

Abstract
The formation of cysteine-sulfinic acid has recently become appreciated as a modification that links protein function to cellular oxidative status. Human DJ-1, a protein associated with inherited parkinsonism, readily forms cysteine-sulfinic acid at a conserved cysteine residue (Cys106 in human DJ-1). Mutation of Cys106 causes the protein to lose its normal protective function in cell culture and model organisms. However, it is unknown whether the loss of DJ-1 protective function in these mutants is due to the absence of Cys106 oxidation or the absence of the cysteine residue itself. To address this question, we designed a series of substitutions at a proximal glutamic acid residue (Glu18) in human DJ-1 that alter the oxidative propensity of Cys106 through changes in hydrogen bonding. We show that two mutations, E18N and E18Q, allow Cys106 to be oxidized to Cys106-sulfinic acid under mild conditions. In contrast, the E18D mutation stabilizes a cysteine-sulfenic acid that is readily reduced to the thiol in solution and in vivo. We show that E18N and E18Q can both partially substitute for wild-type DJ-1 using mitochondrial fission and cell viability assays. In contrast, the oxidatively impaired E18D mutant behaves as an inactive C106A mutant and fails to protect cells. We therefore conclude that formation of Cys106-sulfinic acid is a key modification that regulates the protective function of DJ-1.
Reactive cysteine residues are susceptible to a variety of covalent modifications that are increasingly recognized as a major means of regulating the activities of many proteins (1). Cysteine forms three different species by the direct addition of oxygen; cysteine-sulfenic (-SOH), -sulfinic (-SO2H), and -sulfonic (-SO3H) acid. Because cysteine can be oxidized to three distinct species, each with different structural and chemical properties, cysteine oxidation is a versatile way for reactive oxygen species (ROS)4 to alter the activity of a protein. Of the three oxidation products of cysteine, only cysteine-sulfenic acid is readily reduced to the thiol under physiological conditions. However, enzymes that catalyze the ATP-dependent reduction of overoxidized peroxiredoxins containing cysteine-sulfinic acid to cysteine have been discovered and characterized (2, 3). With reversibility comes the potential for cysteine-sulfinic acid modifications to modulate the function of various target proteins in a redox-dependent manner. Therefore, at least in some proteins, cysteine-sulfinic acid should be regarded as a post-translational modification rather than simply a type of protein damage.
As expected, many of the proteins that are modified by cysteine oxidation are involved in the oxidative stress response or in the maintenance of cellular redox homeostasis. Of these proteins, DJ-1 has special importance in understanding the role of regulatory cysteine oxidation in neuronal survival. Loss of function mutations in DJ-1 are a rare cause of early onset recessive parkinsonism (4, 5), although the exact function of DJ-1 is unclear. The protein is part of the large DJ-1 superfamily with evolutionarily conserved members in bacteria, fungi, plants, and animals (6, 7). A number of activities have been proposed for human DJ-1, including a weak peroxiredoxin-like activity (8), a chaperone activity (9, 10), and translational (11, 12) and transcriptional regulation (13, 14).
The best-established aspect of DJ-1 function is its ability to respond to
oxidative stress. DJ-1 is modified under oxidative stress both in
vitro and in vivo by oxidation of a very highly conserved
cysteine residue (Cys106 in human DJ-1) to form a cysteine-sulfinic
acid (Cys106-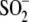 )
(15). Several studies have
shown that of the three cysteine residues in human DJ-1, Cys106 is
the most prone to oxidative modification
(8,
16,
17). In addition,
Cys106 has a low pKa value of 5.4 and therefore
exists almost exclusively as the highly reactive cysteine thiolate anion at
physiological pH (18).
Replacement of Cys106 with other amino acids in DJ-1 results in a
loss of protective activity against oxidative stressors in a number of systems
(15,
19,
20).
)
(15). Several studies have
shown that of the three cysteine residues in human DJ-1, Cys106 is
the most prone to oxidative modification
(8,
16,
17). In addition,
Cys106 has a low pKa value of 5.4 and therefore
exists almost exclusively as the highly reactive cysteine thiolate anion at
physiological pH (18).
Replacement of Cys106 with other amino acids in DJ-1 results in a
loss of protective activity against oxidative stressors in a number of systems
(15,
19,
20).
We have therefore previously suggested that formation of cysteine-sulfinic acid is required for DJ-1 to exert its protective effects (15). However, the substitution of the equivalent cysteine residue in Drosophila melanogaster DJ-1 with aspartic acid (C104D in Drosophila) inactivates DJ-1, suggesting that the simple addition of a negatively charged residue at this position is insufficient to support DJ-1 function (21). As a consequence of the need for direct mutation of Cys106 in these studies, it is unclear whether the cysteine residue itself or its oxidation to a sulfinic acid is critical for the protective activity of DJ-1.
We have previously shown using x-ray crystallography that
Cys106-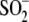 interacts with
nearby residues, most notably the highly conserved Glu18 residue
(15). In reduced DJ-1, the
carboxylic acid side chain of Glu18 is protonated and donates a
hydrogen bond to Cys106, which depresses the thiol
pKa value
(18). Therefore, we
hypothesized that modifying the environment around the side chain of
Cys106 could decouple the oxidation propensity and
pKa of Cys106 without changing the cysteine
residue itself. In the present study, we have tested this approach by
characterizing the effect of Glu18 mutations on the oxidative
propensity and cytoprotective activity of Cys106. Our results show
that the formation of
Cys106-
interacts with
nearby residues, most notably the highly conserved Glu18 residue
(15). In reduced DJ-1, the
carboxylic acid side chain of Glu18 is protonated and donates a
hydrogen bond to Cys106, which depresses the thiol
pKa value
(18). Therefore, we
hypothesized that modifying the environment around the side chain of
Cys106 could decouple the oxidation propensity and
pKa of Cys106 without changing the cysteine
residue itself. In the present study, we have tested this approach by
characterizing the effect of Glu18 mutations on the oxidative
propensity and cytoprotective activity of Cys106. Our results show
that the formation of
Cys106-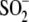 is critical for
DJ-1 to protect cells against mitochondrial damage. In addition, this targeted
mutagenesis strategy could be used to manipulate the oxidation state of other
cysteine redox-regulated proteins of known structure.
is critical for
DJ-1 to protect cells against mitochondrial damage. In addition, this targeted
mutagenesis strategy could be used to manipulate the oxidation state of other
cysteine redox-regulated proteins of known structure.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—Wild-type and mutant DJ-1 variants were cloned between the NdeI and XhoI sites of the bacterial expression vector pET21a and expressed in BL21(DE3) Escherichia coli (Novagen). All proteins were expressed with a noncleavable C-terminal His6 tag (vector-derived sequence LEHHHHHH) for purification by metal affinity Ni2+-nitrilotriacetic acid chromatography. Bacteria were grown in LB medium supplemented with 100 μg/ml ampicillin at 37 °C with shaking. Once the A600 of the culture reached 0.5-0.7, it was equilibrated at 20 °C for 3 h prior to induction of protein expression by the addition of 0.1 mm isopropyl β-d-1-thiogalactopyranoside. The induced culture was incubated at 20 °C with shaking overnight and harvested by centrifugation. Cell pellets were stored at -80 °C until needed.
Recombinant His6-tagged proteins were purified using Ni2+-nitrilotriacetic acid His-Select resin (Sigma). Eluted DJ-1 protein was dialyzed against storage buffer (25 mm HEPES, pH 7.5, 100 mm KCl, 1 mm dithiothreitol (DTT)), loaded onto an equilibrated High Q anion exchange column, and collected in the flow-through, since contaminants bind to the anion exchange resin under these conditions. Purified DJ-1 was concentrated to 1 mm (ε280 = 4000 m-1 cm-1) and ran as a single band on overloaded Coomassie-stained SDS-PAGE. The purified protein was supplemented with 2 mm DTT, snap-frozen on liquid nitrogen, and stored at -80 °C.
