

Abstract
1-Nitropyrene, the most abundant nitro polycyclic aromatic hydrocarbon in diesel emissions, has been found to react with DNA to form predominantly N-(deoxyguanosin-8-yl)-1-aminopyrene (dGAP). This bulky adduct has been shown to induce genetic mutations, which may implicate Y-family DNA polymerases in its bypass in vivo. To establish a kinetic mechanism for the bypass of such a prototype single-base lesion, we employed pre-steady-state kinetic methods to investigate individual nucleotide incorporations upstream, opposite, and downstream from a site-specifically placed dGAP lesion catalyzed by Sulfolobus solfataricus DNA polymerase IV (Dpo4), a model Y-family DNA polymerase. Dpo4 was able to bypass dGAP but paused strongly at two sites: opposite the lesion and immediately downstream from the lesion. Both nucleotide incorporation efficiency and fidelity decreased significantly at the pause sites, especially during extension of the bypass product. Interestingly, a 4-fold tighter binding affinity of damaged DNA to Dpo4 promoted catalysis through putative interactions between the active site residues of Dpo4 and 1-aminopyrene moiety at the first pause site. In the presence of a DNA trap, the kinetics of nucleotide incorporation at these sites was biphasic in which a small, fast phase preceded a larger, slow phase. In contrast, only a large, fast phase was observed during nucleotide incorporation at non-pause sites. Our kinetic studies support a general kinetic mechanism for lesion bypass catalyzed by numerous DNA polymerases.
Environmental pollutants have been shown to impact human health at the molecular level. One detrimental route is the modification of genomic DNA and nucleotides (1). If DNA lesions are not recognized and removed by the cellular DNA repair machinery, they will stall replicative DNA polymerases (2–8). To rescue DNA replication, cells employ lesion bypass DNA polymerases to traverse unrepaired lesions. Most of these enzymes belong to the Y-family of DNA polymerases. The Y-family enzymes possess relatively flexible and solvent-accessible active sites to accommodate bulky DNA lesions (9, 10). However, Y-family DNA polymerases catalyze DNA synthesis over undamaged DNA with low fidelity and poor processivity (6, 10–12). The Y-family DNA polymerases have been identified in all three domains of life, e.g. four in humans (DNA polymerases η, ι, κ, and Rev1), two in Escherichia coli (DNA polymerases IV and V), and one in Sulfolobus solfataricus (Dpo4). Because Dpo4 can be expressed in E. coli and purified with a high yield, it has been extensively studied in vitro as a prototype Y-family enzyme. Dpo4 catalyzes DNA synthesis on an undamaged DNA template with a fidelity of one error per 1,000–10,000 nucleotide incorporations based on pre-steady-state kinetic analysis from 37 to 56 °C (13–15). Dpo4 is capable of bypassing a myriad of DNA lesions including apurinic/apyrimidinic (abasic) sites (16–19), 8-oxo-7,8-dihydro-2′-deoxyguanosine (20, 21), 1,N2-etheno(ε)guanosine (22), cis-syn thymine-thymine dimer (23–25), cisplatin-induced 1,2-intrastrand cross-links with adjacent deoxyguanosines (cisplatin-d(GpG) adducts) (24, 26), benzo[a]pyrene diol epoxide (BPDE)5 on deoxyguanosine (BPDE-dG) or deoxyadenosine (BPDE-dA) (27, 28), and N-2-acetyl-aminofluorene (AAF) on deoxyguanosine (AAF-dG) (24).
So far there are no comprehensive in vitro studies of the bypass of 1-nitropyrene (1-NP)-induced DNA adducts catalyzed by a Y-family DNA polymerase. 1-NP, one of the most abundant polycyclic aromatic hydrocarbons, is a product of incomplete diesel and gasoline combustion (1, 29, 30). There are two known pathways by which 1-NP is metabolized: nitro reduction (Scheme 1) and C-hydroxylation. When an aromatic ring of 1-NP is oxidized into non-DNA-reactive metabolites by P450 enzymes while in the gastrointestinal tract, respiratory system, or skin, the organism can excrete the metabolites through a detoxification process (29–31). However, in a gastrointestinal tract containing bacteria, such as Clostridium paraputrificum, Clostridium clostridiiforme, Eubacterium sp., and Clostridium leptum (32), the majority of 1-NP proceeds through nitro reduction, thereby leading to the production of DNA-reactive metabolites (Scheme 1) (31). The intermediate metabolite, N-hydroxy-1-aminopyrene, is critical for creating an electrophilic nitrenium ion capable of reacting with DNA (Scheme 1). The major product formed from these reactive metabolites is N-(deoxyguanosin-8-yl)-1-aminopyrene (dGAP) that was shown to be mutagenic in bacterial and mammalian cells (30, 31, 33). 1-NP is a potent mutagen and a carcinogen in rodents (34, 35), and the International Agency for Research on Cancer classifies 1-NP as a class 2B carcinogen (31, 33, 36, 37).
SCHEME 1.

Dpo4 has not been shown to bypass the dGAP adduct in vivo for several reasons. First, S. solfataricus, a hyperthermophilic archeon, would have to be able to uptake 1-NP. Because this organism grows optimally at 80 °C and pH 2–4 (38), 1-NP may not be stable under these extreme conditions (39). Second, S. solfataricus would have to encode the necessary enzymes like nitro reductase to metabolize 1-NP and form dGAP (38). We chose to study dGAP bypass catalyzed by Dpo4 because (i) the kinetic mechanism of nucleotide incorporation into undamaged DNA catalyzed by Dpo4 has been previously elucidated by our laboratory (14), (ii) there are many published crystal structures of the Dpo4 ternary complexes which contain various DNA lesions (16, 20, 22, 25, 27, 28, 40–43), (iii) Dpo4 is the only Y-family DNA polymerase encoded by S. solfataricus and, thus, is responsible for most translesion synthesis events in that organism (38), and (iv) we have established kinetic mechanisms and pathways for the bypass of an abasic site (17) and a cisplatin-d(GpG) adduct (26) catalyzed by Dpo4. In addition, both Dpo4 and eukaryotic DNA polymerase κ are DinB (damage-induced protein) homologs (45, 46), and the bypass abilities for a spectrum of DNA lesions are similar to those of eukaryotic DNA polymerase η (24, 47, 48). Thus, Dpo4 is a good model for those eukaryotic Y-family DNA polymerases, and our studies may implicate how eukaryotic Y-family enzymes bypass dGAP. To better understand the mutagenic potential of 1-NP-induced DNA damage, a single-base lesion, dGAP, was placed specifically in a GC-rich region of a synthetic DNA template. Regions composed of repetitive DNA sequences, such as those in oncogenes, have been shown previously to induce more mutations than non-repetitive regions (33, 49, 50). The mechanistic basis of the bypass of this bulky dGAP lesion catalyzed by Dpo4 was comprehensively investigated using pre-steady-state kinetic methods.
EXPERIMENTAL PROCEDURES
Materials—Reagents were purchased from the following companies: OptiKinase from United States Biochemical (Cleveland, OH), [γ-32P]ATP from GE Healthcare, and dNTPs from Invitrogen. The full-length Dpo4 was expressed in E. coli and purified as previously described (15).
Synthetic Oligonucleotides—The DNA template 26-mer dGAP (Table 1) was synthesized and purified as previously described (51). The monoisotopic mass (M-H) 8109.32 of the purified 26-mer-dGAP by electrospray ionization was consistent with the calculated mass of 8109.58. Other DNA substrates listed in Table 1 were purchased from Integrated DNA Technologies (Coralville, IA) and purified by denaturing PAGE. The concentration of each DNA oligomer was determined by the UV absorbance at 260 nm.
TABLE 1.
