6-Bnz-cAMP or 8-Br-cGMP, unlike U-46619, results in increased MYPT1 Ser-695 phosphorylation with respect to pCa 6.2 control. C, quantification of changes in MYPT1 Ser-695 phosphorylation in both the intact and permeabilized cerebral vasculature. Error bars represent mean ± S.E.; *, p < 0.05 with respect to control, n = 4 independent experiments.
cGMP Mediates TXA2-dependent MYPT1 Ser-695 Phosphorylation—To determine the role of the endothelium in modulating MYPT1 Ser-695 phosphorylation in response to TXA2R activation, we examined the effects of U-46619 on cyclic nucleotide concentrations. In intact cerebral vessels treated with the nitric-oxide synthase (NOS) inhibitor NG-nitro-l-arginine methyl ester (l-NAME), we observe a complete elimination of basal MYPT1 Ser-695 and VASP Ser-239/Ser-157 phosphorylation (Fig. 5A), as well as the complete inhibition of U-46619-induced phosphorylation of MYPT1 at Ser-695, suggesting that both basal and TXA2-induced phosphorylations of these serine sites are mediated by NO/cGMP signaling. Treatment with l-NAME increases (2.45 ± 0.36-fold, n = 4, p < 0.05) and potentiates (8.95 ± 1.3-fold, n = 4, p < 0.05) (Fig. 5B, left panel) MYPT1 phosphorylation at Thr-853 in response to U-46619 stimulation. Importantly, force measurements on endothelium-intact nonpermeabilized MCA strips contracted in response to l-NAME, demonstrating the presence of functional endothelial NO/cGMP signaling (Fig. 5D). In addition, l-NAME treatment increased U-46619 maximally induced contractile force by 55 ± 18% (n = 3, p < 0.05) over U-46619 treatment alone (Fig. 5D, inset). l-NAME treatment alone did not significantly increase MYPT1 Thr-696 phosphorylation (Fig. 5B, right panel), indicating that basal MYPT1 Ser-695 phosphorylation does not negatively modulate phosphorylation of MYPT1 at Thr-696. Together these results suggest that (i) endogenous NO/cGMP signaling negatively modulates the phosphorylation of Thr-853 in vivo, which (ii) is more indicative of MLCP activity and contractile force than Thr-696 phosphorylation. In addition, l-NAME pretreatment completely abrogated the U-46619-induced increase in MYPT1 Ser-695 phosphorylation (Fig. 5A), suggesting that TXA2 signaling leads to an increase in the intracellular cGMP concentration. To address this, we next performed direct measurements of cyclic nucleotides in intact cerebral vessels (Fig. 5C). In Fig. 5C, experiment 1, we observe significant increases of 22.6 ± 8.7% (5.35 ± 0.46 pmol/mg protein, n = 3, p < 0.05) in the intracellular cGMP concentration following U-46619 in comparison with control values (4.37 ± 0.32 pmol/mg protein, n = 3). SNP stimulation used as a positive control resulted in an increase of 28.5 ± 9.8% (5.61 ± 0.55 pmol/mg protein, n = 3, p < 0.05). l-NAME significantly reduces cGMP concentrations (0.21 ± 0.07 pmol/mg protein, n = 3, p < 0.05) to ∼5% of control. In Fig. 5C, experiment 2, we observe an even greater 86% increase in cGMP (8.58 ± 0.78 pmol/mg protein, n = 3, p < 0.05) with respect to control (4.61 ± 0.39 pmol/mg protein, n = 3). Although samples from both experiments (n = 3) were run in triplicate showing significant increases in cGMP with U-46619 stimulation, we show them separately to demonstrate the variability in the range of the changes. The effects of TXA2 signaling on cyclic nucleotide concentrations appear to be limited to cGMP, as we observe no significant change in the intracellular cAMP concentration following U-46619 stimulation (607.53 ± 17.40 pmol/mg protein, n = 3) with respect to control (584.22 ± 11.01 pmol/mg protein, n = 3).
FIGURE 5.

In vivo MYPT1 Ser-695 phosphorylation is dependent upon NOS signaling. A, treatment with the NOS inhibitor l-NAME completely abrogates both basal and U-46619-induced MYPT1 phosphorylation at Ser-695 in the intact cerebral vasculature. NOS inhibition results in increased MYPT1 phosphorylation at both Thr-696 and Thr-853. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. B, quantification of changes in MYPT1 Thr-696 (left panel) and Thr-853 (right panel) phosphorylation. Bars represent mean ± S.E.; *, p < 0.05 with respect to control, n = 4. Significant differences between experimental conditions are indicated. C, quantification of changes in cyclic nucleotide concentrations in response to l-NAME, U-46619, SNP, and forskolin. Intact cerebral vessels were treated with 100 μm l-NAME and 25 μm SNP for 60 and 30 min, respectively, or with 20 μm forskolin for 20 min. 300 nm U-46619 was added in the presence of l-NAME for the last 10 min. Significant changes in cGMP (left panel) were observed following stimulation with U-46619 (22%, Experiment 1) and SNP (28%) with respect to control. A separate experiment was performed under identical condition as Experiment 1 in which U-46619 increased cGMP by ∼86%. Treatment with forskolin significantly increased cAMP (right panel) >300-fold as compared with control. Results represent mean ± S.D.; *, p < 0.05 with respect to control, n = 3. D, l-NAME treatment augments U-46619 contractile response. Helical strips with an intact endothelium were cut from the MCA. Force induced by l-NAME treatment alone indicates the presence of an intact endothelium. l-NAME treatment significantly enhanced U-46619-induced contractile force. Results represent mean ± S.D.; *. p < 0.05 with respect to control, n = 3.