Hydrogen Peroxide Titration of DJ-1—The in vitro oxidative susceptibility of Cys106 in DJ-1 was assayed by titration with several molar ratios of H2O2 to protein monomer. Thawed DJ-1 was rapidly exchanged into extensively degassed nanopure water using a centrifugal spin column containing P6-DG desalting resin (Bio-Rad). Control experiments using degassed buffered solutions (10 mm potassium phosphate, pH 7.4) instead of water showed similar oxidative behavior of DJ-1 but gave noisier mass spectrometry data due to the presence of buffer salts. Freshly diluted H2O2 was added to DJ-1 in molar ratios of 0:1, 0.5:1, 1:1, 2.5:1, 5:1, 7.5:1, and 10:1 H2O2/protein monomer and incubated on ice for 30 min. Excess H2O2 was removed using a P6-DG centrifugal desalting column, and the protein samples were immediately supplemented with 5 mm DTT, frozen on liquid nitrogen, and stored at -80 °C. DTT, which cannot reduce cysteine-sulfinic acid, was added to ensure that the sample did not further oxidize during sample handling for mass spectrometric analysis. Previous results have indicated that only Cys106 is oxidized by these conditions in vitro (10).
Mass Spectrometry of Oxidized DJ-1—Intact DJ-1 protein was
analyzed by liquid chromatography-mass spectrometry (LC-MS/MS) with a 4000
Q-trap mass spectrometer (ABS) using a turbo ion spray source probe at the
University of Nebraska Redox Biology Center Mass Spectrometry Core Facility.
Protein samples (20 μl) were loaded onto a C18 reverse phase column using a
PE 200 Autosampler. A SCL-10A high performance liquid chromatography system
(Shimadzu) was used for room temperature gradient elution at a flow rate of
100 ml/min in 5 min by using a linear gradient from 0.3% formic acid in water
(Solvent A) to 0.3% formic acid in acetonitrile (Solvent B). The elution time
for DJ-1 was about 4 min. Data were acquired and processed using Analyst 1.4.1
software in Q1 (quadrupole one)-positive ion mode, and the
m/z range of 880-1120 atomic mass units was scanned in 4 s.
The total run time for each sample was 10 min. The molecular mass of protein
was generated from several multiply charged peaks using the Bayesian Protein
Reconstruct option in BioAnalyst 1.4 software. For all experiments, only two
species were observed: the reduced protein and an adduct at +32 atomic mass
units, corresponding to the
Cys106-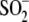 form of DJ-1.
form of DJ-1.
Crystallization, Data Collection, and Processing—For all crystallization experiments, DJ-1 at 1 mm (20 mg/ml) in storage buffer was crystallized using the hanging drop vapor diffusion method with drops containing 2 μl of protein and 2 μl of reservoir solution. Crystals of E18Q DJ-1 in space group P3121 were grown in 2-5 days at room temperature using a reservoir solution of 30% polyethylene glycol 400, 50 mm HEPES, pH 7.5, 125 mm sodium citrate. Crystals of E18D DJ-1 in space group P3121 were grown from 1.3-1.5 m sodium citrate, 50 mm HEPES, pH 7.5. For crystals of E18Q DJ-1, the 30% polyethylene glycol 400 in the mother liquor was sufficient for cryoprotection. E18D DJ-1 crystals were cryoprotected in 2.4 m sodium malonate, pH 7.0 (22). All crystals were transferred to nylon loops and cryocooled by direct immersion into liquid nitrogen.
Diffraction data were collected at the Advanced Photon Source, BioCARS beamline 14BM-C using 13.776 keV (0.9 Å) incident x-rays and an ADSC Q315 detector. Single crystals maintained at 100 K were used for the collection of each data set, and both data sets were collected in separate high and a low resolution passes with differing exposure times, oscillation ranges, and detector distances to record the full dynamic range of the diffraction data. To avoid radiation-induced damage to Cys106, the x-ray beam was attenuated, and the crystal was exposed to x-rays for 5 s or less per 1° oscillation. Diffraction data were integrated and scaled using HKL2000 (23), and final data statistics for each data set are provided in Table 1.
TABLE 1.
Data collection and refinement statistics
| Oxidized E18D DJ-1 | Oxidized E18Q DJ-1 | |
|---|---|---|
| Data collection | ||
| Space group | P3121 | P3121 |
| Cell dimensions | ||
| a = b, c (Å) | 74.75, 74.80 | 74.77, 74.79 |
| Resolution (Å)a | 30-1.20 (1.24-1.20) | 30-1.15 (1.18-1.15) |
| Rmergeb | 0.066 (0.640) | 0.092 (0.379) |
| <I>/<σ(I)> | 32.0 (3.7) | 25.7 (6.9) |
| Completeness (%) | 99.8 (100) | 99.8 (100) |
| Redundancy | 9.9 (9.5) | 10.0 (9.8) |
| Refinement | ||
| Resolution (Å) | 30-1.20 | 30-1.15 |
| No. of reflections | 75,731 | 85,756 |
| Rwork; Rwork for Fo > 4σ(Fo) (%)c | 12.2; 11.0 | 11.6; 10.0 |
| Rfree; Rfree for Fo > 4σ(Fo) (%)d | 15.5; 14.2 | 14.5; 13.6 |
| Rall; Rall for Fo > 4σ(Fo) (%)e | 12.4; 11.1 | 11.6; 10.0 |
| No. of atoms | ||
| Protein | 1458 | 1439 |
| Water | 261 | 288 |
| Beq factors (Å2) | ||
| Protein | 15.0 | 16.6 |
| Water | 34.9 | 32.6 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.014 | 0.015 |
| Bond angle 1-3 distances (Å) | 0.030 | 0.030 |
Values in parentheses are for the highest resolution shell.
Rmerge =
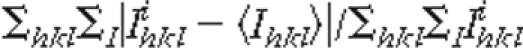 , where i is the
ith observation of a reflection with indices, h, k,
and l, and angle brackets indicate the average over all i
observations.
, where i is the
ith observation of a reflection with indices, h, k,
and l, and angle brackets indicate the average over all i
observations.
Rwork =
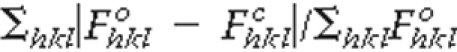 , where
, where
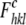 is the calculated structure factor
amplitude with indices h, k, and l.
is the calculated structure factor
amplitude with indices h, k, and l.
Rfree is calculated as Rwork, where
the 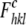 values are taken from a test set
comprising 5% of the data that were excluded from the refinement
(25).
values are taken from a test set
comprising 5% of the data that were excluded from the refinement
(25).
Rall is calculated as Rwork, where
the 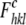 values include all measured data
(including the Rfree test set).
values include all measured data
(including the Rfree test set).
Crystal Structure Refinement—SHELX-97 was used for refinement of coordinates and atomic displacement parameters against a least squares intensity-based residual target function (24). All refinements excluded a test set of 5% of randomly chosen reflections that were sequestered and used for the calculation of the Rfree value (25). For both structures, initial rigid body refinement at 2.5 Å resolution using coordinates for human DJ-1 (Protein Data Bank code 1P5F) (26) was followed by multiple cycles of restrained refinement of coordinates and isotropic B-factors at 1.5 Å resolution using a stepwise increase in resolution (STIR instruction). Manual adjustments to the model were made by inspection of 2mFo - DFc and mFo - DFc electron density maps using the program COOT (27). Additional cycles of conjugate gradient refinement were performed after inclusion of all data to the full resolution limit of each data set. In the later stages of refinement, anisotropic atomic displacement parameters were introduced, resulting in a 3-5% decrease in both R and Rfree. The final cycles of refinement were performed with riding hydrogen atoms (excluding the hydrogen atoms on Oγ of serine, O of tyrosine, Oγ1 of threonine, and Nδ1 of histidine), followed by inclusion of the test set data into the refinement. The models were validated with MolProbity (28) and the validation tools in COOT (27). The only Ramachandran plot outlier is Cys106, which is invariably in marginal or outlying regions of the Ramachandran plot in DJ-1 structures.
Atomic resolution bond length analysis of residue 18 was performed using unrestrained full matrix least-squares refinement in SHELX-97 as previously described (18). Estimated standard uncertainties on coordinates and bond lengths were determined by inversion of the blocked (BLOC 1 instruction) full least squares matrix. Final model statistics are provided in Table 1.
Mouse Embryonic Fibroblasts (MEFs) and Transfections—Fibroblast cultures were established from wild-type and DJ-1 knock-out mice (29). Expression constructs for human DJ-1 containing a C-terminal V5 tag have been described previously (13). Additional mutations (E18N/Q/D and post-V5 stop codon) were generated using QuikChange mutagenesis (Stratagene). Cells were transfected using Lipofectamine 2000 (Invitrogen).
