

Abstract
Systems biology has undergone an explosive growth in recent times. The staggering amount of expression data that can now be obtained from microarray chip analysis and high-throughput in situ screens has lent itself to the creation of large, terabyte-capacity databases in which to house gene expression patterns. Furthermore, innovative methods can be used to interrogate these databases and to link genomic information to functional information of embryonic cells, tissues and organs. These formidable advancements have led to the development of a whole host of online resources that have allowed biologists to probe the mysteries of growth and form with renewed zeal. This review seeks to highlight general features of these databases, and to identify the methods by which expression data can be retrieved.
Key Words: embryo, in situ hybridisation, cDNA micro-array, ontology
Databases are structured repositories. They appeal to both the bioinformatician and the biologist alike by providing raw datasets and semantic information relating to interpretation of biological data. World Wide Web-accessible databases that attempt to link genome sequence to functional information have been developed for many model organisms of development including the slime mould Dictyostelium discoideum,1 the nematode worm Caenorhabditis elegans,2 the fly Drosophila melanogaster3 and chordates (ascidians,4 zebrafish,5 amphibians,6,7 the chick,8 the mouse)9–11 (Table 1). These databases have the capacity to organise organismal data by a range of attributes including genome sequence, developmental stage, levels of gene expression and cell/tissue type. Furthermore, by making data available over the web, these online repositories allow users to browse experimental data, to compare it with data from other sources and to download raw datasets for further scrutiny at a later time.
Table 1.
WWW-Accessible databases of genes and development
| Database | Website | Species | Ontologies | Expression | Spatial Data | ||
| GO | AO | LFO | (xyz) | ||||
| DICTYBASE | http://dictybase.org/ | Dictyostelium discoideum | yes | no* | no* | ESTs | no |
| WORMBASE | http://www.wormbase.org/ | Caenorhabditis elegans | yes | yes | yes | ISH; IHC; R; N; W; RT | no |
| FLYBASE | http://flybase.org/ | Drosophila melanogaster | yes | yes | yes** | ISH; IHC; R; N; W; RT | no |
| ANISEED | http://aniseed-ibdm.univ-mrs.fr/ | Ciona intestinalis | yes | yes | yes | ISH; IHC; R | yes |
| ZFIN | http://fin.org/ | Danio rerio | yes | yes | no | A; ISH; IHC; R; N; W; RT | no |
| fate | |||||||
| map | |||||||
| XENBASE | http://www.xenbase.org/ | Xenopus laevis | yes | no | tool | ISH; IHC; ESTs | no |
| SAL-SITE | http://www.ambystoma.org/ | Ambystoma spp. | no | no | no | A; ESTs | no |
| GEISHA | http://geisha.ariona.edu/geisha/ | Gallus gallus | yes | yes | no | A; ISH; ESTs | no |
| EMAGE | http://genex.hgu.mrc.ac.uk/ | Mus musculus | yes | yes | no | ISH; IHC | yes |
| GUDMAP | http://www.gudmap.org/ | Mus musculus | yes | yes | no | A; ISH | no |
| EUREXPRESS II | http://www.eurexpress.org/ | Mus musculus | yes | yes | no | ISH | yes |
| Gene Expression in Tooth | http://www.bite-it.helsinki.fi/ | Mammals | yes | no*** | no*** | ISH; IHC | no |
| Kidney Development | http://golgi.ana.ed.ac.uk/kidhome.html | Chordates | yes | no*** | no*** | ISH; IHC | no |
| Glandular Organ Development | http://www.ana.ed.ac.uk/anatomy//orghome.html | Mammals | yes | no**** | no**** | ISH; IHC; N; RT | no |
GO, gene ontology terms; AO, anatomy ontology terms; LFO, lineage/fate ontology terms; A, cDNA microarray; ISH, in situ hybridisation; IHC, immunohistochemistry; R, reporter gene; N, Northern blot; W, Western blot; RT, reverse transcriptase PCR; EST, expression sequence tag; *phenotype ontology instead; **lineage for some structures; ***morphological staging instead; ****stage range instead
Ontologies, Fate Maps and Lineage: A Common Framework for Developmental Biology