Spatial Redistribution of MYPT1 in Response to Thromboxane A2 and cGMP Signaling in the Intact Cerebral Vasculature—To determine whether a spatial redistribution of MYPT1, in response to agonist stimulation, occurs at the contractile time points examined in Fig. 1A, we performed confocal microscopy on whole cerebral vessels with an intact endothelium. We observe a profound change in the spatial localization of MYPT1, from uniform localization (control) to accumulation at the cell periphery (stimulus), following 10 min of stimulation with U-46619 (Fig. 6A). Confocal images acquired in the Z direction through the luminal space of cerebrovascular arterioles demonstrate MYPT1 staining in circumferential smooth muscle layer and the absence of MYPT1 staining within the endothelium (supplemental Fig. 6), indicating that the endothelium contributes negligibly to total MYPT1 content in these preparations. To determine whether the phosphorylation state of MYPT1 plays a role in its spatial redistribution, we further examined cerebral vessels labeled with the phospho-specific anti-MYPT1 (Thr-696 and Thr-853) antibodies. Specificity of the phosphospecific anti-MYPT1 antibodies and the absence of fluorescence with secondary antibodies alone are shown in supplemental Fig. 7. Although there are no observable changes in the spatial localization of the MYPT1 phospho-Thr-696 subpopulation following U-46619 with respect to control, there is an observable increase in signal of the MYPT1 phospho-Thr-853 subpopulation localizing at the cell periphery following agonist stimulation (Fig. 6B and supplemental Fig. 8). Together, these results suggest that the subpopulation of MYPT1 phosphorylated at Thr-853 comprises the subpopulation of MYPT1 localizing at the cell periphery following U-46619 stimulation. Interestingly, the absence of MYPT1 phospho-Thr-853 signal under control conditions and the absence of a MYPT1 phospho-Thr-696 signal at the cell periphery following U-46619 stimulation suggest that two distinct subpopulations of phospho-MYPT1 exist within intact cerebrovascular smooth muscle.
FIGURE 6.

Immunofluorescent localization of MYPT1 and phospho-MYPT1 subpopulations in the intact cerebral vasculature. A, profound change in spatial localization of MYPT1 following 10 min of stimulation with U-46619 indicating MYPT1 accumulation at the plasma membrane. B, spatial localization of the phospho-MYPT1 Thr-696 and Thr-853 subpopulations. Changes in the spatial localization of phospho-MYPT1 Thr-853 following U-46619 stimulation are readily observable with respect to control; however, no such changes are detected in the phospho-MYPT1 Thr-696 subpopulation. C, spatial redistribution of MYPT1 following 10 min of U-46619 stimulation (A) is abrogated in the presence of and following 10 min of 8-Br-cGMP pretreatment. Treatment with 8-Br-cGMP alone does not result in any noticeable changes in either the intensity or localization of MYPT1. The images are representative of data from 8 to 12 independent experiments acquired under identical laser and power gain settings. Scale bar, 10 μm.
To correlate our observed changes in MYPT1 spatial localization with our observed 8-Br-cGMP-mediated inhibition in U-46619 contractile force (Fig. 1A), we repeated our pretreatment experimental paradigm (10 min of 8-Br-cGMP pretreatment followed by addition of U-46619) and again performed confocal microscopy. We observed a more uniform localization of MYPT1 (8-Br-cGMP + U-46619; Fig. 6C) similar to that of untreated control (Fig. 6A), with no apparent changes in MYPT1 localization following 8-Br-cGMP treatment alone (Fig. 6C) as compared with control (Fig. 6A), suggesting that cGMP treatment prevents U-46619-induced MYPT1 spatial redistribution rather than altering MYPT1 spatial localization prior to U-46619 treatment. Images shown are representative of those acquired from n = 8–12 (n = 2–3 samples from n = 4 rats) independent experiments for each indirect immunofluorescence localization of total MYPT1, anti-MYPT1 phospho-Thr-696, and phospho-Thr-853. Similar changes in MYPT1 localization, both total and the phospho-Thr-853 subpopulation, were also observed following 10 μm sphingosine 1-phosphate stimulation (data not shown).
Quantification of Membrane-associated MYPT1—To determine whether MYPT1 is membrane-associated, or localized in the cytosol at the cell periphery, we separated cerebral vessels into their cytosolic, detergent-soluble (membrane fraction) and detergent-insoluble (pellet) fractions. Fig. 7A shows Western blots of MYPT1 following membrane fractionation. Treatment with U-46619 results in MYPT1 association with the membrane fraction, consistent with the immunofluorescent studies in Fig. 6, and coincident in time (10 min) with maximal force (Fig. 1A). Following densitometric analysis, we calculate a nearly 6-fold increase in MYPT1 associated with the detergent-soluble membrane fraction following U-46619 stimulation as compared with control. Although the detergent solubility of MYPT1 increases significantly in response to U-46619, only a relatively small percentage of all extracted (total) MYPT1 becomes detergent-soluble (4% of total MYPT1, n = 5, p < 0.05, Fig. 7B) in response to U-46619. Pretreatment with 8-Br-cGMP (identical experimental paradigm as Fig. 1A) abrogated U-46619-induced MYPT1 association with the detergent-soluble fraction in agreement with the immunofluorescent studies of Fig. 6C. Addition of phosphatase inhibitors has been shown to change the subcellular localization of MYPT1 coincident with increases in phosphorylation at the Thr-696/Thr-853 sites (46). Because of this, phosphatase inhibitors were not used in these experiments. Because of the difficulty in preserving the phosphorylation state of MYPT1 during homogenization and centrifugation, we were unable to verify phosphorylated MYPT1 with this assay. As an unknown portion of the detectable detergent-soluble MYPT1 may have been “lost” to the pellet fraction because of our inability to completely preserve MYPT1 phosphorylation, it is likely that our calculations represent an underestimate.
FIGURE 7.

8-Br-cGMP inhibits U-46619-induced MYPT1 association with the detergent-soluble membrane fraction. A, representative Western blots of the subcellular distribution of MYPT1 following U-46619 stimulation with and without 8-Br-cGMP pretreatment. Membranes reprobed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and vimentin indicate minimal contamination of the detergent-soluble (Det. Soluble) fraction from either the cytosolic or pellet (detergent-insoluble) fractions. B, quantification of changes in MYPT1 subcellular distribution indicates U-46619 stimulation significantly increases MYPT1 association with the detergent-soluble fraction. Pretreatment with 8-Br-cGMP completely inhibits the U-46619-induced association of MYPT1 with the detergent-soluble fraction. Rabbit cerebral vessels were used for the fractionation assay because of protein requirements. Error bars represent mean ± S.D.; *, p < 0.05 with respect to control, n = 5.
