

Abstract
Growing new organs in situ by implanting developing animal organ primordia (organogenesis) represents a novel solution to the problem of limited supply for human donor organs that offers advantages relative to transplanting embryonic stem (ES) cells or xenotransplantation of developed organs. Successful transplantation of organ primordia depends on obtaining them at defined windows during embryonic development within which the risk of teratogenicity is eliminated, growth potential is maximized, and immunogenicity is reduced. We and others have shown that renal primordia transplanted into the mesentery undergo differentiation and growth, become vascularized by blood vessels of host origin, exhibit excretory function and support life in otherwise anephric hosts. Renal primordia can be transplanted across isogeneic, allogeneic or xenogeneic barriers. Pancreatic primordia can be transplanted across the same barriers undergo growth, and differentiation of endocrine components only and secrete insulin in a physiological manner following mesenteric placement. Insulin-secreting cells originating from embryonic day (E) 28 (E28) pig pancreatic primordia transplanted into the mesentery of streptozotocin-diabetic (type 1) Lewis rats or ZDF diabetic (type 2) rats or STZ-diabetic rhesus macaques engraft without the need for host immune-suppression. Our findings in diabetic macaques represent the first steps in the opening of a window for a novel treatment of diabetes in humans.
Key Words: β-cell, chronic kidney disease, diabetes mellitus, non-human primates, transplantation, xenotransplantation
Introduction
Dr. Marc R. Hammerman: I am pleased to be able to deliver the inaugural Organogenesis Forum lecture. I am going to speak about organogenesis of the kidney and endocrine pancreas. The term ‘organogenesis’ has a number of definitions as reflected by the wide range of subject matter represented in Organogenesis, the journal. For purposes of this lecture, what I mean by ‘organogenesis’ is a novel technology for growing new organs in situ from transplanted embryonic organ primordia, in the development of which we and a number of other laboratories around the world are engaged.1
Previously, we have reviewed why the timing of primordia retrieval from embryos is important for organogenesis. In fact, there are ‘windows of opportunity’ for teratogenesis-free differentiation, vascularization and reduction of immunogenicity post-transplantation.1 Recently, we have generated data that provide the basis for what, to our knowledge, is the first published report of organogenesis in non-human primates.2 Since the use of non-human primates is a key intermediate step towards the employment of virtually any transplantation technology to replace kidney or endocrine pancreas in humans, our manuscript reflects the opening of a clinical window for organogenesis.2
Organogenesis of Kidney
A shortage of human donor organs limits transplantation therapy for end-stage renal disease. We and others have suggested that theoretical strategies for kidney replacement therapies of the future include those listed in Figure 1: (1) the use of human ES cells; or (2) the use renal-specific precursors obtained from developed human kidneys (‘adult’ renal stem cells) to grow a new kidney; (3) therapeutic cloning; (4) xenotransplantation of developed animal kidneys to humans; (5) transplantation of differentiated kidney cells in lieu of whole kidneys (cell therapies); and (6) generation of new kidneys in situ from embryonic primordia (renal organogenesis).3
Figure 1.

Organ replacement therapies of the future.
The design of strategies to coax ES cells or ‘adult’ stem cells into generating a functional kidney is going to be a very difficult task. This is because renal anatomy is so complex and renal function requires the precise integration in three dimensions of a large number of highly specialized cells vascularized at least in part from outside of the organ.3 ‘Close’ is not good enough to make a functioning kidney. Rather, renal ultrastructure must be perfect to permit glomerular filtration and tubular secretion and reabsorption. Because the kidney is so complicated I don't think ES or ‘adult’ stem cells are good starting material for whole renal organ replacement.
Organ formation in human embryos begins several weeks after implantation. At the present time, in the USA there is considerable controversy about the generation of human ES cells from fertilized ova. I don't believe that it will ever be acceptable to clone and implant human embryos to the point of organ formation, effectively excluding this sort of therapeutic cloning as an organ-replacement therapy.3 Excluding ES and ‘adult’ stem cells and therapeutic cloning from my list, we are left with xenotransplantation of whole kidneys, renal cell therapies or organogenesis as theoretical means by which renal function can be recapitulated in lieu of human allotransplantation (Fig. 1).
The use of pig kidneys instead of human organs makes sense because adult pigs and humans are the same size and weight and have almost identical renal function.4 Unfortunately, the use of pig kidneys in humans is not a viable option because of humoral rejection that occurs post-transplantation of vascularized pig organs into old-world primates. Humoral rejection is complement-mediated and results in large part from the binding of pre-formed circulating anti-bodies directed against galactose-α-1,3-galactose, an antigen present on vascular endothelium of most mammals, but not old world primates and humans.