Sequences of DNA oligonucleotides
| Sequences | |
|---|---|
| Primers | |
| 17-mer | 5′-AACGACGGCCAGTGAAT-3′ |
| 19-mer | 5′-AACGACGGCCAGTGAATTC-3′ |
| 20-mer | 5′-AACGACGGCCAGTGAATTCG-3′ |
| 21-mer | 5′-AACGACGGCCAGTGAATTCGC-3′ |
| 22-mer | 5′-AACGACGGCCAGTGAATTCGCG-3′ |
| 23-mer | 5′-AACGACGGCCAGTGAATTCGCGC-3′ |
| Templates | |
| 26-mer | 3′-TTGCTGCCGGTCACTTAAGCGCGCCC-5′ |
| 26-mer-dGAP | 3′-TTGCTGCCGGTCACTTAAGCGCGCCC-5′ |
| DNA trap | |
| D-1 (21/41-mer) | 5′-CGCAGCCGTCCAACCAACTCA-3′ / |
| 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ |
G designates N-(deoxyguanosin-8-yl)-1-aminopyrene (dGAP)
Labeling and Annealing of the DNA Substrates—Each primer was 5′-32P-labeled by incubating it with OptiKinase and [γ-32P]ATP for 3 h at 37 °C. The 5′-32P-labeled primer was annealed to the unlabeled 26-mer or 26-mer-dGAP at a molar ratio of 1.00:1.15. This mixture was first heat denatured at 75 °C for 2 min and then cooled slowly to room temperature in several hours.
Buffers—All pre-steady-state kinetic assays, if not specified, were performed in optimized reaction buffer R (50 mm HEPES, pH 7.5 at 37 °C, 5 mm MgCl2, 50 mm NaCl, 0.1 mm EDTA, 5 mm dithiothreitol, 10% glycerol, and 0.1 mg/ml bovine serum albumin) (15). All electrophoresis mobility shift assays (EMSA) were performed in buffer S (50 mm Tris-Cl, pH 7.5 at 23 °C, 5 mm MgCl2, 50 mm NaCl, 5 mm dithiothreitol, 10% glycerol, and 0.1 mg/ml bovine serum albumin). All given concentrations were final after mixing all solutions.
Running Start Assay—The running start assay was performed as previously described (17, 18, 26). Briefly, a preincubated solution of 5′-32P-labeled DNA (100 nm) and Dpo4 (100 nm) in buffer R was rapidly mixed with a solution containing all four dNTPs (200 μm each) at 37 °C via a rapid chemical-quench flow apparatus (KinTek). The reaction was quenched with 0.37 m EDTA after various times, and the reaction products were analyzed by denaturing PAGE (17% polyacrylamide, 8 m urea).
EMSA—Dpo4 (0.5–80 nm) was titrated into a solution containing 5′-32P-labeled DNA (5 nm) in buffer S at 23 °C. To separate the binary complex from free DNA, native PAGE was conducted at a constant voltage of 70 V for 35 min at 23 °C using running buffer A (50 mm Tris acetate, pH 7.5, at 23 °C, 0.5 mm EDTA, 5.5 mm Mg(OAc)2). After drying the gel, the bands were quantitated using a PhosphorImager 445 SI (Molecular Dynamics). The dependence of the concentration of the binary complex Dpo4·DNA on the Dpo4 concentration was fit to Equation 1 to yield Kd,DNA the equilibrium dissociation constant for the binary complex (Dpo4·DNA) at 23 °C.
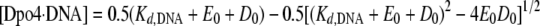 |
(Eq. 1) |
In Equation 1, Eo is the active Dpo4 concentration, and Do is the DNA concentration.
Determination of Substrate Specificity—The dNTP incorporation efficiency (kp/Kd,dNTP) was calculated using the measured maximum dNTP incorporation rate (kp) and equilibrium dissociation constant (Kd,dNTP) of an incoming dNTP. Single-turnover dNTP incorporation assays were employed to obtain the kp and Kd,dNTP as previously described (14, 15, 17, 26). Briefly, a preincubated solution of Dpo4 (120 nm) and 5′-32P-labeled DNA (30 nm) in buffer R was mixed with increasing concentrations of a dNTP. The reactions were terminated after various times using 0.37 m EDTA. Reaction products were analyzed by denaturing PAGE (17% acrylamide, 8 m urea) and quantitated with a PhosphorImager 445 SI. The time course of product formation at each dNTP concentration was fit to a single-exponential equation (Equation 2),
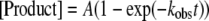 |
(Eq. 2) |
where kobs is the observed reaction rate constant, and A is the reaction amplitude. Next, the plot of the kobs versus the dNTP concentration was fit to a hyperbolic equation (Equation 3),
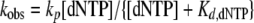 |
(Eq. 3) |
where kp is the maximum dNTP incorporation rate, and Kd,dNTP is the equilibrium dissociation constant for the ternary complex (Dpo4·DNA·dNTP).
Biphasic Kinetic Assay—A preincubated solution of Dpo4 (120 nm) and 5′-32P-labeled DNA (30 nm) in buffer R was rapidly mixed with 5 μm DNA trap D-1 (Table 1) (15) and 1.2 mm correct dNTP in buffer R for various times before being quenched with 0.37 m EDTA. Reaction products were resolved and quantitated as described above. The plot of the product concentration versus reaction time was fit to a double-exponential equation (Equation 4),
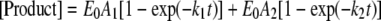 |
(Eq. 4) |
where Eo is the active Dpo4 concentration, A1 and A2 are the reaction amplitudes of the first and second phase, respectively, and k1 and k2 are the rate constants of the first and second phases, respectively.
RESULTS
Bypass of a dGAP Lesion Catalyzed by Dpo4—A running start assay (“Experimental Procedures”) was performed to observe the DNA polymerization pattern of how Dpo4 responded to a 1-aminopyrene (1-AP) adduct in a DNA substrate (17/26-mer-dGAP). As described previously with other normal DNA substrates (17, 26), Dpo4 synthesized the full-length product 26-mer with an undamaged DNA substrate 17/26-mer (Table 1) within 10 s (Fig. 1A). Despite the bulky size of dGAP, Dpo4 was able to bypass this lesion in 17/26-mer-dGAP (Table 1) with the observation of the full-length product after 180 s (Fig. 1B). However, the accumulation of intermediate products 20- and 21-mer signaled that there were two consecutive strong polymerase pause sites. The 20- and 21-mer intermediates corresponded to dNTP incorporation opposite dGAP and extension of the lesion bypass product, respectively. In comparison, Dpo4 did not pause significantly at the comparable sites in Fig. 1A. The product 27-mer in Fig. 1A was likely formed through a blunt-end addition (52). Furthermore, the accumulation of 24- and 25-mer in Fig. 1A and 25-mer in Fig. 1B was likely due to the dC-rich sequence at the 3′-terminus of the 26-mer template (Table 1) which caused polymerase “slippage” via primer realignment. This possibility was supported by the observation that an addition of 5% DMSO in the reaction buffer diminished the accumulation of both the 24- and 25-mer (data not shown).
FIGURE 1.

Running start assays. A preincubated solution of Dpo4 (100 nm) and 5′-32P-labeled DNA (100 nm) was rapidly mixed with all four dNTPs (200 μm each), and the reaction was quenched with 0.37 m EDTA at various time intervals. A, 17/26-mer. B, 17/26-mer-dGAP. Sizes of important products are indicated, and the 21st position marks the location of the dGAP lesion from the 3′-terminus of the DNA template.