DISCUSSION
The major previously undescribed findings of this study are that in nonpermeabilized cerebral arteries with an intact endothelium, (i) MYPT1 phosphorylation, both inhibitory (Thr-696/Thr-853) and the recently identified Ser-695 site (23, 24), is modulated by endothelial NO/cGMP/cGKI signaling; (ii) U-46619 acts on both the endothelium and smooth muscle to elicit increases in phosphorylation at Ser-695, via NO/cGMP/cGKI, as well as at Thr-696 and Thr-853, the latter of which is significantly potentiated in the presence of l-NAME; and (iii) exogenous cGMP reduces both basal and U-46619-induced RLC20 phosphorylation, as well as U-46619-induced RhoA activation and contractile force without the anticipated reductions in MYPT1 phosphorylation at either Thr-696 or Thr-853. In agreement with prior studies, 8-Br-cGMP significantly increased MYPT1 Ser-695 phosphorylation in α-toxin-permeabilized preparations in which the in vivo basal Ser-695 phosphorylation is abolished. Furthermore, we present evidence that basal MYPT1 Ser-695 phosphorylation may be considered a marker for functional endothelial NO/cGMP/cGKI signaling. Finally, we demonstrate the importance of studying intact cerebral vessels to elucidate their regulation; although membrane permeabilization protocols have proven essential for elucidating multiple cellular signaling mechanisms regulating the smooth muscle contractile state, such protocols, by design, destroy endothelium-smooth muscle coupling and smooth muscle membrane polarization potentially eliminating additional regulatory pathways of possible therapeutic interest.
The finding that 8-Br-cGMP, and cyclic nucleotide signaling in general, desensitizes or reduces maximal smooth muscle contractile force is not new, as the effects of cyclic nucleotides on [Ca2+]i (25, 26, 47–49), RhoA activation (14, 15, 19, 30, 32, 50), and MYPT1/CPI-17 phosphorylation (20, 21, 23, 24, 31, 32, 37) have been well documented. Mechanistically, a cGKI-mediated decrease in [Ca2+]i is unlikely to be dominant in our studies of the cerebral vasculature, as contraction mediated by TXA2R activation is predominantly through Ca2+-independent pathways (9–11). Furthermore, 8-Br-cGMP (i) nearly completely relaxed (∼70% reduction, n = 3; data not shown) pCa 6.3 contractile force, and (ii) both inhibited (Fig. 1A) and reversed (Fig. 1B) U-46619-induced contractile force as did the Rho-kinase inhibitor. Together these results suggest that cyclic nucleotide/cGKI-mediated vasorelaxation is dependent upon other mechanisms/targets in addition to the well established reductions in [Ca2+]i.
Results strongly suggest that the kinase(s) responsible for the phospho-inhibition of MLCP are not solely dependent upon RhoA·GTP in cerebrovascular smooth muscle in vivo. A fraction of the TXA2-induced contractile force is insensitive to inhibition of Rho-kinase by Y-27632 (Fig. 1A). Inhibition of basal endothelial NO/cGMP/cGKI signaling with l-NAME significantly increases contractile force (Fig. 5D) and MYPT1 Thr-853 phosphorylation (Fig. 5B), likely involving increased RhoA·GTP (32). This is consistent with observations that phosphorylation of the Thr-853 site is primarily by RhoA/Rho-kinase signaling in vivo (46, 51, 52). However, our data would suggest that phosphorylation of the Thr-853 site by RhoA-independent kinases in vivo is likely; 8-Br-cGMP pretreatment did not alter the ability of U-46619 to induce phosphorylation of MYPT1 at Thr-853 (Fig. 3) despite significantly inhibiting its ability to induce RhoA activation (Fig. 2). The in vivo phosphorylation of the Thr-696 site is more complex as it is known to be phosphorylated by both Rho-kinase and RhoA-independent kinases (8). Integrin-linked kinase and myotonic dystrophy protein kinase, independent of RhoA·GTP, are known to phosphorylate MYPT1 at Thr-696 and additional threonine sites (53, 54). In in vitro assays, MLCP activity is significantly reduced following phosphorylation of MYPT1 at Thr-696 (53, 54) in response to myotonic dystrophy protein kinase and integrin-linked kinase, and Thr-708 in response to integrin-linked kinase (53). Alternatively, TXA2-induced inhibition of the MYPT1 Thr-696/Thr-853 phosphatase(s) could contribute to our observations. Although sensitive to okadaic acid, suggesting type-2 protein phosphatase activity (46), its molecular identity and regulation remain unknown and thus outside the scope of this study. It is likely that multiple kinases are involved in the in vivo phosphorylation of MYPT1 at Thr-696, a subset of which may be spontaneously active and inhibited by cGKI-induced Ser-695 phosphorylation and consistent with the mechanism proposed by Wooldridge et al. (24). We observed spatially distinct intracellular pools of MYPT1 phosphorylated at Thr-696 and Thr-853. It is possible that these pools were differentially phosphorylated at Ser-695 as follows: (i) one pool phosphorylated at Ser-695 would become phosphorylated at Thr-853 but not Thr-696, and (ii) another pool in which MYPT1 is not phosphorylated at Ser-695 and therefore is amenable to Thr-696 phosphorylation. Our Western blot data represents the sum of all phospho-MYPT1 subpopulations, with changes in MYPT1 phosphorylation being the average of these spatially distinct subpopulations. This model would be consistent with the spatial compartmentalization of cAMP signaling because of localized AKAP·PDE complexes (55–57). Similarly, compartmentalization of cGMP signaling is likely because its intracellular concentration is tightly controlled by PDE activity (56).
cGKI has previously been shown to inhibit RhoA activation as follows: (i) cGKI-mediated desensitization of the TXA2R (30), (ii) phosphorylation of RhoA at Ser-188 (14), and (iii) inhibition of the Gα13 switch I region (19). However, the results presented herein demonstrate that the effects of 8-Br-cGMP on TXA2R-mediated contractile force are neither through disinhibition of MLCP (at least phospho-Thr-696/Thr-853) or MYPT1 Ser-695 phosphorylation but may involve additional mediators. Although MYPT1 is phosphorylated, and MLCP activity is inhibited, its enzymatic activity is not abolished (58, 59). Phosphorylation of MYPT1 at Thr-853 has been demonstrated to inhibit MLCP activity by reducing the affinity of MYPT1/MLCP toward myosin (59). We propose that cGKI/MYPT1-interacting proteins (60–62) and or MYPT1 LZ isoform expression may be contributing to our observations. The ratio of MYPT1 LZ+/LZ- isoform expression has been shown to correlate with the sensitivity of smooth muscle to 8-Br-cGMP (63), likely due to a LZ-LZ interaction between cGKI and MYPT1 (20). Consistent with this idea, we observed that the MYPT1 LZ+ isoform is expressed in the cerebral vasculature (supplemental Fig. 3); however, it remains unknown if the expression level of the LZ+ isoform was sufficient to “activate” MLCP activity in response to 8-Br-cGMP in vivo.
