

Abstract
Dopamine D1 receptors play an important role in movement, reward, and learning and are implicated in a number of neurological and psychiatric disorders. These receptors are concentrated in dendritic spines of neurons, including the spine head and the postsynaptic density. D1 within spines is thought to modulate the local channels and receptors to control the excitability and synaptic properties of spines. The molecular mechanisms mediating D1 trafficking, anchorage, and function in spines remain elusive. Here we show that the synaptic scaffolding protein PSD-95 thought to play a role in stabilizing gluta-mate receptors in the postsynaptic density, interacts with D1 and regulates its trafficking and function. Interestingly, the D1-PSD-95 interaction does not require the well characterized domains of PSD-95 but is mediated by the carboxyl-terminal tail of D1 and the NH2terminus of PSD-95, a region that is recognized only recently to participate in protein-protein interaction. Co-expression of PSD-95 with D1 in mammalian cells inhibits the D1-mediated cAMP accumulation without altering the total expression level or the agonist binding properties of the receptor. The diminished D1 signaling is mediated by reduced D1 expression at the cell surface as a consequence of an enhanced constitutive, dynamin-dependent endocytosis. In addition, genetically engineered mice lacking PSD-95 show a heightened behavioral response to either a D1 agonist or the psychostimulant amphetamine. These studies demonstrate a role for a glutamatergic scaffold in dopamine receptor signaling and trafficking and identify a new potential target for the modulation of abnormal dopaminergic function.
The dopamine D1-class receptors (D1 and D5) regulate movement, reward, and learning and are implicated in multiple neurological and psychiatric conditions (1-3). This dopaminereceptor class is positively coupled to Gsα to regulate the PKA/ DARPP-32/PP1 pathway (4–7), leading to the regulation of neuronal excitability, synaptic transmission and plasticity, and gene expression (8). A key site of action for D1, the predominant subtype of the D1-class receptor, is the dendritic spine of glutamatergic synapses, where the receptor is especially concentrated. Ultrastructural studies reveal that the majority of D1 receptors are localized postsynaptically in the neck, head, as well as the postsynaptic density (PSD)2 of medium spiny neurons (9, 10) and cortical pyramidal neurons (11–13). This sub-cellular specialization thus provides a location where D1 can modulate the channels and receptors located at spines to control the excitability and synaptic properties of the (glutamatergic) spines. The molecular and cellular mechanisms that regulate D1 targeting and signaling within spines are poorly understood.
PSD-95 is a prototypical scaffolding protein highly enriched in the PSD and belongs to the membrane-associated guanylate kinase family (14). PSD-95 contains three PSD-95, Dlg, ZO-1 homology (PDZ) domains, a Src homology 3 (SH3) domain, and a guanylate kinase-like (GK) domain, allowing the protein to interact with a host of proteins of diverse functions, most notably N-methyl-D-aspartate (NMDA) receptors, and regulate the functional integration of these receptors into the signaling complexes in PSD (14, 15). Functionally, PSD-95 regulates NMDA receptor internalization, synaptic transmission and plasticity and plays essential roles in learning (16–20).
We previously showed that PSD-95 regulates behavioral responses to the psychostimulant cocaine in mice (21), suggesting that PSD-95 may play a role in dopaminergic signaling. Here, we identify a novel interaction between PSD-95 and D1. We demonstrate that PSD-95, through its NH2terminus (NT), induces robust internalization of the otherwise plasma membrane localized D1 receptors in the absence of agonist stimulation, leading to reduced surface D1 expression and diminished D1 signaling. Consequently, mice lacking PSD-95 exhibits enhanced behavioral responses to a D1 agonist and the psycho-stimulant amphetamine. Our studies establish a new molecular mechanism by which the synaptic delivery of the D1 receptor and thus dopamine signaling can be regulated locally in spines and identify a new potential target in abnormal dopaminergic function.
EXPERIMENTAL PROCEDURES
Mice and Behavioral Tests
All experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of animals and with an approved animal protocol from the Harvard Medical Area Standing Committee on Animals. PSD-95 wild-type (WT) and knock-out (KO) mice (21) were housed at standard lab conditions (12 h light/dark cycle) with food and water provided ad libitum.
Locomotion was evaluated using the automated ENV510 activity monitor (Med Associates Inc.). Age-matched mice were placed in the monitor and their horizontal and vertical activities were recorded at 4- or 5-min intervals. In each experiment, mice were habituated to the chamber environment and their activities monitored for up to 60 min. Each of the mice then received a dose of dopamine agonists or saline and their locomotor activities were measured for up to 120 min. All injections were performed intraperitoneally.
cDNA Constructs, Cell Culture, and Transfections
The full-length human dopamine D1 receptor cloned in pcDNA3 and a green fluorescence protein (GFP) tagged PSD-95 (PSD-95-GFP) in GW1 (22) were used to generate various D1 and PSD-95 constructs. HA-D1 and HA-D1ΔCT lacking amino acids 334–446 were constructed by placing the hemagglutinin (HA) epitope at the NT of the full-length WT D1 and the truncated D1, respectively. D1-FLAG and D1ΔCT-FLAG were constructed by placing the FLAG epitope at the COOH terminus (CT) of the full-length and the truncated D1, respectively. PSD-95-FLAG and PSD-95ΔNT-FLAG lacking amino acids 1–72 were constructed by placing the FLAG epitope at the CT of the full-length and the truncated PSD-95, respectively. Constructs were subcloned into the mammalian expression vector pcDNA5/FRT/TO (Invitrogen) unless otherwise specified. GFP-tagged PSD-95 truncation mutants ΔNT-GFP (lacking amino acids 1–72), ΔGK-GFP (lacking amino acids 533–712), ΔSH3, GK-GFP (lacking amino acids 431–712), and ΔPDZ1-3, SH3, GK-GFP (i.e. NT-GFP, lacking amino acids 73–712) were constructed by deleting the respective amino acid residues or motif(s) from the full-length PSD-95 and subcloned into the pEGFP-N2 vector (Clontech) in-frame. The rat dynamin I WT (Dyn I-WT) and the K44A (Dyn I-K44A) mutant constructs were described previously (23). All DNA constructs were generated by PCR and sequence verified.
Human embryonic kidney (HEK) 293, HEK293T (collectively referred as HEK293(T)), and HEK293 cells stably expressing the rhesus monkey D1 receptor (D1-stable cells) were cultured in a high glucose Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). Cells were grown to 70–80% conflunence and transfected with 1–10 μg of total DNA using TransFectin (Bio-Rad). E14 rat striatal neurons were grown in Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated horse serum, glucose (6.0 mg/ml), 1% penicillin/streptomycin, and 2 mM glutamine (Invitrogen) for 4 days before subsequent experiments.
Immunocytochemistry, Confocal Microscopy, and Immunofluorescence Analysis
Cultured HEK293 cells or primary neurons were briefly fixed with freshly made 4% paraformaldehyde. After three washes with phosphate-buffered saline (PBS), cells were permeabilized with 0.25% Triton X-100 for 5 min and blocked with 10% goat serum (in PBS) for 30 min. Cells were incubated with the following primary antibodies overnight at 4 °C: anti-HA (1:500, Covance), anti-D1 (1:50, a generous gift from Dr. Allan Levey, Emery University; and from Sigma), anti-PSD-95 (1:200, BD Biosciences), and anti-dynamin I (1:100; Affinity BioReagents). Cells were incubated with appropriate Alexa Fluor dyes (Invitrogen) at room temperature for up to 1 h and mounted on glass slides.
