

Abstract
Biolistic transfection is a technique in which subcellular-sized particles coated with DNA are accelerated to high velocity to propel them into cells. This method is applicable to tissues, cells and organelles, and can be used for both in vitro and in vivo transformations; with the right equipment, it is simple, rapid and efficient. Here we provide a detailed protocol for biolistic transfection of plasmids into cultured human embryonic kidney (HEK) 293 cells and organotypic brain slices using a hand-held gene gun. There are three major steps: (i) coating microcarriers with DNA, (ii) transferring the microcarriers into a cartridge to make a ‘bullet’, and (iii) firing the DNA-coated microcarriers into cells using a pulse of helium gas. The method can be readily adapted to other cell types and tissues. The protocol can be completed in 1–2 h.
INTRODUCTION
Genetic material can be delivered to cells using biological, chemical or mechanical methods. Biolistic transfection is a mechanical method that is potentially applicable to a wide variety of cell and tissue types. It involves the high-speed propulsion of subcellular particles coated with DNA into cells, and therefore does not depend on specific biochemical features (such as particular proteins or lipids) of the cell surface or on the growth rate of the cells. It was initially developed as a method of gene transfer into plants, as it allows transfer across cell walls1,2; however, it is now becoming appreciated as a technique that is far more widely applicable and, because it can be used for in vivo as well as in vitro transfections, it has tremendous potential for gene therapy3,4. Finally, co-transfection with two or more different DNA plasmids is easy and efficient. Other popular methods of inserting DNA into cultured cells include the following: injection, which is extremely precise but requires operator skill and is slow; viral infection, which is rapid and efficient, but has considerable safety implications and might change cell phenotype; lipofection, which might be toxic to cells, has some limitations in terms of the DNA it can deliver and is expensive, although it is also simple and efficient; electroporation, which requires cells in suspension, and is dependent on cell type and specific conditions, although it is relatively efficient, especially when transfecting many cells; and calcium phosphate precipitation, which is simple and cheap but inefficient. Most of these methods require rapidly dividing cells and therefore cannot be used on terminally differentiated cells, such as neurons. Few of these methods can be adapted to transfect cells in tissues. Indeed, the gene gun is the only method currently available that allows the transfection of DNA deep into tissues. It has the advantage of being able to overcome physical barriers, (e.g., the stratum corneum of the epidermis), it can be used multiple times on the same sample, it is suitable for co-transfection of two or more DNAs in a single shot, it can be used on large numbers of cells, and it is rapid and simple to use5,6. The major advantage, however, it that it is highly efficient; a recent study in rat brain cultures indicated that this method was 160-fold, 189-fold and 450-fold more effective than lipofection, electroporation and calcium phosphate precipitation, respectively, when assaying luciferase activity7. The major drawback is that the gene gun itself is expensive to purchase, although the consumable costs thereafter are relatively low.
Early biolistic devices required a vacuum chamber; the target sample was placed inside the chamber, the overlying air was evacuated and microcarriers coated with the gene of interest were fired into the target. The size of the sample was therefore limited and the vacuum had the potential to damage the sample, although the technique was reasonably successful in some cases8. While this method is still in use, much more popular today is the hand-held gene gun, which can be used in a wide range of applications, including in bacteria, organelles, culture cells, tissues and whole animals. These devices have been shown to be highly effective in delivering DNA, although there are some problems with cell damage as high gas pressures (routinely 1,379 kPa (200 psi)) have frequently been used9. Recently, however, modifications of the accelerator chamber (Fig. 1) have allowed the use of lower gas pressure (routinely 345 kPa (50 psi)) without loss of transfection efficiency5. In addition this modified gun transfects a smaller area and increases depth penetration, thereby allowing smaller and deeper tissues to be transfected.
Figure 1.