We observed a significant increase in MYPT1 Ser-695 phosphorylation, and intracellular cGMP, in response to U-46619 stimulation, indicating that TXA2R activation is not only coupled to vasoconstrictive pathways but also to the classical vasorelaxant NO/cGMP/cGKI signaling pathway. Nonetheless, U-46619 stimulation results in increased contractile force and RLC20 phosphorylation, suggesting that Ca2+-sensitized force generation overcomes the up-regulation of cGMP. Signaling through Gα12/13 (64), angiotensin II (type-1) receptor (AT1) activation has been shown to stimulate NO production and release in cultured bovine aortic endothelial cells (65) and rat carotid arteries in vivo (66). We observe significant increases in cGMP in intact cerebral vessels following TXA2R activation, likely through NOS activation. Consistent with this idea, l-NAME pretreatment completely abrogated both basal and TXA2R-mediated MYPT1 Ser-695 phosphorylation. Elimination of NO signaling significantly reduced smooth muscle cGMP content, thereby reducing both basal and U-46619-induced cGKI activity. The simplest interpretation of these findings is that activation of TXA2R on endothelial cells increases [Ca2+]i, thus activating NOS, leading to an increase in phosphorylation of MYPT1 at Ser-695 in the underlying smooth muscle through the classical endothelial NO/cGMP/cGKI signaling pathway. However, this results in a small but significant increase (Fig. 3B), and not the expected decrease in Thr-696 phosphorylation previously reported in in vitro studies and permeabilized smooth muscles (23, 24), indicating that MYPT1 Ser-695 phosphorylation is insufficient to inhibit TXA2-induced increases in Thr-696 phosphorylation. On the other hand, l-NAME treatment abolished basal phosphorylation at Ser-695, significantly increased Thr-853 phosphorylation, and markedly enhanced the ability of U-46619 to phosphorylate MYTP1 at Thr-853. However, under identical experimental conditions l-NAME treatment did not result in a significant increase in MYPT1 Thr-696 phosphorylation (Fig. 5B), indicating that endothelial NO/cGMP/cGKI signaling predominantly modulates MLCP inhibitory phosphorylation of MYPT1 at Thr-853, rather than the Thr-696 site. Together, these results suggest that MYPT1 Ser-695 phosphorylation may play an as yet unknown role, dependent upon endothelium-derived NO, in maintaining basal cerebrovascular tone through active Ca2+ desensitization.
In nonpermeabilized cerebral arteries with an intact endothelium, we readily observe basal phosphorylation of MYPT1 at Ser-695 and VASP phosphorylation at Ser-157 and Ser-239 indicating that the cyclic nucleotide-dependent protein kinases that phosphorylate these serine sites are active. Reduction in cGMP following NOS inhibition with l-NAME, or endothelial denudation of intact pulmonary artery, completely abrogated VASP Ser-239 phosphorylation, consistent with prior studies (67, 68). Our results would further suggest that MYPT1 Ser-695 is an additional marker of functional endothelial NO/cGMP signaling, as both l-NAME treatment in nonpermeabilized cerebral vessels and endothelial denudation of pulmonary artery completely abrogate basal Ser-695 phosphorylation. We similarly observe the complete elimination of basal VASP Ser-157 phosphorylation in l-NAME-treated nonpermeabilized cerebral vessels consistent with the cGMP activation of PKA (69), the preferential kinase for VASP Ser-157 (45). However, activation of cGKI by exogenous 8-Br-cGMP and 6-Bnz-cAMP in nonpermeabilized cerebral vessels failed to elicit phosphorylation of both MYPT1 and VASP at the serine sites examined. This is likely due to a combination of (i) compartmentalization of PKA/cGKI activities resulting from the spatial localization of PDEs and AKAPs (55–57) and (ii) insufficient cGKI activity in response to 25 μm 8-Br-cGMP and 6-Bnz-cAMP (unknown intracellular concentration). However, it should be noted that 8-Br-cGMP is resistant to PDE activity and was added far in excess of the apparent cGKI dissociation constant (300 nm) for cGMP (70). Furthermore, the localized intracellular 8-Br-cGMP concentrations were sufficient to significantly reduce RLC20 phosphorylation (Fig. 3B) and to diminish the ability of U-46619 to activate RhoA (Fig. 2). Following membrane permeabilization, both 8-Br-cGMP and 6-Bnz-cAMP induce large increases in phosphorylation of MYPT1 at Ser-695, similar to that observed by others in permeabilized rabbit ileum (24) and femoral artery (23), respectively. In permeabilized cerebral vessels, 8-Br-cGMP and 6-Bnz-cAMP resulted in increased VASP phosphorylation at Ser-239, but surprisingly not Ser-157 in response to 6-Bnz-cAMP (Fig. 4), likely due to PKA sequential VASP phosphorylation (Ser-157 then Ser-239) (71), followed by dephosphorylation of Ser-157 prior to the 10-min time point examined.