Immunocytochemistry and Western Blotting—Mitochondrial fractions were prepared using a commercially available mitochondrial isolation kit (Pierce) as directed. Mitochondrial fractions were then stripped of all loosely associated proteins using 20 μm sodium carbonate in HEPES buffer for 30 min on ice, followed by ultracentrifugation at 60,000 × g for 30 min. Subcellular fractions were Western blotted and probed with a V5-specific antibody (Invitrogen) to visualize transfected DJ-1. Enrichment of mitochondria was confirmed by simultaneously reprobing the same blots with monoclonal antibodies to the voltage-dependent anion channel (VDAC1; Calbiochem clone 31HL, 1:4000) and to cytosolic β-actin (clone AC-15, 1:5000; Sigma). Immunostaining for tagged DJ-1 was performed as described (30). Paraquat and rotenone were purchased from Sigma.
Two-dimensional Gel Electrophoresis—Two-dimensional gel electrophoresis was performed as previously described (15). Samples were obtained from human M17 neuroblastoma cells that were transiently transfected with various DJ-1 Glu18 mutants. For all DJ-1 constructs, stop codons introduced immediately following the V5 tag were used to avoid changes in pI due to the His6 tag. Immobiline DryStrips (11 cm) with linear separation in the pH range 4-7 (GE Healthcare) were used for isoelectric focusing.
Fluorescence Recovery after Photobleaching (FRAP) Measurement of Mitochondrial Fragmentation—FRAP was performed as previously described (31, 32). Cells were transiently transfected with 0.5 mg of mitochondrial matrix-localized yellow fluorescent protein using Fugene and seeded into Lab-Tek borosilicate chambers in phenol red-free Opti-MEM. Circular 25-pixel diameter regions of mitochondria were imaged using an LSM 510 confocal microscope with a Plan-Apochromat ×100/1.4 objective (Zeiss) before and after photobleaching at 100% power with 488- and 514-nm wavelength lasers. Scans were taken in 300-ms intervals, for a total of 40 images over 12 s, and the fluorescence intensity was observed over time. Fluorescence recovery was represented as a fraction of initial fluorescence after normalization to both nonspecific photobleaching and background. Each FRAP curve represented the average of ≥60 measurements over at least two independent experiments. Mobile fractions were calculated as ((FRAPt - background)/FRAPt) × ((NSPBi - background)/NSPBt), where NSPB is the nonspecific photobleaching, the subscript t refers to the signal at time = t and the subscript i refers to the initial signal before photobleaching.
Cell Viability—M17 cells were seeded onto glass coverslips and transiently transfected with V5-tagged DJ-1 variants using Lipofectamine 2000 (Invitrogen) for 48 h and then either left untreated or exposed to rotenone (200 nm) for 24 h. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline, permeabilized with 0.1% Triton X-100, and stained using monoclonal antibody to V5 (1:500; Invitrogen), followed by anti-mouse IgG conjugated to Alexa-Fluor488 (Molecular Probes). Nuclei were counterstained with Hoechst 33342 (Roche Applied Science), and coverslips were mounted using ProLong gold (Molecular Probes). For each experiment, three randomly selected microscope fields (between 26 and 75 cells/field) were counted by an observer blind to the DJ-1 transfection status of the cells. Each experiment was then repeated three times, and statistical analysis was performed on the combined results. Cell viability was expressed as the percentage of transfected (V5-positive) cells that had intact nuclei compared with all transfected cells. For base line viability in the same cultures, we counted three fields of untransfected cells in the same way, where viability was expressed as percentage of visible nuclei per field.
RESULTS
Substitutions at Residue 18 Alter the Oxidation Propensity of
Cys106—Three substitutions were made at residue 18 in
human DJ-1 for this study; E18Q, E18D, and E18N. The 1.15 Å resolution
crystal structure of oxidized E18Q DJ-1 superimposes nearly perfectly
(Cα root mean square deviation = 0.08 Å) with wild-type DJ-1
(Protein Data Bank code 1SOA). In addition, like the wild-type protein
(15), E18Q DJ-1 oxidizes
during crystal growth to form
Cys106-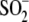 (Fig. 1B). There are a
few small structural differences between E18Q and wild-type DJ-1 near the site
of mutation, the most notable being the lengthening of the hydrogen bond
between residue 18 and
Cys106-
(Fig. 1B). There are a
few small structural differences between E18Q and wild-type DJ-1 near the site
of mutation, the most notable being the lengthening of the hydrogen bond
between residue 18 and
Cys106-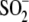 to 2.71 Å
(Fig. 1C). In
wild-type DJ-1, the 2.47-Å hydrogen bond between the protonated
Glu18 carboxylic acid side chain and
Cys106-
to 2.71 Å
(Fig. 1C). In
wild-type DJ-1, the 2.47-Å hydrogen bond between the protonated
Glu18 carboxylic acid side chain and
Cys106-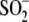 is an unusually
short and presumably a very strong interaction
(15). The 0.24-Å
increase in hydrogen bond length in E18Q
Cys106-
is an unusually
short and presumably a very strong interaction
(15). The 0.24-Å
increase in hydrogen bond length in E18Q
Cys106-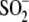 DJ-1 is
accommodated by the correlated displacements of
Cys106-
DJ-1 is
accommodated by the correlated displacements of
Cys106-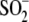 and
Gln18 away from each other (Fig.
1C). Because the E18Q substitution is very structurally
conservative, altered oxidation for Cys106 in E18Q (see below) can
be attributed solely to the changes in the hydrogen bond between residue 18
and Cys106.
and
Gln18 away from each other (Fig.
1C). Because the E18Q substitution is very structurally
conservative, altered oxidation for Cys106 in E18Q (see below) can
be attributed solely to the changes in the hydrogen bond between residue 18
and Cys106.
FIGURE 1.

Structural effects of mutations designed to test the hypothesis that
Cys106-sulfinic acid formation is critical to DJ-1 function.
A, a ribbon representation of the DJ-1 dimer, with one
monomer in brown and the other in green. The dimer 2-fold
axis is perpendicular to the page and indicated by an
ellipse. The oxidationprone cysteine (C106) and the
interacting glutamic acid (E18) are represented in each monomer.
B, electron density for the 1.15 Å resolution structure of E18Q
DJ-1 around Cys106 is shown at the 1σ contour level and
calculated with σA weighted coefficients
2mFo - DFc. In E18Q DJ-1,
Cys106 is oxidized to the cysteine-sulfinic acid, where stabilizing
hydrogen bonds are shown as dotted lines with distances given in
Å. C, a superposition of oxidized E18Q (darker model)
and wild-type DJ-1 (lighter model) shows that the key stabilizing
hydrogen bond between residue 18 and
Cys106-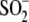 is lengthened in
E18Q DJ-1, weakening this interaction. D, 2mFo -
DFc electron density contoured at 1σ is shown in
blue for the 1.20 Å resolution crystal structure of E18D DJ-1.
Cys106 is oxidized to the easily reduced
Cys106-SO- oxidation product in this variant. In
addition, there is minor electron density that is consistent with either
Cys106-
is lengthened in
E18Q DJ-1, weakening this interaction. D, 2mFo -
DFc electron density contoured at 1σ is shown in
blue for the 1.20 Å resolution crystal structure of E18D DJ-1.
Cys106 is oxidized to the easily reduced
Cys106-SO- oxidation product in this variant. In
addition, there is minor electron density that is consistent with either
Cys106-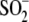 or an alternate
conformation for Cys106-SO-. E, a superposition
of residues in the vicinity of Cys106 in E18D DJ-1 (darker
model) and the corresponding region in oxidized wild-type DJ-1
(lighter model). The E18D substitution results in structural
perturbations at Cys106 that stabilize the
Cys106-SO- oxidation product and hinder further
oxidation. All figures were created using POVscript+
(40).
or an alternate
conformation for Cys106-SO-. E, a superposition
of residues in the vicinity of Cys106 in E18D DJ-1 (darker
model) and the corresponding region in oxidized wild-type DJ-1
(lighter model). The E18D substitution results in structural
perturbations at Cys106 that stabilize the
Cys106-SO- oxidation product and hinder further
oxidation. All figures were created using POVscript+
(40).
