

Abstract
The 5-HT3 receptor is a typical ligand-gated ion channel of the Cys-loop superfamily, which is activated by binding of serotonin (5-HT). Models of the binding site of this protein reveal potential interactions between 5-HT and Tyr143, Tyr153, and Tyr234. Here we describe a series of ab initio calculations, based on density functional theory, to assess the effects of mutating these tyrosine residues on the binding of 5-HT. A series of mutations to these tyrosines, previously studied experimentally, were tested, and the binding energies compared with the available experimental data. Our results show that Tyr153 could form a hydrogen bond with the tertiary amine of 5-HT, and that mutation in this location revealed binding energies broadly in line with experimentally determined EC50s. Tyr143 could also form a hydrogen bond, but as EC50s do not relate to binding energies, it is unlikely that such a bond is formed here. Tyr234 is quite distinct in that it may interact with 5-HT via a mixed hydrogen bond/cation-π interaction.
Introduction
Ligand-gated ion channels (LGICs) are crucial mediators in neuronal transmission. These receptors, located in the cell membrane, are pentameric proteins, with each subunit composed of an extracellular, a transmembrane, and an intracellular domain. The LGICs activation is initiated by binding, in the extracellular domain, of the appropriate neurotransmitter, which triggers a series of conformational changes culminating with the opening of an ion selective channel (gating). The resulting ion flux generates changes in the membrane potential that can drive specific effects depending on the particular receptor.
The 5-hydroxytryptamine type 3 receptor (5-HT3R) is a member of the Cys-loop superfamily of LGICs, which includes nicotinic acetylcholine (nAChR), γ-amino butyric acid A and C (GABAA and GABAC), and glycine receptors (GlyR).1 These proteins are responsible for fast synaptic transmission and are also the target for many neurologicallly active drugs; understanding how molecules bind is therefore of both phisiological and pharmacological importance. Functional receptors are pentamers, and five distinct 5-HT3 receptor subunits (A-E) have been identified, but only homooligomeric (A only) or heterooligomeric (A and B) forms have been characterized.1,2
In vivo 5-HT3R is activated by two or more molecules of 5-HT (5-hydroxytryptamine or serotonin). However, the molecular mechanism of 5-HT3R activation is still unclear, partly due to the limited structural information available. Recently, a model for 5-HT3R extracellular domain has been proposed3 by analogy with the structure of the acetylcholine binding protein (AChBP),4 a soluble protein with a structure resolved to 2.1 Å, whose extracellular domain is homologous to the nAChR. The 5-HT binding site in such a homology model was evaluated with a docking procedure based on a genetic algorithm.3,5 5-HT is in its positively charged state, with the positive charge localized on the amine group. The most likely binding site for 5-HT from the docking procedure and from experimental evidence is shown in Figure 1. The amino acids which are likely to interact with 5-HT have been identified; there are a high proportion of aromatics and, like other Cys-loop receptor binding sites, some of these form an aromatic box around the neurotransmitter. The best characterized aromatic amino acid in the 5-HT3R binding site is Trp183, in magenta in Figure 1: mutagenesis experiments indicated that 5-HT binds here through a cation-π interaction and this was confirmed by correlating the experimental data with theoretical Hartree-Fock calculations.6 However, there are also other important aromatic residues. In particular three tyrosines (Tyr143, Tyr153, and Tyr234, in yellow in Figure 1) have been located in the binding site and may interact with 5-HT: Tyr153 has been proposed to form a hydrogen bond with the 5-HT indole nitrogen, and Tyr143 may also form a hydrogen bond, possibly with the charged amine of 5-HT or another residue; it is not yet clear what type of interaction there is between Tyr234 and 5-HT.7 The role of these three tyrosines has been investigated by using unnatural amino acid mutagenesis experiments.8 These experiments rely on comparison of EC50s: an EC50 (effective concentration half) is the concentration that evokes 50% of the maximal response. Thus receptors with high EC50s either bind ligand with lower affinity and/or are less efficient at channel opening than those with lower EC50s.
Figure 1.
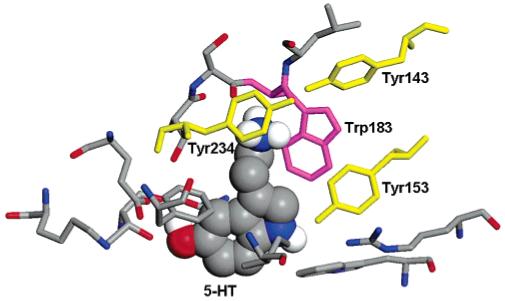
The 5-HT binding site in the 5-HT3R homology model;3 5-HT is shown in a space filling view and the residues within 5 Å with sticks. Tyr153, Tyr143, and Tyr243 are shown in yellow, and Trp183 in magenta.
Here we have performed a series of binding energy calculations using density functional theory (DFT) to disentangle the effects due to mutation purely on the binding of 5-HT to Tyr143, Tyr153, and Tyr234.