To study D1 internalization processes, live cells were incubated with an HA antibody (1:50) at 4 °C for 1 h, washed, and stimulated with or without SKF81297 (10 μM, 30 min, 37 °C). Cells were fixed and blocked before application of Alexa Fluor 647-conjugated secondary antibody to label surface HA-D1. The internalized HA-D1 was recognized by an Alexa Fluor 568-conjugated secondary antibody after permeabilizing the cells with 0.25% Triton X-100.
Confocal images were acquired using a Leica confocal microscope. Alexa Fluor 488 (green), Alexa Fluor 568 (red), and Alexa Fluor 647 (pseudo-colored “blue” as the emission wavelength of this fluorophore is beyond the sensitivity of human vision) were measured at the following excitation/emission wavelengths: 488/519, 568/604, and 647/669 nm, respectively. GFP fluorescence was measured directly in fixed cells without involving any primary and secondary antibody staining using excitation/ emission at 488/508 nm.
For quantification of internalization, the confocal settings for image acquisition were kept the same for all cells. Each acquired image file was a stack of 10 images along the z axis. Image stacks were flattened into a single image using a maximum projection and analyzed with Metamorph (Universal Imaging Corp.). Surface and internal D1 fluorescence intensities were measured as the integrated pixel intensities (i.e. total 8-bit intensity values across all pixels contained in a cell) in the blue and red channels, respectively. Total D1 fluorescence was determined as the sum of the surface (blue) and the internal (red) fluorescence intensities. Internalized receptors were operationally defined as those unambiguously localized inside the cells. These receptors were selected by tracing the cytoplasm 4 μm beneath the cell surface in the blue channel and transferred to the red channel, and the red fluorescence intensity was measured as the level of the internalized receptors. For each cell, the internalization index was defined as the ratio of the internalized fluorescence intensity to the total fluorescence intensity.
Subcellular Fractionation
PSD fraction was isolated from mouse forebrain structures (containing the striatum, hippocampus, and cerebral cortex) according to Carlin et al. (24) using sucrose density gradient centrifugation. Briefly, brain tissues were homogenized in ice-cold 0.32 M sucrose containing 1 mM Hepes, 1 mM MgCl2,1mM NaHCO3and protease inhibitor mixture (Roche Applied Science), and centrifuged at 700 × g for 10 min (P1). The supernatant was centrifuged at 1300 × g for 15 min to yield a crude synaptosome and mitochondria (P2). The P2 pellet was resuspended in ice-cold 0.32 M sucrose containing 1 mM Hepes and 1 mM NaHCO3, loaded onto a sucrose gradient of 0.85, 1.0, and 1.2 M, and centrifuged at 82,000 × g for 2 h. The synaptosome fraction isolated between 1.0 and 1.2 M was centrifuged at 32,800 × g for 30 min. The resulting pellet was resuspended and centrifuged at 201,800 × g for 2 h over a sucrose gradient of 1.0, 1.5, and 2.0 M. The PSD fraction between 1.5 and 2.0 M was obtained after centrifugation at 201,800 × g for 30 min.
Immunoprecipitation and Western Blotting
Mouse fore-brain structures were rapidly dissected and homogenized in ice-cold Tris-HCl (50 mM, pH 7.4) containing protease inhibitor mixture, and centrifuged at 100,000 × g for 30 min. The pellet was solubilized in a DOC buffer (50 mM Tris-HCl, pH 9.0, 1% DOC, 1 mM EDTA, 1 mM EGTA and protease inhibitors) at 37 °C for 45 min and centrifuged at 100,000 × g for 30 min. Isolated PSD proteins were incubated in the solubilization buffer (25 mM Tris-HCl, pH 7.5, containing 1% SDS, 68.5 mM NaCl, 1.35 mM KCl, 2.5 mM EDTA, 2.5 mM EGTA, 25 mM NaF, 50 μM Na vanadate, protease inhibitor mixture, and 1 mM dithiothreitol) at 4 °C for 30 min, supplemented with five volumes of dilution buffer (50 mM Tris-HCl, pH 7.5, containing 2% Triton X-100, 137 mM NaCl, 2.7 mM KCl, 5 mM EDTA, 5 mM EGTA, 50 mM NaF, 100 μM Na vanadate and protease inhibitor mixture), incubated at 4 °C for an additional 2 h, and centrifuged at 13,000 × g for 20 min. Transfected HEK293(T) cells were sonicated in 25 mM Tris-HCl containing 150 mM NaCl, 3 mM KCl, 1 mM EDTA and protease inhibitors and centrifuged at 13,000 × g for 20 min. The protein extracts of the supernatant from brain tissues, PSD, or HEK293(T) cells were used in immunoprecipitation and pulldown assays.
Protein extracts were incubated with an anti-D1 (10 μl, Sigma), an anti-PSD-95 (8 μl, BD Biosciences), an anti-HA (5 μl, Covance), or an anti-FLAG antibody (5 μl, Sigma) at 4 °C overnight with gentle rotation. Immunocomplexes were captured by protein A/G-agarose (Santa Cruz Biotechnology), resolved by SDS-PAGE, and transferred to polyvinylidene difluoride membranes. Blots were immunostained with desired primary antibodies and horseradish peroxidase-conjugated secondary antibodies, and immunoreactive signals were detected with an ECL-based LAS-3000 image system (Fujifilm). Densitometric analysis was carried out within linear range using ImageGauge (Fujifilm).
Glutathione S-Transferase (GST) Fusion Proteins and Pull-down Assay
The coding sequences of the intracellular loop 2 (IL2, amino acids 117–139), loop 3 (IL3, amino acids 218–273), and CT (amino acids 334–446) of the D1 receptor and the NT (amino acids 1–72), PDZ 1 (PDZ1, amino acids 73–167), PDZ 2 (PDZ2, amino acids 168–320), PDZ 3 (PDZ3, amino acids 321–430), SH3 (SH3, amino acids 431–532), and GK (GK, amino acids 533–712) domains of PSD-95 were generated by PCR and cloned into pGEX4T-1 vector in-frame and verified by automated sequencing. The GST fusion protein production was induced by 0.5 mM isopropyl-β-D-thiogalactopyranoside (Promega) for 2 h in BL21 bacterium and immobilized on glutathione-Sepharose 4B-agarose (Amersham Biosciences). Equal amounts of GST fusion proteins were incubated at 4 °C overnight with lysates of HEK293T cells overexpressing PSD-95-GFP or D1-FLAG, or mouse brain extracts, followed by extensive washes with ice-cold PBS containing 0.1% Triton X-100. The pulled down proteins were resolved by SDS-PAGE and analyzed by Western blotting.
cAMP Enzyme Immunoassay
D1-stable HEK293 cells were transiently transfected with mock or PSD-95-GFP cDNA plasmids and split into 24-well plates 12 h after transfection. The cells were serum-starved for 1 h and stimulated by SKF81297 for 30 min. Whole-cell cAMP concentrations were measured using the Direct cAMP Enzyme Immunoassay Kit (Sigma) following the manufacturer’s instructions. Optical density was measured at 405 nm by a microplate reader (PerkinElmer Life Sciences).
