

Abstract
The relative ability of IgG subclasses to cause acute inflammation, and the roles of specific effector mechanisms in this process is not clear. We explored this in an in vivo model of glomerular inflammation in the mouse. TNP was planted on the glomerular basement membrane after conjugation to nephrotoxic antibody. The relative nephritogenicity of anti-TNP switch-variant monoclonal antibodies was then explored and shown to be IgG2a>IgG2b, with no disease caused by IgG1. Using knockout mice, we showed that FcγRIII was necessary for both neutrophil influx and glomerular damage induced by IgG2a and IgG2b. Surprisingly IgG1 did not cause disease although it binds to FcγRIII. Using blocking antibodies, we showed that this was explained by an additional requirement for FcγRIV which does not bind to IgG1. IgG2a or IgG2b induced neutrophil influx was not affected by deficiency of either FcγRI or C3. Bone marrow chimeras were constructed to test the effect of combined deficiency of FcγRI and C3, and there was no effect on IgG2a or IgG2b mediated neutrophil influx. However, IgG2b-induced albuminuria and thrombosis was reduced in C3 deficient mice, showing an additional role for complement in IgG2b-mediated glomerular damage. The results show that IgG2a and IgG2b are the pathogenic subclasses in acute neutrophil-mediated glomerular inflammation, with an indispensible role for both FcγRIII and FcγRIV. In addition complement contributes to IgG2b induced glomerular injury.
Keywords: rodent, antibody, Fc receptor, inflammation
Introduction
Several lines of evidence support a central pathogenic role for immune complexes in systemic autoimmune disease such as systemic lupus erythematosus, rheumatoid arthritis and vasculitis. Many of these systemic diseases have glomerulonephritis as their most serious manifestation, and in addition immune complexes play a role in other forms of primary GN3. Although the relative roles of cell-mediated and humoral immunity in glomerulonephritis continue to be debated, some key observations show a crucial role for immune complexes. Lupus-prone mice deficient in Fc receptors are almost completely protected from glomeruonephritis, despite the deposition of immune complexes (1). Fc receptor-deficient mice are also protected from autologous nephrotoxic nephritis (2). The latter is an induced model of glomerulonephritis in which disease is caused by an autologous response to a foreign antibody planted on the glomerular basement membrane.
The proinflammatory functions of IgG are largely determined by the interaction of the Fc portion of the heavy chain with complement and with Fc receptors. Murine IgG subclasses differ in their ability to activate complement, with IgG2a, IgG2b, but not IgG1 activating complement well (3, 4). Cell membrane receptors for the Fc portion of IgG are widely distributed on macrophages, monocytes, neutrophils, and couple immune complex formation to cellular effector mechanisms (5). Activating Fc receptors comprise FcγRI, FcγRIII and the recently described FcγRIV (6). FcγRI is a high affinity receptor that has been shown in vitro to bind monomeric and complexed IgG2a, with a much lower affinity for IgG2b (7). FcγRIII is a low affinity receptor that binds to IgG2a, IgG1 and IgG2b but not IgG3 (8). FcγRIV binds to IgG2a and IgG2b with little affinity for IgG1 and IgG3 (6).
Particular forms of GN are associated with specific subclasses of IgG in both men and in mice. Human lupus nephritis has been associated with IgG1, 2 and 3, membranous nephropathy with IgG4, mesangiocapillary GN with IgG3, and anti-GBM disease with IgG1 and IgG4 (9-11). In murine lupus, earlier studies suggested that IgG2a was the dominant subclass in glomerular eluates from NZB/W mice (12, 13). In BXSB mice IgG2b was reported be the dominant subclass in glomerular eluates, with IgG2a and IgG2b equally common in MRL/lpr mice (13). A recent report in spontaneous lupus-like disease, in mice lacking the inhibitory receptor FcγRIIb, has also suggested that IgG2a and IgG2b are the dominant subclasses deposited in glomeruli (14). The descriptive finding of particular IgG subclasses in serum, on immunfluorescence staining or in glomerular eluates does not directly demonstrate the pathogenicity of these subclasses. It is quite possible that a subclass deposited in small amounts is highly pathogenic. In addition the relative amounts in serum or glomeruli are difficult to judge in assays that use subclass specific detecting antibodies that have different affinities. In order to directly show the effect of an IgG subclass, the antibody needs to be passively given in vivo in a model of inflammation. Even then, the relative role of IgG subclasses cannot be reliably deduced from studies that compare random panels of monoclonal antibodies. A previous study in rats compared disease that was induced by monoclonal antibodies to type IV collagen in WKY rats (15). However, as well as being of different subclasses, these monoclonal antibodies would have differed in affinity and specificity and this could have lead to differences in pathogenicity. The most rigorous way to assess the role of subclass is through the use of switch variants that have identical heavy and light chain variable regions, and differ only in the heavy chain portion determining subclass.
Previous in vivo studies have explored the role of IgG subclass using switch variants in models such as haemolytic anaemia (16), melanoma therapy and thrombocytopenia (6, 17), and lymphoma treatment (18). In order to assess the role of IgG subclass in acute glomerular inflammation we have developed a model in which disease is passively induced by monoclonal switch variants against trinitrophenol (TNP), after TNP was first planted on the glomerular basement-membrane. We could show that even a high dose IgG1 was unable to induce disease, whereas lower doses of IgG2a and IgG2b were pathogenic, with more severe disease caused by IgG2a. Using knockout mice and blocking antibodies, we explored the effector mechanisms responsible. Our data show that both FcγRIII and FcγRIV were required for all of the disease manifestations caused by IgG2a and IgG2b. In addition, complement contributed to IgG2b-mediated glomerular damage, as reflected by the development of thrombosis and proteinuria.
Materials and Methods
Mice
FcγRI and FcγRIII deficient mice were generated as described (19, 20). FcγRI deficient mice were backcrossed 6 generations onto a C57BL/6 background (from a mixed C57BL/6/BALBc/129 background), and FcγRIII deficient mice were backcrossed 12 generations onto C57BL/6. C3 deficient mice were originally obtained from M Carroll (21) and backcrossed 11 generations onto C57BL/6. Wildtype C57BL/6 mice were obtained from Harlan, UK. Bone marrow chimeras were constructed as described previously (22). Chimerism was assessed as described using real-time PCR for the neomycin resistance gene (present in C3-/- but not FcγRI-/- mice) on genomic DNA from peripheral blood as described (22). C3 levels were measured in the serum of FcγRI-/-→ C3-/- chimeras by ELISA. The coating antibody was sheep anti-mouse C3 (ICN Biomedicals) and the detecting antibody HRP-conjugated goat anti-mouse C3 (Nordic). The standard was serial dilutions of normal mouse serum. Age, sex and weight matched mice were used for all experiments. All experiments were performed according to UK home office regulations.