The crystal structure of oxidized E18N was determined in a previous study
and showed that Cys106 is robustly oxidized to the sulfinic acid,
even when these crystals were grown from solutions containing 10 mm
DTT that favored reduced Cys106 in wild-type DJ-1
(18). Asn18 was
discretely disordered in this structure but made a similar set of hydrogen
bonds with the oxygen atoms of
Cys106-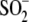 (18). Importantly, we have
never been able to successfully grow crystals of E18N DJ-1 with a reduced
Cys106 residue, and the recombinant protein is difficult to purify
in its reduced form (18),
suggesting very facile oxidation to
Cys106-
(18). Importantly, we have
never been able to successfully grow crystals of E18N DJ-1 with a reduced
Cys106 residue, and the recombinant protein is difficult to purify
in its reduced form (18),
suggesting very facile oxidation to
Cys106-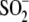 in the E18N DJ-1
mutant.
in the E18N DJ-1
mutant.
The power of using semiconservative substitutions at residue 18 to control
the oxidation state of Cys106 is best illustrated by E18D DJ-1. The
crystal structure of E18D DJ-1 at 1.20 Å resolution reveals that
Cys106 is oxidized predominantly to cysteine-sulfenic acid
(Cys106-SO-) in the crystal rather than the
cysteine-sulfinic acid
(Cys106-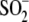 ) that is
observed for wild-type, E18N, and E18Q DJ-1
(Fig. 1D). The oxygen
atom of Cys106-SO- is stabilized by a 2.88-Å
hydrogen bond to the carboxylic acid side chain of Asp18.
Inspection of the 2mFo - DFc electron
density also shows an additional feature near Cys106 that is
consistent with either minor oxidation to
Cys106-
) that is
observed for wild-type, E18N, and E18Q DJ-1
(Fig. 1D). The oxygen
atom of Cys106-SO- is stabilized by a 2.88-Å
hydrogen bond to the carboxylic acid side chain of Asp18.
Inspection of the 2mFo - DFc electron
density also shows an additional feature near Cys106 that is
consistent with either minor oxidation to
Cys106-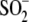 or a second low
occupancy conformation for Cys106-SO-
(Fig. 1D). In
addition, the oxygen atom of Cys106-SO- has an elevated
equivalent isotropic B-factor compared with surrounding residues (32
Å2 for O versus 14 Å2 for
Sγ), indicating reduced occupancy for the oxygen atom due to incomplete
oxidation of Cys106. Unrestrained bond length analysis of
Asp18 (see “Experimental Procedures”) yields a
Cγ-Oδ1 bond length of 1.222 Å (estimated standard
uncertainty = 0.027 Å) and a Cγ-Oδ2 bond length of 1.319
Å (estimated standard uncertainty = 0.024 Å), in good agreement
with standard bond lengths for protonated carboxylic acids
(33). Therefore, like
Glu18 in wild-type DJ-1
(18), Asp18 is
protonated and donates a hydrogen bond to
Cys106-SO-.
or a second low
occupancy conformation for Cys106-SO-
(Fig. 1D). In
addition, the oxygen atom of Cys106-SO- has an elevated
equivalent isotropic B-factor compared with surrounding residues (32
Å2 for O versus 14 Å2 for
Sγ), indicating reduced occupancy for the oxygen atom due to incomplete
oxidation of Cys106. Unrestrained bond length analysis of
Asp18 (see “Experimental Procedures”) yields a
Cγ-Oδ1 bond length of 1.222 Å (estimated standard
uncertainty = 0.027 Å) and a Cγ-Oδ2 bond length of 1.319
Å (estimated standard uncertainty = 0.024 Å), in good agreement
with standard bond lengths for protonated carboxylic acids
(33). Therefore, like
Glu18 in wild-type DJ-1
(18), Asp18 is
protonated and donates a hydrogen bond to
Cys106-SO-.
We propose that the interaction between Asp18 and the initial
Cys106-SO- oxidation product diminishes further
oxidation of Cys106 by displacing Cys106-SO-
toward Asp18, which increases the distance between Sγ of
Cys106 and the Gly75/Ala107 amide pocket
(Fig. 1E). This
structural change disfavors further oxidation to
Cys106-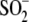 by lengthening
and weakening the hydrogen bonds donated by Gly75 and
Ala107 backbone amides, which would stabilize addition of a second
oxygen atom to form
Cys106-
by lengthening
and weakening the hydrogen bonds donated by Gly75 and
Ala107 backbone amides, which would stabilize addition of a second
oxygen atom to form
Cys106-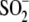 . Because both
Glu18 and the Gly75/Ala107 amide pocket
appear to be important for stabilizing the
Cys106-
. Because both
Glu18 and the Gly75/Ala107 amide pocket
appear to be important for stabilizing the
Cys106-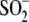 modification in
wild-type DJ-1, subtle structural changes in the relative orientation of
Cys106 have a clearly detrimental impact on
Cys106-
modification in
wild-type DJ-1, subtle structural changes in the relative orientation of
Cys106 have a clearly detrimental impact on
Cys106-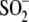 formation. We
note that the more highly oxidized
Cys106-
formation. We
note that the more highly oxidized
Cys106-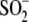 species can be
observed in E18D DJ-1 crystals that have been incubated for multiple weeks
prior to harvesting. Therefore, the E18D substitution results in
Cys106 becoming oxidation-impaired but not completely
oxidation-deficient.
species can be
observed in E18D DJ-1 crystals that have been incubated for multiple weeks
prior to harvesting. Therefore, the E18D substitution results in
Cys106 becoming oxidation-impaired but not completely
oxidation-deficient.
Effects of Glu18 Substitutions on Cys106
Oxidation in Vitro and in Vivo—The ability of each of the mutant
DJ-1 proteins to oxidize at Cys106 in solution was determined using
controlled titration of hydrogen peroxide and electrospray mass spectrometry.
Each mutant oxidizes less robustly than wild-type DJ-1 at higher peroxide
ratios; however, E18N DJ-1 is ∼50% oxidized to
Cys106-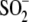 at all molar
ratios of H2O2/DJ-1, including the control with no added
H2O2 (Fig.
2A). Multiple preparations of E18N display this behavior,
even when the protein is freshly purified and immediately used, consistent
with difficulties encountered in purifying and crystallizing reduced E18N
DJ-1. E18Q DJ-1 shows a mild impairment of Cys106 oxidation at
higher peroxide ratios in solution (Fig.
2A), although at low molar ratios of
H2O2/DJ-1 (0.5:1),
Cys106-
at all molar
ratios of H2O2/DJ-1, including the control with no added
H2O2 (Fig.
2A). Multiple preparations of E18N display this behavior,
even when the protein is freshly purified and immediately used, consistent
with difficulties encountered in purifying and crystallizing reduced E18N
DJ-1. E18Q DJ-1 shows a mild impairment of Cys106 oxidation at
higher peroxide ratios in solution (Fig.
2A), although at low molar ratios of
H2O2/DJ-1 (0.5:1),
Cys106-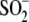 formation was
similar to that of wild-type protein.
formation was
similar to that of wild-type protein.
FIGURE 2.

Substitutions at position 18 of DJ-1 impact
Cys106-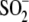 formation. A, oxidation of Cys106 in vitro
to Cys106-
formation. A, oxidation of Cys106 in vitro
to Cys106-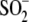 . Mass
spectrometry was used to monitor the oxidation of DJ-1 as a function of
hydrogen peroxide concentration in solution. The fraction of protein oxidized
was calculated as a ratio of the integrated area of the oxidized protein peak
to the total area of both the oxidized and reduced peaks. A comparison of the
oxidation curves of these proteins shows that every substitution at position
18 results in diminished oxidation compared with the wild-type protein,
although the extent of this diminution varies among the three substitutions.
E18D abolishes the ability of Cys106 to be oxidized to
cysteine-sulfinic acid, and E18N oxidizes very easily at low
H2O2 levels. B, oxidation of DJ-1 in
vivo. Human M17 neuroblastoma cells were transfected with V5-tagged
versions of the indicated DJ-1 constructs (wild type (WT), E18N, E18Q, E18D,
and C106A, from top to bottom) and exposed to 300
μm paraquat for 24 h. Protein extracts were separated on
two-dimensional gels and blotted for DJ-1. Estimated pI values for each
isoform are indicated above the blots. Images are
representative of duplicate experiments for each construct.