Radioligand Competition Binding
Cells were rinsed, harvested, and homogenized with a Polytron homogenizer in 5 mM Tris-HCl containing 2 mM EDTA and protease inhibitors and centrifuged at 3400 × g for 30 min at 4 °C. The resulting pellet was resuspended in binding buffer (5 mM Tris containing 8.5 mM Hepes, 120 mM NaCI, 5.4 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4,and5mM glucose, pH 7.4) and protein concentrations were determined by the DC protein assay (Bio-Rad). 200 μgof sample was incubated with 0.4 nM of [3H]SCH23390 (86 Ci/mmol, Amersham Biosciences) in the presence of increasing concentrations of SKF81297 (10-12 to 10-4 M)at 4 °C for 2 h. Nonspecific binding was determined in the presence of 1 μM SCH39166 (25). All experiments were performed in triplicate. Binding data were analyzed by fitting the data with a sigmoidal dose-response curve to derive Bmaxand IC50using Graphpad Prism software.
Cell Surface Biotinylation
Confluent monolayers of HEK293T cells transfected with HA-D1 in the absence and presence of PSD-95-GFP co-transfection were washed three times with PBS and then incubated with gentle agitation for 30 min at 4 °C with 2 ml of 1 mg/ml Sulfo-NHS-SS-biotin (Pierce) in PBS (pH 7.4). The reaction was quenched by incubating the cells for an additional 10 min with 50 mM glycine in PBS. Cells were rinsed twice with a Tris-buffered saline buffer, dissolved in cold radioimmune precipitation assay buffer, and briefly sonicated. Each sample was divided into two aliquots. One aliquot was used for isolation of biotinylated proteins after immobilizing them to NeutrAvidin (Pierce). The second aliquot was used to determine the total proteins. Samples were analyzed by Western blotting.
RESULTS
PSD-95 Interacts with D1 in Vitro and in Vivo
We first investigated whether PSD-95 can interact with D1. HEK293T cells were transfected with PSD-95-GFP, HA-D1, or both. Immunoblot analysis (Fig. 1A) of whole-cell lysates showed that PSD-95-GFP was co-precipitated with an anti-HA antibody only when both proteins were expressed in these cells. Conversely, HA-D1 was co-precipitated with an anti-PSD-95 antibody only when both proteins were expressed, suggesting that D1 and PSD-95 were closely and specifically associated in these cells. To investigate whether PSD-95 may associate with D1 in vivo, we performed immunoprecipitation on protein extracts prepared from WT and PSD-95 KO mouse forebrain structures (Fig. 1B). A rat monoclonal anti-D1 antibody was able to precipitate PSD-95 in WT but not in KO mice. Similarly, an anti-PSD-95 antibody was able to precipitate D1 (Fig. 1B).
FIGURE 1. Interaction between D1 and PSD-95.

A, D1 interaction with PSD-95 in HEK293T cells. Cells were transfected with cDNA constructs encoding HA-D1 or PSD-95-GFP or both. Immunoprecipitation (IP) was performed by incubation of cell lysates with the indicated antibodies followed by immunoblotting (IB). IgG was used as a control in all immunoprecipitation experiments. B, D1 association with PSD-95 in the brain of WT but not PSD-95 KO mice. DOC-extracted mouse forebrain tissues were immunoprecipitated with an anti-D1 antibody or an anti-PSD-95 antibody, and the blots were revealed by PSD-95 and D1 antibodies, respectively. C, subcellular distribution of D1. Subcellular fractionation was performed on mouse forebrain structures. 40 μg of fractionations were loaded to each lane for Western blot analysis. D1 was particularly enriched in the PSD (one Triton) fraction compared with total homogenates (H), nuclear fraction (P1), the crude synaptosome (P2), and synaptosome (SSM). D, D1 association with PSD-95 in the PSD. Solubilized PSD was immunoprecipitated with an anti-D1 antibody, and the blot was revealed by an antibody against PSD-95. E, D1 co-localized with PSD-95 clusters in processes of cultured striatal neurons.
Previous studies suggest that PSD-95 is localized primarily in the PSD of dendritic spines (26). For D1 to interact with PSD-95 in the PSD, the receptor needs to travel to this postsynaptic site of glutamatergic transmission. Consistent with the electron microscopic (9, 10) and biochemical (27) studies, we found that D1 was enriched in the PSD purified from mouse forebrain structures. The high purity of our isolated PSD fraction was confirmed by its enrichment of PSD-95, PSD-93, and NMDA receptor subunits compared with other subcellular fractions, but the lack of the presynaptic protein synaptophysin (Fig. 1C). Moreover, D1 co-precipitated with PSD-95 in the purified PSD (Fig. 1D).
To confirm the immunoprecipitation data, we examined the subcellular localization of PSD-95 and D1 in cultured striatal neurons using immunofluorescence confocal microscopy. As expected, the more diffusive D1 staining co-localized with a subset of discrete clusters immunopositive to PSD-95 along neuronal processes (Fig. 1E). Together, these data demonstrate that PSD-95 interacts with D1 in vivo, possibly in the PSD.
Interaction between PSD-95 and D1 Receptors through the PSD-95 NH2 Terminus and the D1 COOH Terminus
We next mapped the protein domains on PSD-95 and D1 that mediate their interaction using recombinant GST affinity purification assays (Fig. 2, A–D). The intracellular loops and carboxyl tail of D1 contain putative protein-protein interaction domains important for intracellular signaling (28–30). Several domains of PSD-95, including the PDZs, the SH3, and the GK, are well characterized protein-protein interaction motifs (14). Recently, the NT of PSD-95, a region not previously known to be involved in protein-protein interaction, has been shown to interact with Src (31). GST fusion proteins encoding the various domains of D1 and PSD-95 were constructed (Fig. 2, A and B) and used as baits to precipitate associated proteins. Incubation of GST, GST-D1-IL2, GST-D1-IL3, or GST-D1-CT fusion proteins with PSD-95-GFP-expressing HEK293T lysates revealed a co-purification of PSD-95 and GST-D1-CT only. To our surprise, GST-PSD-95-NT, but not other GST-PSD-95 fusion proteins or GST alone, could precipitate solubilized D1 from HEK293T cells expressing the FLAG-tagged D1 (D1-FLAG). These data show that the D1 CT and the PSD-95 NT may mediate the interaction between these proteins.
FIGURE 2. Domain mapping of D1-PSD-95 interaction.

A, D1 CT binds PSD-95 in HEK293T cells. A1, schematic diagram showing D1 domain structures. A2, GST-D1-CT, but not GST-D1-IL2, GST-D1-IL3, or GST alone, was able to precipitate PSD-95 from HEK293T cells overexpressing PSD-95-GFP. GST fusion proteins used in the pull-down assays are shown at the bottom. B, PSD-95 NT binds D1 in HEK293T cells. B1, PSD-95 domain structures. B2, GST-PSD-95-NT, but not other GST-PSD-95 fusion proteins or GST alone, was able to precipitate D1 from HEK293T cells overexpressing D1-FLAG. C, GST-D1-CT, but not other GST-D1 fusion proteins or GST, alone was able to precipitate PSD-95 from mouse forebrain extracts. D, GST-PSD-95-NT, but not other GST-PSD-95 fusion proteins or GST alone, was able to precipitate D1 from mouse forebrain extracts. The GST fusion proteins shown above were used to precipitate brain extracts. E, the D1 truncation mutant D1ΔCT-FLAG failed to interact with PSD-95 in HEK293T cells. Cells were transfected with cDNAs encoding PSD-95-GFP and D1-FLAG or D1ΔCT-FLAG. F, the PSD-95 truncation mutant PSD-95ΔNT-FLAG failed to interact with HA-D1 in HEK293T cells. Cells were transfected with cDNAs encoding HA-D1 and PSD-95-FLAG or PSD-95ΔNT-FLAG. Expression of the truncated PSD-95, compared with the full-length protein, in the cells are shown at the bottom. IP, immunoprecipitation. IB, immunoblotting.