Our observed changes in MYPT1 subcellular distribution in the intact cerebral vasculature following G-protein coupled receptor stimulation by both confocal microscopy and traditional biochemical fractionation methods are in agreement with previously published studies (33, 34). However, our observations suggest that MYPT1 phosphorylation at either Thr-696 or Thr-853 is not a prerequisite for membrane association, contrary to the model proposed by Shin et al. (33) based upon data from isolated ferret portal vein smooth muscle cells. We demonstrate that pretreatment with 8-Br-cGMP fully inhibited U-46619 induced membrane association without inhibiting U-46619-induced MYPT1 phosphorylation at either Thr-696 or Thr-853. This is consistent with the in vitro inhibition of MYPT1 cosedimentation with phospholipid vesicles following PKA phosphorylation (72). The spatial redistribution of MYPT1 and differential localization of phospho-MYPT1 (Thr-696/Thr-853) has been demonstrated in cultured HepG2 cells in response to okadaic acid (46). Our calculated 4% of membrane-associated (detergent-soluble) MYPT1 is likely an underestimate, possibly due to dephosphorylation during the lengthy fractionation procedure. However, the magnitude of sensitized force due to MYPT1 membrane association (redistribution), thus rendering it inaccessible to myosin, in comparison with MLCP inhibition due to MYPT1 Thr-696/Thr-853 phosphorylation remains unknown. Interestingly, our ability to spatially localize MYPT1 within cerebrovascular smooth muscle cells may be dependent upon the phosphorylation state of MYPT1 itself. Our indirect immunofluorescent localizations of the MYPT1 and the MYPT1 phospho-Thr-853 subpopulations following TXA2 stimulation, although virtually indistinguishable, are in apparent disagreement with the differential localization of MYPT1 and the MYPT1 phospho-Thr-696 subpopulations. Although total MYPT1 appears to be exclusively at the cell periphery following TXA2 stimulation, the MYPT1 phospho-Thr-696 subpopulation is localized throughout the cell, suggesting that the MYPT1 epitope (residues 304–374) (73) to which the total MYPT1 monoclonal antibody was generated is inaccessible in the cytosol but exposed at the cell periphery. The difference between the total and phospho-Thr-696 subpopulations would suggest either a phosphorylation-dependent conformational change in MYPT1 (74) or a protein-protein interaction between MYPT1 and an as yet unknown protein; the Thr-696/Thr-853 phosphorylation sites (polyclonal antibody epitopes) remain physically available to the phospho-specific polyclonal antibodies.
Cerebrovascular tone reflects the summation of vasoconstriction and
vasorelaxation signaling pathways. TXA2, secreted from platelets
(75) and from cerebrovascular
endothelial cells (76),
positively modulates vasoconstriction, whereas endothelium-derived NO and
prostaglandin I2
(77) positively modulate
vasorelaxation. Luminal perfusion of TXA2 in rabbit afferent
arterioles induced potent vasoconstrictions that were abolished with the
superoxide dismutase mimetic 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl
(tempol), and potentiated with l-NAME
(78), suggesting that
endothelial TXA2R activation induces
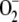 -mediated vasoconstrictions that are
“buffered” with NO. Intact rat cerebrovascular arteries, with
intact endothelia, have intrinsic NOS activity, based upon the ability of
l-NAME to decrease cGMP content, increase MCA contractile force
(Fig. 5D), and to
change MYPT1 phosphorylation at Ser-695/Thr-696/Thr-853. Active
RhoA·GTP as well as phospho-MYPT1 (Ser-695/Thr-696/Thr-853) are
detectable in the basal state, and 8-Br-cGMP relaxes basal tone in cerebral
arteries with decreased RLC20 phosphorylation. Thus, both
Ca2+ desensitization and sensitization pathways are primed in this
vascular bed; the Ca2+ desensitization pathways are more
predominant in the cerebral arteries than in other vessels, such as the
pulmonary artery. We propose that the primed states of the NO- and
Ca2+-sensitizing pathways place the basal contractile state on the
early phases of their sigmoid response curves, with respect to TXA2
signaling, such that changes in either vasoconstrictive or vasorelaxant
signaling can produce rapid, but subtly buffered, contractile or relaxant
responses; thus, maintaining the highly responsive homeostatic cerebral blood
flow within a narrow range in the face of changes is systemic blood pressure
(79). Interestingly,
cerebrovascular TXA2
(80) and NO
(76) production are greater
than in other arteries, such as the aorta, lending further support that the
basal cerebrovascular contractile state is tightly controlled.
-mediated vasoconstrictions that are
“buffered” with NO. Intact rat cerebrovascular arteries, with
intact endothelia, have intrinsic NOS activity, based upon the ability of
l-NAME to decrease cGMP content, increase MCA contractile force
(Fig. 5D), and to
change MYPT1 phosphorylation at Ser-695/Thr-696/Thr-853. Active
RhoA·GTP as well as phospho-MYPT1 (Ser-695/Thr-696/Thr-853) are
detectable in the basal state, and 8-Br-cGMP relaxes basal tone in cerebral
arteries with decreased RLC20 phosphorylation. Thus, both
Ca2+ desensitization and sensitization pathways are primed in this
vascular bed; the Ca2+ desensitization pathways are more
predominant in the cerebral arteries than in other vessels, such as the
pulmonary artery. We propose that the primed states of the NO- and
Ca2+-sensitizing pathways place the basal contractile state on the
early phases of their sigmoid response curves, with respect to TXA2
signaling, such that changes in either vasoconstrictive or vasorelaxant
signaling can produce rapid, but subtly buffered, contractile or relaxant
responses; thus, maintaining the highly responsive homeostatic cerebral blood
flow within a narrow range in the face of changes is systemic blood pressure
(79). Interestingly,
cerebrovascular TXA2
(80) and NO
(76) production are greater
than in other arteries, such as the aorta, lending further support that the
basal cerebrovascular contractile state is tightly controlled.
Supplementary Material
Acknowledgments
We thank Dr. Andra Stevenson for introducing the techniques for the isolation and imaging of the cerebral vessels. We are grateful to Drs. David Brautigan and Robert Nakamoto for stimulating and helpful discussions and Drs. Tim Haystead and Frank Brozovich for generously sharing reagents.
This work was supported, in whole or in part, by National Institutes of Health Grants PO1 HL19242, PO1 HL48807 (to A. V. S.), RO1 HL83261, and PO1 HL48807. This work was also supported by a Pennsylvania Department of Health CURE Grant (to M. E.) and grants from the medical faculty of the University of Cologne (to G. P. and L. T. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–8.