Effect of a dGAP Lesion on DNA Binding to Dpo4—The accumulation of intermediate products in Fig. 1B suggested that the presence of the bulky lesion may weaken the binding of DNA to Dpo4, thereby reducing the extension of intermediates. To measure the binding affinity (1/Kd,DNA) of DNA to Dpo4, EMSA was performed for DNA substrates containing either the control (26-mer) or damaged (26-mer-dGAP) DNA templates (Table 1). The binary complex (Dpo4·DNA) was separated from free DNA using native PAGE (Fig. 2A). As a representative example, the plot of the concentration of Dpo4·20/26-mer-dGAP against the total concentration of Dpo4 (Fig. 2B) was fit to Equation 1 (“Experimental Procedures”) to obtain a Kd, DNA of 1.0 ± 0.1 nm. EMSAs were repeated for other DNA substrates, and the Kd,DNA values are listed in Table 2. As expected, Dpo4 bound to undamaged DNA substrates with similar affinity (3.1–4.0 nm). In comparison, Dpo4 bound to damaged DNA substrates with a larger range of Kd, DNA values (1.0–4.4 nm, Table 2). Interestingly, a 4-fold tighter binding affinity (Table 2) was observed with 20/26-mer-dGAP, the DNA substrate at the first pause site in Fig. 1B. This suggested that the 1-AP moiety may interact directly with the active site residues of Dpo4. However, the tighter binding of 20/26-mer-dGAP to Dpo4 should facilitate processive polymerization and, thus, cannot be used to account for the accumulation of 20-mer in the first strong pause site (Fig. 1B). Other kinetic studies have shown that the accumulation of intermediates in the vicinity of a DNA lesion is a strong indication that certain microscopic kinetic parameters, such as maximum dNTP incorporation rate (kp) and ground-state dNTP binding affinity (1/Kd,dNTP), were altered (17, 26, 53). Thus, we suspected that alterations in kp and Kd,dNTP of an incoming dNTP by dGAP were the kinetic reasons for polymerase pausing in Fig. 1B.
FIGURE 2.

Measurement of Kd, DNA at the first pause site. Various amounts of Dpo4 (0.5–80 nm) were titrated into a solution containing 5′-32P-labeled 20/26-mer-dGAP (5 nm). The binary complex of Dpo4·DNA was separated from free DNA by native PAGE. A, gel image of titration. B, the plot of the binary complex concentration versus the total concentration of Dpo4. The data were fit to Equation 1 (“Experimental Procedures”), which yielded a Kd, DNA of 1.0 ± 0.1 nm.
TABLE 2.
Binding affinity of Dpo4 to damaged and control DNA substrates at 23 °C
Effect of a dGAP Lesion on the Kinetics of dNTP Incorporation—To determine kp and Kd,dNTP, we performed single dNTP incorporation assays under single-turnover reaction conditions. A representative example is shown in Fig. 3. First, a preincubated solution of Dpo4 (120 nm) and 5′-32P-labeled 22/26-mer-dGAP were rapidly mixed with dCTP (25–1500 μm) and quenched with 0.37 m EDTA at various times. The products were resolved by denaturing PAGE. The product concentration was plotted against time (Fig. 3A), and the data were fit to Equation 2 (“Experimental Procedures”) to determine the observed reaction rate (kobs). The dependence of kobs on the dCTP concentration was plotted and fit to Equation 3 (“Experimental Procedures”), which yielded a kp of 6.3 ± 0.3 s-1 and a Kd, dCTP of 682 ± 80 μm (Fig. 3B). This assay was repeated for the series of DNA substrates representing the progression of Dpo4 as it approached, encountered, and bypassed the dGAP lesion in template 26-mer-dGAP, and these kinetic data are listed in Table 3. For comparison, we used the same kinetic assay to determine the kinetic parameters (supplemental Table 1) for the corresponding dNTP incorporations with the control template 26-mer (Table 1). Although sequence-dependent, these control kinetic parameters for both correct and incorrect dNTP incorporations were similar to our previously published results with a different undamaged template (15).
FIGURE 3.

Kinetics of dCTP incorporation into 22/26-mer-dGAP. A, a preincubated solution of Dpo4 (120 nm) and 5′-32P-labeled 22/26-mer-dGAP (30 nm) was rapidly mixed with increasing concentrations of dCTP (25 μm, ▪; 50 μm, ○; 100 μm, ▴; 250 μm, ⋄; 500 μm, •; 1000 μm, □; 1500 μm, ♦) for various time intervals. Each time course was fitted to Equation 2 to yield a kobs. B, the plot of kobs values against dCTP concentrations was fit to Equation 3 to produce a kp of 6.3 ± 0.3 s-1 and a Kd,dCTP of 682 ± 80 μm.
TABLE 3.
Kinetic parameters for single dNTP incorporation opposite template 26-mer-dGAP
| dNTP | Kd, dNTP | kp | (kp/Kd, dNTP)damaged | Efficiency ratioa,b | Fidelityc | Probabilityd |
|---|---|---|---|---|---|---|
| μm | s–1 | μm–1 s–1 | ||||
| 19/26-mer-dGAP | ||||||
| dGTP | 187 ± 36 | 4.3 ± 0.3 | 2.3 × 10–2 | 1.1 | 99.8 | |
| dATP | 373 ± 81 | (6.3 ± 0.5) × 10–3 | 1.7 × 10–5 | 0.8 | 7.4 × 10–5 | 0.1 |
| dCTP | 227 ± 23 | (6.4 ± 0.2) × 10–3 | 2.8 × 10–5 | 5 | 1.2 × 10–3 | 0.1 |
| dTTP | 1180 ± 190 | (1.0 ± 0.1) × 10–2 | 8.7 × 10–6 | 1.0 | 3.8 × 10–4 | 0.0 |
| 20/26-mer-dGAPe | ||||||
| dCTP | 167 ± 15 | 1.03 ± 0.03 | 6.2 × 10–3 | 9.2 | 98.4 | |
| dATP | 856 ± 184 | (7.6 ± 0.8) × 10–2 | 8.9 × 10–5 | 0.2 | 1.4 × 10–2 | 1.4 |
| dGTP | 955 ± 160 | (2.0 ± 0.2) × 10–3 | 2.1 × 10–6 | 21 | 3.3 × 10–4 | 0.0 |
| dTTP | 557 ± 36 | (5.5 ± 0.1) × 10–3 | 9.9 × 10–6 | 8.4 | 1.6 × 10–3 | 0.2 |
| 21/26-mer-dGAPe | ||||||
| dGTP | 674 ± 231 | (2.8 ± 0.3) × 10–2 | 4.2 × 10–5 | 88 | 88.6 | |
| dATP | 886 ± 145 | (2.7 ± 0.2) × 10–3 | 3.1 × 10–6 | 0.8 | 6.8 × 10–2 | 6.5 |
| dCTP | 328 ± 79 | (6.8 ± 0.5) × 10–4 | 2.1 × 10–6 | 15 | 4.7 × 10–2 | 4.4 |
| dTTP | 2300 ± 461 | (4.9 ± 0.7) × 10–4 | 2.2 × 10–7 | 8.2 | 5.1 × 10–3 | 0.5 |
| 22/26-mer-dGAP | ||||||
| dCTP | 682 ± 80 | 6.3 ± 0.3 | 9.3 × 10–3 | 3.7 | 99.8 | |
| dATP | 826 ± 85 | (4.2 ± 0.2) × 10–3 | 5.0 × 10–6 | 0.8 | 5.4 × 10–4 | 0.1 |
| dGTP | 502 ± 98 | (4.3 ± 0.3) × 10–3 | 8.6 × 10–6 | 0.8 | 9.3 × 10–4 | 0.1 |
| dTTP | 1540 ± 257 | (4.7 ± 0.5) × 10–3 | 3.1 × 10–6 | 3.6 | 3.3 × 10–4 | 0.0 |
| 23/26-mer-dGAP | ||||||
| dGTP | 62 ± 18 | 1.9 ± 0.1 | 3.1 × 10–2 | 0.8 | 98.9 | |
| dATP | 668 ± 173 | (3.7 ± 0.4) × 10–2 | 5.5 × 10–5 | 0.3 | 1.8 × 10–3 | 0.2 |
| dCTP | 1130 ± 161 | (9.0 ± 0.7) × 10–3 | 7.9 × 10–6 | 0.2 | 2.6 × 10–4 | 0.0 |
| dTTP | 1290 ± 218 | (3.4 ± 0.3) × 10–2 | 2.7 × 10–4 | 0.01 | 8.7 × 10–4 | 0.9 |
Calculated as (kp/Kd, dNTP)control/(kp/Kd, dNTP)damaged
Values for (kp/Kd, dNTP)control are listed in supplemental Table 1
Calculated as (kp/Kd, incorrect dNTP)damaged/(kp/Kd, correct dNTP)damaged + (kp/ Kd, incorrect dNTP)damaged
Calculated as ((kp/Kd, dNTP)damaged/(Σ(kp/Kd, dNTP)damaged)) × 100
Denotes pause sites
From the measured kp and Kd,dNTP values with 26-mer-dGAP, we further calculated dNTP incorporation efficiency (kp/Kd,dNTP), efficiency ratio (relative to undamaged DNA), fidelity, and probability (Table 3). At non-pause sites, the kp/Kd,dNTP values for correct dNTP incorporation were within 1–4-fold of those with control 26-mer (supplemental Table 1) and were 100–4000-fold greater relative to misincorporations (Table 3). For 20/26-mer-dGAP and 21/26-mer-dGAP, the correct dNTP incorporation efficiencies, respectively, decreased by 9- and 88-fold (Table 3 and Fig. 4A) in comparison to those values with control 20/26- and 21/26-mer (supplemental Table 1). In addition, these catalytic efficiencies were up to 740-fold lower than those at non-pause sites (Table 3 and Fig. 4A). Thus, the presence of dGAP unfavorably impacted correct dNTP incorporation at two discrete locations, opposite the lesion and extension of the bypass product.
