

Abstract
Diolistic labeling is a highly efficient method for introducing dyes into cells using biolistic techniques. The use of lipophilic carbocyanine dyes, combined with particle-mediated biolistic delivery using a hand-held gene gun, allows non-toxic labeling of multiple cells in both living and fixed tissue. The technique is rapid (labeled cells can be visualized in minutes) and technically undemanding. Here, we provide a detailed protocol for diolistic labeling of cultured human embryonic kidney 293 cells and whole brain using a hand-held gene gun. There are four major steps: (i) coating gold microcarriers with one or more dyes; (ii) transferring the microcarriers into a cartridge to make a bullet; (iii) preparation of cells or intact tissue; and (iv) firing the microcarriers into cells or tissue. The method can be readily adapted to other cell types and tissues. This protocol can be completed in less than 1 h.
INTRODUCTION
Dyes have been used for many decades to visualize cells. They are, however, particularly important for understanding brain function, as this depends primarily on the organization and interconnections between neurons. Neuronal dyes have been used since the Golgi stain became available over a hundred years ago, and today fluorescent dyes are the most popular methods of labeling neuronal tissue1. There are a wide range of recently developed fluorescent tracer dyes, and some that have been around for many years, such as Lucifer Yellow, are still popular. However, many dyes, including Lucifer Yellow, must be microinjected into cells. This is both technically demanding and time consuming, and thus it is only practical to label relatively few cells in a preparation, limiting biochemical analyses or electron microscope studies. It is also possible to label only certain types of cells, e.g., adherent cultured cells or those that are surface accessible in tissues. An alternative method of introducing dyes is where the “label” is loaded into or coupled to a DNA or RNA carrier that can be transfected into the cell2. The most widely used of these in neuronal labeling is the use of green fluorescent protein (GFP) and its variants. These proteins have been used successfully to label neurons in transgenic animals3,4, and in cell cultures and brain slices5. For the latter, however, there may be difficulties with transfection, and there is also the problem of time: there are considerable structural and functional changes in the hours after brain slice preparation, and as GFP expression takes at least 6 h, cell structures may be compromised.
The use of lipophilic carbocyanine dyes is able to circumvent many of these problems. Carbocyanine dyes are weakly fluorescent in water, but highly fluorescent when incorporated into membranes6. The dyes are non-toxic and do not seem to interfere with the growth or properties of the cells (e.g., the electrophysiological characteristics of labeled neurons); they are therefore proving to be extremely useful as neuronal markers and also for a variety of other applications such as pathway tracing6-10. The most frequently used carbocyanine dyes to date are two of the 18-carbon chain dyes, 1,1-dioctadecyl-3,3,3,3-tetramethylindocarbocyanine perchlorate, DiI-C18-(3) (“DiI”), and 3,3-dioctadecyloxacarbocyanine perchlorate, DiO-C18-(3) (“DiO”), which exhibit distinct red and green fluorescence, respectively; fluorescence excitation and emission maxima are 549 and 565 nm for membrane-bound DiI, and 484 and 501 nm for DiO, respectively. Once in the membrane, the dyes diffuse with the mobility of native lipids: in living tissue, speeds of 6 mm per day have been reported. As a consequence, even when the dye is applied to only a portion of a neuron's surface, the entire cell surface rapidly becomes brightly labeled6,11. Thus, this approach permits the examination of both individual neurons and/or their processes, and the use of multicolored carbocyanine dyes in different combinations has allowed the differential labeling of different neurons, allowing exploration of interactions between different types of neurons8. The carbocyanine dyes can also be used to label neurons in fixed tissue, where they still have the ability to diffuse, although at a slower rate than in living tissue (~0.2–0.6 mm per day)10. As they spread by diffusion rather than by active transport, they are reliable tracers as long as the cell membranes remain intact. They therefore have considerable potential to map neuronal projections in samples that can only be explored using fixed tissue, such as the human brain12.
Introduction of these dyes to the areas under investigation can be achieved by classical techniques such as microinjection, but these have a number of technical difficulties as described above. Attempts have been made to circumvent these by sprinkling dye crystals on neuronal preparations13, but as dye crystal size and density are difficult to control, this approach has led to highly variable results. The use of biolistic delivery, using the gene gun, can overcome these limitations and allow rapid and efficient labeling of the target tissue. This technique is known as diolistics and, combined with the ability of the dyes to label living tissue, has particular potential for dynamic live cell imaging; it has already been used to follow cell migration and to examine growth and remodeling in neuronal dendrites8,14.