Footnotes
The abbreviations used are: NO, nitric oxide; NOS, nitric-oxide synthase; MLCP, myosin light chain phosphatase; RLC, regulatory light chain; cGKI, cyclic GMP-dependent kinase; MCA, middle cerebral artery; TXA2, thromboxane A2; TXA2R, TXA2 receptor; l-NAME, NG-nitro-l-arginine methyl ester; PKA, protein kinase A; LZ, leucine zipper; MLCK, myosin light chain kinase; PDE, phosphodiesterase; SNP, sodium nitroprusside; VASP, vasodilator-stimulated protein; 6-Bnz-cAMP, N6-benzoyl-cAMP; 8-Br-cGMP, 8-bromo-cGMP.
References
- 1.Nishizawa, S., and Laher, I. (2005) Trends Cardiovas. Med. 15 24-34 [DOI] [PubMed] [Google Scholar]
- 2.Hansen-Schwartz, J., Vajkoczy, P., Macdonald, R. L., Pluta, R. M., and Zhang, J. H. (2007) Trends Pharmacol. Sci. 28 252-256 [DOI] [PubMed] [Google Scholar]
- 3.Shimokawa, H., and Takeshita, A. (2005) Arterioscler. Thromb. Vasc. Biol. 25 1767-1775 [DOI] [PubMed] [Google Scholar]
- 4.Huang, J. S., Ramamurthy, S. K., Lin, X., and Le Breton, G. C. (2004) Cell. Signal. 16 521-533 [DOI] [PubMed] [Google Scholar]
- 5.Ellis, E. F., Oelz, O., Roberts, L. J., II, Payne, N. A., Sweetman, B. J., Nies, A. S., and Oates, J. A. (1976) Science 193 1135-1137 [DOI] [PubMed] [Google Scholar]
- 6.Wilson, D. P., Susnjar, M., Kiss, E., Sutherland, C., and Walsh, M. P. (2005) Biochem. J. 389 763-774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou, X., Gobeil, F., Jr., Peri, K., Speranza, G., Marrache, A. M., Lachapelle, P., Roberts, J., II, Varma, D. R., Chemtob, S., and Ellis, E. F. (2000) Stroke 31 516-525 [DOI] [PubMed] [Google Scholar]
- 8.Somlyo, A. P., and Somlyo, A. V. (2003) Physiol. Rev. 83 1325-1358 [DOI] [PubMed] [Google Scholar]
- 9.Bradley, A. B., and Morgan, K. G. (1987) J. Physiol. (Lond.) 385 437-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Himpens, B., Kitazawa, T., and Somlyo, A. P. (1990) Pfluegers Arch. 417 21-28 [DOI] [PubMed] [Google Scholar]
- 11.Nobe, K., and Paul, R. J. (2001) Circ. Res. 88 1283-1290 [DOI] [PubMed] [Google Scholar]
- 12.Dimopoulos, G. J., Semba, S., Kitazawa, K., Eto, M., and Kitazawa, T. (2007) Circ. Res. 100 121-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murthy, K. S. (2006) Annu. Rev. Physiol. 68 345-374 [DOI] [PubMed] [Google Scholar]
- 14.Sauzeau, V., Le Jeune, H., Cario-Toumaniantz, C., Smolenski, A., Lohmann, S. M., Bertoglio, J., Chardin, P., Pacaud, P., and Loirand, G. (2000) J. Biol. Chem. 275 21722-21729 [DOI] [PubMed] [Google Scholar]
- 15.Sawada, N., Itoh, H., Yamashita, J., Doi, K., Inoue, M., Masatsugu, K., Fukunaga, Y., Sakaguchi, S., Sone, M., Yamahara, K., Yurugi, T., and Nakao, K. (2001) Biochem. Biophys. Res. Commun. 280 798-805 [DOI] [PubMed] [Google Scholar]
- 16.MacDonald, J. A., Walker, L. A., Nakamoto, R. K., Gorenne, I., Somlyo, A. V., Somlyo, A. P., and Haystead, T. A. (2000) FEBS Lett. 479 83-88 [DOI] [PubMed] [Google Scholar]
- 17.Walker, L. A., MacDonald, J. A., Liu, X., Nakamoto, R. K., Haystead, T. A., Somlyo, A. V., and Somlyo, A. P. (2001) J. Biol. Chem. 276 24519-24524 [DOI] [PubMed] [Google Scholar]
- 18.Wu, X., Haystead, T. A., Nakamoto, R. K., Somlyo, A. V., and Somlyo, A. P. (1998) J. Biol. Chem. 273 11362-11369 [DOI] [PubMed] [Google Scholar]
- 19.Manganello, J. M., Huang, J. S., Kozasa, T., Voyno-Yasenetskaya, T. A., and Le Breton, G. C. (2003) J. Biol. Chem. 278 124-130 [DOI] [PubMed] [Google Scholar]
- 20.Surks, H. K., Mochizuki, N., Kasai, Y., Georgescu, S. P., Tang, K. M., Ito, M., Lincoln, T. M., and Mendelsohn, M. E. (1999) Science 286 1583-1587 [DOI] [PubMed] [Google Scholar]
- 21.Surks, H. K., and Mendelsohn, M. E. (2003) Cell. Signal. 15 937-944 [DOI] [PubMed] [Google Scholar]
- 22.Michael, S. K., Surks, H. K., Wang, Y., Zhu, Y., Blanton, R., Jamnongjit, M., Aronovitz, M., Baur, W., Ohtani, K., Wilkerson, M. K., Bonev, A. D., Nelson, M. T., Karas, R. H., and Mendelsohn, M. E. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 6702-6707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura, K., Koga, Y., Sakai, H., Homma, K., and Ikebe, M. (2007) Circ. Res. 101 712-722 [DOI] [PubMed] [Google Scholar]
- 24.Wooldridge, A. A., MacDonald, J. A., Erdodi, F., Ma, C., Borman, M. A., Hartshorne, D. J., and Haystead, T. A. (2004) J. Biol. Chem. 279 34496-34504 [DOI] [PubMed] [Google Scholar]