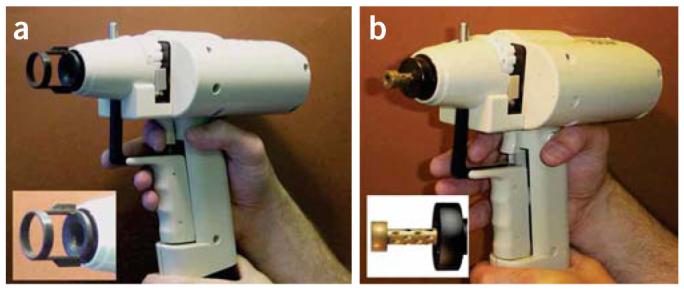
Helios gene gun. (a) The gene gun is loaded and ready for discharge. Here, it is displayed with the original accelerator chamber and associated spacer (inset). The spacer defines the minimum distance between the target and the accelerator channel cone. The cone is designed to spread the gold/DNA particles superficially over a wide area. (b) The gene gun with the modified chamber. The spacer has been eliminated, and the cone-shaped barrel is replaced with an external barrel with a reduced diameter (inset). This reduces the spread of microcarriers over the target site and allows the apparatus to be placed closer to tissue targets.
This protocol requires a hand-held gene gun, although either the original or modified acceleration chamber is suitable. We use the Bio-Rad Helios gene gun with the original chamber for HEK293 cells and the modified chamber for tissue slices. Briefly, plasmids encoding a fluorescent protein or a fluorescent protein tag (usually enhanced yellow fluorescent protein (EYFP), although we have also used cyan fluorescent protein (CFP) and red fluorescent protein (RFP)), are precipitated onto the appropriate microcarriers. We use gold particles, although tungsten particles are also suitable. Gold particles are preferred by many because they are more uniform in shape and size, and DNA on gold particles is less susceptible to catalytic degradation; in addition, tungsten particles might acidify the culture medium and cause cell death10. These microcarriers are attached to the inside of a length of tubing, which is then cut to the appropriate size to form a cartridge, and must be constructed from an inert, tough and flexible material. A widely used example is Tetzel tubing (Bio-Rad), but similar tubing can also be used. The cartridges are loaded into the cartridge holder. Firing of the gun (Fig. 2) involves releasing a propellant gas, which must be of sufficient velocity to strip the microcarriers from the inner surface of a cartridge and to propel them into the sample, but must not damage the target tissue. The propellant used must be inert, diffusible and of low density to ensure the particles can achieve high velocities, thus helium is the gas of choice. The Helios gene gun depends on the ‘Coanda phenomenon’, whereby a pulse of helium fired into the gene gun rapidly expands several fold and pulls the gold/DNA particles from the microcarrier tube. The expanding gas causes the gold/DNA particles to spiral down the accelerating channel with an increasing velocity. As soon as the helium leaves the cone barrel, it follows the contours of the outer surface, which allows the gold/DNA particles to accelerate out of the barrel liner in a continuing outward spiral motion until they reach the sample.
Figure 2.

Using the gene gun. The photograph shows the gene gun being used to transfect cell cultures.
Experimental design
Cell health and DNA quality are as important for efficient biolistic transfection as they are for other transfection methods. In addition, the size of the microcarriers, the amounts of DNA they carry and the quantities delivered should be optimized. The amount of DNA loaded per mg of microcarriers is referred to as the DNA-loading ratio (DLR). Typical DLRs range between 1 and 5 μg DNA per mg gold. The amount of microcarriers delivered per target is referred to as the microcarrier loading quantity (MLQ). Typical MLQs range from 0.1 to 0.5 mg per cartridge.
The final parameters that affect transfection efficiency are the distance the particles travel before striking the target cell and the pressure of the helium gas used as a propellant. The longer the distance traveled, the greater the spread of the particles over the target area and the less pronounced the helium shock wave that strikes the cells, which results in less cell damage and higher transfection efficiency. However, a longer distance results in reduced particle speed and a decreased likelihood of target penetration, thereby decreasing the transfection efficiency. Both the distance and the pressure, therefore, need to be optimized for each transfection. For cell cultures we use the unmodified gene gun, which delivers microcarriers superficially over a relatively wide area. For tissue slices and in vivo work we recommend the modified gene gun, which has a restricted target area and increased depth penetration even at low helium pressures, resulting in higher transfection efficiencies. The modifications have been previously described5. Briefly, the accelerator channel and its distal cone have been replaced by a modified accelerator channel and external barrel; the latter has a number of holes drilled in it, each at an angle of 30°. The barrel and accelerator channel are fabricated from brass and stainless steel, respectively; they are held in a Deldrin support and mounted into the gene gun via a screw thread.