Methods
In recent years, DFT9 has been demonstrated to be a powerful technique to study, from first principles, systems not only of materials science, but also of biological interest.10-13 In fact, its favorable ratio between accuracy and computational cost allows us to study relatively complex systems. It is not yet possible to study whole biomolecules from first principles, but we can perform calculations on the portion of the system where quantum mechanical effects are relevant, or where force-field parameters are not readily available, as for some mutated amino acids.
The docked 5-HT in the 5-HT3 homology model shown in Figure 1 forms a hydrogen bond with Tyr153: we carved from the homology model3 the model structure to study the interaction of 5-HT with Tyr153. While the location of the docked 5-HT also suggests a potential hydrogen bond with Tyr143 and a cation-π interaction with Tyr234, the corresponding distances are too large for bonds to be present in the homology model. Hence, we performed preliminary classical molecular dynamics simulations and selected two structures where 5-HT formed a hydrogen bond with Tyr143 and a cation-π interaction with Tyr234, respectively, which we used to carve out the models for the DFT calculations. These model structures are shown in Figure 2. The model for Tyr153 (Model A) contains 5-HT, Tyr153, and part of the two adjacent residues (Lys154 and Asp152) to mimic the link to the receptor. Similarly the model for Tyr143 (Model B) includes part of the adjacent valine residues Val142 and Val144, and the model for Tyr234 (Model C) includes Ala235 and Ser233. All three models contain 53 atoms each in the cases of nonmutated (wild type, WT) tyrosines, and are positively charged (due to the charge state of 5-HT). In the models of Figure 2, 5-HT is in three different conformers characterized by different torsional angles defining the position of the side chain with respect to the indole plane;14 the lowest energy conformer was that of Model A, followed by Model C and then Model B. We verified that the 5-HT conformers did not affect the calculated binding energies, which are energy differences corresponding to the same conformer. Hence, we would have predicted similar energy trends even if 5-HT would have been in the same conformer in all three models. We modeled these systems in orthorhombic simulation cells of size 15 × 19.5 × 26 Å3. For comparison and to analyze size effects, we also tested smaller systems, including in particular a reduced model of 5-HT + Tyr234 in a simulation cell of size 12 × 13 × 15 Å3, which will be discussed later.
Figure 2.
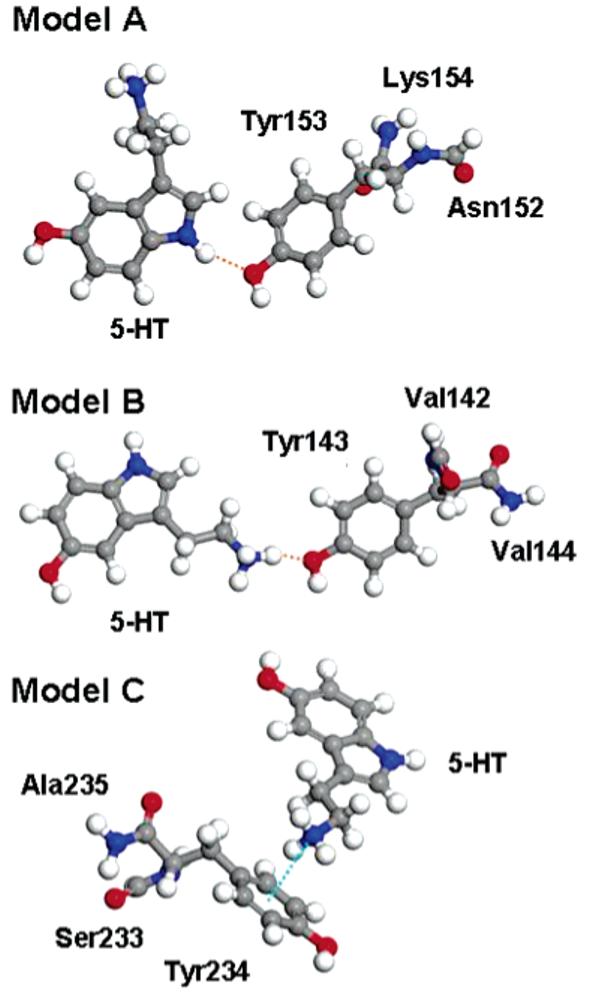
Models used in the density functional theory calculations. In Model A (top) Tyr153 forms a hydrogen bond (orange dashed line) with the 5-HT indole nitrogen. In Model B (center) Tyr143 forms a hydrogen bond (orange dashed line) with the 5-HT charged amine group. In Model C (bottom) Tyr234 forms a cation-π interaction (cyan dashed line) with the 5-HT charged amine group.
The binding energies between 5-HT and, in turn, the three tyrosines were calculated by DFT,9 using the CPMD code.15 We used norm conserving Martins-Troulliers pseudopotentials to describe the interaction between the ion cores and the valence electrons.16 The electronic wavefunctions were expanded in a plane wave basis set with a kinetic energy cutoff of 70 Ry. The exchange-correlation energies were calculated by using the PBE gradient correction functional.17 We used the method proposed by Hockney for the Poisson solver, to avoid spurious interactions between neighboring cells.18 The geometries of the selected structures were relaxed by using the direct inversion iterative subspace method (DIIS) for both the electronic and the ionic degrees of freedom.19 Positional constraints were imposed on the carboxylic carbons of the tyrosines and of the adjacent residues (i.e., Lys154, Val142, and Ser233), in order to reproduce the constrains due to the links to the protein. Binding energies, Eb, were calculated as the difference between the sum of the total energies of the independently relaxed monomers (5-HT and of the protein fragment) and the total energy of the relaxed dimer (5-HT + the protein fragment).