Monoclonal anti-TNP switch variant and anti-FcγRIV antibodies
Hybridomas secreting IgG1, IgG2a and IgG2b anti-TNP switch variants were a gift from Lucien Aarden (Sanquin, Amsterdam). An IgG1 hybridoma Hy2.15 derived from BALBc mice was originally a gift from George Kohler. The switch variants were obtained from this hybridoma by sequential sublining in combination with isotype-specific ELISAs according to previously published methods (23). Anti-TNP hybridomas were grown at standard cell density in serum free medium, and purified using protein G chromatography from concentrated supernatant under identical conditions. Purity was confirmed at greater than 95% by SDS gel electrophoresis. After purification, the affinity of the monoclonal antibodies were compared by ELISA. Maxisorb ELISA (Nunc) plates were coated with Trinitrophenol conjugated sheep IgG (prepared as below) at 10μg/ml. After blocking with 1% BSA, various dilutions of monoclonal anti-TNP were applied. The detecting antibody was alkaline phosphatase conjugated polyclonal anti-kappa light chain (Southern Biotech). FcγRIV receptor-blocking antibody 9E9 has been described previously (6) and was purified with protein G chromatography. Flow cytometric analysis 9E9 specificity was performed as described (6). Normal hamster IgG (Jackson’s Immunoresearch) was used as a control.
Induction of glomerulonephritis
Nephrotoxic antibody was prepared by immunising sheep with a kidney extract prepared as previous described (24). IgG was purified using DEAE sepharose, and was more than 90% pure on SDS gel electrophoresis. This was conjugated with TNP by incubation with trinitrofluorobenzene in 0.2 M Bicarbonate buffer pH9. After dialysis into PBS, the conjugate was characterised spectrophotometrically using a described formula (25). Conjugates used had a ratio of 11 moles TNP per mole of IgG. Disease was induced by injecting 1.3 mg of conjugate intravenously, followed 24 hours later by anti-TNP monoclonal antibody. LPS was from Escherichia coli 0111:B4 (Sigma, catalogue number L3024).
Assessment of disease severity
Kidney was fixed in Bouin’s solution, and paraffin embedded sections stained with periodic acid-Schiff. Neutrophils were counted and thrombosis scored as described (22, 26). Urine was collected from mice that were housed in metabolic cages for 24 hours immediately following the injection of monoclonal antibody, and albuminuria was measured by radial immunodiffusion as described (22) The sensitivity of the assay was 25μg/ml. Peripheral blood neutrophil numbers were measured as described (22).
Immunofluorescence staining
This was performed on material snap-frozen in isopentane. Direct immunofluorescence for TNP was performed using the IgG1 monoclonal anti-TNP, conjugated with FITC using standard techniques. Indirect immunofluorescence for complement components was performed using rat monoclonal anti C1q, C3 or C4 (all from Cedar lane), and FITC mouse anti-rat IgG (Jacksons Immunoresearch). For assessment of complement staining, image analysis techniques could not be used due to the mesangial deposition of complement seen in normal mice. The basement membrane staining was therefore assessed by the observer (with no knowledge if sample identity) on a scale 0-4, disregarding the mesangial staining. For FcγRIV staining, 9E9 was used with detection by goat anti-hamster IgG (Jacksons Immunoresearch), followed by Alexa fluor 488 goat anti-FITC (Molecular Probes).
Statistics
Statistics were performed using Graphpad Prism (Graphpad Software). A student’s t test was used where two groups were compared and a one-way ANOVA with either Tukey’s or Dunnett’s post-test where there were more than two groups. Data were analysed after a logarithmic transformation if the variances were significantly different.
Results
Induction of glomerular inflammation by monoclonal anti-TNP antibody
We first established a model in which glomerular inflammation was induced by anti-TNP monoclonal antibody. TNP-conjugated nephrotoxic antibody was injected, followed 24 hours later by monoclonal anti-TNP antibody. Immunofluorescence staining verified that TNP was deposited on the glomerular basement membrane, along with mouse IgG, as shown in figure 1A and B. Preliminary experiments established that endotoxin was necessary as a co-factor, and that 15mg/kg of IgG2a was needed to cause robust disease. When 100ng of endotoxin was given with 15mg/kg IgG2a anti-TNP monoclonal as a single intravenous injection, a significant neutrophil influx was seen, as shown in figure 1C. When either monoclonal antibody or endotoxin alone was given, only a mild neutrophil influx was seen, as shown in Figure 1D. These results established a requirement for endotoxin, and this was given with monoclonal antibody at a dose of 100ng per mouse in all subsequent experiments. As we planned to compare disease induced by the switch variants, it was important to verify identical binding to TNP of purified antibody. This was confirmed by ELISA as shown in figure 1E.
Figure 1.
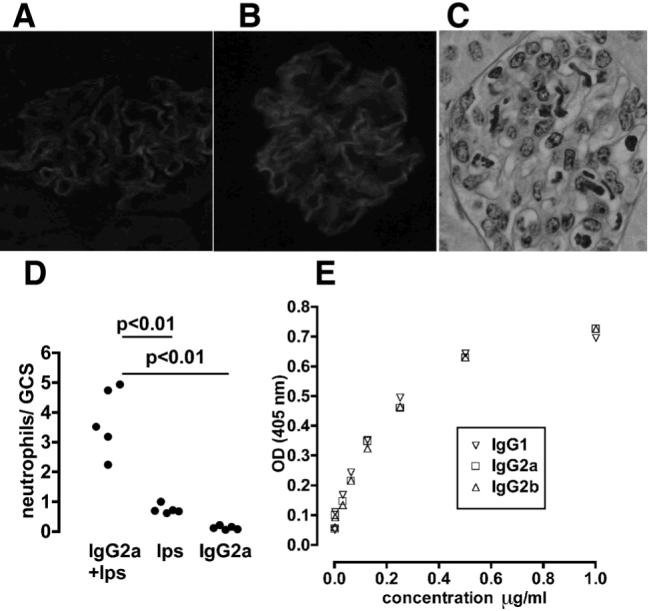
Glomerular inflammation induced by switch variant monoclonal antibodies. A. Immunofluorescence staining for trinitrophenol in mice given nephrotoxic antibody conjugated with TNP. This shows a linear glomerular basement membrane pattern. B. Immunofluorescence staining for mouse IgG in mice given anti-TNP monoclonal antibody 24 hours after an injection of TNP conjugated nephrotoxic antibody, again showing a linear glomerular basement membrane pattern. C. Histological changes observed 2 hours after the injection of anti-TNP monoclonal antibody given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. This shows a prominent neutrophil influx (identified as irregular lobulated nuclei). D. Endotoxin is required for the neutrophil influx. A significant neutrophil influx is seen when both endotoxin (100ng) and 15mg/kg IgG2a anti-TNP monoclonal are given together, but not when either is given alone. E. Switch variant anti-TNP monoclonals have identical binding to TNP as shown by ELISA.