. Mass
spectrometry was used to monitor the oxidation of DJ-1 as a function of
hydrogen peroxide concentration in solution. The fraction of protein oxidized
was calculated as a ratio of the integrated area of the oxidized protein peak
to the total area of both the oxidized and reduced peaks. A comparison of the
oxidation curves of these proteins shows that every substitution at position
18 results in diminished oxidation compared with the wild-type protein,
although the extent of this diminution varies among the three substitutions.
E18D abolishes the ability of Cys106 to be oxidized to
cysteine-sulfinic acid, and E18N oxidizes very easily at low
H2O2 levels. B, oxidation of DJ-1 in
vivo. Human M17 neuroblastoma cells were transfected with V5-tagged
versions of the indicated DJ-1 constructs (wild type (WT), E18N, E18Q, E18D,
and C106A, from top to bottom) and exposed to 300
μm paraquat for 24 h. Protein extracts were separated on
two-dimensional gels and blotted for DJ-1. Estimated pI values for each
isoform are indicated above the blots. Images are
representative of duplicate experiments for each construct.
In contrast, mass spectrometry of E18D DJ-1 shows no oxidized protein, even at a 10:1 molar ratio of H2O2/DJ-1 (Fig. 2A). Because E18D DJ-1 forms the easily reduced Cys106-SO- species, we propose that the DTT quenching step in the oxidation reaction (see “Experimental Procedures”) may reduce the Cys106-SO- observed in the crystal structure of E18D DJ-1 (Fig. 1D). Therefore, the E18D substitution stabilizes an easily reduced oxidation product at Cys106 that is expected to revert to the thiol in the reducing environment of the cytoplasm.
To evaluate the oxidation of these DJ-1 variants in an in vivo system, we transfected the same DJ-1 mutants into M17 neuroblastoma cells, subjected them to oxidative stress using 300 μm paraquat, and monitored DJ-1 mobility on two-dimensional gels. Using V5-tagged DJ-1 to facilitate electrophoretic separation from endogenous DJ-1, we observed a pattern of multiple pI isoforms that is similar to that found for the endogenous protein (15) but with a pI increase of ∼0.4 units due to the V5 tag on DJ-1. Wild-type DJ-1 showed two major isoforms and other minor oxidized species (Fig. 2B). E18N and E18Q DJ-1 showed a greater amount of the 6.2 pI isoform (corresponding to the endogenous DJ-1 pI of 5.8) compared with the wild-type protein, suggesting increased sulfinic acid formation in these mutants. In contrast, both E18D and C106A showed primarily a single unoxidized pI isoform, with a small proportion of protein at pI = 6.5 (corresponding to the endogenous 6.1 isoform). These data support the in vitro finding that E18D is largely resistant to Cys106 oxidation, whereas E18N and E18Q are somewhat more sensitive to oxidation than wild-type DJ-1.
Relative Cytosolic and Mitochondrial Pools of DJ-1 Are Influenced by Cys106 Modification—DJ-1 is present in nuclear, cytoplasmic, and mitochondrial pools (8). In previous studies, DJ-1 has been shown to protect against mitochondrial damage, which is a major source of oxidative stress in eukaryotic cells (15, 21). We have previously suggested that the mitochondrial pool of DJ-1 responds to oxidative stress and that the oxidative response of DJ-1 correlates with recruitment of the protein to the mitochondria (15), although this has not been confirmed in other studies (34). Therefore, we sought to establish how mutation of Glu18 impacted recruitment of DJ-1 to mitochondria. We transfected DJ-1 variants into M17 neuroblastoma cells and exposed these cells to the oxidant paraquat at a concentration (300 μm) that did not affect cell viability. Using subcellular fractionation, we saw that a pool of wild-type DJ-1 was present in mitochondria and the amount of mitochondrially associated DJ-1 increased under oxidative stress (Fig. 3A).
FIGURE 3.

DJ-1 mutants are present in mitochondrial and cytosolic pools. A, M17 neuroblastoma cells were transfected with V5-tagged WT (lane 1), E18N (lane 2), E18Q (lane 3), E18D (lane 4), C53A (lane 5), or C106A (lane 6) DJ-1 variants. Cells were also subjected to oxidative stress by exposure to 300 μm paraquat (PQ) for 24 h as indicated. Cytosolic fractions (left, top two blots) or mitochondrial proteins retained after carbonate extraction (right, top two blots) were probed for V5-DJ-1. Mitochondrial enrichment was confirmed by simultaneously reprobing the same blots with monoclonal antibodies to VDAC1 and to cytosolic β-actin, as indicated. B, subcellular localization of the V5-tagged DJ-1 variants was verified using transiently transfected M17 neuroblastoma cells that were stained for V5 (green) and mitochondria using Mitotracker (red). Upper panels, untreated cells; middle panels, cells treated with 300 μm paraquat (PQ); lower panels, cells treated with 100 nm rotenone. The scale bar in the lower right panel of the merged images represents 10 μm and applies to all. To show mitochondrial morphology, a higher magnification view of the boxed areas of the rotenone-treated cells is shown in black and white below each set. Although cells transfected with WT, E18N, or E18Q DJ-1 maintained elongated and connected mitochondria in the presence of rotenone, cells transfected with E18D or C106A DJ-1 showed mitochondrial fragmentation.
We confirmed the mitochondrial enrichment of these fractionated samples by Western blot for VDAC1 and β-actin as markers for the mitochondria and cytosol, respectively (Fig. 3A). The relative mitochondrial enrichment of all samples was consistent between DJ-1 variants both in the presence or absence of oxidative stress. However, there is some residual contamination of mitochondrial fractions with the abundant cytosolic β-actin, which is common for cytoskeletal proteins that tend to associate with various organelles. Mitochondrial contamination of cytosolic fractions was minimal based on the relative VDAC1 staining (Fig. 3A).
Both E18N and E18Q DJ-1 were basally associated with mitochondria to a larger extent than the wild-type protein and maintained that association under oxidative stress (Fig. 3A). In contrast, although there is a small pool of E18D and C106A DJ-1 associated with mitochondria, this localization does not respond to paraquat-induced oxidative stress. A recent study showed that some DJ-1 can associate with mitochondria even when Cys106 is mutated to a nonoxidizable serine residue, supporting these observations (35). Considered together, these results indicate that mitochondrial localization of DJ-1 is enhanced by Cys106 oxidation, although oxidation of this residue is not an absolute requirement for DJ-1 to associate with the mitochondria.
We confirmed the mitochondrial localization of DJ-1 using confocal microscopy (Fig. 3B). Exposure of cells to oxidative stress induced by exposure to either 300 μm paraquat or 100 nm rotenone caused an accumulation of wild-type DJ-1 in mitochondria. E18N and E18Q DJ-1 were clearly associated with mitochondria under basal conditions and remained so under oxidative stress. In contrast, E18D and C106A DJ-1 remained largely cytoplasmic under all conditions tested. In addition, during these experiments, we saw that treatment with rotenone and, to a lesser extent, paraquat was associated with a shorter, fragmented mitochondrial morphology (enlarged black and white images in Fig. 3B).
Cys106-Sulfinic Acid Formation Is Required for Protection against Mitochondrial Fragmentation and Cell Death—Oxidative and mitochondrial stressors can result in fragmentation of mitochondria due to stimulation of mitochondrial fission. The above experiments examining mitochondrial localization of DJ-1 provided some evidence that cells expressing wild-type but not E18D or C106A DJ-1 might protect against mitochondrial fission, presumably as a consequence of lowering cellular oxidative stress. Therefore, we investigated the ability of wild-type and Glu18 DJ-1 mutants to diminish mitochondrial fragmentation using FRAP of mitochondrially directed yellow fluorescent protein, which recovers well after photobleaching if the mitochondria are highly connected. We used DJ-1 knock-out MEFs for these experiments to avoid potential interference from endogenous DJ-1 protein. DJ-1 knock-out MEFs have grossly normal mitochondria (Fig. 4A) but lower FRAP mobile fraction values, indicating reduced mitochondrial connectivity (Fig. 4, B and C). Mitochondrial connectivity was rescued by stable reexpression of human wild-type DJ-1, showing that fragmentation is due only to the absence of DJ-1 in these MEFs.