The PBE gradient correction for the exchange and correlation energy has an accuracy of about 1 kcal/mol in the evaluation of hydrogen bond strengths, for “linear” hydrogen bonds (i.e., with donor-hydrogen-acceptor bond angle larger than 130°). This is the case for all the hydrogen bonds in this study. For nonlinear hydrogen bonds the error increases up to 1.5 kcal/mol, due to the difficulty of PBE functional in describing dispersion forces, which become predominant, with respect to the electrostatic forces, for nonlinear hydrogen bonds.20 To compare our results with other gradient-corrected functionals, we repeated some of the calculations using the BLYP gradient-corrected functional,21,22 another popular choice for DFT calculations of biological and chemical systems. The results show a decrease in the hydrogen bond energies with respect to PBE by less than 1 kcal/mol, as previously reported in the literature.23 However, the trend in the change of binding energies upon mutation, which we will compare with the available experimental data, remained the same.
Results and Discussion
Tyr153 Mutations
The location of Tyr153 with respect to the docked 5-HT ligand suggests a potential hydrogen bond with the 5-HT indole nitrogen; this was supported by mutagenesis experiments, which showed that the substitution of Tyr153 with amino acids which do not permit or can only form weak hydrogen bonding resulted in an increase in EC50.
We calculated binding energies for Model A in Figure 2 and mutated analogues, where we substituted Tyr153 with 4-F-Phe, 4-MeO-Phe, mTyr, 4-Me-Phe, and F5-Phe, the unnatural amino acids in Figure 3 which where studied experimentally.8 Such energies are shown in Table 1, with the experimental EC50s and the structural characteristics of the hydrogen bonds.
Figure 3.

Side chains of the unnatural amino acids studied in this paper and experimentally.8
TABLE 1. Binding Energies (Eb) and Binding Energy Differences (ΔEb) with Respect to WT-Tyr153 for Model A and Its Mutations. N-H···A Hydrogen Bond Distances and Angles, Where the Acceptor A Can Be Either Oxygen or Fluorinea.
| Model A |
||||||
|---|---|---|---|---|---|---|
| Tyr153 | E b | Δ E b | d H···A | d N···A | ∠NHA | EC508 |
| WT | 5.8 | 2.10 | 3.09 | 161 | 1.15 ± 0.03 | |
| 4-MeO-Phe | 6.0 | 0.2 | 2.03 | 3.03 | 163 | 6.6 ± 0.27 |
| 4-F-Phe | 3.5 | -2.3 | 2.14 | 3.11 | 158 | 19.7 ±1.22 |
| F5-Phe | 1.8 | -4.0 | 2.29 | 3.26 | 158 | >500 |
| 4-Me-Phe | 1.7 | -4.1 | 18.2 ± 0.48 | |||
| mTyr | 0.7 | -5.1 | >500 | |||
Binding energies in kcal/mol. Distances in Å. Angles in deg. EC50 in μM. Experimental EC50s from ref 8.
4-MeO-Phe forms the strongest hydrogen bond with 5-HT followed by WT, 4-F-Phe, and F5-Phe. WT tyrosine and 4-MeO-Phe have almost identical binding energies (6.0 and 5.8 kcal/mol). The substitution of the Tyr153 hydroxyl with a fluorine weakens the hydrogen bond, lowering the binding energy by 2.3 kcal/mol with respect to WT. A more dramatic reduction of 4.0 kcal/mol with respect to WT was obtained by substituting all the hydrogen atoms in the tyrosine phenyl ring with fluorine atoms (F5-Phe), a procedure that reduces the aromaticity of the ring. 4-Me-Phe and mTyr do not form hydrogen bonds (in the case of mTyr assuming that the relative orientation between 5-HT and mTyr is the same as for WT); the corresponding binding energies with 5-HT decrease, with respect to WT, by 4.1 and 5.1 kcal/mol, respectively.
To further characterize the hydrogen bonds with the mutated amino acids, we analyzed the corresponding electron localization functions (ELFs), shown in Figure 4, to illustrate where the electrons are maximally localized. WT and 4-MeO-Phe oxygen lone pairs are oriented in the direction of hydrogen, while the fluorine lone pairs (in 4-F-Phe and F5-Phe) surround the atom. This results in a greater directionality of the first two hydrogen bonds, which could in part explain their greater strength.
Figure 4.
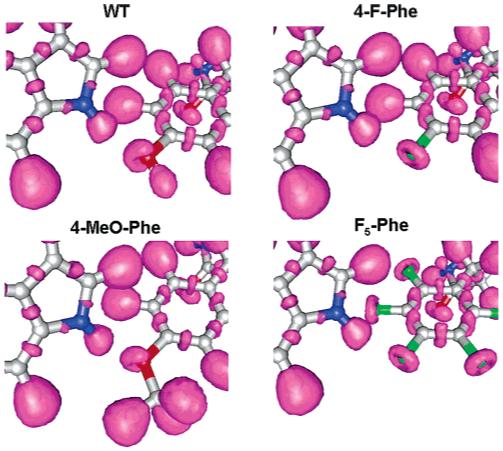
Electron localization function isosurfaces (0.85) for WT-Tyr153 (top, left), 4-MeO-Phe (bottom, left), 4-F-Phe (top, right), and F5-Phe (bottom, right) in Model A.