Relative pathogenicity of switch variant monoclonal antibodies
As we had established that 15mg/kg of IgG2a was necessary in order to induce disease, we compared disease induced by this dose of IgG1, IgG2a and IgG2b anti-TNP switch variants. Results at 2 hours are shown in figure 2A, with representative histology in figure 3. IgG2a induced a greater neutrophil influx into glomeruli than IgG2b or IgG1. IgG2b induced a greater neutrophil influx into glomeruli than IgG1. IgG1 did not cause a neutrophil influx, since neutrophil numbers in glomeruli after IgG1 administration were not significantly different to those seen after endotoxin alone. Note that in this and later experiments, the control group given endotoxin alone were given TNP-conjugated nephrotoxic antibody 24 hours earlier, as were the other mice. We then assessed whether a higher dose of IgG1 could induce a neutrophil influx. Even 100mg/kg of IgG1 did not result in a neutrophil influx greater than that seen after endotoxin alone. IgG2a induced more severe albuminuria than IgG2b as shown in figure 2B. Even after 100mg/kg of IgG1, albuminuria was seen only at trace levels (< 70μ□/24 hours in all mice). At 24 hours, assessment of glomerular thrombosis gave results that mirrored those for albuminuria as shown in figure 2B with representative histology in figure 3. There was significantly more thrombosis after IgG2a than IgG2b with none seen even after 100mg/kg of IgG1. These data established the relative pathogenicity of IgG subclasses as IgG2a > IgG2b ≫ IgG1.
Figure 2.

The relative pathogenicity of switch variant monoclonal antibodies. A. Numbers of neutrophils infiltrating glomeruli at 2 hours in mice given the same intravenous dose of switch variant anti-TNP monoclonal antibodies, with endotoxin 100ng, 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. The lps group are mice given endotoxin 100ng alone in the second injection. Numbers for IgG2a were significantly greater than for IgG2b, and numbers for IgG2b were significantly greater than for IgG1. IgG1 did not cause a neutrophil influx above that seen with endotoxin alone. B. Even a higher dose of IgG1 did not cause a neutrophil influx above that seen with endotoxin alone. C. Albuminuria in the first 24 hours after anti-TNP monoclonal given with endotoxin 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. IgG2a induced a greater albuminuria than IgG2b. Even after a much higher dose of IgG1, albuminuria was minimal. D. Quantitation of glomerular thrombosis, with results that mirror those for albuminuria.
Figure 3.
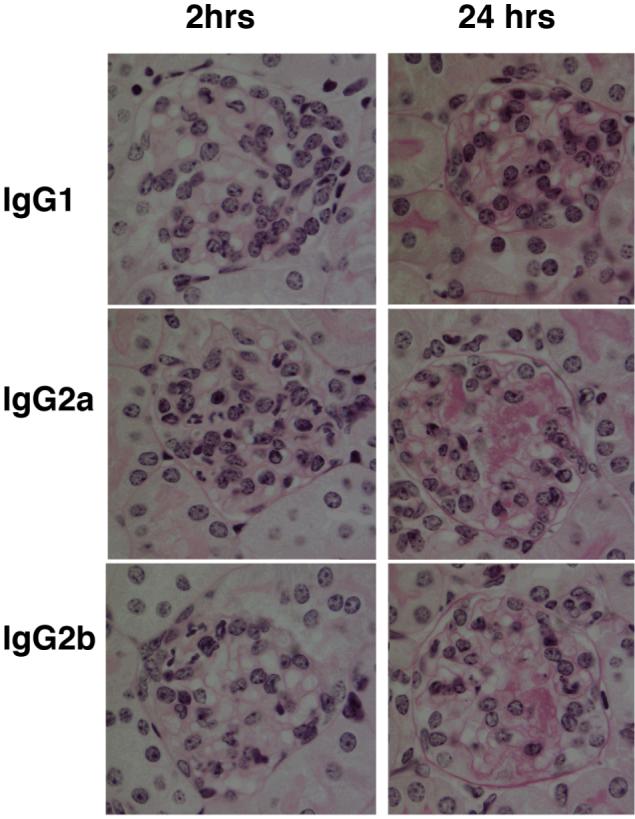
The relative pathogenicity of switch variant monoclonal antibodies. Histology after anti-TNP monoclonal, given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. The left hand column showns representative glomeruli 2 hours after 15mg/kg anti-TNP monoclonal antibody. Neutrophils are identified by lobulated nuclei, with a greater number seen after IgG2a than IgG2b, and none shown after IgG1. The right hand column shows representative glomeruli 24 hours after anti-TNP monoclonal antibody. There is glomerular thrombosis after IgG2a, with less after IgG2b and none after IgG1.
Complement activation in vivo due to switch variant monoclonal antibodies
In order to assess the relative ability of the switch variants to activate complement we stained kidney sections for C1q, C4 and C3. This was performed on samples from the experiment shown in figure 2A in which 15mg/kg of IgG1, IgG2a or IgG2b had been given. C1q was detected in a linear pattern on the glomerular capillary wall after IgG2a and IgG2b but not after IgG1. In mice that were given endotoxin alone and no monoclonal antibody, faint mesangial C3, and strong mesangial C4 staining was seen, at similar levels to those seen in untreated mice. However, superimposed on this pattern, there was linear capillary wall staining after IgG2a and IgG2b but not after IgG1. Capillary wall staining for both C3 and C4 was stronger for IgG2b than for IgG2a. Representative glomeruli are shown in figure 4A and data in figure 4B. These data established that IgG2b was a stronger activator of the classical pathway than IgG2a, with no activation by IgG1. They also established that IgG1 did not bind C1q.
Figure 4.