FIGURE 4.

Quantitative effects of a dGAP lesion on correct dNTP incorporation catalyzed by Dpo4. Extracted kinetic parameters from Table 3 were plotted against DNA substrates. A, the dNTP incorporation efficiency ratio. B, the kp ratio. C, the Kd,dNTP ratio.
In Table 3, the polymerase fidelity both upstream and downstream of the pause sites is in the range of 10-3 to 10-5, which is similar to the fidelity range obtained with control DNA (supplemental Table 1). In contrast, the fidelity at the pause sites (10-2–10-4), especially at the 2nd pause site, was lowered by 10–100-fold compared with non-pause sites (Table 3) and with control DNA (supplemental Table 1). Interestingly, the correct dNTP incorporation probability was above 98% at all sites tested except for the extension step, whereby it dropped to 89% (Table 3). Based on the dNTP incorporation efficiency values in Table 3, Dpo4 catalyzed the insertion of dNTPs with the following selection preference: dCTP >> dATP > dTTP, dGTP at the first pause site and dGTP > dATP, dCTP > dTTP at the second pause site.
Biphasic Kinetics of dNTP Incorporation at the Pause Sites— Our previous studies have shown that dNTP incorporation at pause sites follows biphasic kinetics (17, 26). Such multiple phase kinetics, which were hidden in the single-turnover dNTP incorporation assay, can be deconvoluted by including a DNA trap. For this assay 5 μm concentration of undamaged D-1 (Table 1) was used as the trap. The effectiveness of this trap was examined and confirmed to be sufficient (supplemental Fig. 1). To investigate the kinetics of dNTP incorporation at pause sites, a preincubated solution of Dpo4 (120 nm) and 5′-32P-labeled DNA (30 nm) was rapidly mixed with a solution of correct dNTP (1.2 mm) and unlabeled D-1 (5 μm) for various times before termination with 0.37 m EDTA. The time courses (Fig. 5) of correct dNTP incorporation into 20/26-mer-dGAP and 21/26-mer-dGAP were both biphasic and were fit to Equation 4 (“Experimental Procedures”), which yielded the biphasic kinetic parameters listed in Table 4. With both damaged substrates, the rate (k1) of the first phase was significantly faster than the k2 of the second phase, whereas the reaction amplitude of the first phase (A1) was much smaller than the amplitude of the second phase (A2). The total amplitudes (A1 + A2) were 66.7% with 20/26-mer-dGAP and 22% with 21/26-mer-dGAP, which were much less than 100%. In contrast, similar DNA trap assays with their control DNA substrates (20/26-mer and 21/26-mer) revealed only a single, fast phase (data not shown). Moreover, correct dGTP incorporation into damaged 19/26-mer-dGAP at an upstream, non-pause site and into the control substrate 19/26-mer in the presence of a DNA trap also exhibited monophasic kinetics (data not shown). The time courses of product formation were fit to Equation 2 (“Experimental Procedures”) to yield the kinetic parameters listed in Table 4. For the time courses exhibiting monophasic kinetics, the reaction amplitudes of dNTP incorporation were all about 90%, whereas the reaction rates were 2.5–4.6 s-1 (Table 4). Taken together, these DNA trap assay experiments demonstrated that the dGAP lesion altered only the kinetics of dNTP incorporation at the two critical steps of translesion synthesis.
FIGURE 5.

Biphasic kinetics of correct dNTP incorporation in the presence of a DNA trap. A preincubated solution of Dpo4 (120 nm) and 5′-32P-labeled 20/26-mer-dGAP (▪, 30 nm) or 21/26-mer-dGAP (•, 30 nm) was mixed rapidly with 21/41-mer D-1 (5 μm) and dCTP (▪, 1.2 mm) or dGTP (•, 1.2 mm). The reaction was quenched with 0.37 m EDTA after various times. The product concentration was plotted as a function of reaction time for each DNA substrate which was then fit to Equation 4. For 20/26-mer-dGAP, the fast phase had a reaction amplitude of 2.0 ± 0.7 nm and a reaction rate of 11 ± 6 s-1, whereas the slow phase had a reaction amplitude of 18 ± 1 nm and a reaction rate of 0.7 ± 0.1 s-1. For 21/26-mer-dGAP, the fast phase had a reaction amplitude of 0.9 ± 0.1 nm and a reaction rate of 1.9 ± 0.3 s-1, whereas the slow phase had a reaction amplitude of 5.7 ± 0.2 nm and a reaction rate of 0.031 ± 0.003 s-1.
TABLE 4.
Biphasic kinetic parameters for correct dNTP incorporation into 5′-32P-labeled DNA (30 nm) in the presence of a DNA trap (5 μm) at 37 °C
| DNA substrate | Correct dNTP | A1 | k1 | A2 | k2 |
|---|---|---|---|---|---|
| nm | s–1 | nm | s–1 | ||
| 19/26-mer | dGTP | 27 ± 1 (91%)a | 3.2 ± 0.4 | ||
| 20/26-mer | dCTP | 26 ± 1 (86%)a | 4.6 ± 0.5 | ||
| 21/26-mer | dGTP | 27.0 ± 0.4 (86%)a | 2.3 ± 0.1 | ||
| 19/26-mer-dGAP | dGTP | 27.0 ± 0.4 (89%)a | 2.5 ± 0.2 | ||
| 20/26-mer-dGAP | dCTP | 2.0 ± 0.7 (6.7%)a | 11 ± 6 | 18 ± 1 (60%)a | 0.7 ± 0.1 |
| 21/26-mer-dGAP | dGTP | 0.9 ± 0.1 (3%)a | 1.9 ± 0.3 | 5.7 ± 0.2 (19%)a | 0.031 ± 0.003 |
Calculated as (reaction amplitude/30 nm) × 100
DISCUSSION
The full-length product in Fig. 1B indicated that Dpo4 was able to bypass a site-specifically placed dGAP. However, the initial formation of 26-mer was slower with the damaged template 26-mer-dGAP (180 s) than with the control template 26-mer (10 s) (Fig. 1). Moreover, the accumulation of intermediates 20- and 21-mer indicated that Dpo4 paused significantly when incorporating dNTP opposite dGAP and extending the bypass product. Interestingly, the second pause site was stronger than the first, which has been reported previously with AAF-dG (24). To mechanistically understand how a Y-family DNA polymerase traverses a single-base lesion like dGAP, here we utilized EMSA and pre-steady-state kinetic methods to investigate the kinetic impact of this lesion on DNA binding to Dpo4 and dNTP incorporations at positions upstream, opposite, and downstream from the dGAP lesion.