Further insight into the hydrogen bonds was obtained by studying how the electronic charge density redistributes in response to the hydrogen bond formation. We calculated the difference between the electronic density of the whole relaxed system and the sum of densities of the single monomers (with the structure they had when forming the bond). In Figure 5 the purple isosurfaces represent an electronic (i.e., negative) charge gain, while the orange isosurfaces represent an electronic charge depletion: the occurrence of the hydrogen bond involves a charge rearrangement in the region between the hydrogen bond donor and acceptor. A qualitative correlation between the extent of these charge rearrangements and the hydrogen bond strength can be observed: WT tyrosine and 4-MeO-Phe show the most evident charge oscillations, followed by 4-F-Phe, while F5-Phe does not show significant charge transfer, in agreement with its weaker bond.
Figure 5.
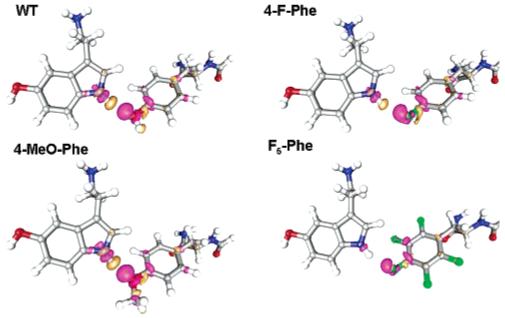
Electronic density differences due to the hydrogen bond formation for WT-Tyr153 (top, left), 4-MeO-Phe (bottom, left), 4-F-Phe (top, right), and F5-Phe (bottom, right) in Model A. Purple isosurfaces correspond to ρ = 0.0015 e/Å3 (electronic charge gain) and orange isosurfaces to ρ = -0.0015 e/Å3 (electronic charge depletion).
The cases of 4-F-Phe and F5-Phe might seem quite peculiar at first inspection: fluorine is more electronegative than oxygen and nitrogen, thus it would be expected to form strong hydrogen bonds. However, the substitution of a hydroxyl group with a fluorine atom decreases the hydrogen bond strength. Organic fluorine (e.g., fluorine bonded to a carbon) hardly ever accepts hydrogen bonds if a better acceptor is present;24,25 only in cases where the approach of the hydrogen atom to better acceptors is sterically hindered will it act as a weak hydrogen bond acceptor. Its low polarizability and tightly contracted lone pairs, as evident from Figure 4, show why fluorine is unable to compete with stronger hydrogen bond acceptors such as oxygen or nitrogen.
The experimental EC50s show a relatively small change in the receptor response upon mutation to 4-MeO-Phe, while the mutations with F5-Phe and mTyr lead to considerably less efficient receptors, and mutations to 4-F-Phe and 4-Me-Phe had intermediate effects.8 The linear correlation coefficient, between ln(EC50(mutant)/EC50(WT)) for the available finite EC50s and the binding energies, is 0.75. An anomalous result was the similar EC50s for 4-F-Phe, where we predict a weak hydrogen bond, and 4-Me-Phe, which cannot form a hydrogen bond, but overall the comparison of the calculated binding energy with the experimental data suggests that the formation of a hydrogen bond between Tyr153 and 5-HT has a direct influence on the activity of the channel.
The lack of a strong correlation between the binding energies and EC50s could indicate that this residue also plays a role in gating, as previously proposed.8 However, it is also possible that the assumptions we have made are not entirely accurate. First, we are assuming that 5-HT would bind in the same way to the mutated amino acid, which might not be the case, especially if a more convenient binding partner was available (e.g., for the fluorinated amino acids or for 4-Me-Phe), and second, our calculations are performed on a small model system, which does not account for the effects of the environment. The former may be a significant problem, but the latter probably not, as our goal was to estimate the trend that the mutation has on the binding energy and the effect of the environment is likely to be similar in all mutated systems. Moreover, as shown in the work of Morozov et al.,26 quantum mechanical calculations, even if performed in vacuum taking into account only the groups involved in the hydrogen bonds, give a better description of hydrogen bonds in proteins than that obtained by molecular mechanics force fields.
Tyr143 Mutations
The docking procedure identified a potential hydrogen bond between the 5-HT positively charged amine group, acting as donor, and the hydroxyl group of Tyr143, whose oxygen acts as acceptor. Mutagenesis experiments highlighted the importance of this amino acid for 5-HT3R functionality by measuring no receptor response after its substitution with a range of amino acids.