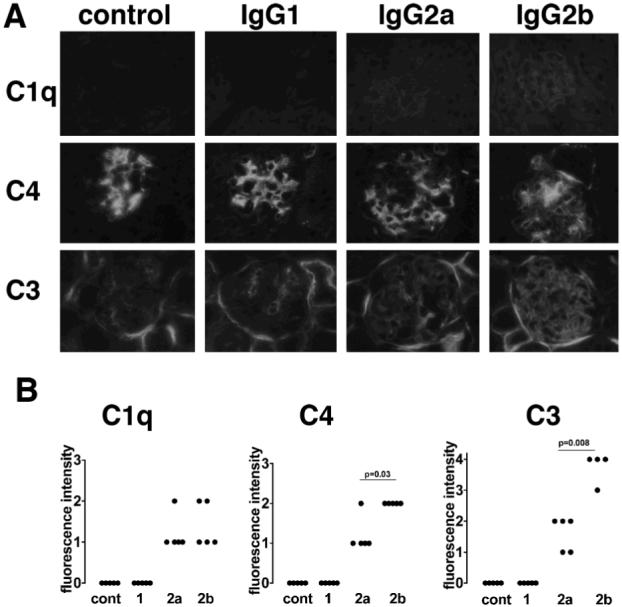
Complement activation by switch variant monoclonal antibodies. A. Immunofluorescence staining of glomeruli for C1q, C4 and C3 after 15mg/kg anti-TNP monoclonals with endotoxin, or endotoxin alone (control), given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. In the control group there was no C1q present, with sparse mesangial C3 and prominent mesangial C4. After IgG2a or IgG2b linear capillary wall C1q, C3 and C4 staining was present. In the case of C3 and C4, this was superimposed on the mesangial staining, with more C3 and C4 after IgG2b. There was no C1q, C3 or C4 deposited after injection of IgG1 anti-TNP. B. Quantitative data confirming the above findings.
Effector mechanisms causing IgG2a and IgG2b mediated disease
We then proceeded to explore the effector mechanisms responsible for disease induced by IgG2a and IgG2b using knockout mice. We compared disease induced by 15mg/kg IgG2a or IgG2b in wildtype mice with that seen in FcγRI, FcγRIII and C3 deficient mice. Figures 5A and 6A show that FcγRIII is absolutely required for the neutrophil influx due to IgG2a and IgG2b respectively. In the absence of FcγRIII, neutrophil numbers were the same as those seen after endotoxin alone in both cases. We did not demonstrate any role for FcγRI or complement in the neutrophil influx due to either IgG2a or IgG2b, as neutrophil numbers in FcγRI and C3-deficient mice were equivalent to those seen in wild type mice in both cases. As shown in figure 5B, albuminuria due to IgG2a was significantly decreased in FcγRIII deficient mice but not in C3 deficient mice compared to wildtype controls. However IgG2b induced significantly less albuminuria in both FcγRIII and C3 deficient mice than in wildtype controls, as shown in figure 6B. The results for glomerular thrombosis mirrored these data as shown in figure 5C and 6C. These data established that FcγRIII, but not FcγRI or C3, was indispensable for the glomerular neutrophil influx due to either IgG2a or IgG2b. However glomerular damage (as shown by thrombosis and albuminuria) due to either IgG2a or IgG2b depended on interaction with FcγRIII with an additional role for complement in the case of IgG2b but not IgG2a.
Figure 5.

Effector mechanisms of IgG2a induced disease. A. Numbers of neutrophils infiltrating glomeruli 2 hours after IgG2a anti-TNP monoclonal with endotoxin, given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. The WT control groups are wildtype mice given endotoxin alone in the second injection. There were significantly less neutrophils in FcγRIII -/- mice, with neutrophil numbers reduced to those seen after endotoxin alone. There was no significant difference in neutrophil numbers for wildtype compared to FcγRI -/- or C3-/- mice. B. Albuminuria in the first 24 hours after IgG2a with endotoxin given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. There was significantly less albuminuria in FcγRIII -/- mice compared to wildtypes. There was no significant difference in albuminuria for wildtype compared to C3 -/- mice. C. Quantitation of the glomerular thrombosis, with results that mirror those for albuminuria.
Figure 6.

Effector mechanisms of IgG2b-induced disease. A. Numbers of neutrophils infiltrating glomeruli 2 hours after IgG2b anti-TNP monoclonal with endotoxin given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. The WT control groups are wildtype mice given endotoxin alone in the second injection. There were significantly less neutrophils in FcγRIII -/- mice, with neutrophil numbers reduced to those seen after endotoxin alone. There was no significant difference in neutrophil numbers for wildtype compared to FcγRI-/- or C3-/- mice. B. Albuminuria in the first 24 hours after IgG2b with endotoxin was significantly less in FcγRIII-/- and C3-/- mice compared to wildtypes. C. Quantitation of the glomerular thrombosis, with results that mirror those for albuminuria.
FcγRIV is also required with no role for combined C3 and FcγRI deficiency
Our experiments so far had shown that IgG2a and IgG2b but not IgG1 was pathogenic with a dominant role for FcγRIII. This presented a paradox since IgG1 is known to interact with FcγRIII. It remained possible that FcγRI and C3 were important for the neutrophil influx in IgG2a and IgG2b-mediated disease, but that this was not apparent in mice deficient in only one of these molecules. In order to explore the effect of a combined deficiency, we generated bone marrow chimeras by transplanting FcγRI deficient marrow into C3 deficient mice. These animals lacked FcγRI on neutrophils, and circulating C3. Analysis of genomic DNA from peripheral blood in 8 chimeric mice showed 94.89±5.11 % donor-derived bone marrow cells. We confirmed that circulating levels of C3 were less than 1% of those found in wildtype mice in all 7 chimeras tested. These animals were compared with sham chimeric mice (wildtype donor and recipient). As shown in figure 7, there were no differences in neutrophil numbers when disease was induced by 15mg/kg IgG2a or IgG2b (with endotoxin) in these mice. These data established that FcγRI and C3 did not play a role in the neutrophil influx in this model, even in mice rendered doubly deficient. A further possible explanation for the lack of a pathogenic role of IgG1 is that the development of disease requires not only activation of FcγRIII but also of the recently described FcγRIV which does not bind IgG1. In order to explore this we used the blocking monoclonal antibody 9E9. A previous publication has shown specificity data for another monoclonal 9G8.1 (6), and in figure 8A we show similar data for 9E9. Data showing that cells expressed the relevant transfected Fc receptor in these experiments is in this previous publication (figure 2B). We also performed immunofluorescence staining using 9E9. As shown in figure 8B, this confirmed that FcγRIV was expressed in glomeruli of mice with disease and a glomerular neutrophil influx, but not in glomeruli from normal mice. FcγRIV was also expressed in the intersitium but this was also present in normal mice and probably represents resident dendritic cells or macrophages.