To investigate whether the D1 CT and the PSD-95 NT are responsible for D1-PSD-95 interaction in the brain, we performed GST pulldown assays on brain protein extracts using the above GST fusion proteins. GST-D1-CT, but not other GST-D1 fusion proteins or GST alone, was able to pull down PSD-95 from mouse forebrain extracts (Fig. 2C). Similarly, GST-PSD-95-NT, but not other GST-PSD-95 fusion proteins or GST alone, was able to pull down D1 from brain extracts (Fig. 2D). These data support our finding that the D1 CT and the PSD-95 NT may mediate the interaction seen between these two proteins in the brain.
To further confirm the interaction domains involved, we generated truncation mutants that lack the PSD-95 NT or the D1 CT. Immunoprecipitation analysis in HEK293T cells demonstrated that these mutations completely abolished the interaction between PSD-95 and D1 (Fig. 2, E and F). Thus, we conclude that the interaction between PSD-95 and D1 is mediated by PSD-95 NT and D1 CT.
PSD-95 Inhibits D1-mediated cAMP Production
We investigated how PSD-95 may regulate D1 function by measuring D1-mediated cAMP production in response to agonist stimulation in a HEK293 cell line stably expressing the rhesus macaque dopamine D1 receptor that shares all but two amino acid residues with its human homolog (referred as D1 stable cells) (Fig. 3). Basal cAMP levels were minimally detectable in these cells in the absence of agonist stimulation. However, stimulation of D1 by the full agonist SKF81297 (10 μM) produced a robust accumulation of cAMP in these cells (Fig. 3A). This effect was blocked by the D1 antagonist SCH23390, suggesting that the cAMP accumulation was mediated specifically by D1. Overexpression of PSD-95-GFP in these cells significantly inhibited the SKF81297-induced whole-cell cAMP accumulation (Fig. 3A). To evaluate whether PSD-95 overexpression may alter the ability of SKF81297 to stimulate cAMP production, we determined the dose-response curve of cAMP production in response to increasing concentrations of SKF81297 (Fig. 3B). PSD-95 co-expression decreased the maximal cAMP levels (Bmax) induced by saturating doses of SKF81297 but did not significantly alter the EC50of SKF81297 (D1 stable + mock: 4.95 ± 0.64 × 10-8 M; D1 stable + PSD-95: 4.45 ± 1.41 × 10-8 M; p = 0.72, Student’s t tests).
FIGURE 3. Inhibition of D1-mediated cAMP accumulation by PSD-95.

A, PSD-95 co-expression diminishes SKF81297-induced cAMP accumulation in D1 stable HEK293 cells. PSD-95-GFP- or mock-transfected cells were treated with saline or SKF81297 (10 μM) in the presence or absence of SCH23390 (10 μM) for 30 min. cAMP accumulation was measured with an enzyme immunoassay. **, p < 0.01, Student’s t test. B, dose-response curves of cAMP production in mock- or PSD-95-GFP-transfected D1 stable cells. Cells were stimulated with increasing concentrations of SKF81297 for 30 min. The maximum cAMP accumulation (Bmax) was reduced by PSD-95 co-expression. C, unaltered competition binding curves for mock- or PSD-95-GFP-transfected D1 stable cells. D, PSD-95 co-expression did not alter IC50or the total binding determined at the saturating doses. The data represent the mean ± S.E. of three independent experiments. Competition binding experiments were carried out using 400 pM [3H]SCH23390 and nine concentrations of competing SKF81297 ranging from 1 pM to 100μM. Each data point represents the mean ± S.E. of three replications. All experiments were repeated at least three times with essentially identical results.
To test whether PSD-95 may modulate the agonist binding properties of the D1 receptor, radioligand competition binding experiments were performed to quantify the number as well as agonist binding parameters of D1 in both mock- and PSD-95-GFP-transfected D1 stable cells using the ligand [3H]SCH23390 and increasing concentrations of SKF81297 (Fig. 3C). PSD-95 overexpression had no effect on either the total number of D1 receptors expressed or the IC50of SKF81297 (Fig. 3D). These results indicate that the inhibition of D1 activity by PSD-95 co-expression was not due to a decrease in the whole-cell levels of the receptor or altered D1 pharmacological properties.
PSD-95 Reduces Surface but Increases Intracellular D1 Expression
One mechanism by which PSD-95 inhibits D1 signaling is to reduce the surface expression of the receptor. To examine this possibility, we performed surface biotinylation experiments using Sulfo-NHS-SS-biotin on HEK293T cells transfected with an extracellularlly tagged HA-D1 and PSD-95-GFP or the HA-D1 and an empty vector (Fig. 4, A and B). Sulfo-NHSSS-biotin binds to free amino groups of proteins and, because it is membrane impermeable, can be used to distinguish between cell surface and intracellular proteins. Membrane-bound proteins labeled with Sulfo-NHS-SS-biotin were isolated with NeutrAvidin beads and analyzed by Western blotting using an anti-HA antibody. Total levels of D1 receptors expressed in these cells were determined by Western blotting on whole-cell lysates. We found that the expression of D1 on the cell membrane was decreased when PSD-95 was co-expressed. In contrast, total D1 levels were similar in cells expressing D1 and cells expressing both D1 and PSD-95 (Fig. 4, A and B).
FIGURE 4. PSD-95 reduces D1 surface expression and promotes intracellular D1 localization.
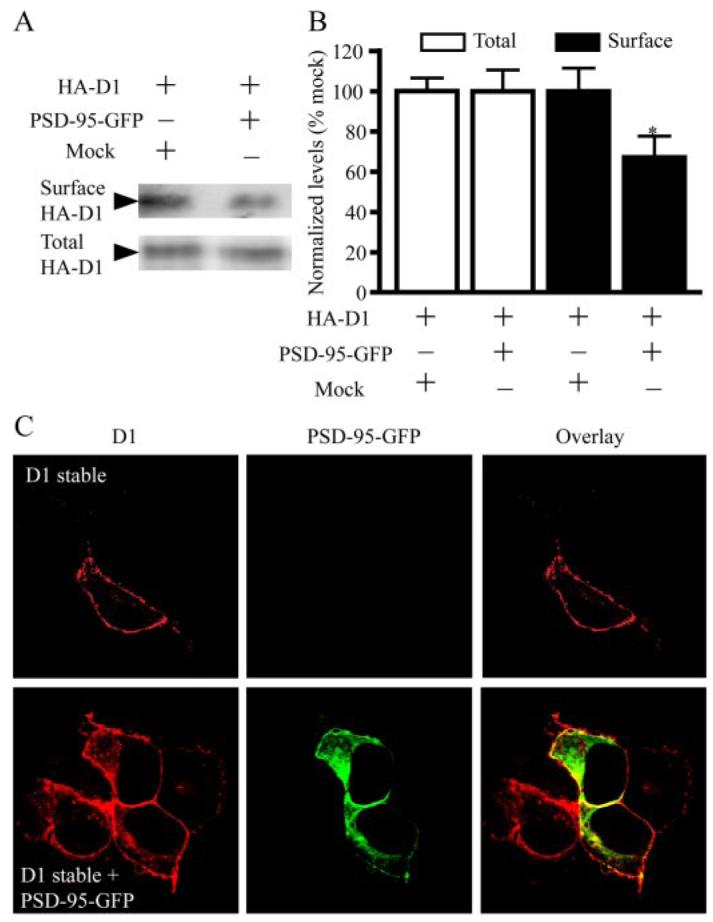
A, reduced surface D1 expression by PSD-95. Cell surface biotinylation experiments were performed on HEK293T cells transiently trancfected with HA-D1 and an empty vector or HA-D1 and PSD-95-GFP. Biotin-labeled surface D1 and total D1 were analyzed by Western blots. B, quantification of surface and total D1 levels. Mean ± S.E. represents three independent experiments. *, p < 0.05, Student’s t test. C, increased presence of intracellular D1 in PSD-95-GFP co-expressing cells. D1 stable HEK293 cells were transiently transfected with a mock vector or PSD-95-GFP. Following immunostaining, fluorescence was visualized by confocal microscopy. For this and the following figures, confocal images shown were single focal plane scannings through the middle of cells.