The model used for the binding energy calculations is shown in Figure 2 (Model B). Here 5-HT, which can exist in several conformers,14 is in a metastable conformation, of higher energy than its lower energy conformers in Models A and C. In this metastable structure, the 5-HT side chain is in, rather than perpendicular to, the indole plane. This difference in the 5-HT conformer does not influence the binding energy values, since the difference in the absolute energy between the two conformers is cancelled out in the binding energy calculation: tests showed that the binding energy of Tyr143 with the two different 5-HT conformers differed by less than 0.1 kcal/mol. We also verified that the valine residues included in Model B affect the value of binding energies only mildly (<1 kcal/mol); in fact, calculations on a smaller model, where Tyr143 was approximated by its phenyl ring and the hydroxyl headgroup (leading to a system, including 5-HT, containing 39 atoms), gave a binding energy of 14.1 kcal/mol to compare to 13.3 kcal/mol for the larger model. Most importantly the binding energy differences between WT and its mutation to 4-F-Phe were very similar, -3.7 kcal/mol for the small model and -3.8 kcal/mol for the model containing the valine residues. This demonstrates that the binding energy differences are unaffected by the residues attached to the tyrosine and are therefore representative for the hydrogen bond interaction of any tyrosine with the charge amine tail of 5-HT.
The binding energies between 5-HT and Tyr143, shown in Table 2, are larger than those for Tyr153 because the 5-HT group involved in the hydrogen bond is positively charged. WT tyrosine forms the strongest hydrogen bond with 5-HT, followed by 4-MeO-Phe and 4-F-Phe. The stronger character of the hydrogen bond is also evident from the shorter donor-acceptor distances in Table 2. Also here, the substitution of the Tyr143 hydroxyl with a methoxyl group does not significantly change the binding energy, while the substitution with a fluorine weakens the hydrogen bond, lowering the binding energy by 3.8 kcal/mol compared to WT. No hydrogen bond is formed with mTyr, which has consequently substantially lower binding energy.
TABLE 2. Binding Energies (Eb) and Binding Energy Differences (ΔEb) with Respect to WT-Tyr143 for Model B and Its Mutations. N-H···A Hydrogen Bond Distances and Angles, Where the Acceptor A Can Be Either Oxygen or Fluorinea.
| Model B |
||||||
|---|---|---|---|---|---|---|
| Tyr143 | E b | Δ E b | d H···A | d N-A | ∠NHA | EC508 |
| WT | 13.3 | 1.69 | 2.75 | 168 | 1.15 ± 0.03 | |
| 4-MeO-Phe | 12.9 | -0.4 | 1.70 | 2.72 | 158 | NR |
| 4-F-Phe | 9.5 | -3.8 | 1.78 | 2.77 | 156 | NR |
| mTyr | 2.1 | -11.2 | NR | |||
Binding energies in kcal/mol. Distances in Å. Angles in deg. EC50 in μM. Experimental EC50s from ref 8; NR indicates no response.
The ELFs show a behavior similar to those of Tyr153, with the oxygen lone pairs of WT and 4-MeO-Phe oriented in the hydrogen direction, and the fluorine lone pairs of 4-F-Phe tightly bound to the fluorine atom. The charge density variations due to the hydrogen bond formation also have a behavior similar to those of Tyr153, although they are more evident due to the stronger character of these hydrogen bonds.
No relationship is found between the EC50s and the binding energies upon the mutation of WT tyrosine to 4-MeO-Phe, 4-F-Phe, and mTyr.8 Hence the experimentally observed variation in the EC50s cannot be explained purely on binding energy effects. It is possible that the hydrogen bond with 4-F-Phe was simply not formed because 5-HT was able to find better acceptors elsewhere; if this were the case, then Tyr143 could be important for the binding. However, the results for 4-MeO-Phe would still be puzzling, since the hydrogen bond strength is very similar to that of WT. This inconsistency between experimental data and the calculated binding energies suggests that this residue does not play a role in binding but may be important for the conformational change that results in channel opening.
Our DFT results do not explain why a hydrogen bond between 5-HT and Tyr143 does not occur. Further information could be obtained by performing classical molecular dynamics or QMMM (Quantum Mechanics/Molecular Mechanics) simulations providing a dynamical description of the binding site. For example, if the 5-HT charged amine group happens to interact already with both Trp183 and Tyr234, there might be no space for an additional hydrogen bonding with Tyr143.
Tyr234 Mutations
Tyr234 is important in 5-HT3R function: mutagenesis experiments show that substitution of Tyr234 with unnatural amino acids led, for all but one mutation (i.e., 4-Me-Phe), to increased EC50s.8 However, how Tyr234 interacts with 5-HT is still unclear.
We first investigated a potential cation-π interaction with the 5-HT positively charged amine group, as suggested by the docking procedure. The model we used, Model C, is shown in Figure 2. Table 3 show the binding energies for several mutations, together with structural parameters characterizing the cation-π interaction, i.e., the distance between the phenyl ring center of mass and the 5-HT amine nitrogen (dN-CoM) and the angle (θ) between the normal to the phenyl ring and the vector pointing from its center of mass to the 5-HT amine nitrogen. No value of θ is shown for Cha, since its ring is not planar. 4-Me-Phe forms the strongest bond with 5-HT, followed, in turn, by mTyr, WT, 4-F-Phe, F3-Phe, Cha, and F5-Phe. The coefficient of linear correlation between binding energies and EC50s is only 0.17, suggesting no correlation. However, this number should be taken with caution since the variations in binding energies for the first four mutations in Table 3 are small, within the calculation error. A drop in binding energy is observed for 4-F-Phe, which still has a measurable EC50, and more substantial drops for F3-Phe, Cha, and F5-Phe, which show no response in the experiments. This is a different situation than what was observed for Trp1836 in 5-HT3R and Trp149 in nAChR27 where a large range of EC50s was measured, providing a reliable linear correlation coefficient between EC50s and the cation-π binding energies, which was found to be high (0.99).