Documenting the being, becoming, and lineage of various embryonic cells/tissues has become an invaluable framework for organising spatiotemporal developmental data. The use of controlled vocabularies, or ontologies, to describe anatomical structures has been incorporated into a range of atlas projects.2–5,8–10 Important design features of these anatomy ontologies are that they are structured hierarchically, enabling easy navigation of substructures, and expandable, allowing anatomical terms of increasing resolution to be added at later times. Anatomy ontologies are routinely used to archive gene expression patterns and other experimental findings. An example of a database that uses anatomy ontology effectively in this way is ZFIN.5 ZFIN, “The Zebrafish Model Organism Database,” allows users to find an assortment of embryo data (images, annotated records of in situ hybridization gene expression patterns, microarray data) by browsing an annotated ontology describing zebrafish anatomy at distinct stages of development. The number of positive hits associated with each anatomical term is shown in parentheses as a hyperlink. Clicking on these links takes you to the original data records that describe the experiment.
In model organisms where each cell type can be rigorously identified, ontologies can be extended to include the lineage and fate of distinct cell types. Complete fate maps have famously been established for the nematode C. elegans whereby every cell division and differentiation event have been rigorously pursued through development.12 In the C. elegans database Wormbase,2 interactive hierarchial ontologies allow users to browse cell lineage (up the tree) and cell fate (down the tree) and thus to obtain data relating to ancestral and future gene expression patterns in specific cells. Consequently, by using Wormbase it is possible to obtain descriptions of changing patterns of expression through development. In other organisms, such as the African-clawed toad Xenopus laevis, fate mapping studies demonstrate the contribution of early embryo cells (blastomeres) to future tissues.13–15 Xenbase,6 a Xenopus web resource, allows users to browse the fated tissue distributions of specific blastomeres using a graphical user interface. In addition, a reverse map allows users to query the blastula origins of later-stage tissues (germ layers and tissues derived thereof). The latter uses a controlled vocabulary of 27 terms. By the fate map interface being both simple and graphical, it invites the non-Xenopus specialist to investigate further.
Gene products also have ontological descriptions.16 The Gene Ontology (GO) project describes gene products in a species-independent manner by using a controlled vocabulary that seeks to define cellular components, molecular functions, and associated biological processes. Sequence alignment methods can be used to ascribe GO terms to novel transcripts. As such, they are an important method for inferring function to poorly characterized gene products, such as expression sequence tags (ESTs). GO terms linked to terms from an anatomical ontology provide invaluable descriptions of the spatial location of in situ hybridization gene expression. For example, they provide detail as to whether gene expression is epithelial or mesenchymal, and whether the expression pattern is limited to only a few cell types. In addition, by mapping gene function to organs and tissue, it may be possible to recognize evolutionary conserved mechanisms of development. It is noteworthy that, GO provides a common, cross-species framework for investigating the relationship between genes and function. By doing so, it provides a potent resource for linking queries across a range of model organisms.
How to Archive Expression Data
Submitting data to gene expression databases is a relatively straightforward process and for a simple submission (e.g., a single in situ hybridization study) can be achieved using online forms. For in situ hybrisation data, the information required should include the sequence of the probe (if available), ontological descriptions of where the gene is expressed (supplemented with images if appropriate), details on how the experiment was conducted, and contact details of the submitter. In addition, it should be indicated whether this data has been published elsewhere. Following submission, the data presented in these forms ideally undergo a curation process. This would involve an email correspondence (or similar) between the curatorial staff and the submitter to resolve possible conflicts or ambiguities in the annotation details of the submitted data. Following curation, the expression data is submitted to the WWW-accessible database where it is ascribed an ID that allows both the submitter and other users to access this data easily.