A Kinetic Basis for the Pausing of Dpo4 Caused by a dGAP— Fig. 1B showed that several intermediate products accumulated (i.e. 20-, 21-, and 25-mer). The accumulation of 25-mer was likely due to polymerase slippage at the dC-rich region (“Results”). A comparison of the catalytic efficiencies at the four other positions (Table 3) revealed the kinetic basis for the remaining incorporation profile. For example, correct dGTP incorporation into 21/26-mer-dGAP is 150-fold less efficient than correct dCTP into 20/26-mer-dGAP, whereas the latter is 4-fold less efficient than correct dGTP into 19/26-mer-dGAP. These inefficiencies led to the accumulation of 20- and 21-mer with the latter accumulating more than the former in Fig. 1B. In contrast, correct dCTP incorporation into 22/26-mer-dGAP was 220-fold more efficient than correct dGTP incorporation into 21/26-mer-dGAP but was 3-fold less efficient than correct dGTP incorporation into 23/26-mer-dGAP. These resulted in the non-accumulation of 22- and 23-mer. Taken together these data suggested the following kinetic pattern; (i) if an intermediate accumulates at a polymerase pause site, its elongation is less efficient than its production, and the larger the difference in incorporation efficiency, the stronger the accumulation of the intermediate; (ii) the contrary is true for intermediate species which do not accumulate at a polymerase non-pause site. Consistently, this kinetic pattern has been observed in the bypass of an abasic site (17) and a cisplatin-d(GpG) adduct (26) catalyzed by Dpo4.
The analysis of efficiency ratios showed that dGAP significantly altered the kinetics of elongating the species at the strong pause sites in Fig. 1B (Table 3 and Fig. 4A). To determine which kinetic parameters were affected, the kp and Kd,dNTP ratios were plotted against the template positions (Fig. 4). As displayed in Fig. 4B, the kp was significantly affected only at the two pause sites (11-fold for 20/26-mer-dGAP and 58-fold for 21/26-mer-dGAP). In contrast, the Kd,dNTP ratios were within 5-fold, with the largest increase in Kd,dNTP at a non-pause site (Fig. 4C). Thus, the inefficient elongation of 20- and 21-mer in Fig. 1B was primarily due to slow kp values for correct dNTP incorporation.
These slow kp values may be due to DNA being trapped in
nonproductive complexes with Dpo4. This hypothesis was supported by the
biphasic kinetics of nucleotide incorporations in the presence of a DNA trap
whereby a small, fast phase (A1 and
k1) preceded a large, slow phase (A2
and k2) (Fig.
5 and Table 4).
Opposite dGAP, the contribution of the fast phase (11
s-1 × 6.7% of reaction amplitude) and the slow phase (0.7
s-1 × 60%) yielded an overall dCTP incorporation rate of 1.2
s-1. This value was close to the calculated
kobs 0.9 s-1) estimated using
Equation 3, 1.2 mm
dCTP in Fig. 5, and measured
Kd,dNTP and kp values
(Table 3). Similarly, analysis
of the biphasic rates for dGTP incorporation into 21/26-mer-dGAP
(Table 4) agreed with the
single-turnover rate in Table
3. In contrast, the same DNA trap experiments with
19/26-mer-dGAP revealed only the fast phase kinetics of dGTP
incorporation with a reaction amplitude of ∼90%
(Table 4), whereas the slow
phase was not observed. Despite the molar excess of Dpo4, the remaining 10% of
19/26-mer-dGAP was never elongated (data not shown), which was not
caused by dGAP, for similar reaction amplitudes were also observed
with the three control DNA substrates in the presence of the DNA trap
(Table 4). Observing reaction
amplitudes less than 100% could be due to experimental errors, incomplete
annealing of the DNA duplexes, Dpo4 bound to DNA in an inactive mode, and Dpo4
binding at the blunt end rather than the staggering end of DNA. Notably,
correct nucleotide incorporation into 19/26-mer-dGAP and the three
control DNA substrates all followed monophasic kinetics with similar kinetic
parameters (Table 4). This
suggested that the dGAP lesion did not affect nucleotide
incorporation at a non-pause site, and 19/26-mer-dGAP was bound by
Dpo4 as a productive complex
(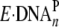 ) similar to
control DNA. In comparison, the small fast phases with both
20/26-mer-dGAP and 21/26-mer-dGAP occurred with similar
reaction rates (k1,
Table 4) as the single-turnover
rates observed with the control DNA substrates (supplemental Table 1) and
likely represented the same kinetic process. Thus, small percentages
(A1) of 20/26-mer-dGAP and
21/26-mer-dGAP were bound productively by Dpo4
(
) similar to
control DNA. In comparison, the small fast phases with both
20/26-mer-dGAP and 21/26-mer-dGAP occurred with similar
reaction rates (k1,
Table 4) as the single-turnover
rates observed with the control DNA substrates (supplemental Table 1) and
likely represented the same kinetic process. Thus, small percentages
(A1) of 20/26-mer-dGAP and
21/26-mer-dGAP were bound productively by Dpo4
(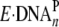 ) in the fast
phase. In the slow phase, large percentages (A2) of these
two damaged substrates must be bound in a less catalytically competent mode by
Dpo4 (
) in the fast
phase. In the slow phase, large percentages (A2) of these
two damaged substrates must be bound in a less catalytically competent mode by
Dpo4 ( ), as they
were elongated with much slower rates (k2) than in the
fast phase (k1). Moreover, the elongation of
), as they
were elongated with much slower rates (k2) than in the
fast phase (k1). Moreover, the elongation of
 occurred in a
single binding event, suggesting a slow conversion of
occurred in a
single binding event, suggesting a slow conversion of
 to
to
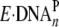 (k2) before a rapid extension (k1).
The total reaction amplitudes observed with both 20/26-mer-dGAP
(66.7%) and 21/26-mer-dGAP (22%) were much smaller than the highest
possible amplitudes (∼90%) observed with 19/26-mer-dGAP and
control DNA substrates (Table
4). The kinetic partitioning between the dissociation of
(k2) before a rapid extension (k1).
The total reaction amplitudes observed with both 20/26-mer-dGAP
(66.7%) and 21/26-mer-dGAP (22%) were much smaller than the highest
possible amplitudes (∼90%) observed with 19/26-mer-dGAP and
control DNA substrates (Table
4). The kinetic partitioning between the dissociation of
 and the
conversion from
and the
conversion from  to
to 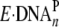 led to the
reduction of A2 by a factor of
k2/(k2 + koff),
whereas the koff is the DNA dissociation rate from
Dpo4·DNA. The koff of D-1
(Table 1) has been previously
determined to be 0.02 s-1
(14). Because both
20/26-mer-dGAP and 21/26-mer-dGAP bound to Dpo4 with
tighter or similar affinities as control DNA
(Table 2), we assumed their
koff to be 0.02 s-1. On the basis of the
koff, the k2, A1,
and A2 values (Table
4), and the above factor, we further estimated that 62% of
20/26-mer-dGAP and 31% of 21/26-mer-dGAP are in the form
of
led to the
reduction of A2 by a factor of
k2/(k2 + koff),
whereas the koff is the DNA dissociation rate from
Dpo4·DNA. The koff of D-1
(Table 1) has been previously
determined to be 0.02 s-1
(14). Because both
20/26-mer-dGAP and 21/26-mer-dGAP bound to Dpo4 with
tighter or similar affinities as control DNA
(Table 2), we assumed their
koff to be 0.02 s-1. On the basis of the
koff, the k2, A1,
and A2 values (Table
4), and the above factor, we further estimated that 62% of
20/26-mer-dGAP and 31% of 21/26-mer-dGAP are in the form
of  and that
21.3% of 20/26-mer-dGAP and 56% of 21/26-mer-dGAP were
never elongated. Moreover, only 1% of 20/26-mer-dGAP and 4.7% of
21/26-mer-dGAP were calculated to be free in solution based on
Kd, DNA values in
Table 2 and Dpo4 and DNA
concentrations in Fig. 5, Thus,
significant amounts of 20/26-mer-dGAP and 21/26-mer-dGAP
bound by Dpo4 were catalytically incompetent
(
and that
21.3% of 20/26-mer-dGAP and 56% of 21/26-mer-dGAP were
never elongated. Moreover, only 1% of 20/26-mer-dGAP and 4.7% of
21/26-mer-dGAP were calculated to be free in solution based on
Kd, DNA values in
Table 2 and Dpo4 and DNA
concentrations in Fig. 5, Thus,
significant amounts of 20/26-mer-dGAP and 21/26-mer-dGAP
bound by Dpo4 were catalytically incompetent
(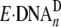 ). Together,
these DNA trap experiments suggest a kinetic mechanism for bypassing
dGAP as shown in Fig.