The latter lack an enzyme α(1,3-galactosyl-transferase) required for galactose-α-1,3-galactose antigen formation.4–6 Humoral rejection of pig kidneys transplanted into non-human primates can be prevented by using kidneys from transgenic swine that express an enzyme on their vascular endothelium that deactivates complement, human decay accelerating factor5 or in which the gene for α-1,3-galactosyl-transferase is deleted.6 However, the intensity of cellular rejection of kidney transplants from such transgenic swine is such that the immune suppressive regimens required for engraftment in primate hosts result in complications that could never be tolerated in humans.5,6
Renal cell therapy of sorts has been carried out via implantation of sectioned7 or whole8 embryonic kidney primordia into renal parenchyma7 or beneath the kidney capsule.8 Under these conditions renal primordia differentiate into new nephrons in host kidneys. Unfortunately, the collecting systems of newly-developed donor nephrons and host kidneys do not connect, precluding excretion of urine and therefore clearance of filtered solutes. Also, transplantation into renal parenchyma or beneath the renal capsule is confining for newly-grown nephrons. As a result, obstruction occurs early during development and is manifest by formation of urine-containing cysts on the surface of host kidneys.8
In contrast to what occurs after implantation in renal parenchyma or beneath the renal capsule, renal primordia transplanted into a host rodent's fold of mesentery undergo differentiation and growth in hosts (renal organogenesis) that is not confined anatomically.8–16 We8 and others11,12 have shown experimentally that growth is enhanced if one of the host's kidneys is removed at the time of implantation, and that timing of renal primordia retrieval is important [embryonic day (E)15 works best for transplanted rat primordia].1 If implanted into an adult rat with its ureteric bud attached, the renal primordium enlarges and becomes kidney-shaped within three weeks.8 The ureteric bud differentiates into a ureter that can be anastomosed to the ureter of the host (ureteroureterostomy).8,9,13–16 Ureteroureterostomy turns out to be a key procedure for preserving the function of transplanted primordia. This is because the ureter must be freed from adhesions that often form as primordia develop in the mesentery. The adhesions obstruct the ureteric bud and prevent excretion of glomerular/tubular filtrate into the mesentery. As is the case for obstructed native kidneys during embryogenesis16 or for renal primordia implanted in renal parenchyma or beneath the renal capsule,7,8 the obstructed mesenteric transplant does not undergo normal renal differentiation, but rather atrophies and becomes fibrotic.16 Ureteroureterosomy preserves urine flow as the transplant differentiates. In our hands (rat-to-rat) it is not possible before 17 days post-transplantation because the ureter of the transplant is too friable. If ureteroureterostomy is not performed by 21 days post-transplantation, obstruction results. Therefore this procedure must be performed between 17–21 days after transplantation of metanephroi from rat embryos into adult rats if optimal differentiation is to occur.16 We have transplanted as many as four metanephroi at the same time (rat to rat) each of which undergoes growth and development in the mesentery. In our hands only one ureteroureterostomy is possible in the rat because of the small size of transplant and host ureters.16 However Marshall et al. are able to connect ureters from two transplants into the ureter of a host rat kidney.13
In contrast to transplanted developed kidneys that undergo acute rejection post transplantation into non-immunosuppressed hosts,8 rat renal primordia differentiate into small, but ultrastructurally normal kidneys after allotransplantation in the mesentery. The new kidneys become vascularized via arteries that originate from the superior mesenteric artery of hosts and veins that originate from the host mesentery.14 Developed renal primordia produce urine that is excreted via the bladder following ureteroureterostomy between transplant and host. Levels of renal function in transplanted renal primordia [glomerular filtration rate (GFR)] can be determined by measuring inulin clearance in otherwise anephric rats. In our initial experiments GFRs were very low.8 However, incubation of renal primordia with growth factors prior to implantation increased GFRs more than 100-fold compared to those in rats with non growth factor-incubated renal primordia implanted concurrently. GFRs in growth factor treated renal primordia are about 6% of normal.14 Others have reported even higher levels of GFR in rat-to-rat transplants.9 Renal plasma flow can be measured in transplanted renal primordia by calculating P-aminohippurate (PAH) clearances. The ratio of GFR/PAH clearance (filtration fraction) is 0.6, comparable to filtration fractions measured in rats with reduced renal function. Urine flow rates in transplanted rats are about 12% of the inulin clearance (GFR) measured in growth factor-treated renal primordia. The UV/GFR of 0.12 demonstrates that developed primordia can concentrate urine.14
Hemodialysis provides renal failure patients with GFRs that are about 10% of normal.3 Therefore, 6% of normal approximates a level of renal function that would be expected to preserve life. Indeed, life can be prolonged in otherwise anephric rat hosts by prior transplantation and ureteroureterostomy of a single renal primordium15 and survival can be extended if two primordia are connected to the host's ureter.13 If the ureteroureterostomy is severed prior to removing all native renal mass from the host rat, survival is not prolonged.15 Therefore, metabolic functions of the transplant alone, are insufficient to preserve life in the absence of intact excretory function.