The major practical advantages of the diolistic approach are the following: (i) preparation of the dye-loaded cartridges and firing them into the sample is straightforward and can be achieved in less than an hour; (ii) the cartridges can be stored for months without a major loss of fluorescence; (iii) labeling of cells is rapid; they can be visualized in minutes to hours after transfection; (iv) the labeling technique involves passive dye transfer and therefore diffusion is independent of gene transcription and protein synthesis; and (v) diffusion, and therefore cell labeling, occurs in both fixed and living tissues.
This protocol requires a hand-held gene gun, although either the original or the modified acceleration chamber is suitable. We used the Bio-Rad Helios gene gun with the original chamber for human embryonic kidney 293 (HEK293) cells and the modified barrel for tissue slices. For microcarriers, we used gold particles, although tungsten particles are also suitable. Gold particles are the preferred choice of many because they are more uniform in shape and size; in addition, tungsten particles may acidify the culture medium and cause cell death. Dyes are dried onto the microcarriers; these are then attached to the inside of a length of tubing, which is then cut into the appropriate size to form a cartridge. The cartridges are loaded into the gene gun and the microcarriers are propelled into the sample using a pressurized gas. This gas must be of sufficient velocity to strip the microcarriers from the inner surface of a cartridge and propel them into the sample, but must not be of sufficient velocity to damage the target tissue. The propellant used must be inert, diffusible and of low density to ensure that particles can achieve high velocities, and helium is the gas of choice.
The method we describe has been optimized for HEK293 cells, and fish and mammalian brain tissue15,16, but would also work, with minimal modification, for any cultured cells or tissue slices. It is also applicable, with appropriate optimization, to a range of other tissues.
Experimental design
Many of the important parameters for diolistics are similar to those for biolistic transfection17. Briefly, the size of microcarriers, the amount of dye they carry and the number of microcarriers delivered per cartridge should be optimized for each sample. In addition, the distance the particles travel before striking the target cell and the pressure of the helium gas used as a propellant also need to be optimized18. The longer the distance traveled, the greater the spread of the particles over the target area and the less pronounced the helium shock wave that strikes the cells, which results in less cell damage and higher transfection efficiency. However, a longer distance results in reduced particle speed and a decreased likelihood of target penetration, thereby decreasing transfection efficiency. For cell cultures we used the unmodified gene gun, which delivers microcarriers superficially over a relatively wide area. For whole brain and tissue slices, we recommend the modified gene gun, which has a restricted target area and an increased depth penetration even at low helium pressures, resulting in higher transfection efficiencies and less cell damage. The modifications have been previously described and are illustrated in Figure 116. Briefly, the accelerator channel and its distal cone have been replaced by a modified accelerator channel and an external barrel; the latter has a number of holes drilled in it, each at an angle of 30°. The barrel and accelerator channel are fabricated from brass and stainless steel, respectively; they are held in a Deldrin support and mounted into the gene gun via a screw thread. The final critical parameter is cell health, which must be good for efficient labeling. In addition, labeling in the brain is age dependent, being most effective in cortical slices from animals less than 3 weeks old. As myelination increases with age, the labeling of axons in some adult neurons becomes less complete1.
Figure 1.

Helios gene gun with the original accelerator chamber and associated spacer (boxed, and its replacement in the modified version). The spacer defines the minimum distance between the target and the accelerator channel cone. The cone is designed to spread the gold/DNA particles superficially over a wide area. The modified chamber is shown in the inset. The spacer has been eliminated, and the cone-shaped barrel is replaced with an external barrel with a reduced diameter. This allows deeper tissue penetration at lower gas pressures over a reduced area16.
MATERIALS
REAGENTS
Experimental animals and/or cultured cells (e.g., HEK293) ! CAUTION All experiments must be performed in accordance with the relevant authorities ethical and safety guidelines and regulations.
Gold microcarriers (Bio-Rad)
S22 polyvinylpyrollidone (PVP; Bio-Rad): prepare a 75 μg ml−1 solution in ethanol (BDH)
D7566 methylene chloride (Sigma) ! CAUTION Toxic; prepare in a fume cupboard and wear appropriate protective clothing
D282 DiI (Molecular Probes; http://www.invitrogen.com)
D275 DiO (Molecular Probes)
Paraformaldehyde (BDH): prepare 4% wt/vol solution in phosphate buffer (50 mM, pH 7.4) ! CAUTION Toxic; prepare in a fume cupboard and wear appropriate protective clothing.