6.
). Together,
these DNA trap experiments suggest a kinetic mechanism for bypassing
dGAP as shown in Fig.
6.
FIGURE 6.

Proposed kinetic mechanism for the bypass of dGAP catalyzed
by Dpo4. The kinetic parameters of the observed fast phase
(A1, k1) and slow phase
(A2, k2) in the presence of a DNA trap
are labeled. E, polymerase; DNAn, DNA substrate;
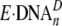 , dead-end binary complex;
, dead-end binary complex;
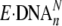 , nonproductive binary
complex;
, nonproductive binary
complex; 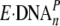 , productive
binary complex; DNAn+1, extended DNA product by
a base; PPi, pyrophosphate.
, productive
binary complex; DNAn+1, extended DNA product by
a base; PPi, pyrophosphate.
There is structural evidence to support this lesion bypass mechanism. For
example, the combined NMR molecular mechanics computational studies reveal
that the 1-AP moiety of an embedded dGAP:dC base pair in an 11-mer
duplex is only intercalated into the DNA helix between adjacent Watson-Crick
base pairs (54). Moreover, the
sugar of the modified dG has a syn glycosidic torsion angle, whereas
both bases of the dGAP:dC base pair are displaced into the major
groove (54). If
dGAP at the pause sites possesses the same conformation, then 1-AP
will occupy the position of the incoming dNTP, thereby blocking catalysis.
Such binary complexes would not be elongated without undergoing dramatic
structural changes and likely represent the form of
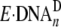 . Interestingly,
an energy minimization study suggests the presence of other conformers in
which 1-AP is either quasi-intercalative or externally bound
(51). These minor conformers
could be stabilized by the interactions between damaged DNA and the active
site residues of Dpo4, which has been observed in comparisons of structures of
polycyclic aromatic hydrocarbons without and with DNA polymerases
(55). The existence of these
conformers is supported by a molecular modeling and simulation study
(5) and two x-ray
crystallographic studies (27,
28). In the first study the
AAF of damaged DNA is situated in the Dpo4 major groove open pocket, and an
anti glycosidic torsion with C1′-exo deoxyribose
conformation allows AAF-dG to be Watson-Crick-paired to dCTP with modest
polymerase perturbation (5).
Such conformation would hinder the translocation of Dpo4 little finger, based
on a series of crystal structures of Dpo4 during DNA polymerization
(10,
21,
56). In the crystal ternary
complexes of Dpo4, DNA containing BPDE and a dNTP
(27,
28), there are two
conformations of the BPDE, one intercalated between base pairs and another
flipped out of the DNA helix into a structural gap between the little finger
and core domains. Additionally, the distance between the 3′-OH of the
primer and the α-phosphate of the incoming dNTP is 9.0 Å if the
bulky BPDE is in the former conformation and 3.9 Å if it is in the
latter conformation. This distance is close to 3.4 Å, the optimum
catalytic distance (28,
57). Thus, if 1-AP is flipped
out as the aforementioned AAF and BPDE at the Dpo4 active site, those
molecules of Dpo4·20/26-mer-dGAP will be in the form of
. Interestingly,
an energy minimization study suggests the presence of other conformers in
which 1-AP is either quasi-intercalative or externally bound
(51). These minor conformers
could be stabilized by the interactions between damaged DNA and the active
site residues of Dpo4, which has been observed in comparisons of structures of
polycyclic aromatic hydrocarbons without and with DNA polymerases
(55). The existence of these
conformers is supported by a molecular modeling and simulation study
(5) and two x-ray
crystallographic studies (27,
28). In the first study the
AAF of damaged DNA is situated in the Dpo4 major groove open pocket, and an
anti glycosidic torsion with C1′-exo deoxyribose
conformation allows AAF-dG to be Watson-Crick-paired to dCTP with modest
polymerase perturbation (5).
Such conformation would hinder the translocation of Dpo4 little finger, based
on a series of crystal structures of Dpo4 during DNA polymerization
(10,
21,
56). In the crystal ternary
complexes of Dpo4, DNA containing BPDE and a dNTP
(27,
28), there are two
conformations of the BPDE, one intercalated between base pairs and another
flipped out of the DNA helix into a structural gap between the little finger
and core domains. Additionally, the distance between the 3′-OH of the
primer and the α-phosphate of the incoming dNTP is 9.0 Å if the
bulky BPDE is in the former conformation and 3.9 Å if it is in the
latter conformation. This distance is close to 3.4 Å, the optimum
catalytic distance (28,
57). Thus, if 1-AP is flipped
out as the aforementioned AAF and BPDE at the Dpo4 active site, those
molecules of Dpo4·20/26-mer-dGAP will be in the form of
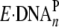 and be rapidly
elongated to Dpo4·21/26-mer. If 1-AP is in the quasi-intercalative
conformation (51), the binary
complex Dpo4·20/26-mer-dGAP requires subtle to mild
structural changes for efficient catalysis and is likely in the form of
and be rapidly
elongated to Dpo4·21/26-mer. If 1-AP is in the quasi-intercalative
conformation (51), the binary
complex Dpo4·20/26-mer-dGAP requires subtle to mild
structural changes for efficient catalysis and is likely in the form of
 . To verify these
structural speculations, we are currently attempting to solve the x-ray
crystal structure of Dpo4·DNA -dGAP.
. To verify these
structural speculations, we are currently attempting to solve the x-ray
crystal structure of Dpo4·DNA -dGAP.
Potential Origin of Enhanced DNA Binding—Notably, Table 2 shows that the binding of Dpo4·20/26-mer-dGAP is about 4-fold tighter than Dpo4·21/26-mer-dGAP, and Dpo4·20/26-mer is the only binary complex affected by 1-AP. This suggests that the 1-AP in Dpo4·20/26-mer-dGAP likely interacted with the residues in the little finger domain of Dpo4, as depicted in structures of Dpo4 with BPDE-dG (27). The binding effect of 1-AP is surprising because DNA lesions usually distort DNA structure and weaken the binding of DNA to a DNA polymerase (17, 26, 58). The tighter binding caused by the interactions between 1-AP and Dpo4 likely promoted catalysis, as 20/26-mer-dGAP was elongated with ∼150-fold higher efficiency than 21/26-mer-dGAP (Table 3). A similar tight binding effect has been observed with human polymerase η and a 3-ring polycyclic aromatic hydrocarbon (59).