FIGURE 4.

DJ-1 Glu18 substitutions affect mitochondrial function in living cells. A-C, living mouse embryonic fibroblasts were imaged after transfection with mitochondrial matrix-localized yellow fluorescent protein (A) to reveal mitochondrial morphology. FRAP was used to measure mitochondrial connectivity. B, time course in wild-type (WT; brown) or DJ-1 knock-out (KO; black) mouse embryonic fibroblasts and knock-out fibroblasts stably expressing human DJ-1 (green). Three independent experiments were performed, each with 30 cells measured; hence, overall n = 90. C, mobile fractions were calculated from FRAP time course experiments. Horizontal lines, median values; boxes, upper and lower quartiles. Range bars show 10-90% intervals. Individual values outside of the range are shown as single dots. Differences were analyzed by one-way ANOVA; *, p < 0.05; **, p < 0.01 by Newman-Kuell's post hoc tests. D and E, mutations around the oxidation sensor residue Cys106 have varied impact on the ability of human DJ-1 to rescue mouse DJ-1 deficiency phenotypes. DJ-1 knock-out fibroblasts were transfected with vector alone (black) or with the indicated human DJ-1 variants (green, WT; magenta, E18N; cyan, E18Q; blue, E18D; red, C106A). Mitochondrial connectivity was assessed by FRAP. Time course is shown in D, and mobile fractions are shown in E. Each point is the average of 60 cells combined from two independent experiments. Differences were analyzed comparing the indicated constructs with WT DJ-1; *, p < 0.05; ***, p < 0.01 by one-way ANOVA with Newman-Kuell's post hoc tests. ns, not significant.
We used the FRAP assay to assess the ability of Glu18 mutations
to increase mitochondrial connectivity using transient DJ-1 transfections of
these MEFs. As before, wild-type DJ-1 improved FRAP recovery compared with
empty vector (Fig.
4D). The Glu18 mutations showed variable
ability to rescue the lack of endogenous DJ-1 in this assay, with E18N and
E18Q being closer to wild-type DJ-1 than vector alone. In contrast, E18D (or
C106A) DJ-1 transfectants did not rescue mitochondrial fragmentation and
actually had a slightly lower mobile fraction value than vector alone. These
results indicate that the Glu18 variants that support
Cys106-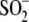 formation (E18N
and E18Q) can substitute for wild-type DJ-1, whereas those that do not support
sulfinic acid formation at Cys106 (E18D and C106A) are
inactive.
formation (E18N
and E18Q) can substitute for wild-type DJ-1, whereas those that do not support
sulfinic acid formation at Cys106 (E18D and C106A) are
inactive.
Several studies have reported that DJ-1 can protect cells against mitochondrial toxins in a variety of in vitro and in vivo models (15, 20). We have shown previously that human dopaminergic M17 neuroblastoma cells show enhanced resistance to complex I inhibitors (15) when transfected with wild-type DJ-1 but not the oxidation-deficient C106A mutant. We investigated the ability of Glu18 DJ-1 variants to protect against rotenone-induced cell death using nuclear morphology as a measure of cell viability. M17 neuroblastoma cells that were transiently transfected with wild-type or Glu18 mutant DJ-1 were stained, visually assayed, and classified as either having intact rounded nuclei or having a shrunken and fragmented nuclear morphology (Fig. 5A). Using these criteria, the basal viability of untransfected, untreated cells was 84.6 ± 2.7% (mean ± S.D., n = 9 fields). We counted transfected cells for all DJ-1 variants in the presence or absence of rotenone and saw statistically significant differences in viability across all groups (p < 0.0001 by one-way ANOVA; n = 9 observations per group combined from three independent experiments). Cells transfected with wild-type DJ-1 had a viability similar to that of untransfected cells without treatment (85.1 ± 7.9%). Cell viability was slightly decreased to 77.0 ± 6.8% after exposure to 200 nm rotenone for 24 h, although this difference did not reach statistical significance (p > 0.05 by Student-Newman Kuell's post hoc test comparing untreated versus treated wild-type DJ-1 transfected cells). Similarly, E18N DJ-1 did not affect basal cell viability (84.5 ± 5.5%), and cells transfected with E18N displayed greater resistance to rotenone toxicity than wild-type DJ-1 (82.8 ± 5.1%, p > 0.05 compared with untreated by post hoc tests, n = 9). E18Q DJ-1 had a moderate effect on basal cell viability (77.3 ± 10.3%), and rotenone decreased viability further, to 67.6 ± 8.2% (p < 0.05 compared with untreated by post hoc tests, n = 9). Interestingly, the two oxidation-impaired DJ-1 mutants, E18D and C106A, had negative effects on basal cell viability, which were decreased to 66.5 ± 6.5 and 70.8 ± 4.4%, respectively. Furthermore, rotenone treatment had a statistically significant (p < 0.05 by post hoc test comparing untreated and treated cells for each variant, n = 9) effect on viability for E18D and C106A DJ-1, which were 57.5 ± 7.1 and 61.5 ± 6.4%, respectively. These results show that although wild-type and E18N DJ-1 are capable of protecting cells against rotenone-induced toxicity, the E18D and C106A mutants cannot and are therefore both loss of function variants.
FIGURE 5.

DJ-1 Glu18 substitutions affect cellular resistance to rotenone-induced toxicity. A, nuclear morphology as a marker of rotenone-induced loss of cell viability. M17 neuroblastoma cells were transiently transfected with V5-tagged WT, E18N, E18Q, E18D, or C106A DJ-1 constructs, as indicated, and either left untreated (upper panels) or exposed to 200 nm rotenone for 24 h (lower panels). Cells were stained for V5 (green) and counterstained with Hoechst 33342 (blue) and scored as having intact nuclei (arrowheads) or fragmented/shrunken nuclei (arrows). The insets show examples of nuclei from the blue channel at higher magnification. The scale bar in the bottom right panel represents 20 μm and applies to all images. B, cells were transfected as in A with WT (green), E18N (magenta), E18Q (blue), E18D (cyan), and C106A (red) DJ-1 variants. Cell viability is expressed as the percentage of transfected (V5-positive) cells that had intact nuclei compared with all transfected cells. Each box plot represents data from three randomly selected microscope fields (between 26 and 75 cells/field) counted in each of three independent experiments for an overall n = 9 per construct. Horizontal lines, median values; boxes, upper and lower quartiles; range bars, the range of percentage viabilities for all fields counted. The dotted line represents mean viability counted in untransfected cells from the same cultures, with the shaded box indicating one S.D. value (84.6 ± 2.7% viability, n = 9 fields, mean of 28 cells/field). Differences were analyzed comparing the indicated untreated and rotenone-treated cells for the same construct; *, p < 0.05 by one-way ANOVA (p < 0.001 overall) with Newman-Kuell's post hoc tests. ns, not significant.
DISCUSSION
In this study, we have examined the role that the specific formation of a cysteine-sulfinic acid has on the mitochondrial and cellular protective function of DJ-1, a protein involved in certain forms of rare familial parkinsonism. Our results show that the ability of Cys106 to be oxidized to cysteine-sulfinic acid is required for the protective activity of DJ-1. Importantly, by altering the dominant oxidation state of Cys106 without mutating Cys106 itself, we have been able to decouple the effect of Cys106 oxidation from other possible roles of this residue. Therefore, we can use this approach to independently interrogate the relative importance of Cys106 oxidation for the function of DJ-1. The use of engineered substitutions to modulate the redox state of important cysteine residues may be applicable to other systems that contain readily oxidized cysteine residues and where the structure of the protein is known.