It is noteworthy that individual database projects may have preferred methods for dealing with large expression datasets (high-throughput in situ hybridization screens, microarray data). Consequently, it is prudent for submitters to contact the respective database curatorial staff before attempting to submit large datasets using these online forms.
How to Query Expression Data
A major use of gene expression databases is to find records of in situ hybridization patterns from text-based queries. Data from in situ hybridization screens may include expression patterns of novel marker genes, or alternatively novel domains of expression that may coincide with local inductive influences. To find expression patterns of interest, search queries involving stage range, anatomical location, and gene name are used by various databases.2–5,8–10 In principle, queries of this form can be used to retrieve both in situ hybridization and cDNA microarray expression data (Fig. 1). Entering alternative search terms can be used to refine database queries. For example, by using a coarse anatomical term from higher up the ontology tree one may find more expression patterns. Alternatively, extending the stage range, or including lineage/fate terms (if available) may also allow users to find more patterns of interest. An additional and useful feature of some databases is that users can add patterns of interest to a collection that can be accessed at later times.10–11 These collections can be browsed and curated by the user, and can be used as the basis of further searches.
Figure 1.
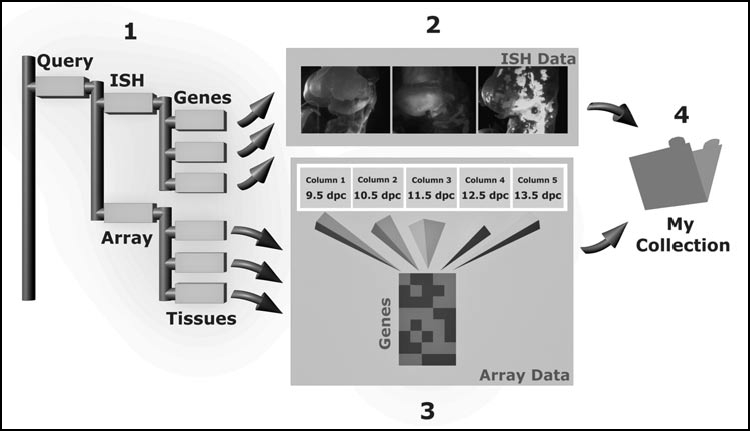
A search interface that uses ontologies to find gene expression patterns. A search query retrieves both in situ hybridisation (ISH) and cDNA microarray gene expression patterns that can be browsed using a hierarchial ontology tree (1). In this example, ISH entries are ordered by genes (gene ontology terms), whilst array data is ordered by tissue type (anatomy ontology terms). Whilst original ISH images (2) are screened visually using a slide sorter, array data (3) is used to determine gene expression trends between samples. In this example, the array series represents a time series of organ development and could be used to verify ISH findings. ISH and array datasets of interest can be saved in a folder entitled ‘My Collections’ (4) and can be accessed at a later time.
A powerful use of gene expression databases is to link spatio-temporal domains of gene expression to WWW-available microarray data describing global gene expression. When combined with in situ hybridization findings, array data can be used to determine whether signaling pathways are up or downregulated during organ development (Fig. 1). The annotation of microarray data using ontological terms to describe the cDNA array and the tissue analyzed greatly facilitates finding this data from text-based queries. Monitoring changes using cDNA microarray requires rigorous pairwise comparisons between samples, and hierarchial clustering methods to structure the data in a meaningful way. Online resources are available which can perform many of these processes. For example, pairwise comparisons can be achieved on cDNA microarray data series using downloadable software, such as FSPMA (Friendly Statistics Toolbox for Microarray Analysis)17 and/or applications from the MathWorks Bioinformatics Toolbox.18 Other online resources, such as GUD-MAP,10 provide pairwise comparisons of microarray series data and tools to segment and visualise this data. Data segmentation involves using hierarchial clustering methods to generate correlations between samples, whereby the correlations can be quantified using a range of numerical measures. The choice of measure should consider features of the array chip (e.g., the Pearson correlation coefficient is better suited for short oligo arrays) and researchers wishing to analyze microarray datasets are advised to consider the appropriateness of specific clustering algorithms (an excellent review of such methods is provided by ref. 19). Hierarchial clusters can be visualized as a red(up)/green(down) heatmap. Cluster data is linked to other details (e.g., GO terms) of the array data. As such, the findings of cluster analysis can be used as the basis of further queries and offer other methods of probing the relationship between genes and development.