Figure 7.

The effect of combined FcγRI and C3 deficiency in IgG2a and IgG2b induced disease. FcγRI-/-→C3-/- bone marrow chimeras were constructed and compared to WT→WT sham chimera controls. A. Numbers of neutrophils infiltrating glomeruli 2 hours after IgG2a anti-TNP monoclonal with endotoxin, given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody. The WT→WT control groups are WT→WT mice given endotoxin alone in the second injection. There were no significant differences. B Shows a similar experiment in which disease was induced by IgG2b anti-TNP.
Figure 8.
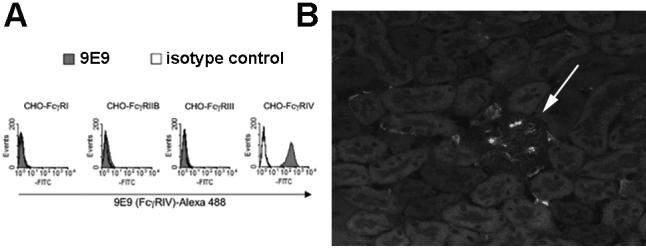
Specificity of 9E9 and localisation of FcγRIV in diseased kidney. A. Characterisation of the FcγRIV-specific monoclonal antibody 9E9. Chinese Hamster ovary cells were stably transfected with the indicated Fc receptor (which was FLAG tagged). In the case of FcγRI, FcγRIIII and FcγRIV the common γ chain was cotransfected. Shaded histograms are 9E9 staining and open histograms are isotype matched control antibody staining. Data showing similar expression of all Fc receptors, using a FLAG-specific antibody, is in reference (6). B. Immunofluorescence staining using 9E9 to show FcγRIV in inflamed kidney. A representative section from a mouse in which a neutrophil influx was induced 2 hours previously is shown. The arrow indicated a glomerulus (darker then the surrounding tubules) with positive staining that is not seen in normal mice. The interstitium has some positive cells which are also seen in normal mice.
We performed experiments in which 20mg/kg FcγRIV blocking antibody or control was given to mice 90 minutes before disease induction. As shown in figure 9A and 9B, there were significantly less neutrophils infiltrating glomeruli after FcγRIV blockade in both IgG2a and IgG2b-mediated disease. Levels were similar to those seen after endotoxin alone (with mice given TNP-conjugated nephrotoxic antibody, then control hamster IgG, followed by endotoxin alone in this control group). Albuminuria (figure 9C and 9D) and thrombosis (figure 9E and 9F) were also significantly decreased after FcγRIV blockade. These data established that, in addition to FcγRIII, FcγRIV was required for both glomerular neutrophil influx and glomerular damage in both IgG2a and IgG2b mediated disease.
Figure 9.

The role of FcγRIV in IgG2a and IgG2b induced disease. A. Numbers of neutrophils infiltrating glomeruli 2 hours after IgG2a anti-TNP monoclonal with endotoxin, given 24 hours after an intravenous injection of TNP conjugated nephrotoxic antibody, and 90 minutes after 20mg/kg of anti-FcγRIV or control antibody. The control group (left group of each graph) is wildtype mice given normal hamster IgG in the second injection and endotoxin alone in the third injection. B. Shows a similar experiment in which disease was induced by IgG2b anti-TNP. For both IgG2a and IgG2b induced disease, there were significantly less neutrophils in mice given anti-FcγRIV antibody compared to control, with neutrophil numbers reduced to those seen after endotoxin alone. C. Albuminuria in disease induced by IgG2a with endotoxin, was significantly less after anti-FcγRIV compared to control antibody. D. Albuminuria in disease induced by IgG2b with endotoxin was significantly less after anti-FcγRIV compared to control antibody. E. Quantitation of the glomerular thrombosis induced by IgG2a, with results that mirror those for albuminuria. F. Thrombosis was also decreased by anti-FcγRIV antibody in IgG2b-induced disease.
Peripheral blood neutrophils are similar in all groups of mice
It was possible that differences in glomerular neutrophil numbers simply reflected differences in peripheral blood neutrophils. To explore this possibility, we measured peripheral blood neutrophil numbers at two hours as this was the time at which we assessed glomerular neutrophil numbers. As shown in table I, no differences were seen between groups in experiments with either IgG2a or IgG2b. Our findings regarding the role of FcγRIII, FcγRI, FcγRIV and C3 in IgG2a or IgG2b-mediated disease could therefore not be explained by differences in peripheral blood neutrophil numbers.
Table I.
Peripheral blood neutrophil counts two hours after disease induction
Data are shown for the experiments in which 15mg/kg IgG2a or IgG2b anti-TNP antibody, given 24 hours after TNP-conjugated nephrotoxic antibody, was given to mice as shown in figures 5, 6, 7 and 8. Peripheral blood neutrophil numbers were assessed at 2 hours after disease induction. The first three columns are from experiments in which IgG2a or IgG2b was given to knockout or wildtype mice. Values for wildtype (WT) mice from each experiment are shown in brackets beneath values for the knockout mice (mean±SEM) in each case. The fourth column shows data from experiments in which FcγRI -/→C3 -/- bone marrow chimeras were compared with WT→WT controls (the latter shown in brackets). The fifth column shows data from experiments in which mice given anti- FcγRIV blocking antibody were compared with control treated mice (shown in brackets). There were no significant differences in peripheral blood neutrophil counts at two hours in any of these experiments.
| FcγRIII -/- (WT) | FcγRI -/- (WT) | C3 -/- (WT) | FcγRI -/→C3 -/- chimeras (WT→WT) | anti-FcγRIV (control IgG) | |
| IgG2a | 266±115 (355±170) | 340±69 (355±170) | 169±29 (222±49) | 337±87 (305±42) | 179±30 (199±77) |
| IgG2b | 425±100 (378±88) | 346±39 (311±50) | 443±48 (459±11) | 181±21 (255±34) | 187±47 (133±11) |
Discussion
The major finding in these studies is that IgG2a and IgG2b are the pathogenic IgG subclasses in acute glomerular inflammation (IgG2a>IgG2b) and that IgG1 is not pathogenic. Both FcγRIII and FcγRIV are required for neutrophil influx and glomerular damage mediated by IgG2a or IgG2b. We further demonstrated an additional role for complement in proteinuria and thrombosis due to IgG2b but not IgG2a.