The selective reduction of the surface, but not the total, D1 expression by PSD-95 suggests that PSD-95 should increase the intra-cellular localization of D1. To directly test this, we examined the subcellular localization of D1 receptors in D1 stable cells in the presence and the absence of PSD-95-GFP overexpression. Consistent with previous reports (29, 32), the stably expressing D1 receptors were predominantly localized on the plasma membrane (Fig. 4C). The overex-pressed PSD-95 co-localized with the stably expressed D1 receptors at the plasma membrane and, to a lesser extent, in the cytosol (Fig. 4C). Significantly, co-expression with PSD-95 resulted in a redistribution of D1 receptors evident by the substantially increased levels of intracellular D1. Together, our data suggest that PSD-95 diminishes surface D1 expression by promoting the internalization of D1 receptors.
PSD-95 Facilitates Constitutive D1 Internalization
We next investigated whether the increased intracellular D1 localization in PSD-95 co-expressing cells may be due to an enhanced constitutive endocytosis of D1. We specifically labeled the surface D1 receptors with an anti-HA antibody and followed the internalization of these receptor-antibody complexes in live cells for 30 min (Fig. 5). The unique extracellular position of the HA epitope at the NH2terminus of D1 allowed the labeling of the surface receptors under non-permeabilized or live conditions followed by intracellular staining under permeabilized conditions for the same cell. We observed that, when expressed alone, HA-D1 displayed little constitutive endocytosis, consistent with previous reports (32). In contrast, robust D1 internalization occurred in cells co-expressing D1 and PSD-95 (Fig. 5A), suggesting that PSD-95 promotes the constitutive endocytosis of D1 receptors.
FIGURE 5. PSD-95 promotes constitutive D1 internalization in HEK293 cells.
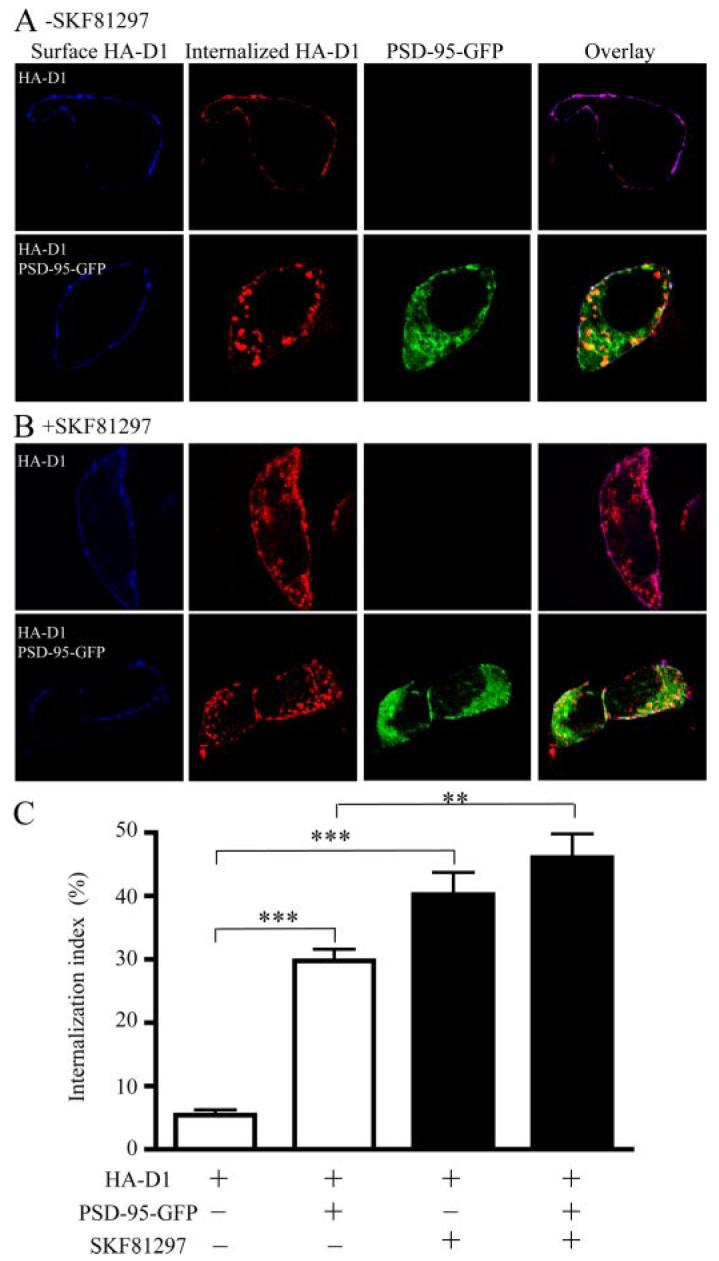
A, constitutive D1 endocytosis in HEK293 cells. B, SKF81297-stimulated D1 endocytosis in HEK293 cells. C, quantification of constitutive and agonist-induced D1 internalization. HEK293 cells were transfected with cDNAs encoding HA-D1 in the presence or absence of PSD-95-GFP co-transfection. Surface receptors were live-conjugated with an anti-HA antibody and were allowed for endocytosis (37 °C, 30 min) in the absence or presence of SKF81297 (10 μM), and processed for differential staining of remaining surface (before permeabilization) and internalized (after permeabilization) receptors. Internalization index, defined as the ratio of internalized to total fluorescence intensities, was presented as mean ± S.E. for 15 cells in each condition. **, p < 0.01; ***, p < 0.0001 versus controls, Student’s t tests.
Agonist-stimulated receptor internalization, a process in which activated receptors move from the cell surface into intra-cellular vesicles, is a common feature of G protein-coupled receptors important for regulation of the receptor responsiveness (33). D1 undergoes agonist-dependent internalization (32, 34, 35) mediated by residues in the carboxyl tail (36). Because PSD-95 selectively binds the D1 CT, we examined the degree to which co-expression of PSD-95 with D1 might affect the agonist-induced D1 internalization process. In agreement with previous reports (32, 34, 35), SKF81297 induced robust D1 internalization in HA-D1 transfected HEK293 cells (Fig. 5B)or D1 stable cells (data not shown), regardless of the PSD-95 over-expression. Quantification of the SKF81297-induced D1 internalization in HA-D1 transfected HEK293 cells indicated similar degrees of D1 internalization in the presence and absence of PSD-95 co-expression in these cells (Fig. 5C). It is interesting to note that, while the agonist stimulation caused a large portion (∼40% induced compared with ∼5% basal) of surface D1 receptors to internalize in cells transfected with HA-D1 alone, the same stimulation only induced an additional 15% (45% induced compared with 30% basal) of D1 receptors to internalize in cells overex-pressing both HA-D1 and PSD-95-GFP. Thus, the constitutive and agonist-induced endocytic processes may act on the same pool of surface receptors, and PSD-95 may be a regulatory component of the agonist-induced D1 endocytosis pathway.