TABLE 3. Binding Energies (Eb) and Binding Energy Differences (ΔEb) with Respect to WT-Tyr234 for Model C and Its Mutations. Distances (dN-CoM) Between the 5-HT Amine Nitrogen and the Center of Mass of the Phenyl Ring and Angles (θ) Between the Normal to the Phenyl Ring and the Vector Pointing From Its Center of Mass to the 5-HT Amine Nitrogena.
| Model C |
|||||
|---|---|---|---|---|---|
| Tyr234 | E b | Δ E b | d N-CoM | θ | EC508 |
| 4-Me-Phe | 13.2 | 0.3 | 3.04 | 6 | 1.13 ± 0.05 |
| mTyr | 13.4 | 0.5 | 3.09 | 8 | 7.42 ± 0.32 |
| WT | 12.9 | 3.06 | 7 | 1.24 ± 0.05 | |
| Phe | 12.5 | -0.4 | 3.11 | 7 | 10.22 ± 0.08 |
| 4-F-Phe | 10.2 | -2.7 | 3.08 | 8 | 4.11 ± 0.2 |
| F3-Phe | 5.9 | -7.0 | 3.15 | 6 | NR |
| Cha | 4.1 | -8.8 | 3.81 | NR | |
| F5-Phe | 2.5 | -10.4 | 3.17 | 6 | NR |
Binding energies in kcal/mol. Distances in Å. Angles in deg. EC50 in μM. Experimental EC50s from ref 8; NR indicates no response.
Even disregarding the linear correlation coefficient, it is difficult to explain the experimental data only in term of alleged cation-π interaction. For example WT tyrosine and Phe have similar binding energy while the EC50 varies by a factor of 10. Also 4-F-Phe, which has a lower binding energy than Phe, shows a smaller value of EC50 than mTyr and Phe which can form stronger cation-π interaction.
The Hartree Fock binding energies for Trp1836 in 5-HT3R and Trp149 in nAChR27 were calculated for a simplified model system in which 5-HT (or ACh) was substituted by a sodium cation, assuming that trends in cation-π binding energy across a series of aromatics are independent of the identity of the cation. Our model system allows us to explore not only potential cation-π interactions but also hydrogen bonds (involving for example the tyrosine hydroxyl) in case they occur in combination. Hence, we performed further analysis on reduced models, shown in Figure 6: in Model D Tyr234 interacts with 5-HT with a pure cation-π interaction as in Model C, while in Model E a hydrogen bond component is also present. The binding energies for the reduced Model D show a very similar trend to the binding energies of the larger Model C, with a linear correlation coefficient between the two binding energy sets larger than 0.99, although the Eb values for Model D are larger; hence, we considered this model representative of the larger one. In Model D, the charge variations due to the interaction of 5-HT with Tyr234 are on the ring, confirming the cation-π nature of the bond for this structure. Similar to Model C, the binding energies show no correlation with the experimentally determined EC50s, with a linear correlation coefficient of 0.26.
Figure 6.
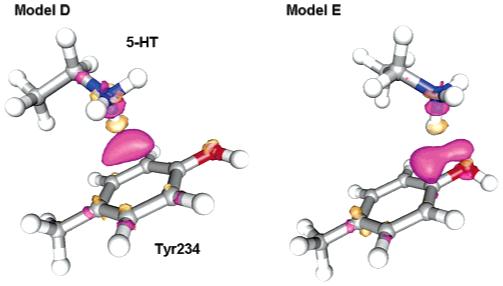
Reduced models for the interaction of 5-HT with Tyr234. In Model D (left) Tyr234 experiences a pure cation-π interaction with the 5-HT charged amine, while in Model E (right) Tyr234 experiences a mixed hydrogen bond/cation-π interaction with the 5-HT charged amine. Electronic density differences due the interaction of Tyr234 with 5-HT are also shown: purple isosurfaces correspond to ρ = 0.003 e/Å3 (electronic charge gain) and orange isosurfaces to ρ = -0.003 e/Å3 (electronic charge depletion).