Visual Language: Alternate Methods of Archiving and Querying Expression Data
The use of spatial databases to supplement existing text-based ontological databases is currently being explored by a number of groups.4,9,11,20–21 Whereas conventional search engines can be used to query textual ontologies, a thorough mining of an image bank requires tabled values describing the image data. One method to do this, employed by EMAGE,9 involves painting domains of gene expression, as determined from images of in situ hybridization-stained specimens, onto a range of Theiler-staged mouse embryo models. Values relating to these painted domains are tied with other information about the embryo including location and strength of gene expression as described by an ontology. To derive these painted domains, submitters to EMAGE overlay and ‘warp’ their original image data onto an image obtained from the EMAP atlas of Theiler-staged embryos. Threshold bars are used to determine cut-off points of levels of expression (strong, moderate, weak, not detected). Spatial queries that involve painting search domains on a web-based interface retrieve records of in situ hybridization images, gene expression as defined by an ontology, and other details relating to the probes, the specimen, and the in situ hybridization experiment. They further link to other databases, such as the EMAP anatomy ontology,22 the GXD,23 GO16 and OMIM.24 In doing so, this type of spatial search offers different ways into these databases.
An alternative method of archiving/querying spatial data is to use measurements of form. The ascidian database ANISEED (Ascidian Network for In Situ Expression and Embryological Data)4 includes tables of ‘biometric data’ that describe specific physical features of the cells of early-stage embryos, such as sphericity, flatness, convexity and surface/volume ratio. A graphical user interface (GUI) allows users to query gene expression in specific cells of staged ascidian embryos and to correlate gene expression patterns at specific embryonic stages with both identifiable cell types and morphometric data about those cell types. ANISEED thus offers a framework for correlating the morphology of cells (e.g. sphericity, flatness) with the expression of genes (e.g. adhesion genes).
Archiving morphometric data in this way could be a useful way of quantifying phenotypes. For example, by archiving measurements of µMRI data of mutant mouse embryos, it may be possible to order embryos by virtue of phenotypic penetrance. These can then be compared with more conventional text-based phenotype descriptions. Text-based phenotype ontologies do exist for a range of model organisms at the moment.1,25–27 However, genotype-phenotype relationships are notoriously complex and may vary significantly between strains within a species.28 It is possible that by correlating ontological (text-based) terms with physical (numerical) descriptions of cells, tissues and organs, a more robust description of phenotype may be arrived at. Such a framework would provide a platform for exploring genotype-phenotype relationships across phyla. In addition, it would offer a means of identifying gene networks/signalling pathways attenuated in anatomically abnormal structures (i.e., from phenotype to genotype), and of predicting physical consequences of mis-expression of genes.
In summary, this review reports on WWW-resources used to assist the developmental biologist in their studies of genes and development. Hierarchially structured ontologies are a powerful tool for organising biological data that can be browsed and queried quickly. These ontologies can be used to link gene and anatomical descriptions to gene expression patterns. Relationships between cDNA microarray datasets can be found using hierarchial clustering methods and differentially expressed genes found in this way can provide the basis of further searches. Spatial search engines provide alternative ways into databases that are distinct from ontology-based queries.
Acknowledgements
I wish to thank Dr. Jane Brennan for critically reading the manuscript and Dr. Jane Armstrong for helpful discussions.