PSD-95 NT Mediates Constitutive D1 Internalization
To investigate whether the D1-PSD-95 interaction mediated by PSD-95 NT was responsible for the constitutive D1 internalization, we generated a series of PSD-95 truncation mutants and analyzed their effects on constitutive D1 internalization in co-transfected HEK293 cells (Fig.6). Removing the GK and SH3-GK domains of PSD-95 (Fig. 6A) still induced robust constitutive D1 internalization not distinguishable from that induced by the full-length PSD-95. In contrast, deletion of PSD-95 NT completely abolished the PSD-95 induced constitutive D1 internalization. Significantly, PSD-95 NT, the 72-amino acid peptide alone in the absence of the three PDZ, SH3, and GK domains, while displaying altered subcellular distribution compared with the full-length PSD-95, retained the full potential in inducing constitutive D1 endocytosis (Fig. 6, B and C). These data demonstrate that the constitutive D1 internalization in cells expressing both D1 and PSD-95 is entirely mediated by PSD-95 NT, with little contribution from other regions of the protein.
FIGURE 6. PSD-95 promotes constitutive D1 internalization via PSD-95 NT.

A, diagrams illustrating PSD-95 truncation constructs. B, deletion of PSD-95 NT, but not other motifs, abolished the constitutive D1 internalization in HEK293 cells. Note that the GFP-tagged NH2-terminal domain (NT-GFP) by itself is sufficient in inducing constitutive D1 internalization when co-expressed with HA-D1 in HEK293 cells. C, summary graph showing the effects of overexpressing the various PSD-95 constructs on D1 internalization. HEK293 cells were transfected with HA-D1 or HA-D1 and the various PSD-95 constructs. Receptor internalization was performed and quantified as in Fig. 5. Data were presented as mean ± S.E. (n = 15–17 cells for each group). ***, p < 0.0001 versus control, Student’s t tests.
PSD-95 Promotion of Constitutive D1 Internalization is Dynamin-dependent
D1 is known to internalize via the dynamin-dependent, clathrin-coated vesicle-mediated endocytic pathway (32). Dynamin, a GTPase, catalyzes the GTP-dependent pinching off of endocytic vesicles from the plasma membrane (37, 38). The Lys-44 dynamin mutants defective in GTPase activity can block endogenous dynamin function and inhibit clathrin-dependent endocytosis in many cell types (39), thus these dominant negative mutants serve as powerful tools in determining whether a process is dynamin-dependent. We investigated whether the PSD-95-elicited constitutive D1 internalization was mediated by the dynamin-dependent endocytic pathway in HEK293 cells (Fig. 7). Consistent with previous studies, the neuronal isoform dynamin I (40) was not detectable in HEK293 cells (data not shown), which presumably express the ubiquitous dynamin II as the endogenous dynamin (41). Transfection of the WT (Dyn I-WT) and the dominant negative K44A mutant (Dyn I-K44A) of dynamin I constructs (23) resulted in their expression in HEK293 cells (Fig. 7A). Co-expression of Dyn I-WT in cells expressing HA-D1 and PSD-95-GFP did not affect the intracellular localization of D1 in these cells. However, Dyn I-K44A overexpression abolished the constitutive D1 endocytosis, as D1 receptors remained associated with the plasma membrane in these cells.
FIGURE 7. PSD-95-mediated constitutive D1 internalization is dynamin-dependent.

A, representative images showing that overexpression of Dyn I-K44A, but not Dyn I-WT, abolished the intracellular D1 localization in HEK293 cells co-expressing HA-D1 and PSD-95-GFP. B, constitutive and SKF81297-stimulated D1 internalization in HEK293 cells expressing HA-D1 or HA-D1/PSD-95-GFP. Sur., surface; Inter., internalized. C, overall lack of effect of Dyn I-WT overexpression on the constitutive and stimulated D1 internalization in HEK293 cells. D, Dyn I-K44A overexpression abolished the constitutive and stimulated D1 internalization in HEK293 cells. E, summary of K44A effect on D1 internalization from (B–D). HEK293 cells were transfected with combinations of cDNAs encoding HA-D1, PSD-95-GFP, Dyn I-WT, or Dyn I-K44A. Receptor internalization shown in B–E was performed and quantified as in Fig. 5. Data were presented as mean ± S.E. (n = 15– 41 cells for each group). **, p < 0.01; ***, p < 0.0001 versus corresponding Dyn I-WT transfection controls; ###, p < 0.0001 versus corresponding no Dyn I transfection controls, Student’s t tests.
To quantify the effect of K44A on D1 endocytosis, we performed a series of experiments on HA-D1-expressing or HA-D1/PSD-95-GFP-co-expressing cells in the presence or absence of Dyn I-WT or Dyn I-K44A co-transfection using the above D1 internalization assay. In the absence of Dyn I-WT or Dyn I-K44A transfection, D1 showed little constitutive endocytosis in HA-D1 expressing cells but displayed robust constitutive internalization when PSD-95 is present, and SKF81297 stimulation induced D1 internalization regardless of PSD-95 overexpression (Fig. 7B; also see Figs. 5 and 6). Overexpression of Dyn I-WT (Fig. 7, C and E) only slightly affected the constitutive and agonist-induced D1 internalization profiles compared with the no Dyn I transfection control. In contrast, over-expression of Dyn I-K44A (Fig. 7, D and E) essentially blocked the agonist-induced D1 internalization and the PSD-95-induced constitutive D1 internalization, compared with the no-Dyn I (Fig. 7B) or the Dyn I-WT (Fig. 7C) controls. It is worthy noting that our D1 internalization assay precluded a simultaneous monitoring of the dynamin I expression with the surface D1, internal D1, and PSD-95 expression in the same cells, thus the expression level of transfected Dyn I-K44A in individual cells could not be confirmed. However, our independent experiments evaluating the transfection efficiency of dynamin I suggest that greater than 85% of cells expressed the transfected constructs at significant levels (data not shown). Thus, our quantitative analysis based on a large number of randomly sampled cells (Fig. 7E) represents an underestimate of the K44A effect on D1 internalization. Taken together, these data demonstrate that PSD-95 promotes D1 internalization through a dynamin-dependent mechanism.
Enhanced Behavioral Responses to D1 Agonists in PSD-95 KO Mice
To explore the in vivo physiological significance of PSD-95 regulation of the D1 receptor, we analyzed D1-mediated behaviors in mice deficient in PSD-95 (Fig. 8). Locomotor activation in response to the selective D1 agonist SKF81297 was compared between WT and PSD-95 KO mice. Consistent with our previous report (21), PSD-95 KO mice displayed reduced locomotor activity in the open field (Fig. 8, A–C). However, administration of SKF81297 at 3 and 4 mg/kg (intraperitoneal) elicited substantially larger and more prolonged locomotor activation in PSD-95 KO mice at both doses compared with their WT littermates (Fig. 8, A, B, and D).
FIGURE 8. Enhanced D1-mediated behavioral responses in PSD-95 KO mice.

A and B, time courses of the effect of SKF81297 (3 and 4 mg/kg, intraperitoneal) on locomotion, measured as the distance traveled. C, time courses of the effect of amphetamine (3 mg/kg, intraperitoneal) on locomotion. D, quantification of total distance traveled by the mice for 60 min after SKF81297 (45–105 min) and amphetamine (120–180 min) administration. PSD-95 KO and WT littermates (n = 6) were habituated in a locomotor activity monitor for 60 min or until the mice were fully habituated. The mice were then injected (arrows) with dopamine receptor agonists at the doses indicated. Horizontal activity, measured as the distance traveled, was continuously recorded in blocks of 4 or 5 min. All data are mean ± S.E. *, p < 0.05 versus WT mice, Student’s t test.