Figure 7 shows the electrostatic potential of the mutated amino acids, with the structures adopted while bonded to 5-HT, mapped onto an electron density isosurface. Although the cation-π interaction is more complex than a purely electrostatic effect, qualitatively the more negative the electrostatic potential in the ring, the stronger is the interaction with the cation:6,27,28 this is reflected in our calculated binding energies. It is clear from Figure 7 that for WT tyrosine and some mutants there are other areas, beside the ring, that might attract the positively charged amine, such as the oxygen and fluorine atoms attached to the phenyl ring, which could act as hydrogen bond acceptors. In such cases Tyr234 might form, rather than a purely cation-π interaction, a combined hydrogen bond/cation-π interaction with 5-HT, or even a pure hydrogen bond. To probe such a possibility, we recalculated the binding energies starting from an initial configuration where the 5-HT charged amine lies in-between the tyrosine phenyl ring and the hydroxyl head or its mutated counterpart (see Figure 6, Model E, and Table 4). The geometries for Model E and its mutations show, except for 4-Me-Phe where they basically remained the same, an increment in both the distance dN-CoM and the angle θ, corresponding to a decrement in the cation-π interaction strength. Whenever an electronegative atom (e.g., oxygen or fluorine) was attached to the phenyl ring, a relaxed structure was found characterized by a combined hydrogen bond/cation-π interaction. This can be seen in Figure 6, where Model E displays charge oscillations due to bond formation, which are less localized with respect to the hydrogen bonds previously studied for Tyr153 and Tyr143 (see for comparison Figure 5 for Model A), and extend toward the tyrosine phenyl ring, besides involving the 5-HT charged amine and the tyrosine hydroxyl. For the fluorinated amino acids, which in Model D did not form strong cation-π interactions, larger binding energies than in Model D were found. For the amino acids where oxygen acts as a hydrogen bond acceptor, which in Model D formed strong cation-π interactions, the relaxed structures were local minima, less strongly bonded. The hydrogen bond distances and angles show that 4-F-Phe, F3-Phe, and F5-Phe formed hydrogen bonds characterized by a higher degree of linearity and shorter donor-acceptor distances than in the cases involving oxygen: the weaker cation-π interaction experienced by fluorinated amino acids opposed a weaker competition to the hydrogen bond interaction than when oxygen was present. Phe was also trapped in a higher energy local minimum configuration, in which 5-HT lies farther from the center and closer to the border of the ring with respect to Model D, while both Cha and 4-Me-Phe had binding energies similar to those of Model D.
Figure 7.
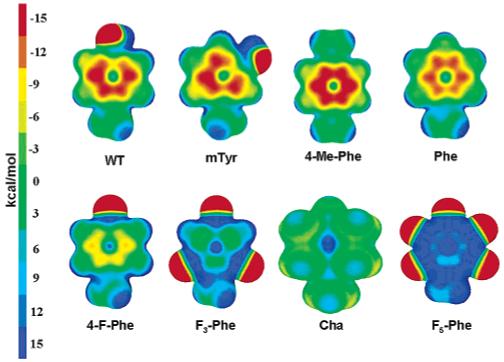
Electrostatic potential for Tyr234 as modeled in Models D and E and its mutations mapped onto an electronic density isosurface of 0.01 e/Å3. Red indicates negative potential, while blue indicates positive potential.
TABLE 4. Binding Energies (Eb) and Binding Energy Differences (ΔEb) with Respect to WT-Tyr234 for Model D and Model E and Their Mutationsa,b.
| Model D |
Model E |
||||
|---|---|---|---|---|---|
| Tyr234 | E b | Δ E b | E b | Δ E b | EC508 |
| 4-Me-Phe | 16.9 | 0.7 | 16.9 | 2.9 | 1.13 ± 0.05 |
| mTyr | 16.7 | 0.5 | 14.4 | 0.4 | 7.42 ± 0.32 |
| WT | 16.2 | 14.0 | 1.24 ± 0.05 | ||
| Phe | 15.6 | -0.6 | 12.6 | -1.4 | 10.22 ± 0.08 |
| 4-F-Phe | 12.8 | -3.4 | 13.3 | -0.6 | 4.11 ± 0.2 |
| F3-Phe | 8.6 | -7.6 | 9.1 | -4.9 | NR |
| Cha | 6.6 | -9.6 | 6.5 | -7.5 | NR |
| F5-Phe | 4.3 | -11.6 | 7.3 | -6.7 | NR |
Binding energies in kcal/mol. EC50 in μM.
Experimental EC50 from ref 8; NR indicates no response.
The binding energies for Model E show a better correlation with the experimental EC50s, with a linear correlation coefficient of 0.69. On the contrary, correlating the highest of the binding energies of Models D and E (in boldface in Table 4) gives a low correlation coefficient of 0.28. Even if reasoning on the linear correlation coefficient needs to be taken with caution as previously explained, the comparison of our calculations with the available experimental data suggests a mixed hydrogen bond/cation-π interaction between 5-HT and Tyr234 rather than a cation-π interaction.
Conclusions
We have performed a series of density functional theory binding energy calculations on model systems, derived from a homology model, to study how mutations on three specific tyrosines (Tyr143, Tyr153, and Tyr234) affect the 5-HT binding to the 5-HT3 receptor.
Our results suggest the presence of a hydrogen bond between 5-HT and Tyr153, but not between 5-HT and Tyr143; they thus support and extend available information data from experimental studies.8 The data also suggest a novel mixed hydrogen bond/cation-π interaction between Tyr234 and 5-HT.
Our density functional calculations are limited to small although realistic model systems, but they are able to capture the essential trends in binding energy variation upon mutation, assuming the binding takes place as in wild type. They would benefit from complementary classical and QMMM molecular dynamics simulations that can provide further dynamical information on the stability of the homology model, the binding modes, and the effect of the environment.