The relative potency of switch variants of IgG subclasses has been assessed in studies of passively induced cellular depletion or cytotoxicity such as haemolytic anaemia (16, 27), B cell depletion (18), thrombocytopenia and melanoma therapy (17). These studies agreed that IgG2a and IgG2b were the most pathogenic in experimental situations where high affinity antibodies are used. A recent further study documented that FcγRI, III and IV all contributed to IgG2a mediated anaemia with FcγRIII and FcγRIV important for IgG2b-mediated disease (28). Our study differs from these previous experiments in that we employ switch variants to explore the role of IgG subclass in a model of tissue-specific immune-complex mediated inflammation. This is the first such in vivo study.
A previous study in the heterologous nephrotoxic nephritis model has suggested an important role for FcγRIII (29). Our experimental system is derived from this model, in which acute glomerular inflammation is induced by a single dose of foreign polyclonal antibody. Our finding of a dominant role for FcγRIII is consistent with this previous work, and extends it by showing that FcγRIII is essential regardless of the IgG subclass initiating disease. IgG1 is known to bind to FcγRIII, although with a lower affinity than IgG2a and IgG2b (6). This lower affinity for FcγRIII may contribute to the lack of disease due to IgG1. However a more convincing explanation is given by the additional requirement that we have shown for FcγRIV which does not significantly bind to IgG1 (6). Our experiments did not explore the contribution of binding to the inhibitory receptor FcγRIIb in this model. Recent data have shown that pathogenicity is determined by the relative balance of interactions with activating and inhibiting Fcγ receptors (17). IgG2b has a greater affinity than IgG2a for FcγRIIb, and also a lower affinity for FcγRIV (6). These differences may explain the lesser pathogenicity of IgG2b compared to IgG2a. IgG1 binds to FcγRIIb with higher affinity than IgG2a and a similar affinity to IgG2b. This would also tip the balance towards a lack of pathogenicity for IgG1.
There are at present limited in vivo data on the role of FcγRIV. In addition to the role in IgG2a or IgG2b-mediated anaemia described above (28), FcγRIV blockade inhibited IgG2a or IgG2b-mediated thrombocytopenia. In the anaemia study, the effect was seen in wildtype mice only for IgG2b-mediated disease. For IgG2a-mediated disease, FcγRIV blockade only had an effect in mice deficient in FcγRIII (28). In the thrombocytopenia studies the effect of FcγRIV blockade was moderate and led to a partial inhibition of platelet reduction (6, 17). FcγRIV is also important for tumour destruction though again the effect of FcγRIV blockade was partial (17). The autologous phase of nephrotoxic nephritis is a model in which progressive glomerulonephritis is mediated by a humoral and cellular immune response to a foreign antibody planted on the glomerular basement membrane. One study suggested a dominant role for FcγRIV in this model (30), though other studies have supported a role for FcγRI and FcγRIII (31). This model is a different experimental system from the current study however, and interactions of Fc receptors on both neutrophils and macrophages may be important. In summary, previous data have shown a contribution of FcγRIV to tissue injury, but in none has there been an absolute requirement for this receptor, as shown in the experiments presented here. The requirement for both FcγRIII and FcγRIV is similar to that seem when haemolytic anaemia is induced by IgG2b (28). This suggests that in both of these experimental models, ligation of both of these receptors is required in order to reach the threshold required for disease.
In the current work and these other studies, the role of FcγRIV has been explored using blocking antibodies. It is unlikely that our data are complicated by an interaction of the Fc portion of 9E9 with FcγRIII. Data in figure 8A shows no interaction of 9E9 with FcγRIII. In addition previous published work shows that 9E9 does not block IgG1 immune complex activity and hence does not bind to FcγRIII in vivo (17). The current data extend the in vivo role of FcγRIV to a model of neutrophil-mediated inflammation. They show a complete requirement for FcγRIV in disease due to either IgG2a or IgG2b. A recent study concluded that FcγRIII and FcγRIV both contributed to murine neutrophil activation by immobilised immune complexes in vitro (32). A moderate effect was seen with deficiency or blockade of one of these receptors, with complete abrogation of responses if both are deficient or blocked. Our in vivo study is consistent with this in vitro work in showing a role for both FcγRIII and FcγRIV. However it differs from the in vitro findings in showing that both receptors are required, and that deficiency in either one is sufficient to prevent disease.
We also demonstrated a role for complement in IgG2b but not IgG2a mediated glomerular damage. This is consistent with our immunofluorescence findings that IgG2b is a stronger activator of the classical pathway than IgG2a. Complement activation due to different IgG subclasses was assessed by immunofluorescence of frozen tissue sections. Glomerular C1q staining was detectable at similar levels in mice given IgG2a and IgG2b following TNP-conjugated nephrotoxic antibody. No deposition of C1q, C4 or C3 was seen in our study after IgG1. In vitro studies have suggested that IgG1 does not activate complement, but have not identified differences between the other subclasses (3, 4, 27). In the current study however, we have shown a difference between IgG2b and IgG2a, with greater complement activation for the former. We found similar fluorescence staining for C1q with IgG2a and IgG2b, with greater C4 and C3 staining for IgG2b. This could suggest that both subclasses bind C1q similarly, but that IgG2b is better able than IgG2a to activate C4 and the rest of the classical pathway. However, immunofluorescence staining may not differentiate between biologically important differences in C1q binding, and this must be interpreted with caution. We found that IgG1 was not able to activate complement, as there was no glomerular C3 or C4 deposition. This could have resulted from two possibilities. Firstly, IgG1 may have been unable to bind C1q, and secondly, IgG1 may have been unable to activate the remainder of the classical pathway after C1q binding. Our demonstration that C1q is not deposited in the glomerulus with IgG1 shows that the first option is the case. It should be noted that recent data has shown that C5 may be activated independently of C3 (33). This means that the lack of effect due to C3 deficiency in IgG2a-mediated disease does not rule out the possibility of a role for terminal complement components.