Footnotes
Previously published online as an Organogenesis E-publication: http://www.landesbioscience.com/journals/organogenesis/article/4942
References
- 1. [Accessed May 29, 2007]. dictyBase website, http://dictybase.org.
- 2. [Accessed May 29, 2007]. WormBase website, http://www.wormbase.org, release WS175.
- 3. [Accessed May 29, 2007]. FlyBase website, http://flybase.org.
- 4. [Accessed May 29, 2007]. ANISEED website, http://aniseed-ibdm.univ-mrs.fr/
- 5. [Accessed May 29, 2007]. ZFIN website, http://zfin.org.
- 6. [Accessed May 29, 2007]. Xenbase website, http://xenbase.org.
- 7. [Accessed May 29, 2007]. Sal-Site website, http://ambystoma.org.
- 8. [Accessed May 29, 2007]. GEISHA website, http://geisha.arizona.edu/geisha.
- 9. EMAGE website, http://genex.hgu.mrc.ac.u. Accessed May 29, 2007.
- 10. [Accessed May 29, 2007]. GUDMAP website, http://gudmap.org.
- 11. [Accessed May 29, 2007]. EUREXPRESS II website, http://www.eurexpress.org.
- 12.Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhadditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 13.Moody SA. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Dev Biol. 1987;119:560–578. doi: 10.1016/0012-1606(87)90059-5. [DOI] [PubMed] [Google Scholar]
- 14.Moody SA. Fates of the blastomeres of the 32-cell stage Xenopus embryo. Dev Biol. 1987;122:300–319. doi: 10.1016/0012-1606(87)90296-x. [DOI] [PubMed] [Google Scholar]
- 15.Bauer DV, Huang S, Moody SA. The cleavage stage origin of Spemann's Organizer: Analysis of the movements of blastomere clones before and during gastrulation in Xenopus. Development. 1994;120:1179–1189. doi: 10.1242/dev.120.5.1179. [DOI] [PubMed] [Google Scholar]
- 16. [Accessed May 29, 2007]. Gene Ontology website, http://www.geneontology.org.
- 17.Sykacek P, Furlong RA, Micklem G. A friendly statistics package for microarray analysis. Bioinformatics. 2005;21:4069–4070. doi: 10.1093/bioinformatics/bti663. [DOI] [PubMed] [Google Scholar]
- 18. [Accessed May 29, 2007]. The MathWorks -Bioinformatics Toolbox website, http://www.mathworks.com/products/bioinfo/functionlist.html.
- 19.Datta S, Datta S. Evaluation of clustering algorithms for gene expression data. BMC Bioinformatics. 2006;7:S17. doi: 10.1186/1471-2105-7-S4-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. [Accessed May 29, 2007]. The Caltech Mouse Atlas Project website, http://mouseatlas.caltech.edu.
- 21. [Accessed May 29, 2007]. Allen Brain Atlas website, http://brainatlas.org.
- 22. [Accessed May 29, 2007]. EMAP website, http://genex.hgu.mrc.ac.uk.
- 23. [Accessed May 29, 2007]. The Gene Expression Database (GXD) website, http://www.informatics.jax.org/mgihome/GXD/aboutGXD.shtml.
- 24. [Accessed May 29, 2007]. Online Mendelian Inheritance in Man (OMIM) website http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM.
- 25. [Accessed May 29, 2007]. Mouse Phenome Database (MPD) website http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=docs/home.
- 26. [Accessed May 29, 2007]. C. elegans RNAi Phenome Database website http://omicspace.riken.jp/Ce/rnai/jsp/index.jsp.
- 27. [Accessed May 29, 2007]. PhenomicDB http://217.91.40.111/
- 28.Bogue MA, Grubb SC, Maddatu TP, Bult CJ. Mouse Phenome Database (MPD) Nucleic Acids Res. 2007;35:D643–D649. doi: 10.1093/nar/gkl1049. [DOI] [PMC free article] [PubMed] [Google Scholar]