As suggested previously
(15,
18,
36) and as the current data
confirm, the pocket around Cys106 in human DJ-1 contains a number
of residues that modulate Cys106 reactivity toward reactive oxygen
species. Glu18 forms a hydrogen bond with
Cys106-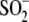 that is important
for the stabilization of the modified residue. Interestingly, both
Glu18 and Cys106 are very highly conserved in members of
the DJ-1 family. This includes ancient homologues, such as YajL from E.
coli and YDR533c from Saccharomyces cerevisiae, where we have
previously shown that the structurally equivalent cysteine residue is subject
to similar oxidative modifications that are stabilized by a conserved glutamic
acid (37,
38). Thus, Cys106
and Glu18 (or their equivalent residues in other DJ-1 superfamily
proteins) are probably important for a conserved and ancient function of this
protein family.
that is important
for the stabilization of the modified residue. Interestingly, both
Glu18 and Cys106 are very highly conserved in members of
the DJ-1 family. This includes ancient homologues, such as YajL from E.
coli and YDR533c from Saccharomyces cerevisiae, where we have
previously shown that the structurally equivalent cysteine residue is subject
to similar oxidative modifications that are stabilized by a conserved glutamic
acid (37,
38). Thus, Cys106
and Glu18 (or their equivalent residues in other DJ-1 superfamily
proteins) are probably important for a conserved and ancient function of this
protein family.
By mutating Glu18 to other residues, we were able to influence
the ability of Cys106 to oxidize to sulfinic acid without changing
the cysteine residue itself. Two variants (E18N and E18Q) allow sulfinic acid
formation and, in fact, slightly enhance oxidation in a cellular context. In
contrast, the moderately conservative E18D substitution, which shortens the
side chain of residue 18 by a single methylene group, dramatically impairs
Cys106-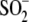 formation and
results in constitutively reduced Cys106 in the cellular
environment. By comparing the behavior of E18D with C106A DJ-1, we can
distinguish whether it is the cysteine residue per se or the
cysteine-sulfinic acid that is important for DJ-1 function.
formation and
results in constitutively reduced Cys106 in the cellular
environment. By comparing the behavior of E18D with C106A DJ-1, we can
distinguish whether it is the cysteine residue per se or the
cysteine-sulfinic acid that is important for DJ-1 function.
Our cell-based mitochondrial fission assay shows that although E18N and E18Q can functionally substitute for wild-type protein, neither C106A nor E18D can do so. The power of this approach is enhanced by the known pKa values for Cys106 in each of these mutants (18). E18N and E18D DJ-1 have identical Cys106 pKa values of 6.1 but very different oxidative and in vivo protective capabilities in the present study. Therefore, we conclude that it is the oxidative propensity rather than the nucleophilicity or general reactivity of Cys106 that is required for the mitochondrial protective activity of DJ-1.
We also confirmed that the ability of Glu18 to support oxidative modification of Cys106 is required for DJ-1 to protect cells against loss of viability due to exposure to the mitochondrial complex I inhibitor rotenone. This is consistent with our previous data (15) and similar results from other laboratories (21) but again emphasizes that the ability of Cys106 to undergo facile oxidative modification is the critical determinant for the cytoprotective effects of DJ-1. In these experiments, we also noted a generally detrimental effect of the oxidation-deficient DJ-1 variants on M17 neuroblastoma cell viability, which we speculate may represent a dominant negative effect resulting from the formation of functionally compromised DJ-1 heterodimers of E18D (or C106A) DJ-1 with endogenous wild-type protein.
In this study, a combination of the FRAP mitochondrial fission assay and rotenone-induced cellular toxicity was used because of the potential relevance of mitochondrial dysfunction to Parkinson disease, although it may also be important for other activities of DJ-1 in aerobic situations. Supporting a role for DJ-1 in the maintenance of redox homeostasis, a recent study has shown that DJ-1 deficiency is associated with increased generation of H2O2 and diminished activity of mitochondrial iron-sulfur proteins (8). The loss of mitochondrial connectedness seen here in DJ-1-deficient MEFs is therefore probably related to increased oxidative stress, which is known to trigger mitochondrial fission (39). The observation that E18N and E18Q are largely mitochondrial suggests that this pool of protein contributes significantly to the observed effects on mitochondrial fission, supporting the results of a recent study that showed that intentionally targeting DJ-1 to the mitochondria enhances its protective activity (35). Furthermore, the ability of DJ-1 to be recruited to mitochondria is tightly correlated with its propensity for oxidation, confirming a previous suggestion that cysteine-sulfinic acid formation at Cys106 is important for mitochondrial localization (15).
The precise function of DJ-1 that protects cells against oxidative damage
is unclear, and several previous studies have suggested different possible
biochemical activities (8,
10-14).
However, in the context of the oxidative stress response, a mitochondrial
activity for DJ-1 is likely responsible for the protective effect of the
protein. The ability of DJ-1 to scavenge reactive oxygen species could be
important by itself, since this may decrease cellular levels of ROS. However,
there are many more abundant antioxidant molecules with a greater ability to
scavenge ROS in the cell. For example, cells have millimolar levels of
glutathione under normal circumstances and low micromolar amounts of DJ-1.
Furthermore, the observed second order rate constant of DJ-1 oxidation by
H2O2 in vitro is ∼0.56
m-1 · s-1, or 105 to
108 lower than the better-characterized peroxiredoxins
(8). In addition, the oxidation
of glutathione and peroxiredoxins is reversible and thus can detoxify
superstoichiometric quantities of ROS, whereas there is no known mechanism by
which Cys106-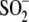 DJ-1 can be
reduced. Therefore, since there are more efficient systems to remove ROS in
the cell, sacrificial irreversible autoxidation of DJ-1 is unlikely to
contribute significantly to its antioxidant status.
DJ-1 can be
reduced. Therefore, since there are more efficient systems to remove ROS in
the cell, sacrificial irreversible autoxidation of DJ-1 is unlikely to
contribute significantly to its antioxidant status.
Furthermore, although a redox-sensitive chaperone activity toward
α-synuclein has been demonstrated for
Cys106-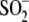 DJ-1
(10), this does not account
for the established protective role of DJ-1 homologues in nonvertebrate
species, such as D. melanogaster, that lack an α-synuclein
homologue but where the same cysteine oxidation has been shown to be important
(21). We have proposed that
DJ-1 may bind selectively to mitochondrial and antioxidant mRNA species
(12), which is broadly
consistent with the results presented here in supporting a major role for the
mitochondrial pool of DJ-1 in protection against oxidative stress. This is
formally difficult to assess, however, since DJ-1 oxidation influences its RNA
binding activity, and thus mutations that affect Cys106 oxidation
will also be predicted to influence RNA binding. Defining the activity of DJ-1
that is responsible for cellular protection against oxidative stress will
require further studies that integrate structural, biochemical, cellular, and
organismal approaches.
DJ-1
(10), this does not account
for the established protective role of DJ-1 homologues in nonvertebrate
species, such as D. melanogaster, that lack an α-synuclein
homologue but where the same cysteine oxidation has been shown to be important
(21). We have proposed that
DJ-1 may bind selectively to mitochondrial and antioxidant mRNA species
(12), which is broadly
consistent with the results presented here in supporting a major role for the
mitochondrial pool of DJ-1 in protection against oxidative stress. This is
formally difficult to assess, however, since DJ-1 oxidation influences its RNA
binding activity, and thus mutations that affect Cys106 oxidation
will also be predicted to influence RNA binding. Defining the activity of DJ-1
that is responsible for cellular protection against oxidative stress will
require further studies that integrate structural, biochemical, cellular, and
organismal approaches.
Overall, these data strengthen previous suggestions that oxidative stress and mitochondrial dysfunction are critical contributors to the etiology of various forms of parkinsonism. Furthermore, they identify cysteine-sulfinic acid as a critical post-translational modification that has powerful effects on DJ-1 function in vivo. Additional studies that use a similar strategy could be applied to other redox-sensitive proteins that contain oxidized cysteine residues.
Acknowledgments
We thank the staff of BioCARS 14-BMC at the Advanced Photon Source (Argonne, IL) for beamline support. Use of the Advanced Photon Source was supported by United States Department of Energy Contract W-31-109-Eng-38, and use of BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under Grant RR07707.
The atomic coordinates and structure factors (codes 3EZG and 3F71) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant P20RR17675 (to M. A. W.) and by the Intramural Research Program of the National Institutes of Health, NIA, Project Z01-AG000953 (to M. R. C.). This work was also supported in part by the American Parkinson's Disease Association. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; ANOVA, analysis of variance; DTT, dithiothreitol; FRAP, fluorescence recovery after photobleaching; LC-MS/MS, liquid chromatography-mass spectrometry; MEF, mouse embryonic fibroblast; VDAC1, voltage-dependent anion channel 1; WT, wild type.