Previous work in the model of heterologous nephrotoxic nephritis has given conflicting results on the role of complement in acute antibody-mediated glomerular inflammation. In this model disease is induced by a single pathogenic dose of foreign nephrotoxic antibody. Some studies have shown a role for complement in both neutrophil influx and proteinuria, some a role in proteinuria but not neutrophil influx, and others a role in neither one nor the other (34-37). The current data suggest a possible reason for these discrepancies. The polyclonal antibodies used in these studies would have differed in subclass composition with differing abilities to activate mouse complement and bind to mouse Fc receptors. We have shown that complement activation may be required for proteinuria with some but not other subclasses. In addition we did not show a role for complement in causing neutrophil influx in any circumstances. However it is possible that with heterologous antibody that activates complement well, but binds Fc receptors poorly, such an effect may occur. In the current model and also in heterologous nephrotoxic nephritis, LPS is required as a cofactor with antibody in order to induce disease. We have previously shown that stimulation of TLR4 (the LPS receptor) on both renal cells and leukocytes is required for disease induction in heterologous nephrotoxic nephritis (38). The mechanism of the effects on neutrophils in these models could include modulation of Fc receptor expression. Indeed previous data has shown that both FcγRIII and FcγRIV are upregulated by LPS on myeloid cells (6) (39).
In the model used in the current study, disease develops within hours of deposition of antibody on the glomerular basement membrane, and is characterised by a glomerular neutrophil influx. Therefore our findings concerning Fc receptors can be considered to apply to neutrophil Fc receptors in the context of acute glomerular inflammation, and to reflect the early stages of immune complex mediated glomerulonephritis. However in recent years, a neutrophil glomerulitis has been recognised as a feature of humorally-mediated renal transplant rejection (40). Although the antibodies in this context will be against HLA molecules, the principles regarding the mechanisms by which antibody recruits and activates neutrophils in the glomerulus may be similar. This will therefore widen the potential relevance of our findings. In many forms of glomerular inflammation, humoral and cell-mediated immunity combine to cause glomerular inflammation. We have isolated the component due to humoral immunity in a simplified passive model in order to study in detail IgG effector mechanisms. In an active immune response the processes we have described will occur in parallel with cell-mediated mechanisms. It has been proposed that proliferative crescentic glomerulunephritis is largely mediated by Th1 type immune responses, though the relative roles of humoral and cell-mediated processes is not clear (41). In agreement with this, we have shown that IgG2a and IgG2b subclasses, associated with Th1 type immune response, are most pathogenic. It is likely that the immune complex-mediated acute inflammation increases T cell recruitment to the glomerulus, and subsequent cell-mediated processes. The manner in which humoral and cell-mediated processes interact in glomerular and other inflammatory diseases is an area in need of further study.
Acknowledgements
We are grateful to Lucien Aarden for providing hybridomas for the switch variants, to Ram Abuknesha for help with TNP conjugation and to Mike Carroll for C3 deficient mice.
Footnotes
This work was funded by the Wellcome Trust.
Glomerulonephritis
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Disclosures
The authors have no conflicting financial interests.
References
- 1.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 2.Park SY, Ueda S, Ohno H, Tanaka S, Shiratori T, Yamazaki T, Arase H, Karasawa A, Sato S, Ledernann B, Kondo Y, Okumura K, Ra C, Saito T. Resistance of Fc Receptor deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102:1229–1238. doi: 10.1172/JCI3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neuberger MS, Rajewsky K. Activation of mouse complement by monoclonal mouse antibodies. Eur J Immunol. 1981;11:1012–1016. doi: 10.1002/eji.1830111212. [DOI] [PubMed] [Google Scholar]
- 4.Dangl JL, Wensel TG, Morrison SL, Stryer L, Herzenberg LA, Oi VT. Segmental flexibility and complement fixation of genetically engineered chimeric human, rabbit and mouse antibodies. Embo J. 1988;7:1989–1994. doi: 10.1002/j.1460-2075.1988.tb03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 6.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 7.Sears DW, Osman N, Tate B, McKenzie IF, Hogarth PM. Molecular cloning and expression of the mouse high affinity Fc receptor for IgG. J Immunol. 1990;144:371–378. [PubMed] [Google Scholar]
- 8.Weinshank RL, Luster AD, Ravetch JV. Function and regulation of a murine macrophage-specific IgG Fc receptor, Fc gamma R-alpha. J Exp Med. 1988;167:1909–1925. doi: 10.1084/jem.167.6.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noel LH, Aucouturier P, Monteiro RC, Preud’Homme JL, Lesavre P. Glomerular and serum immunoglobulin G subclasses in membranous nephropathy and anti-glomerular basement membrane nephritis. Clin Immunol Immunopathol. 1988;46:186–194. doi: 10.1016/0090-1229(88)90181-x. [DOI] [PubMed] [Google Scholar]
- 10.Weber M, Lohse AW, Manns M, Meyer zum Buschenfelde KH, Kohler H. IgG subclass distribution of autoantibodies to glomerular basement membrane in Goodpasture’s syndrome compared to other autoantibodies. Nephron. 1988;49:54–57. doi: 10.1159/000184986. [DOI] [PubMed] [Google Scholar]
- 11.Imai H, Hamai K, Komatsuda A, Ohtani H, Miura AB. IgG subclasses in patients with membranoproliferative glomerulonephritis, membranous nephropathy, and lupus nephritis. Kidney Int. 1997;51:270–276. doi: 10.1038/ki.1997.32. [DOI] [PubMed] [Google Scholar]
- 12.Lambert PH, Dixon FJ. Pathogenesis of the glomerulonephritis of NZB/W mice. J Exp Med. 1968;127:507–522. doi: 10.1084/jem.127.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slack JH, Hang L, Barkley J, Fulton RJ, D’Hoostelaere L, Robinson A, Dixon FJ. Isotypes of spontaneous and mitogen-induced autoantibodies in SLE-prone mice. J Immunol. 1984;132:1271–1275. [PubMed] [Google Scholar]
- 14.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohda T, Okada S, Hayashi A, Kanzaki S, Ninomiya Y, Taki M, Sado Y. High nephritogenicity of monoclonal antibodies belonging to IgG2a and IgG2b subclasses in rat anti-GBM nephritis. Kidney Int. 2004;66:177–186. doi: 10.1111/j.1523-1755.2004.00719.x. [DOI] [PubMed] [Google Scholar]