- 25.Sausbier, M., Schubert, R., Voigt, V., Hirneiss, C., Pfeifer, A., Korth, M., Kleppisch, T., Ruth, P., and Hofmann, F. (2000) Circ. Res. 87 825-830 [DOI] [PubMed] [Google Scholar]
- 26.Schlossmann, J., Ammendola, A., Ashman, K., Zong, X., Huber, A., Neubauer, G., Wang, G. X., Allescher, H. D., Korth, M., Wilm, M., Hofmann, F., and Ruth, P. (2000) Nature 404 197-201 [DOI] [PubMed] [Google Scholar]
- 27.Tang, K. M., Wang, G. R., Lu, P., Karas, R. H., Aronovitz, M., Heximer, S. P., Kaltenbronn, K. M., Blumer, K. J., Siderovski, D. P., Zhu, Y., and Mendelsohn, M. E. (2003) Nat. Med. 9 1506-1512 [DOI] [PubMed] [Google Scholar]
- 28.Hofmann, F., Feil, R., Kleppisch, T., and Schlossmann, J. (2006) Physiol. Rev. 86 1-23 [DOI] [PubMed] [Google Scholar]
- 29.Lincoln, T. M., Dey, N., and Sellak, H. (2001) J. Appl. Physiol. 91 1421-1430 [DOI] [PubMed] [Google Scholar]
- 30.Reid, H. M., and Kinsella, B. T. (2003) J. Biol. Chem. 278 51190-51202 [DOI] [PubMed] [Google Scholar]
- 31.Etter, E. F., Eto, M., Wardle, R. L., Brautigan, D. L., and Murphy, R. A. (2001) J. Biol. Chem. 276 34681-34685 [DOI] [PubMed] [Google Scholar]
- 32.Seko, T., Ito, M., Kureishi, Y., Okamoto, R., Moriki, N., Onishi, K., Isaka, N., Hartshorne, D. J., and Nakano, T. (2003) Circ. Res. 92 411-418 [DOI] [PubMed] [Google Scholar]
- 33.Shin, H. M., Je, H. D., Gallant, C., Tao, T. C., Hartshorne, D. J., Ito, M., and Morgan, K. G. (2002) Circ. Res. 90 546-553 [DOI] [PubMed] [Google Scholar]
- 34.Bolz, S. S., Vogel, L., Sollinger, D., Derwand, R., de Wit, C., Loirand, G., and Pohl, U. (2003) Circulation 107 3081-3087 [DOI] [PubMed] [Google Scholar]
- 35.Gong, M. C., Fujihara, H., Somlyo, A. V., and Somlyo, A. P. (1997) J. Biol. Chem. 272 10704-10709 [DOI] [PubMed] [Google Scholar]
- 36.Ren, X. D., Kiosses, W. B., and Schwartz, M. A. (1999) EMBO J. 18 578-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lubomirov, L. T., Reimann, K., Metzler, D., Hasse, V., Stehle, R., Ito, M., Hartshorne, D. J., Gagov, H., Pfitzer, G., and Schubert, R. (2006) Circ. Res. 98 1159-1167 [DOI] [PubMed] [Google Scholar]
- 38.Somlyo, A. V., Horiuti, K., Trentham, D. R., Kitazawa, T., and Somlyo, A. P. (1992) J. Biol. Chem. 267 22316-22322 [PubMed] [Google Scholar]
- 39.Khromov, A. S., Wang, H., Choudhury, N., McDuffie, M., Herring, B. P., Nakamoto, R., Owens, G. K., Somlyo, A. P., and Somlyo, A. V. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 2440-2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitazawa, T., Gaylinn, B. D., Denney, G. H., and Somlyo, A. P. (1991) J. Biol. Chem. 266 1708-1715 [PubMed] [Google Scholar]
- 41.Gray, D. W., and Marshall, I. (1992) Br. J. Pharmacol. 107 691-696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Senba, S., Eto, M., and Yazawa, M. (1999) J. Biochem. (Tokyo) 125 354-362 [DOI] [PubMed] [Google Scholar]
- 43.Eto, M., Kitazawa, T., Matsuzawa, F., Aikawa, S., Kirkbride, J. A., Isozumi, N., Nishimura, Y., Brautigan, D. L., and Ohki, S. Y. (2007) Structure (Lond.) 15 1591-1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choudhury, N., Khromov, A. S., Somlyo, A. P., and Somlyo, A. V. (2004) J. Muscle Res. Cell Motil. 25 657-665 [DOI] [PubMed] [Google Scholar]
- 45.Krause, M., Dent, E. W., Bear, J. E., Loureiro, J. J., and Gertler, F. B. (2003) Annu. Rev. Cell Dev. Biol. 19 541-564 [DOI] [PubMed] [Google Scholar]
- 46.Lontay, B., Kiss, A., Gergely, P., Hartshorne, D. J., and Erdodi, F. (2005) Cell. Signal. 17 1265-1275 [DOI] [PubMed] [Google Scholar]
- 47.Ammendola, A., Geiselhoringer, A., Hofmann, F., and Schlossmann, J. (2001) J. Biol. Chem. 276 24153-24159 [DOI] [PubMed] [Google Scholar]
- 48.Fritsch, R. M., Saur, D., Kurjak, M., Oesterle, D., Schlossmann, J., Geiselhoringer, A., Hofmann, F., and Allescher, H. D. (2004) J. Biol. Chem. 279 12551-12559 [DOI] [PubMed] [Google Scholar]
- 49.Robertson, B. E., Schubert, R., Hescheler, J., and Nelson, M. T. (1993) Am. J. Physiol. 265 C299-C303 [DOI] [PubMed] [Google Scholar]
- 50.Lang, P., Gesbert, F., Delespine-Carmagnat, M., Stancou, R., Pouchelet, M., and Bertoglio, J. (1996) EMBO J. 15 510-519 [PMC free article] [PubMed] [Google Scholar]
- 51.Muranyi, A., Derkach, D., Erdodi, F., Kiss, A., Ito, M., and Hartshorne, D. J. (2005) FEBS Lett. 579 6611-6615 [DOI] [PubMed] [Google Scholar]