Kinetic Effect of dGAP on dNTP Incorporation at Adjacent Sites—Interestingly, Table 3 shows that dGAP did not kinetically affect dNTP incorporations at any downstream positions of the pause sites. In contrast, both an abasic site and a cisplatin-d(GpG) adduct kinetically affected six to seven downstream dNTP incorporations during translesion synthesis catalyzed by Dpo4 (17, 26). These downstream effects have also been observed for the Klenow fragment of E. coli DNA polymerase I replicating through AAF-dG and 8-oxo-dG lesions (60). Such a difference may be a reflection of how each lesion distorts the DNA structure within a DNA polymerase active site.
General Kinetic Mechanism for DNA Lesion Bypass—Similar
biphasic kinetics of dNTP incorporation at pause sites has been observed in
the bypass of an abasic site catalyzed by Dpo4
(17) as well as a
cisplatin-d(GpG) adduct catalyzed by Dpo4
(26) and human
immunodeficiency virus-1 reverse transcriptase
(53). Like the bypass of
dGAP, the total reaction amplitude in each of these cases is much
less than the reaction amplitude obtained with either control DNA or a DNA
substrate at a non-pause site, indicating the existence of
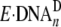 . Some of these
dead-end binary complexes can bind a nucleotide and form dead-end ternary
complexes. Previously, such dead-end ternary complexes have been proposed as
human immunodeficiency virus-1 reverse transcriptase and T7 DNA polymerase
encounter N2-methylguanine
(61),
O6-benzylguanine
(44), and
O6-methylguanine
(44). Because replicative DNA
polymerases have more stringent active sites, the conversion of
. Some of these
dead-end binary complexes can bind a nucleotide and form dead-end ternary
complexes. Previously, such dead-end ternary complexes have been proposed as
human immunodeficiency virus-1 reverse transcriptase and T7 DNA polymerase
encounter N2-methylguanine
(61),
O6-benzylguanine
(44), and
O6-methylguanine
(44). Because replicative DNA
polymerases have more stringent active sites, the conversion of
 to
to
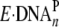 should be
extremely difficult. Thus, the majority of
E·DNAn likely exists in the form of
should be
extremely difficult. Thus, the majority of
E·DNAn likely exists in the form of
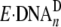 . For example,
the exonuclease-deficient T7 DNA polymerase inefficiently bypasses a
cisplatin-d(GpG) adduct with total reaction amplitudes below 5% at each of the
three consecutive pause sites
(53). Thus,
Fig. 6 is a general mechanism
for DNA lesion bypass catalyzed by several DNA polymerases.
. For example,
the exonuclease-deficient T7 DNA polymerase inefficiently bypasses a
cisplatin-d(GpG) adduct with total reaction amplitudes below 5% at each of the
three consecutive pause sites
(53). Thus,
Fig. 6 is a general mechanism
for DNA lesion bypass catalyzed by several DNA polymerases.
In summary, Dpo4 was shown to be capable of traversing a model single-base lesion dGAP in a kinetically inefficient and error-prone manner. The extension step rather than the insertion opposite dGAP was more challenging. A kinetic mechanism for the dGAP lesion bypass was established via pre-steady-state kinetic analysis.
Supplementary Material
This work was supported, in whole or in part, by National Science Foundation Career Award Grant MCB-0447889 (to Z. S.). This work was also supported by National Institutes of Health Grant ES-09127 (to A. K. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Fig. 1.
Footnotes
The abbreviations used are: BPDE, benzo[a]pyrene diol epoxide; BPDE-dA, benzo[a]pyrene 7,8-diol 9,10-epoxide-derived adduct at the N6 position of 2′-deoxyadenosine; BPDE-dG, benzo[a]pyrene 7,8-diol 9,10-epoxide-derived adduct at the N2 position of 2′-deoxyguanosine; AAF, N-acetyl-2-aminofluorene; AAF-dG, N-acetyl-2-aminofluorene adduct at the C8 position of deoxyguanosine; 1-AP, 1-aminopyrene; cisplatin-d(GpG), cis-[Pt(NH3)2{d(GpG)-N7(1),-N7(2)}] intrastrand cross-link; Dpo4, Sulfolobus solfataricus DNA polymerase IV; dGAP, N-(deoxyguanosin-8-yl)-1-aminopyrene; EMSA, electrophoretic mobility shift assay; 1-NP, 1-nitropyrene.
References
- 1.Pohjola, S. K., Savela, K., Kuusimaki, L., Kanno, T., Kawanishi, M., and Weyand, E. (2004) Polycycl. Aromat. Compd. 25 451-465 [Google Scholar]
- 2.Liu, Y., Yang, Z., Utzat, C. D., Geacintov, N. E., Basu, A. K., and Zou, Y. (2005) Biochem. J. 385 519-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou, Y., Shell, S. M., Utzat, C. D., Luo, C., Yang, Z., Geacintov, N. E., and Basu, A. K. (2003) Biochemistry 42 12654-12661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duvauchelle, J. B., Blanco, L., Fuchs, R. P., and Cordonnier, A. M. (2002) Nucleic Acids Res. 30 2061-2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang, L., and Broyde, S. (2006) Nucleic Acids Res. 34 785-795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunkel, T. A. (2004) J. Biol. Chem. 279 16895-16898 [DOI] [PubMed] [Google Scholar]
- 7.Freisinger, E., Grollman, A. P., Miller, H., and Kisker, C. (2004) EMBO J. 23 1494-1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hogg, M., Wallace, S. S., and Doublie, S. (2004) EMBO J. 23 1483-1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizukami, S., Kim, T. W., Helquist, S. A., and Kool, E. T. (2006) Biochemistry 45 2772-2778 [DOI] [PubMed] [Google Scholar]
- 10.Ling, H., Boudsocq, F., Woodgate, R., and Yang, W. (2001) Cell 107 91-102 [DOI] [PubMed] [Google Scholar]
- 11.Fowler, J. D., and Suo, Z. (2006) Chem. Rev. 106 2092-2110 [DOI] [PubMed] [Google Scholar]
- 12.Kokoska, R. J., Bebenek, K., Boudsocq, F., Woodgate, R., and Kunkel, T. A. (2002) J. Biol. Chem. 277 19633-19638 [DOI] [PubMed] [Google Scholar]
- 13.Fiala, K. A., Sherrer, S. M., Brown, J. A., and Suo, Z. (2008) Nucleic Acids Res. 36 1990-2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiala, K. A., and Suo, Z. (2004) Biochemistry 43 2116-2125 [DOI] [PubMed] [Google Scholar]
- 15.Fiala, K. A., and Suo, Z. (2004) Biochemistry 43 2106-2115 [DOI] [PubMed] [Google Scholar]
- 16.Ling, H., Boudsocq, F., Woodgate, R., and Yang, W. (2004) Mol. Cell 13 751-762 [DOI] [PubMed] [Google Scholar]
- 17.Fiala, K. A., Hypes, C. D., and Suo, Z. (2007) J. Biol. Chem. 282 8188-8198 [DOI] [PubMed] [Google Scholar]
- 18.Fiala, K. A., and Suo, Z. (2007) J. Biol. Chem. 282 8199-8206 [DOI] [PubMed] [Google Scholar]
- 19.Kokoska, R. J., McCulloch, S. D., and Kunkel, T. A. (2003) J. Biol. Chem. 278 50537-50545 [DOI] [PubMed] [Google Scholar]