The method we describe here has been optimized for HEK293 cells and tissue slices, but would also work with minimal modification for any cultured cells or tissue slices (Figs. 3-5). It is also applicable, with appropriate optimization, to whole tissues and, at the other extreme, organelles and bacteria.
Figure 3.
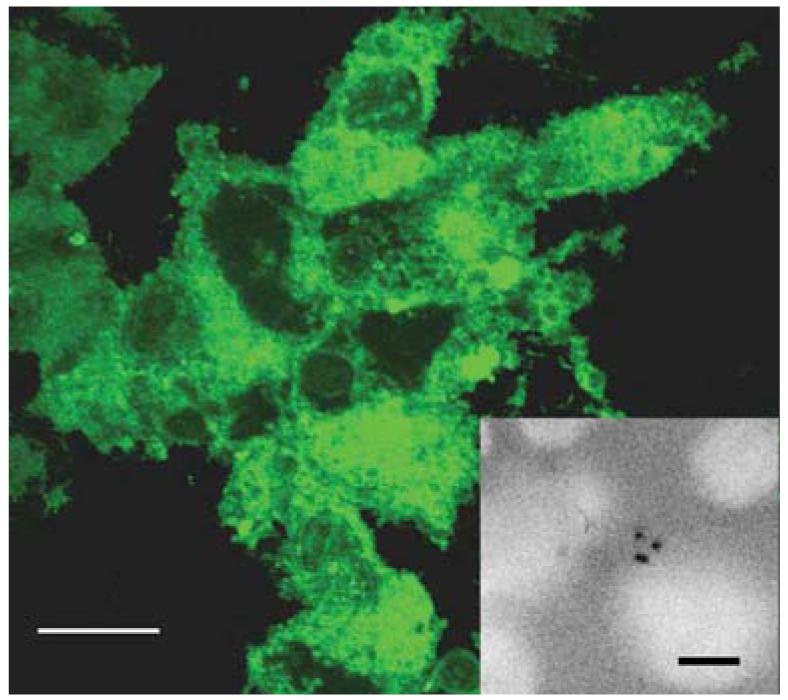
HEK293 cells expressing EYFP. Cells were transfected with the original barrel on the gene gun and observed 24 h post-transfection. In this sample, approximately 80% of the cells were transfected. The inset shows the location of bullets in a sample stained with Hoechst stain to identify nuclei5. Scale bar, 10 μm.
Figure 5.
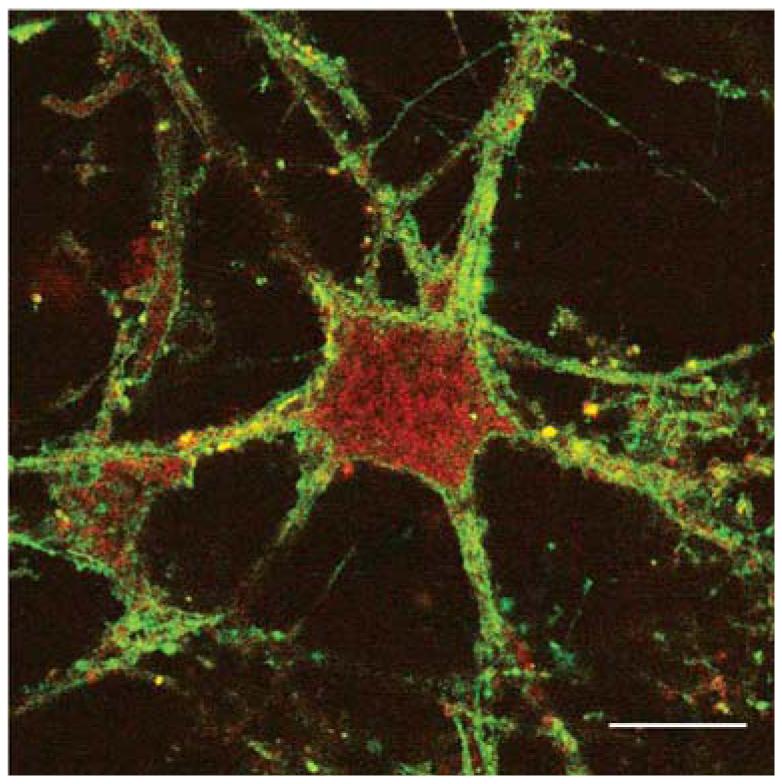
A neuron in a brain slice expressing two protein subunits. Brain slices were transfected using the modified gene gun with two 5-HT3 subunits, labeled with EYFP (subunit A) or RFP (subunit B). Functional 5-HT3 receptors are pentamers of subunit A only, or subunit A plus subunit B, as the latter will only reach the plasma membrane when co-assembled with the former. The image shows significant red labeling in the cytoplasm, where subunit B is retained if not ‘rescued’ by subunit A, but both yellow and orange labeling on the cell surface indicating subunit A- and subunit A plus B-containing receptors. Scale bar, 5 μm.