References
- 1.Reddie, K. G., and Carroll, K. S. (2008) Curr. Opin. Chem. Biol. 12 746-754 [DOI] [PubMed] [Google Scholar]
- 2.Biteau, B., Labarre, J., and Toledano, M. B. (2003) Nature 425 980-984 [DOI] [PubMed] [Google Scholar]
- 3.Jonsson, T. J., Murray, M. S., Johnson, L. C., Poole, L. B., and Lowther, W. T. (2005) Biochemistry 44 8634-8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Annesi, G., Savettieri, G., Pugliese, P., D'Amelio, M., Tarantino, P., Ragonese, P., La Bella, V., Piccoli, T., Civitelli, D., Annesi, F., Fierro, B., Piccoli, F., Arabia, G., Caracciolo, M., Cirò Candiano, I. C., and Quattrone, A. (2005) Ann. Neurol. 58 803-807 [DOI] [PubMed] [Google Scholar]
- 5.Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., Dekker, M. C., Squitieri, F., Ibanez, P., Joosse, M., van Dongen, J. W., Vanacore, N., van Swieten, J. C., Brice, A., Meco, G., van Duijn, C. M., Oostra, B. A., and Heutink, P. (2003) Science 299 256-259 [DOI] [PubMed] [Google Scholar]
- 6.Bandyopadhyay, S., and Cookson, M. R. (2004) BMC Evol. Biol. 4 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucas, J. I., and Marin, I. (2007) Mol. Biol. Evol. 24 551-561 [DOI] [PubMed] [Google Scholar]
- 8.Andres-Mateos, E., Perier, C., Zhang, L., Blanchard-Fillion, B., Greco, T. M., Thomas, B., Ko, H. S., Sasaki, M., Ischiropoulos, H., Przedborski, S., Dawson, T. M., and Dawson, V. L. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 14807-14812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shendelman, S., Jonason, A., Martinat, C., Leete, T., and Abeliovich, A. (2004) PLoS Biol. 2 1764-1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou, W., Zhu, M., Wilson, M. A., Petsko, G. A., and Fink, A. L. (2006) J. Mol. Biol. 356 1036-1048 [DOI] [PubMed] [Google Scholar]
- 11.Hod, Y., Pentyala, S. N., Whyard, T. C., and El-Maghrabi, M. R. (1999) J. Cell Biochem. 72 435-444 [PubMed] [Google Scholar]
- 12.van der Brug, M. P., Blackinton, J., Chandran, J., Hao, L. Y., Lal, A., Mazan-Mamczarz, K., Martindale, J., Xie, C., Ahmad, R., Thomas, K. J., Beilina, A., Gibbs, J. R., Ding, J., Myers, A. J., Zhan, M., Cai, H., Bonini, N. M., Gorospe, M., and Cookson, M. R. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 10244-10249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong, N., Kim, C. Y., Rizzu, P., Geula, C., Porter, D. R., Pothos, E. N., Squitieri, F., Heutink, P., and Xu, J. (2006) J. Biol. Chem. 281 20940-20948 [DOI] [PubMed] [Google Scholar]
- 14.Zhou, W., and Freed, C. R. (2005) J. Biol. Chem. 280 43150-43158 [DOI] [PubMed] [Google Scholar]
- 15.Canet-Avilés, R. M., Wilson, M. A., Miller, D. W., Ahmad, R., McLendon, C., Bandyopadhyay, S., Baptista, M. J., Ringe, D., Petsko, G. A., and Cookson, M. R. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 9103-9108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito, G., Ariga, H., Nakagawa, Y., and Iwatsubo, T. (2006) Biochem. Biophys. Res. Commun. 339 667-672 [DOI] [PubMed] [Google Scholar]
- 17.Kinumi, T., Kimata, J., Taira, T., Ariga, H., and Niki, E. (2004) Biochem. Biophys. Res. Commun. 317 722-728 [DOI] [PubMed] [Google Scholar]
- 18.Witt, A. C., Lakshminarasimhan, M., Remington, B. C., Hasim, S., Pozharski, E., and Wilson, M. A. (2008) Biochemistry 47 7430-7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aleyasin, H., Rousseaux, M. W., Phillips, M., Kim, R. H., Bland, R. J., Callaghan, S., Slack, R. S., During, M. J., Mak, T. W., and Park, D. S. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 18748-18753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meulener, M., Whitworth, A. J., Armstrong-Gold, C. E., Rizzu, P., Heutink, P., Wes, P. D., Pallanck, L. J., and Bonini, N. M. (2005) Curr. Biol. 15 1572-1577 [DOI] [PubMed] [Google Scholar]
- 21.Meulener, M. C., Xu, K., Thomson, L., Thompson, L., Ischiropoulos, H., and Bonini, N. M. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 12517-12522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holyoak, T., Fenn, T. D., Wilson, M. A., Moulin, A. G., Ringe, D., and Petsko, G. A. (2003) Acta Crystallogr. Sect. D Biol. Crystallogr. 59 2356-2358 [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 307-326 [DOI] [PubMed]
- 24.Sheldrick, G. M. (2008) Acta Crystallogr. A 64 112-122 [DOI] [PubMed] [Google Scholar]
- 25.Brunger, A. T. (1992) Nature 355 472-475 [DOI] [PubMed] [Google Scholar]
- 26.Wilson, M. A., Collins, J. L., Hod, Y., Ringe, D., and Petsko, G. A. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 9256-9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 28.Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., III, Snoeyink, J., Richardson, J. S., and Richardson, D. C. (2007) Nucleic Acids Res. 35 W375-W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandran, J. S., Lin, X., Zapata, A., Höke, A., Shimoji, M., Moore, S. O., Galloway, M. P., Laird, F. M., Wong, P. C., Price, D. L., Bailey, K. R., Crawley, J. N., Shippenberg, T., and Cai, H. (2008) Neurobiol. Dis. 29 505-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blackinton, J., Ahmad, R., Miller, D. W., van der Brug, M. P., Canet-Avilés, R. M., Hague, S. M., Kaleem, M., and Cookson, M. R. (2005) Brain Res. Mol. Brain Res. 134 76-83 [DOI] [PubMed] [Google Scholar]
- 31.Karbowski, M., Norris, K. L., Cleland, M. M., Jeong, S. Y., and Youle, R. J. (2006) Nature 443 658-662 [DOI] [PubMed] [Google Scholar]
- 32.Szabadkai, G., Simoni, A. M., Bianchi, K., De Stefani, D., Leo, S., Wieckowski, M. R., and Rizzuto, R. (2006) Biochim. Biophys. Acta 1763 442-449 [DOI] [PubMed] [Google Scholar]
- 33.Engh, R. A., and Huber, R. (1991) Acta Crystallogr. Sect. A 47 392-400 [Google Scholar]
- 34.Zhang, L., Shimoji, M., Thomas, B., Moore, D. J., Yu, S. W., Marupudi, N. I., Torp, R., Torgner, I. A., Ottersen, O. P., Dawson, T. M., and Dawson, V. L. (2005) Hum. Mol. Genet. 14 2063-2073 [DOI] [PubMed] [Google Scholar]
- 35.Junn, E., Jang, W. H., Zhao, X., Jeong, B. S., and Mouradian, M. M. (2008) J. Neurosci. Res. 87 123-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei, Y., Ringe, D., Wilson, M. A., and Ondrechen, M. (2007) PLoS Comput. Biol. 3 120-126 [DOI] [PubMed] [Google Scholar]
- 37.Wilson, M. A., Ringe, D., and Petsko, G. A. (2005) J. Mol. Biol. 353 678-691 [DOI] [PubMed] [Google Scholar]
- 38.Wilson, M. A., St Amour, C. V., Collins, J. L., Ringe, D., and Petsko, G. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 1531-1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheung, E., McBride, H. M., and Slack, R. S. (2007) Apoptosis 12 979-992 [DOI] [PubMed] [Google Scholar]
- 40.Fenn, T., Ringe, D., and Petsko, G. A. (2003) J. Appl. Crystallogr. 36 944-947 [Google Scholar]