To evaluate the impact of PSD-95 deletion on the overall responsiveness of dopamine system to dopamine, we tested the locomotor responses to the psychostimulant amphetamine, an indirect dopamine agonist that raises extracellular dopamine levels, in WT and PSD-95 KO mice. Amphetamine (3 mg/kg, intraperitoneal) elicited substantial locomotor activation in WT mice (Fig. 8C). Notably, PSD-95 KO littermates displayed a robust prolongation and marked enhancement in their loco-motor responses to amphetamine. Taken together, our data define an inhibitory role for PSD-95 in D1 function and establish an important role for PSD-95 in mediating the locomotor stimulant effects of both amphetamine (this study) and cocaine (21).
DISCUSSION
The dopamine D1 receptor plays essential roles in reward, working memory, and motor coordination and altered D1 functions are implicated in addiction, schizophrenia, and neurode-generative disorders (1–3). In this study we provide evidence that D1 interacts in vitro and in vivo with PSD-95, a postsynaptic scaffolding protein involved in synaptic plasticity and learning. The interaction is mediated by the COOH-terminal tail of D1 and the NH2terminus of PSD-95. PSD-95 regulates the surface expression and the intracellular trafficking of D1 via a dynamin-dependent process and inhibits D1-mediated signaling in mammalian cells. When PSD-95 is deleted in mice, D1-mediated behavioral responses are enhanced in the mutant animals. Our studies identify a novel molecular mechanism by which dopamine signaling in dendritic spines and glutamatergic synapses can be regulated.
The inhibition of D1-mediated cAMP accumulation by PSD-95 co-expression is likely mediated by a reduced surface D1 expression, rather than an altered pharmacological profile of the receptor to ligands. Consistent with this interpretation, the decrease (Fig. 4B) of surface D1 receptors when co-expressed with PSD-95 is sufficient to account for the decrease (Fig. 3, A and B) of D1-mediated maximum cAMP production under similar transfection conditions. However, we cannot rule out the possibility that PSD-95 may regulate G protein coupling to the receptor leading to cAMP production and/or uncoupling from activated receptor resulting in desensitization. For example, recent evidence suggests that PSD-95 interacts with a Gγ subunit and may affect G protein signaling (42).
Previous studies have established an inhibitory role of PSD-95 in the agonist-induced internalization of a number of receptors, including NMDA (16), the β1 adrenergic (43), and the serotonin 5HT2Areceptors (44). These inhibitory effects are believed to result from the PDZ interaction-dependent stabilization of these receptors at the cell surface. In this regard, our finding that PSD-95 facilitates, rather than hinders, D1 internalization, which is likely mediated by a novel (see below) NH2terminus-dependent interaction, demonstrates a previously unrecognized role for PSD-95 in receptor trafficking. In the absence of agonists, D1 undergoes little internalization (32). However, upon agonist binding, D1 is quickly internalized following the classical G protein-coupled receptor endocytic pathway (33) that involves receptor phosphorylation by G protein-coupled receptor kinases, β-arrestin-mediated recruitments of clathrin and the AP-2 complex to activated receptors, and subsequent clathrin-mediated endocytosis. The mechanisms of the PSD-95-promoted, dynamin-dependent constitutive D1 endocytosis are unknown but may conceivably involve regulation of one or more steps of this pathway at the plasma membrane. Future studies will also be needed to test whether PSD-95 may retard receptor trafficking to the cell surface, perhaps by associating with newly synthesized receptors.
Despite the mounting evidence of D1 enrichment in the dendritic spines (9–13) and the identifications of a number of D1-interacting proteins that include NMDA receptor subunits (27, 30), calcyon (28), DRiP78 (45), and neurofilament-M (29), the molecular processes mediating the targeting, anchoring, and signaling of D1 within spines remain elusive. Our findings that D1 associates with PSD-95, a signature postsynaptic scaffold in the PSD and dendritic spines, and that PSD-95 can regulate D1 endocytosis and signaling, suggest a mechanism that may mediate D1 trafficking and function within spines. Previous studies show that the NR1 and NR2A type NMDA receptors may be required for the targeting of D1 to the cell surface in vitro (27). Additionally, the association of PSD-95 with NMDA receptors and the functional significance of these PDZ-mediated interactions have been well demonstrated (46, 47). Thus, a tertiary protein complex of D1-NMDA receptor-PSD-95 may exist in the brain that plays a role in the sorting and synaptic delivery of D1 within spines.
Emerging evidence reveals a positive feedback loop between the NMDA receptor and D1 (48), both of which are concentrated in the heads and PSDs of spines, where most of the glutamatergic synapses are formed. Specifically, D1 activation stimulates adenylate cyclases, increases cAMP production, and activates PKA leading to potentiation of the NMDA receptor via the PKA/DARPP-32/PP1 cascade (6). On the other hand, recent studies suggest that NMDA receptor activation enhances D1-mediated cAMP accumulation by recruiting more D1 to the plasma membrane (49). Consequently, a positive feedback loop is created that, if not controlled, can result in concomitant overactivation of both D1 and NMDA receptor systems, triggering NMDA receptor- and D1-dependent neurotoxicity and cell death (50, 51). Our results suggest that D1-PSD-95 interaction may serve as a mechanism to antagonize the NMDA-dependent surface delivery/anchorage of D1, thus preventing excessive D1 insertions to the synapse.
Two alternative NH2-terminal domains define two distinct PSD-95 isoforms, the predominant α-isoform and a small portion of the β-isoform (52). The NH2terminus of the PSD-95 α-isoform studied here is best recognized for its role in the membrane trafficking and clustering of PSD-95 following palmitoylation on the conserved NH2-terminal cysteins (22). The three PDZ, SH3, and GK domains of PSD-95 are well known to interact with proteins of diverse functions, such as the NMDA receptor, signaling enzymes, kinase scaffolds (14), and other monoamine receptors (43, 44). In contrast, the 72 amino acids of the PSD-95 NT lack well defined protein interaction motifs and had not been implicated in protein-protein interaction until a recent report showing that this domain is involved in PSD-95-Src interaction to regulate Src kinase activity (31). Our present findings represent the second case where this domain is implicated in protein-protein interaction. This novel molecular interaction may bring the dopamine system to the vicinity of a delicate signaling network organized by PSD-95 at the synapse.
In summary, we identify a novel molecular mechanism by which dopamine signaling mediated by the D1 receptor can be regulated within dendritic spines. This molecular interaction between PSD-95 and D1 has functional significance in vivo since its disruption enhances D1-mediated behaviors, which may explain the supersensitivity to cocaine and amphetamine, two major drugs of abuse, in the mutant mice. In lieu of the “synaptic triads” formed among the postsynaptic dendritic spines and the afferent dopaminergic and the glutamatergic terminals in cortical pyramidal neurons and striatal medium spiny neurons (53), further investigation of this interaction should facilitate our understanding of the mechanism mediating the functional interaction between dopamine and gluta-mate transmission. Our findings are relevant for the treatments of addiction, schizophrenia, and motor deficits associated with neurological diseases.
Acknowledgments
We thank Dr. Allan Levey for the rat monoclonal anti-D1 antibody. We thank Dr. Jae U. Jung for providing reagents and the New England Primate Research Center Division of Pathology for help with sample processing. We thank Drs. Roger Spealman, James Rowlett, and Tai-Xiang Xu for helpful discussions.