MATERIALS
REAGENTS
Experimental animals (mice or rats) ! CAUTION All experiments must be performed in accordance with the relevant authorities' ethical and safety guidelines and regulations
HEK293 cells and organotypic brain slices
Gold microcarriers (Bio-Rad)
S22 polyvinylpyrollidone (PVP; Bio-Rad): 75 μg ml−1 solution in ethanol (BDH)
S-0266 spermidine: 0.05 M solution (Sigma)
CaCl2: 1 M solution (Sigma)
Plasmid DNA (1 mg ml−1)
EQUIPMENT
Helios gene gun (Bio-Rad)
Tubing-preparation station (Bio-Rad)
165-2425 nitrogen regulator (Bio-Rad)
165-2441 Tefzel tubing (Bio-Rad)
Tubing cutter (Bio-Rad) or equivalent
Modified barrel (see John O'Brien's web page: www2.mrc-lmb.cam.ac.uk/personal/job/)
Helium gas cylinder (BOC)
Helium regulator (Gas Safety)
Nitrogen gas cylinder (BOC)
Plasmid Midi Kit (Qiagen) or equivalent
Culture inserts: 24 mm diameter, 8 μm pore size (e.g., Costar, Corning)
Glass cover slips: 22 mm diameter (e.g., VWR)
PROCEDURE
Cell/tissue culture
1| Prepare a target sample for biolistic transfection: HEK293 cells should be grown on 22 mm diameter glass cover slips as previously described11, and brain organotypic slices from mouse cerebellum on culture inserts should be prepared and maintained as previously described12.
DNA preparation
2| Prepare DNA (1 mg ml−1) using a Qiagen Plasmid Midi Kit (or equivalent) according to the manufacturer's instructions.
Preparation of microcarriers
3| Place 25 mg of 1.0 μm diameter gold microcarriers in a 1.5-ml microfuge tube.
▲ CRITICAL STEP The gold particles come in a variety of sizes, and need to be optimized for each particular instrument and biological system under investigation.
4| Add 50 μl spermidine (0.05 M) and 50 μl DNA (1 mg ml−1). The DLR and MLQ need to be optimized for each particular instrument and biological system under investigation (see Experimental design).
▲ CRITICAL STEP DNA concentration must be appropriate for efficient transfection and must not be too low or too high; the latter can cause agglomeration of the gold particles, probably as a result of DNA binding to more than one particle.
? TROUBLESHOOTING
5| Vortex the tube while adding 50 μl CaCl2 (1 M) in 10–15 μl drops. Allow to stand for 5 min with brief (1–2 s) vortexing every 30 s to precipitate the DNA.
6| Centrifuge briefly (10 s) in a microfuge (1,000–2,000g) at room temperature (20–25 °C) and remove supernatant.
7| To estimate efficiency of coating the DNA content, a sample of the supernatant can be examined in comparison with the previous sample by running on an agarose gel. We routinely observe close to 100% efficiency of coating.