EQUIPMENT
Helios gene gun (Bio-Rad)
Tubing preparation station (Bio-Rad)
165-2425 nitrogen regulator (Bio-Rad)
165-2441 Tefzel tubing (Bio-Rad)
Tubing cutter (Bio-Rad) or equivalent
Glass slides (25×77 mm) (e.g., VWR)
Culture inserts: 24 mm diameter, 8 μm pore size (Costar; Corning)
Glass coverslips (22 mm diameter) (e.g., VWR)
Modified barrel (available from the LMB/MRC; see John O'Brien's web page: http://www2.mrc-lmb.cam.ac.uk/personal/job/)
Helium gas cylinder (BOC)
Helium regulator (Gas Safety)
Nitrogen gas cylinder (BOC)
PROCEDURE
Cell culture
1| Prepare a target sample for biolistic transfection: HEK293 cells should be grown on 22-mm-diameter glass coverslips as previously described, and fish and/or mouse brains should be prepared and maintained as described previously15,16.
Preparation of microcarriers
2| Spread gold microcarriers (50 mg; 1 μm diameter) evenly on a glass slide and add 0.125 mg of dye dissolved in 100 μl of methylene chloride directly to the slide.
▲ CRITICAL STEP The gold particles come in a variety of sizes, and the size needs to be optimized for each particular instrument and biological system under investigation.
3| Allow the methylene chloride to evaporate (2–5 min).
4| Scrape the dye-coated particles gently from the slide on to a small piece of filter paper with a clean razorblade and then put them in a test tube. Resuspend the particles in 3 ml of sterile distilled water.
▲ CRITICAL STEP The particles should be a fine dry golden-colored powder. If the particles appear sticky and/or dark brown, wait for further drying or start again.
5| Dry a 75-cm piece of Tefzel tubing with a flow of nitrogen from the nitrogen cylinder through the nitrogen regulator (0.3–0.4 l min−1 for 10 min) and then insert the PVP solution into the tubing using the tubing preparation station.
6| Leave for 2 min and then remove using a syringe attached to the end of the tubing.
7| Allow the tubing to air-dry (5–10 min).
Lipophilic dye bullet preparation
8| Sonicate the dye slurry for 5 min to prevent the formation of large clusters.
9| Vortex briefly (to ensure an even suspension of gold slurry) and then use the tubing preparation station to insert the gold suspension into the Tefzel tubing. To do this, attach a 10-ml syringe to the end of the tubing and then insert the tubing into the tubing preparation station with the syringe still attached. Remove the supernatant at a constant velocity using the syringe, leaving a strip of gold behind.
10| Rotate the tubing for 3–4 min. This is to ensure an even spread of gold in the tubing.
▲ CRITICAL STEP Labeling must be even for efficient transfection. Uneven labeling of the gold/dye suspension inside the tube may be due to the PVP solution, which should be replaced every 4–6 weeks.
11| Cut the tubing to the desired length, e.g., with a tubing cutter (Bio-Rad), to create cartridges.
■ PAUSE POINT Store desiccated at room temperature (20–25 °C) for up to 3 months.
Bombardment (gun firing)
12| Insert the cartridges into the cartridge holder and place in the gene gun.
! CAUTION Dyes may stain the cartridge holder and thereby might “leach” into the sample in subsequent firings. We recommend a different cartridge holder for each color of the dye.
13| Attach the gene gun to the helium cylinder through the helium regulator.
14| Remove media from cultured cells or place intact brain on membrane support.
! CAUTION Do not allow cells or tissue to dry out.
15| Place the gene gun at appropriate distances from the target tissue (approximately 1 cm for HEK293 cells and 3 cm for intact tissue), set the gas pressure (120 psi for HEK293 cells using the original barrel and 75 psi for intact tissue using the modified barrel) and discharge the gun (Gene gun “in action”, see John O'Brien's web page http://www2.mrc-lmb.cam.ac.uk/personal/job/).
! CAUTION Eye and ear protection are advisable.
▲ CRITICAL STEP The gas pressure needs to be optimized for each particular instrument and biological system under investigation.
16| Place fresh media on the culture cells or place intact brain on a membrane support with media and return to normal growth conditions.
17| After the appropriate time interval (HEK293 cells: 5–10 min; mouse brain: 30 min; fish brain: 2 h), fix cells17 or prepare brain slices. Observe using a fluorescent microscope.