- 16.Fossati-Jimack L, Ioan-Facsinay A, Reininger L, Chicheportiche Y, Watanabe N, Saito T, Hofhuis FM, Gessner JE, Schiller C, Schmidt RE, Honjo T, Verbeek JS, Izui S. Markedly different pathogenicity of four immunoglobulin G isotype-switch variants of an antierythrocyte autoantibody is based on their capacity to interact in vivo with the low-affinity Fcgamma receptor III. J Exp Med. 2000;191:1293–1302. doi: 10.1084/jem.191.8.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 18.Kaminski MS, Kitamura K, Maloney DG, Campbell MJ, Levy R. Importance of antibody isotype in monoclonal anti-idiotype therapy of a murine B cell lymphoma. A study of hybridoma class switch variants. J Immunol. 1986;136:1123–1130. [PubMed] [Google Scholar]
- 19.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, Schmidt RE, Sandor M, Capel PJ, Daeron M, van de Winkel JG, Verbeek JS. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 20.Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, Sedlik C, da Silveira SA, Gerber J, de Jong YF, Roozendaal R, Aarden LA, van den Berg WB, Saito T, Mosser D, Amigorena S, Izui S, van Ommen GJ, van Vugt M, van de Winkel JG, Verbeek JS. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 21.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995;92:11490–11494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown HJ, Lock HR, Sacks SH, Robson MG. TLR2 stimulation of intrinsic renal cells in the induction of immune-mediated glomerulonephritis. J Immunol. 2006;177:1925–1931. doi: 10.4049/jimmunol.177.3.1925. [DOI] [PubMed] [Google Scholar]
- 23.Boot JH, Geerts ME, De Groot ER, Aarden LA. Murine monoclonal isotype switch variants. Detection with rat monoclonal antibodies in ELISA and isolation by sequential sublining. J Immunol Methods. 1988;106:195–202. doi: 10.1016/0022-1759(88)90197-4. [DOI] [PubMed] [Google Scholar]
- 24.Robson MG, Cook HT, Botto M, Taylor PR, Busso N, Salvi R, Walport MJ, Davies KA. Accelerated nephrotoxic nephritis is exacerbated in C1q-deficient mice. J. Immunol. 2001;166:620–628. doi: 10.4049/jimmunol.166.11.6820. [DOI] [PubMed] [Google Scholar]
- 25.Good AH, Wofsy L, Henry C, Kimura J. In: Selected Methods in Cellular Immunology. Mishell BB, Shiigi SM, editors. W. H. Freeman & Co; San Francisco, CA, USA: 1980. pp. 343–350. [Google Scholar]
- 26.Brown HJ, Sacks SH, Robson MG. Toll-like receptor 2 agonists exacerbate accelerated nephrotoxic nephritis. J Am Soc Nephrol. 2006;17:1931–1939. doi: 10.1681/ASN.2005111167. [DOI] [PubMed] [Google Scholar]
- 27.Azeredo da Silveira S, Kikuchi S, Fossati-Jimack L, Moll T, Saito T, Verbeek JS, Botto M, Walport MJ, Carroll M, Izui S. Complement activation selectively potentiates the pathogenicity of the IgG2b and IgG3 isotypes of a high affinity anti-erythrocyte autoantibody. J Exp Med. 2002;195:665–672. doi: 10.1084/jem.20012024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baudino L, Nimmerjahn F, Azeredo da Silveira S, Martinez-Soria E, Saito T, Carroll M, Ravetch JV, Verbeek JS, Izui S. Differential contribution of three activating IgG Fc receptors (FcgammaRI, FcgammaRIII, and FcgammaRIV) to IgG2a- and IgG2b-induced autoimmune hemolytic anemia in mice. J Immunol. 2008;180:1948–1953. doi: 10.4049/jimmunol.180.3.1948. [DOI] [PubMed] [Google Scholar]
- 29.Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G, Luscinskas FW, Mayadas TN. Fc gamma RIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 2001;14:693–704. doi: 10.1016/s1074-7613(01)00150-9. [DOI] [PubMed] [Google Scholar]
- 30.Kaneko Y, Nimmerjahn F, Madaio MP, Ravetch JV. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med. 2006;203:789–797. doi: 10.1084/jem.20051900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tarzi RM, Davies KA, Claassens JW, Verbeek JS, Walport MJ, Cook HT. Both Fcgamma receptor I and Fcgamma receptor III mediate disease in accelerated nephrotoxic nephritis. Am J Pathol. 2003;162:1677–1683. doi: 10.1016/s0002-9440(10)64302-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jakus Z, Nemeth T, Verbeek JS, Mocsai A. Critical but overlapping role of FcgammaRIII and FcgammaRIV in activation of murine neutrophils by immobilized immune complexes. J Immunol. 2008;180:618–629. doi: 10.4049/jimmunol.180.1.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 34.Sheerin NS, Springall T, Carroll MC, Hartley B, Sacks SH. Protection against anti-glomerular basement membrane (GBM)-mediated nephritis in C3- and C4-deficient mice. Clin Exp Immunol. 1997;110:403–409. doi: 10.1046/j.1365-2249.1997.4261438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang T, Rosenkranz A, Assmann KJM, Goodman MJ, Gutierrez Ramos JC, Carroll MC, Cotran RS, Mayadas TN. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186:1853–1863. doi: 10.1084/jem.186.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hebert MJ, Takano T, Papayianni A, Rennke HG, Minto A, Salant DJ, Carroll MC, Brady HR. Acute nephrotoxic serum nephritis in complement knockout mice: relative roles of the classical and alternate pathways in neutrophil recruitment and proteinuria. Nephrol Dial Transplant. 1998;13:2799–2803. doi: 10.1093/ndt/13.11.2799. [DOI] [PubMed] [Google Scholar]
- 37.Robson MG, Cook HT, Pusey CD, Walport MJ, Davies KA. Antibody-mediated glomerulonephritis in mice: the role of endotoxin, complement and genetic background. Clin Exp Immunol. 2003;133:326–333. doi: 10.1046/j.1365-2249.2003.02233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown HJ, Lock HR, Wolfs TG, Buurman WA, Sacks SH, Robson MG. Toll-Like Receptor 4 Ligation on Intrinsic Renal Cells Contributes to the Induction of Antibody-Mediated Glomerulonephritis via CXCL1 and CXCL2. J Am Soc Nephrol. 2007;18:1732–1739. doi: 10.1681/ASN.2006060634. [DOI] [PubMed] [Google Scholar]
- 39.Hirano M, Davis RS, Fine WD, Nakamura S, Shimizu K, Yagi H, Kato K, Stephan RP, Cooper MD. IgEb immune complexes activate macrophages through FcgammaRIV binding. Nat Immunol. 2007;8:762–771. doi: 10.1038/ni1477. [DOI] [PubMed] [Google Scholar]
- 40.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007;18:1046–1056. doi: 10.1681/ASN.2007010073. [DOI] [PubMed] [Google Scholar]
- 41.Huang XR, Tipping PG, Shuo L, Holdsworth SR. Th1 responsiveness to nephritogenic antigens determines susceptibility to crescentic glomerulonephritis in mice. Kidney Int. 1997;51:94–103. doi: 10.1038/ki.1997.12. [DOI] [PubMed] [Google Scholar]