▲ CRITICAL STEP Efficiency must be high for successful transfection. Problems with efficiency might be due to poor spermidine or DNA; the former needs to be replaced every 2–3 months, even when stored at −20 °C.
? TROUBLESHOOTING
DNA-bullet preparation
8| Resuspend the gold pellet in 200 μl PVP solution (75 μg ml−1 in ethanol) from a 3.5-ml aliquot, and then transfer the suspension to a 5-ml polypropylene tube.
▲ CRITICAL The PVP concentration needs to be optimized for each particular instrument and biological system under investigation.
9| Repeat this transfer with 200 μl aliquots of PVP until all the gold particles have been transferred, and then add the remaining PVP to the 5-ml polypropylene tube.
10| Dry a 75-cm piece of Tefzel tubing with a flow of nitrogen from the nitrogen cylinder through the nitrogen regulator (0.3–0.4 l min−1 for 10 min).
11| Vortex briefly (to ensure an even suspension of gold), and then use the tubing-preparation station to insert the gold suspension into the Tefzel tubing. To do this, attach a 10-ml syringe to the end of the tubing, and then insert the tubing into the tubing-preparation station with the syringe still attached. Remove the supernatant at a constant velocity using the syringe, leaving the gold behind.
12| Rotate the tubing for 30–40 s. This is to ensure an even spread of gold in the tubing.
▲ CRITICAL STEP Labeling must be even for efficient transfection. Uneven labeling of the gold/DNA suspension inside microcarriers might be due to the PVP solution, which should be replaced every 4–6 months.
13| Dry the gold/DNA in the tubing using a flow of nitrogen (as above).
14| Cut the tubing to the desired length. We use a tubing cutter to obtain lengths of 1 cm, but this is not essential.
■ PAUSE POINT Store desiccated at 4 °C until required. We routinely use the DNA bullets within 1 month, but have stored them satisfactorily for up to 6 months.
Bombardment (gun firing)
15| Attach the gene gun to the helium cylinder through the helium regulator.
16| Remove media from cultured cells or organotypic slices.
17| Place the gene gun at the appropriate distance from the target tissue, set the gas pressure and discharge the gun. Settings for HEK293 cells are 1 cm distance and 1,379 kPa (200 psi) pressure, whereas settings for brain slices are 3 cm distance and 345 kPa (50 psi) pressure.
! CAUTION Eye and ear protection are advisable.
▲ CRITICAL STEP The gas pressure needs to be optimized for each particular instrument and biological system under investigation.
18| Place fresh media on the cells or tissue slices and return them to normal growth conditions.
■ PAUSE POINT Cells and tissue slices can be left for 8–48 h, although we routinely observe them 12–24 h post-transfection.
19| After the appropriate time interval (usually 1–2 d), fix the HEK293 cells, fix and section the brain slices, and observe using a fluorescent microscope.
● TIMING
Preparation of cells or tissue slices: 1–2 h; 1–5 d before transfection.
Preparation of DNA: 2–4 h; any time before transfection if stored frozen.
Preparation of microcarriers: 15–20 min.
Preparation of bullets: 30–45 min.