- 20.Zang, H., Irimia, A., Choi, J. Y., Angel, K. C., Loukachevitch, L. V., Egli, M., and Guengerich, F. P. (2006) J. Biol. Chem. 281 2358-2372 [DOI] [PubMed] [Google Scholar]
- 21.Rechkoblit, O., Malinina, L., Cheng, Y., Kuryavyi, V., Broyde, S., Geacintov, N. E., and Patel, D. J. (2006) PLoS Biol. 4 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zang, H., Goodenough, A. K., Choi, J. Y., Irimia, A., Loukachevitch, L. V., Kozekov, I. D., Angel, K. C., Rizzo, C. J., Egli, M., and Guengerich, F. P. (2005) J. Biol. Chem. 280 29750-29764 [DOI] [PubMed] [Google Scholar]
- 23.Johnson, R. E., Prakash, L., and Prakash, S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 12359-12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boudsocq, F., Iwai, S., Hanaoka, F., and Woodgate, R. (2001) Nucleic Acids Res. 29 4607-4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ling, H., Boudsocq, F., Plosky, B. S., Woodgate, R., and Yang, W. (2003) Nature 424 1083-1087 [DOI] [PubMed] [Google Scholar]
- 26.Brown, J. A., Newmister, S. A., Fiala, K. A., and Suo, Z. (2008) Nucleic Acids Res. 36 3867-3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer, J., Xing, G., Yagi, H., Sayer, J. M., Jerina, D. M., and Ling, H. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 14905-14910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling, H., Sayer, J. M., Plosky, B. S., Yagi, H., Boudsocq, F., Woodgate, R., Jerina, D. M., and Yang, W. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 2265-2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pohjola, S. K., Lappi, M., Honkanen, M., Rantanen, L., and Savela, K. (2003) Mutagenesis 18 429-438 [DOI] [PubMed] [Google Scholar]
- 30.Mitchelmore, C. L., Livingstone, D. R., and Chipman, J. K. (1998) Biomarkers 3 21-33 [Google Scholar]
- 31.Sabbioni, G., and Jones, C. R. (2002) Biomarkers 7 347-421 [DOI] [PubMed] [Google Scholar]
- 32.Rafil, F., Franklin, W., Heflich, R. H., and Cerniglia, C. E. (1991) Appl. Environ. Microbiol. 57 962-968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malia, S. A., Vyas, R. R., and Basu, A. K. (1996) Biochemistry 35 4568-4577 [DOI] [PubMed] [Google Scholar]
- 34.Hirose, M., Lee, M. S., Wang, C. Y., and King, C. M. (1984) Cancer Res. 44 1158-1162 [PubMed] [Google Scholar]
- 35.El-Bayoumy, K., Hecht, S. S., Sackl, T., and Stoner, G. D. (1984) Carcinogenesis 5 1449-1452 [DOI] [PubMed] [Google Scholar]
- 36.Malia, S. A., and Basu, A. K. (1995) Biochemistry 34 96-104 [DOI] [PubMed] [Google Scholar]
- 37.Hatanaka, N., Yamazaki, H., Oda, Y., Guengerich, F. P., Nakajima, M., and Yokoi, T. (2001) Mutat. Res. 497 223-233 [DOI] [PubMed] [Google Scholar]
- 38.She, Q., Singh, R. K., Confalonieri, F., Zivanovic, Y., Allard, G., Awayez, M. J., Chan-Weiher, C. C., Clausen, I. G., Curtis, B. A., De Moors, A., Erauso, G., Fletcher, C., Gordon, P. M., Heikamp-de Jong, I., Jeffries, A. C., Kozera, C. J., Medina, N., Peng, X., Thi-Ngoc, H. P., Redder, P., Schenk, M. E., Theriault, C., Tolstrup, N., Charlebois, R. L., Doolittle, W. F., Duguet, M., Gaasterland, T., Garrett, R. A., Ragan, M. A., Sensen, C. W., and Van der Oost, J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 7835-7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan, P. (1996) Toxic Rep. Ser. 34 1-D2 [PubMed] [Google Scholar]
- 40.Rechkoblit, O., Malinina, L., Chen, Y., Kuryavyi, V., Broyde, S., Geacintov, N. E., and Patel, D. J. (2006) PLOS Biol. 4 25-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zang, H., Chowdhury, G., Angel, K. C., Harris, T. M., and Guengerich, F. P. (2006) Chem. Res. Toxicol. 19 859-867 [DOI] [PubMed] [Google Scholar]
- 42.Eoff, R. L., Irimia, A., Egli, M., and Guengerich, F. P. (2007) J. Biol. Chem. 282 1456-1467 [DOI] [PubMed] [Google Scholar]
- 43.Eoff, R. L., Angel, K. C., Egli, M., and Guengerich, F. P. (2007) J. Biol. Chem. 282 13573-13584 [DOI] [PubMed] [Google Scholar]
- 44.Woodside, A. M., and Guengerich, F. P. (2002) Biochemistry 41 1039-1050 [DOI] [PubMed] [Google Scholar]
- 45.Ohashi, E., Bebenek, K., Matsuda, T., Feaver, W. J., Gerlach, V. L., Friedberg, E. C., Ohmori, H., and Kunkel, T. A. (2000) J. Biol. Chem. 275 39678-39684 [DOI] [PubMed] [Google Scholar]
- 46.Zhang, Y., Yuan, F., Xin, H., Wu, X., Rajpal, D. K., Yang, D., and Wang, Z. (2000) Nucleic Acids Res. 28 4147-4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masutani, C., Kusumoto, R., Yamada, A., Dohmae, N., Yokoi, M., Yuasa, M., Araki, M., Iwai, S., Takio, K., and Hanaoka, F. (1999) Nature 399 700-704 [DOI] [PubMed] [Google Scholar]
- 48.Levine, R. L., Miller, H., Grollman, A., Ohashi, E., Ohmori, H., Masutani, C., Hanaoka, F., and Moriya, M. (2001) J. Biol. Chem. 276 18717-18721 [DOI] [PubMed] [Google Scholar]
- 49.Wooster, R., Cleton-Jansen, A. M., Collins, N., Mangion, J., Cornelis, R. S., Cooper, C. S., Gusterson, B. A., Ponder, B. A., von Deimling, A., Wiestler, O. D., Cornelisse, C. J., Deville, P., and Stratton, M. R. (1994) Nat. Genet. 6 152-156 [DOI] [PubMed] [Google Scholar]
- 50.Watt, D. L., Utzat, C. D., Hilario, P., and Basu, A. K. (2007) Chem. Res. Toxicol. 20 1658-1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nolan, S. J., Vyas, R. R., Hingerty, B. E., Ellis, S., Broyde, S., Shapiro, R., and Basu, A. K. (1996) Carcinogenesis 17 133-144 [DOI] [PubMed] [Google Scholar]
- 52.Fiala, K. A., Brown, J. A., Ling, H., Kshetry, A. K., Zhang, J., Taylor, J. S., Yang, W., and Suo, Z. (2007) J. Mol. Biol. 365 590-602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suo, Z., Lippard, S. J., and Johnson, K. A. (1999) Biochemistry 38 715-726 [DOI] [PubMed] [Google Scholar]
- 54.Gu, Z., Gorin, A., Krishnasamy, R., Hingerty, B. E., Basu, A. K., Broyde, S., and Patel, D. J. (1999) Biochemistry 38 10843-10854 [DOI] [PubMed] [Google Scholar]
- 55.Broyde, S., Wang, L., Zhang, L., Rechkoblit, O., Geacintov, N. E., and Patel, D. J. (2008) Chem. Res. Toxicol. 21 45-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong, J. H., Fiala, K. A., Suo, Z., and Ling, H. (2008) J. Mol. Biol. 379 317-330 [DOI] [PubMed] [Google Scholar]
- 57.Batra, V. K., Beard, W. A., Shock, D. D., Krahn, J. M., Pedersen, L. C., and Wilson, S. H. (2006) Structure 14 757-766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Einolf, H. J., and Guengerich, F. P. (2001) J. Biol. Chem. 276 3764-3771 [DOI] [PubMed] [Google Scholar]
- 59.Choi, J. Y., and Guengerich, F. P. (2005) J. Mol. Biol. 352 72-90 [DOI] [PubMed] [Google Scholar]
- 60.Miller, H., and Grollman, A. P. (1997) Biochemistry 36 15336-15342 [DOI] [PubMed] [Google Scholar]
- 61.Choi, J. Y., and Guengerich, F. P. (2004) J. Biol. Chem. 279 19217-19229 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.