Footnotes
This work was supported by National Institutes of Health Grants RR00168 (to the New England Primate Research Center), by NINDS/National Institutes of Health Grant P50 NS39793 and the Stern, Orchard, and Anti-aging Foundations (to O. I.), and by a National Alliance for Research on Schizophrenia and Depression Young Investigator Award (to W.-D. Y.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article was selected as a Paper of the Week.
- PSD
- postsynaptic density
- PDZ
- PSD-95, Dlg, ZO-1
- SH3
- Src homology 3
- GK
- guanylate kinase-like
- NMDA
- N-methyl-D-aspartate
- GFP
- green fluorescent protein
- HA
- hemagglutinin
- NT
- NH2terminus
- CT
- COOH terminus
- IL
- intracellular loop
- GST
- glutathione S-transferase
- PBS
- phosphate-buffered saline
- Dyn I
- dynamin I
- HEK
- human embryonic kidney
- WT
- wild-type
- KO
- knock-out
REFERENCES
- 1.Graybiel AM, Aosaki T, Flaherty AW, Kimura M. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- 2.Goldman-Rakic PS. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 3.Berke JD, Hyman SE. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 4.Lachowicz JE, Sibley DR. Pharmacol. Toxicol. 1997;81:105–113. doi: 10.1111/j.1600-0773.1997.tb00039.x. [DOI] [PubMed] [Google Scholar]
- 5.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Physiol. Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 6.Greengard P. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- 7.Bibb JA. Cell. 2005;122:153–155. doi: 10.1016/j.cell.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Nicola SM, Surmeier J, Malenka RC. Annu. Rev. Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 9.Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ. Proc. Natl. Acad. Sci. U. S. A. 1993;90:8861–8865. doi: 10.1073/pnas.90.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI. J. Neurosci. 1995;15:5222–5237. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. Proc. Natl. Acad. Sci. U. S. A. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS. J. Neurosci. 1995;15:7821–7836. doi: 10.1523/JNEUROSCI.15-12-07821.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sesack SR, Carr DB, Omelchenko N, Pinto A. Ann. N. Y. Acad. Sci. 2003;1003:36–52. doi: 10.1196/annals.1300.066. [DOI] [PubMed] [Google Scholar]
- 14.Kim E, Sheng M. Nat. Rev. Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy MB. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- 16.Roche KW, Standley S, McCallum J, Ly C. Dune, Ehlers MD, Wenthold RJ. Nat. Neurosci. 2000;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- 17.Stein V, House DR, Bredt DS, Nicoll RA. J. Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehrlich I, Malinow R. J. Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 20.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O’Dell TJ, Grant SG. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 21.Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Neuron. 2004;41:625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- 22.Topinka JR, Bredt DS. Neuron. 1998;20:125–134. doi: 10.1016/s0896-6273(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Ferguson SS, Barak LS, Menard L, Caron MG. J. Biol. Chem. 1996;271:18302–18305. doi: 10.1074/jbc.271.31.18302. [DOI] [PubMed] [Google Scholar]
- 24.Carlin RK, Grab DJ, Cohen RS, Siekevitz P. J. Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jassen AK, Yang H, Miller GM, Calder E, Madras BK. Mol. Pharmacol. 2006;70:71–77. doi: 10.1124/mol.105.021998. [DOI] [PubMed] [Google Scholar]
- 26.Hunt CA, Schenker LJ, Kennedy MB. J. Neurosci. 1996;16:1380–1388. doi: 10.1523/JNEUROSCI.16-04-01380.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiorentini C, Gardoni F, Spano P, Di Luca M, Missale C. J. Biol. Chem. 2003;278:20196–20202. doi: 10.1074/jbc.M213140200. [DOI] [PubMed] [Google Scholar]
- 28.Lezcano N, Mrzljak L, Eubanks S, Levenson R, Goldman-Rakic P, Bergson C. Science. 2000;287:1660–1664. doi: 10.1126/science.287.5458.1660. [DOI] [PubMed] [Google Scholar]
- 29.Kim OJ, Ariano MA, Lazzarini RA, Levine MS, Sibley DR. J. Neurosci. 2002;22:5920–5930. doi: 10.1523/JNEUROSCI.22-14-05920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Cell. 2002;111:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- 31.Kalia LV, Pitcher GM, Pelkey KA, Salter MW. EMBO J. 2006;25:4971–4982. doi: 10.1038/sj.emboj.7601342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vickery RG, von Zastrow M. J. Cell Biol. 1999;144:31–43. doi: 10.1083/jcb.144.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Annu. Rev. Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- 34.Tiberi M, Nash SR, Bertrand L, Lefkowitz RJ, Caron MG. J. Biol. Chem. 1996;271:3771–3778. doi: 10.1074/jbc.271.7.3771. [DOI] [PubMed] [Google Scholar]
- 35.Jiang D, Sibley DR. Mol. Pharmacol. 1999;56:675–683. [PubMed] [Google Scholar]
- 36.Lamey M, Thompson M, Varghese G, Chi H, Sawzdargo M, George SR, O’Dowd BF. J. Biol. Chem. 2002;277:9415–9421. doi: 10.1074/jbc.M111811200. [DOI] [PubMed] [Google Scholar]
- 37.Hinshaw JE, Schmid SL. Nature. 1995;374:190–192. doi: 10.1038/374190a0. [DOI] [PubMed] [Google Scholar]
- 38.Takei K, McPherson PS, Schmid SL, De Camilli P. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- 39.Cook TA, Urrutia R, McNiven MA. Proc. Natl. Acad. Sci. U. S. A. 1994;91:644–648. doi: 10.1073/pnas.91.2.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sontag JM, Fykse EM, Ushkaryov Y, Liu JP, Robinson PJ, Sudhof TC. J. Biol. Chem. 1994;269:4547–4554. [PubMed] [Google Scholar]
- 41.Altschuler Y, Barbas SM, Terlecky LJ, Tang K, Hardy S, Mostov KE, Schmid SL. J. Cell Biol. 1998;143:1871–1881. doi: 10.1083/jcb.143.7.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Benard O, Margolskee RF. J. Biol. Chem. 2006;281:11066–11073. doi: 10.1074/jbc.M600113200. [DOI] [PubMed] [Google Scholar]
- 43.Hu LA, Tang Y, Miller WE, Cong M, Lau AG, Lefkowitz RJ, Hall RA. J. Biol. Chem. 2000;275:38659–38666. doi: 10.1074/jbc.M005938200. [DOI] [PubMed] [Google Scholar]
- 44.Xia Z, Gray JA, Compton-Toth BA, Roth BL. J. Biol. Chem. 2003;278:21901–21908. doi: 10.1074/jbc.M301905200. [DOI] [PubMed] [Google Scholar]
- 45.Bermak JC, Li M, Bullock C, Zhou QY. Nat. Cell Biol. 2001;3:492–498. doi: 10.1038/35074561. [DOI] [PubMed] [Google Scholar]
- 46.Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 47.Niethammer M, Kim E, Sheng M. J. Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cepeda C, Levine MS. Sci. STKE. 2006;333:pe20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- 49.Pei L, Lee FJ, Moszczynska A, Vukusic B, Liu F. J. Neurosci. 2004;24:1149–1158. doi: 10.1523/JNEUROSCI.3922-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi DW. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 51.Bozzi Y, Borrelli E. Trends Neurosci. 2006;29:167–174. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 52.Schluter OM, Xu W, Malenka RC. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Goldman-Rakic PS, Leranth C, Williams SM, Mons N, Geffard M. Proc. Natl. Acad. Sci. U. S. A. 1989;86:9015–9019. doi: 10.1073/pnas.86.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]