Bombardment: 15–30 min.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| PROBLEM | POSSIBLE REASON | SOLUTION |
|---|---|---|
| Microcarriers agglomerate after coating with DNA. |
DNA concentration is too high. | Reduce the DNA concentration. |
| DNA is impure. | Check the purity of the DNA by running a gel. | |
| Gold does not spread evenly in the tubing once the tube-preparation station is switched on. |
Old PVP solution. | Make a new stock of PVP in ethanol; use a fresh bottle of ethanol. |
| Insufficiently dry tube before loading. | Flush tubing with nitrogen for 5–10 min prior to loading with DNA/gold microcarrier solution. |
|
| Poor or no expression. | Poor cell or tissue health. | Use young healthy cells or tissue. |
| DNA concentration too low. | Increase the DNA concentration. | |
| DNA is impure. | Check the DNA purity by running a gel. | |
| Spermidine needs replacing. | Make a new solution of spermidine. | |
| Contamination of cell cultures. | Poor culture conditions. | Improve culture conditions. |
| Gene gun is contaminated. | Wash the end of the barrel with 70% (vol/vol) ethanol and change the nylon mesh. |
|
| Cell death. | Poor cell or tissue health. | Use young healthy cells or tissue. |
| Gas pressure too high. | Reduce gas pressure. | |
| Microcarriers too large. | Use a smaller size. |
ANTICIPATED RESULTS
Fluorescently labeled cells should be visible 1–2 d after transfection, although we have observed some labeling in less than 6 h. Under the conditions that we use, HEK293 cells show a high efficiency of transfection (up to 80%) in localized regions (Fig. 4), although overall it is usually 30–40%. HEK293 cells are useful for examining neuronal proteins as they have been shown to have the properties of neuronal lineage cells rather than those of more typical kidney cells13. We have observed similar levels of transfection in HeLa cells (30–40%) and primary neurons (20–35%). Thus, the protocol that we have used here can readily be extrapolated to other ‘classic’ neuronal cells. Using the modified gene gun on brain slices, we observed good labeling throughout the slice (typical transfected cells are shown in Fig. 4). Based on 4,6-diamidino-2-phenylindole (DAPI) labeling of nuclei to define numbers of cells, we have observed efficiencies of up to 30% in localized areas in organotypic slices, which compare favorably with the values of 0.6–0.8% previously reported using an unmodified barrel6,14. The technique is easy to adapt for two or more plasmids, as these can be co-precipitated onto the microcarriers. In our experience, the transfection efficiencies using two plasmids are approximately one-third to one-half of those using a single plasmid (e.g., in organotypic slices, we have observed efficiencies of up to 13%). Data from co-transfection are shown in Figure 5.
Figure 4.
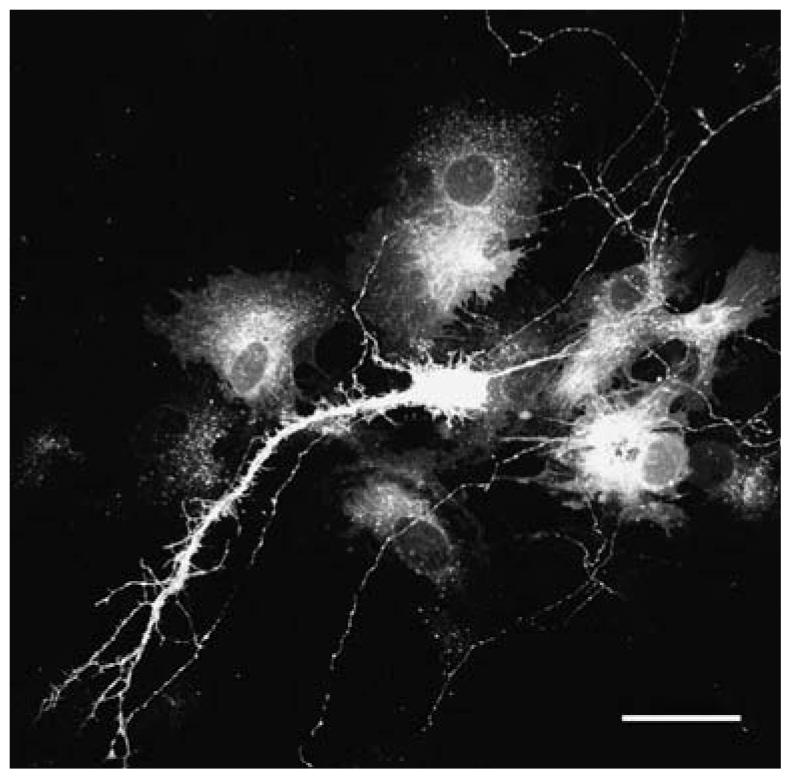
A neuron in a cerebellar brain slice labeled with EYFP. The slice was transfected using the gene gun, and fixed and sectioned after 48 h. Scale bar, 10 μm.