● TIMING
Preparation of tissue: 5–30 min
Preparation of microcarriers: 15–20 min
Preparation of bullets: 25–35 min
Bombardment: 10–15 min
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Problem | Possible reason | Solution |
|---|---|---|
| Cell death. | Poor cells or tissue preparation. | Use younger healthy culture cells; change vibrotome settings when working with intact tissue. |
| Clumps of bullets hitting the tissue. | Place a culture insert over the cells before gun discharge. | |
| Gas pressure too high. | Reduce gas pressure or use culture insert as above. | |
| Microcarriers too large. | Use smaller gold particles. | |
| Microcarriers agglomerate after coating with dye. |
Dye concentration is too high. | Reduce dye concentration. |
| Dye is impure. | Use new methylene chloride. | |
| Tubing is dirty. | Use new tubing. Blast nitrogen through the tube before making microcarriers. |
|
| Gold does not spread evenly in the tubing. |
PVP solution is old. | Make a new stock of PVP in ethanol. Use a fresh bottle of ethanol. |
| Insufficiently dry tube before loading. | Flush tubing with nitrogen for 5–10 min before loading. | |
| Poor or no fluorescence. | Dye concentration is too low. | Increase dye concentration. |
| Dye not coated well enough on the gold. | Make new gold/dye microcarriers. | |
| Dyes “bleaches” within tissue (fluorescence becomes weaker under observation). |
Lower the laser power and increase the gain to limit susceptibility to light damage. |
|
| Contamination of cell cultures. |
Gene gun is contaminated. | Wash the end of the barrel with 70% ethanol (vol/vol); change the nylon mesh. |
| Poor culture conditions. | Improve culture conditions. |
ANTICIPATED RESULTS
Fluorescent labeling should be visible in cultured cells 5–10 min after transfection, although we have observed some labeling in less than 2 min. Labeling efficiency is high: using 80–90% confluent HEK293 cells, we usually observe 95–100% fluorescent in a 35-mm dish (Fig. 2). HEK293 cells are not neurons but, as they have been shown to have the properties of neuronal lineage cells and not the properties of more typical kidney cells18, the protocol we have used here can readily be extrapolated to other “classic” neuronal cells. Using the modified gene gun on intact brain, we observe labeling in the range 50–90% throughout the brain, depending partly on the time allowed before observation; we estimate that the dye can penetrate up to 750 μm h−1 (ref. 12). Modifications to the gene gun barrel have enhanced its accuracy by limiting the spread of gold microcarriers, and have increased its tissue penetration while allowing it to operate at a reduced helium pressure, which also lessens cell damage. These enhancements make the hand-held gene gun particularly effective for use on intact tissue, which requires deeper penetration for effective labeling.
Figure 2.
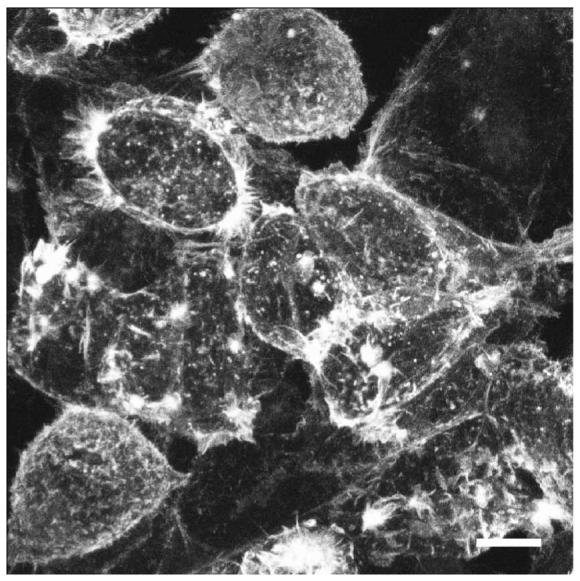
Example of HEK293 cells shot with DiO/gold particles using the unmodified gene gun with gas pressure set at 120 psi. Scale bar, 10 μm.
The diolistic approach, which allows labeling of neurons in their entirety in living tissue with minimal cell damage (Fig. 3), is likely to gain increasing popularity owing to its ease of use and rapid results. The technique provides an unparalleled opportunity to examine features of neuronal cells at high spatial resolution in a complex three-dimensional tissue environment (Fig. 4). For example, this technique has revealed that spines on dendritic projections in the brain are highly ordered in a helical formation15.Some caution, however, must be applied in observing samples, as cells labeled with carbocyanine dyes are more susceptible to light damage compared to those labeled with some other fluorescent dyes such as GFP. One way to reduce bleaching is to use no more light than is absolutely necessary to image a sample (e.g., in the confocal microscope, lower the laser power and increase the photodetector gain to achieve a bright image) and to store samples in the dark when not in use (Table 1).
Figure 3.

Examples of neurons in mouse (a and b)and fish (c) cerebellum shot with DiO/gold particles using the modified gene gun with gas pressure at 50 psi. Scale bars, 25 μm.