- 52.Ren, X. D., Wang, R., Li, Q., Kahek, L. A., Kaibuchi, K., and Clark, R. A. (2004) J. Cell Sci. 117 3511-3518 [DOI] [PubMed] [Google Scholar]
- 53.Muranyi, A., MacDonald, J. A., Deng, J. T., Wilson, D. P., Haystead, T. A., Walsh, M. P., Erdodi, F., Kiss, E., Wu, Y., and Hartshorne, D. J. (2002) Biochem. J. 366 211-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muranyi, A., Zhang, R., Liu, F., Hirano, K., Ito, M., Epstein, H. F., and Hartshorne, D. J. (2001) FEBS Lett. 493 80-84 [DOI] [PubMed] [Google Scholar]
- 55.Seino, S., and Shibasaki, T. (2005) Physiol. Rev. 85 1303-1342 [DOI] [PubMed] [Google Scholar]
- 56.Rybalkin, S. D., Yan, C., Bornfeldt, K. E., and Beavo, J. A. (2003) Circ. Res. 93 280-291 [DOI] [PubMed] [Google Scholar]
- 57.Wong, W., and Scott, J. D. (2004) Nat. Rev. 5 959-970 [DOI] [PubMed] [Google Scholar]
- 58.Feng, J., Ito, M., Ichikawa, K., Isaka, N., Nishikawa, M., Hartshorne, D. J., and Nakano, T. (1999) J. Biol. Chem. 274 37385-37390 [DOI] [PubMed] [Google Scholar]
- 59.Velasco, G., Armstrong, C., Morrice, N., Frame, S., and Cohen, P. (2002) FEBS Lett. 527 101-104 [DOI] [PubMed] [Google Scholar]
- 60.Wooldridge, A. A., Fortner, C. N., Lontay, B., Akimoto, T., Neppl, R. L., Facemire, C., Datto, M. B., Kwon, A., McCook, E., Li, P., Wang, S., Thresher, R. J., Miller, S. E., Perriard, J. C., Gavin, T. P., Hickner, R. C., Coffman, T. M., Somlyo, A. V., Yan, Z., and Haystead, T. A. (2008) J. Biol. Chem. 283 11850-11859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koga, Y., and Ikebe, M. (2005) J. Biol. Chem. 280 4983-4991 [DOI] [PubMed] [Google Scholar]
- 62.Surks, H. K., Richards, C. T., and Mendelsohn, M. E. (2003) J. Biol. Chem. 278 51484-51493 [DOI] [PubMed] [Google Scholar]
- 63.Khatri, J. J., Joyce, K. M., Brozovich, F. V., and Fisher, S. A. (2001) J. Biol. Chem. 276 37250-37257 [DOI] [PubMed] [Google Scholar]
- 64.Maguire, J. J., and Davenport, A. P. (2005) Trends Pharmacol. Sci. 26 448-454 [DOI] [PubMed] [Google Scholar]
- 65.Suzuki, H., Eguchi, K., Ohtsu, H., Higuchi, S., Dhobale, S., Frank, G. D., Motley, E. D., and Eguchi, S. (2006) Endocrinology 147 5914-5920 [DOI] [PubMed] [Google Scholar]
- 66.Boulanger, C. M., Caputo, L., and Levy, B. I. (1995) Hypertension 26 752-757 [DOI] [PubMed] [Google Scholar]
- 67.Ibarra-Alvarado, C., Galle, J., Melichar, V. O., Mameghani, A., and Schmidt, H. H. (2002) Mol. Pharmacol. 61 312-319 [DOI] [PubMed] [Google Scholar]
- 68.Oelze, M., Mollnau, H., Hoffmann, N., Warnholtz, A., Bodenschatz, M., Smolenski, A., Walter, U., Skatchkov, M., Meinertz, T., and Munzel, T. (2000) Circ. Res. 87 999-1005 [DOI] [PubMed] [Google Scholar]
- 69.Worner, R., Lukowski, R., Hofmann, F., and Wegener, J. W. (2007) Am. J. Physiol. 292 H237-H244 [DOI] [PubMed] [Google Scholar]
- 70.Uhler, M. D. (1993) J. Biol. Chem. 268 13586-13591 [PubMed] [Google Scholar]
- 71.Butt, E., Abel, K., Krieger, M., Palm, D., Hoppe, V., Hoppe, J., and Walter, U. (1994) J. Biol. Chem. 269 14509-14517 [PubMed] [Google Scholar]
- 72.Ito, M., Feng, J., Tsujino, S., Inagaki, N., Inagaki, M., Tanaka, J., Ichikawa, K., Hartshorne, D. J., and Nakano, T. (1997) Biochemistry 36 7607-7614 [DOI] [PubMed] [Google Scholar]
- 73.Okubo, S., Ito, M., Takashiba, Y., Ichikawa, K., Miyahara, M., Shimizu, H., Konishi, T., Shima, H., Nagao, M., Hartshorne, D. J., and Nakano, T. (1994) Biochem. Biophys. Res. Commun. 200 429-434 [DOI] [PubMed] [Google Scholar]
- 74.Tan, I., Ng, C. H., Lim, L., and Leung, T. (2001) J. Biol. Chem. 276 21209-21216 [DOI] [PubMed] [Google Scholar]
- 75.Hamberg, M., Svensson, J., and Samuelsson, B. (1975) Proc. Natl. Acad. Sci. U. S. A. 72 2994-2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miyamoto, A., Hashiguchi, Y., Obi, T., Ishiguro, S., and Nishio, A. (2007) Vasc. Pharmacol. 46 85-90 [DOI] [PubMed] [Google Scholar]
- 77.Davidge, S. T. (2001) Circ. Res. 89 650-660 [DOI] [PubMed] [Google Scholar]
- 78.Schnackenberg, C. G., Welch, W. J., and Wilcox, C. S. (2000) Am. J. Physiol. 279 F302-F308 [DOI] [PubMed] [Google Scholar]
- 79.Van Lieshout, J. J., Wieling, W., Karemaker, J. M., and Secher, N. H. (2003) J. Appl. Physiol. 94 833-848 [DOI] [PubMed] [Google Scholar]
- 80.Shirahase, H., Fujiwara, M., Usui, H., and Kurahashi, K. (1987) Blood Vessels 24 117-119 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.