Modifications to the gene gun barrel have enhanced its accuracy by limiting the spread of gold microcarriers, and have also increased its tissue penetration whilst allowing it to operate at a reduced helium pressure. These enhancements make the hand-held gene gun more effective for use both on cell cultures (where tissue damage in the centre of the target can be found) and on intact tissue, which requires deeper penetration for effective transfection.
The improved modified barrel should be cleaned in 70% (vol/vol) ethanol after each use. The tip of the barrel contains a circular nylon mesh, which should be replaced regularly to retain optimum performance. To change the mesh, unscrew the tip, remove the spent mesh and insert a new one over the O-ring. Once the tip is screwed back in place, the hand-held gene gun is operational. For more information, refer to John O'Brien's web page (http://www2.mrc-lmb.cam.ac.uk/personal/job/).
ACKNOWLEDGMENTS
This work was supported by the Medical Research Council and the Wellcome Trust. S.C.R.L. holds a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science.
Footnotes
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Klein TM, Wolf ED, Wu R, Sanford JC. High-velocity microprojectiles for delivering nucleic acids into living cells. Nature. 1987;327:70–73. [PubMed] [Google Scholar]
- 2.Christou P, McCabe DE, Swain WF. Stable transformation of soybean callus by DNA-coated gold particles. Plant Physiol. 1988;87:671–674. doi: 10.1104/pp.87.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin M. The gene gun: current applications in cutaneous gene therapy. Int. J. Dermatol. 2000;39:161–170. doi: 10.1046/j.1365-4362.2000.00925.x. [DOI] [PubMed] [Google Scholar]
- 4.Yang CH, et al. Seeing the gene therapy: application of gene gun technique to transfect and decolour pigmented rat skin with human agouti signalling protein cDNA. Gene Ther. 2004;11:1033–1039. doi: 10.1038/sj.gt.3302264. [DOI] [PubMed] [Google Scholar]
- 5.O'Brien JA, et al. Modification to the hand-held gene gun: improvements for in vitro biolistic transfection of organotypic neuronal tissue. J. Neurosci. Methods. 2001;112:57–64. doi: 10.1016/s0165-0270(01)00457-5. [DOI] [PubMed] [Google Scholar]
- 6.Jiao S, Cheng L, Wolff JA, Yang N-S. Particle bombardment mediated gene transfer and expression in rat brain tissues. Biotechnology. 1993;11:497–502. doi: 10.1038/nbt0493-497. [DOI] [PubMed] [Google Scholar]
- 7.Wirth MJ, Wahle P. Biolistic transfection of organotypic cultures of rat visual cortex using a handheld device. J. Neurosci. Methods. 2003;125:45–54. doi: 10.1016/s0165-0270(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 8.Gerber H-P, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl2 and A1 in vascular endothelial cells. J. Biol. Chem. 1998;273:13313–13306. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 9.Wellmann H, Kaltschmidt B, Kaltschmidt C. Optimized protocol for biolistic transfection of brain slices and dissociated cultured neurons with a hand-held gene gun. J. Neurosci. Methods. 1999;92:55–64. doi: 10.1016/s0165-0270(99)00094-1. [DOI] [PubMed] [Google Scholar]
- 10.Sanford JC, Smith FD, Russell JA. Optimizing the biolistic process for different biological applications. Methods Enzymol. 1993;217:483–509. doi: 10.1016/0076-6879(93)17086-k. [DOI] [PubMed] [Google Scholar]
- 11.Hargreaves AC, Lummis SC, Taylor CW. Ca2+ permeability of cloned and native 5-hydroxytryptamine type-3 receptors. Mol. Pharmacol. 1994;46:1120–1126. [PubMed] [Google Scholar]
- 12.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 13.Shaw G, Morse S, Ararat M, Graham FL. Preferential transformations of human neuronal cells by human adenoviruses and the origin of HEK-293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- 14.Wirth MJ, Patz S, Wahle P. Transcellular induction of neuropeptide Y expression by NT4 and BDNF. Proc. Natl. Acad. Sci. USA. 2005;102:3064–3069. doi: 10.1073/pnas.0404712102. [DOI] [PMC free article] [PubMed] [Google Scholar]