Figure 4.

Examples of fish Purkinje cells in cerebellum (Gnathonemus petersii) shot with DiO/gold particles using the modified gene gun with gas pressure at 50 psi. (a) A planer “palisade” pattern of straight unbranched Purkinje dendrites; scale bar, 200 μm. (b) Individual Purkinje cell displaying dendrites; scale bar, 25 μm. (c) Detailed architecture of dendritic spines shot with DiO gold particles; scale bar, 10 μm.
ACKNOWLEDGMENTS
This work was supported by the Medical Research Council and the Wellcome Trust. S.C.R.L. holds a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science.
Footnotes
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Magrassi L, Purves D, Lichtman JW. Fluorescent probes that stain living nerve terminals. J. Neurosci. 1987;7:1207–1214. doi: 10.1523/JNEUROSCI.07-04-01207.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stephens DJ, Pepperkok R. The many ways to cross the plasma membrane. Proc. Natl. Acad. Sci. USA. 2001;98:4295–4298. doi: 10.1073/pnas.081065198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 4.van den Pol AN, Ghosh PK. Selective neuronal expression of green fluorescent protein with cytomegalovirus promoter reveals entire neuronal arbor in transgenic mice. J. Neurosci. 1998;18:10640–10651. doi: 10.1523/JNEUROSCI.18-24-10640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lo DC, McAllister AK, Katz LC. Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron. 1994;13:1263–1268. doi: 10.1016/0896-6273(94)90412-x. [DOI] [PubMed] [Google Scholar]
- 6.Honig MG, Hume RI. Fluorescent carbocyanine dyes allow living neurons of identified origin to be studied in long-term cultures. J. Cell Biol. 1986;103:171–187. doi: 10.1083/jcb.103.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honig MG, Hume RI. Formation of synapses by sympathetic preganglionic neurons. Soc. Neurosci. Abstr. 1987;13:425. [Google Scholar]
- 8.Gan WB, Grutzendler J, Wong WT, Wong ROL, Lichtman JW. Multicolor “diolistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- 9.Gan WB, Lichtman JW. Synaptic segregation at the developing neuromuscualr junction. Science. 1998;282:1508–1511. doi: 10.1126/science.282.5393.1508. [DOI] [PubMed] [Google Scholar]
- 10.Godement P, Vanselow J, Thanos S, Bonhoeffer FA. Study in developing visual systems with a new method of staining neurons and their processes in fixed tissue. Development. 1987;4:697–713. doi: 10.1242/dev.101.4.697. [DOI] [PubMed] [Google Scholar]
- 11.Liu DW, Westerfield M. The formation of terminal fields in the absence of competitive interactions among primary motorneurons in the zebrafish. J. Neurosci. 1990;12:3947–3959. doi: 10.1523/JNEUROSCI.10-12-03947.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burkhalter A, Bernardo KL. Organization of corticocortical connections in human visual cortex. Proc. Natl. Acad. Sci. USA. 1989;86:1071–1075. doi: 10.1073/pnas.86.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cline HT, Edwards JE, Rajan I, Wu GY, Zou DJ. In: Imaging Neurons, A Laboratory Manual. Yuste R, Lanni F, Konnerth A, editors. Cold Spring Harbor Press; Cold Spring Harbor, New York: 2000. [Google Scholar]
- 14.Pearson RA, Luneborg NL, Becker DL, Mobbs P. Gap junctions modulate interkinetic nuclear movement in retinal progenitor cells. J. Neurosci. 2005;25:10803–10814. doi: 10.1523/JNEUROSCI.2312-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Brien J, Unwin N. Organization of spines on the dendrites of Purkinje cells. Proc. Natl. Acad. Sci. USA. 2006;103:1575–1580. doi: 10.1073/pnas.0507884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Brien JA, Holt M, Whiteside G, Lummis SC, Hastings MH. Modifications to the hand-held gene gun: improvements for in vitro biolistic transfection of organotypic neuronal tissue. J. Neurosci. Methods. 2001;112:57–64. doi: 10.1016/s0165-0270(01)00457-5. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien JA, Lummis SCR. Biolistic transfection of neuronal cultures using a hand-held gene gun. Nat. Protocols. 2006;1:977–981. doi: 10.1038/nprot.2006.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanford JC, Smith FC, Russell JA. Optimizing the biolistic process for different biological applications. Methods. 1993;217:483–509. doi: 10.1016/0076-6879(93)17086-k. [DOI] [PubMed] [Google Scholar]