TABLE 5. Distances (dN-CoM) between the 5-HT Amine Nitrogen and the Center of Mass of the Phenyl Ring and Angles (θ) between the Normal to the Phenyl Ring and the Vector Pointing from Its Center of Mass to the 5-HT Amine Nitrogen for Models D and E and Their Mutations. N-H···A Hydrogen Bond Distances and Angles, Where the Acceptor A Can Be Either Oxygen or Fluorine, for Model Ea.
| Model D |
Model E |
|||||
|---|---|---|---|---|---|---|
| Tyr234 | d N-CoM | θ | d N-CoM | θ | d N···A | ∠NHA |
| 4-Me-Phe | 3.00 | 3 | 2.98 | 2 | ||
| mTyr | 3.06 | 8 | 3.73 | 42 | 3.17 | 109 |
| WT | 3.10 | 10 | 3.94 | 41 | 2.98 | 139 |
| Phe | 3.04 | 4 | 3.94 | 46 | ||
| 4-F-Phe | 3.11 | 7 | 4.60 | 56 | 2.75 | 174 |
| F3-Phe | 3.13 | 7 | 4.60 | 55 | 2.80 | 172 |
| Cha | 3.73 | 4.15 | ||||
| F5-Phe | 3.26 | 8 | 4.75 | 55 | 2.90 | 174 |
Binding energies in kcal/mol. Distances in Å. Angles in deg.
References and Notes
- (1).Reeves DC, Lummis SCR. Mol. Membr. Biol. 2002;19:11. doi: 10.1080/09687680110110048. [DOI] [PubMed] [Google Scholar]
- (2).Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, Kirkness EF. Nature. 1999;397:359. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- (3).Reeves DC, Sayed MFR, Chau P-L, Price KL, Lummis SCR. Biophys. J. 2003;84:2338. doi: 10.1016/S0006-3495(03)75039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brejc K, Dijk WJV, Klassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Nature. 2001;411:269. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- (5).Morris GM, Goodsell DS, Halliday R, Huey R, Hart WE, Belew RK, Olson AJ. J. Comput. Chem. 1998;19:1639. [Google Scholar]
- (6).Beene DL, Brandt GS, Zhong W, Zacharias NM, Lester HA, Dougherty DA. Biochemistry. 2002;41:10262. doi: 10.1021/bi020266d. [DOI] [PubMed] [Google Scholar]
- (7).Price KL, Lummis SCR. J. Biol. Chem. 2004;279:23294. doi: 10.1074/jbc.M314075200. [DOI] [PubMed] [Google Scholar]
- (8).Beene DL, Price KL, Lester HA, Dougherty DA, Lummis SCR. J. Neurosci. 2004;24:9097. doi: 10.1523/JNEUROSCI.2429-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kohn W. Rev. Mod. Phys. 1999;71:1253. [Google Scholar]
- (10).Parrinello M. Solid State Commun. 1997;102:107. [Google Scholar]
- (11).Andreoni W, Curioni A, Mordasini T. IBM J. Res. Dev. 2001;45:397. [Google Scholar]
- (12).Segall MD. J. Phys.: Condens. Matter. 2002;14:2957. [Google Scholar]
- (13).Carloni P, Rothlisberger U, Parrinello M. Acc. Chem. Res. 2002;35:455. doi: 10.1021/ar010018u. [DOI] [PubMed] [Google Scholar]
- (14).van Mourik T, Emson L. Phys. Chem. Chem. Phys. 2002;4:5863. [Google Scholar]
- (15).CPMD V3.9 Copyright IBM Corp, 1990-2001; Copyright MPI für Festkörperforshung Stuttgart, 1997-2001.
- (16).Troullier N, Martins JL. Phys. Rev. B. 1991;43:1993. doi: 10.1103/physrevb.43.1993. [DOI] [PubMed] [Google Scholar]
- (17).Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- (18).Hockney RW. Methods Comput. Phys. 1970;9:136. [Google Scholar]
- (19).Hutter J, Lüthi HP, Parrinello M. Comput. Mater. Science. 1994;2:244. [Google Scholar]
- (20).Ireta J, Neugebauer J, Scheffler M. J. Phys. Chem. A. 2004;108:5692. [Google Scholar]
- (21).Becke AD. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- (22).Lee CL, Parr R. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- (23).Tuma C, Boese AD, Handy NC. Phys. Chem. Chem. Phys. 1999;1:3939. [Google Scholar]
- (24).Dunitz JD. ChemBioChem. 2004;5:614. doi: 10.1002/cbic.200300801. [DOI] [PubMed] [Google Scholar]
- (25).Dunitz JD, Taylor R. Chem. Eur. J. 1997;3:89. [Google Scholar]
- (26).Morozov A, Kortemme T, Tsemekhman K, Baker D. Proc. Natl. Acad. Sci. 2004;101:6946. doi: 10.1073/pnas.0307578101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. Proc. Natl. Acad. Sci. 1998;95:12088. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mecozzi S, West AP, Jr., Dougherty DA. Proc. Natl. Acad. Sci. 1996;93:10566. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]