There are four theoretical reasons why the use of developing renal primordia for transplantation might be advantageous relative to mature kidneys in terms of generating a reduced cellular rejection response and obviating humoral rejection. First, if developing renal primordia are obtained at sufficiently early stage, antigen-presenting cells (APCs) that mediate ‘direct’ host recognition of alloantigen or xenoantigen are absent.17–19 Second, donor antigens such as MHC Class I and II are not expressed on developing renal primordia to the extent they are expressed on mature renal tissue.20–22 Third, the immune response to transplanted fetal renal tissue is ameliorated relative to the response to adult tissue.23 Fourth, transplanted renal primordia are supplied by blood vessels of host origin.10,11,24,25
We transplanted renal primordia from E15 Lewis rat embryos across a concordant xenogeneic barrier into the mesentery of 10-week-old C57Bl/6J mice.24 In mice that receive immunosuppression, but not in its absence, transplanted rat renal primordia undergo differentiation and growth in situ.24
We next transplanted E28 pig renal primordia into C57Bl/6J mice26 or Lewis rats.25,27 The differentiation of E28 pig renal primordia is comparable to that of E15 rat renal primordia.25–27 Pig renal primordia undergo growth and differentiation in immunosuppressed rodents, but not in the absence of host immune suppression. Figure 2 illustrates E28 pig renal primordia prior to transplantation (Fig. 2A and B) and seven weeks post-transplantation into immunosuppressed rat hosts (Fig. 2C–F). The maintenance immunosuppression regimen we used in rats.27 (Tacrolimus 2 mg/kg daily intramuscularly post-implantation) is one that could be tolerated in humans.
Figure 2.
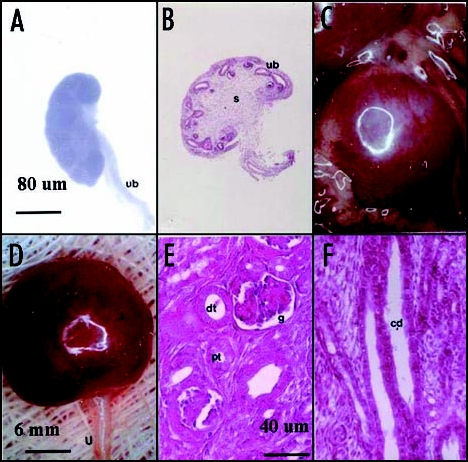
Photomicrographs (A, C and D) and photographs (B, E and F) of pig renal primordia. (A and B) E28 primordia (s, stroma; ub, ureteric bud); (C–F) Pig renal primordia seven weeks post transplantation in a rat mesentery (C) Developed primordium in situ; (D) Primordium after removal from the mesentery (u, ureter) (E) cortex with a glomerulus (g) proximal tubule (pt) and distal tubule (dt) labeled. (F) Medulla with collecting ducts (cd) labeled. Magnifications are shown for (A and B) in (A); (C and D) in (D); for (E and F) in (E) Reproduced with permission (ref. 40).
Pig renal primordia transplanted in rats grow to a size larger than native rat kidneys in rat hosts.27 This suggests that the hypoplasia may be more characteristic of transplanted rat renal primordia than of pig renal primordia.16 Hypoplasia can result from excessive cell death in metanephric blastema.28,29 Rapidly-dividing blastema cells in renal primordia could be placed at risk during the time of relative hypoperfusion that occurs between dissection from donor embryos and re-vascularization in situ. It may be that pig renal primordia, cells in which divide more slowly over a longer gestation period, are at reduced risk for apoptosis relative to rat primordia during the time of relative hypoperfusion.16 Our finding of more complete differentiation (kidneys larger than native rat kidneys) following pig-to-rat xenotransplantation relative to rat-to-rat allotransplantation27 is consistent with this possibility. Experiments currently in progress will determine whether transplantation of pig kidneys in rats or larger animals will prolong the recipient's life long-term. If so, embryonic pig kidneys may prove a suitable source for replacement of human renal function.
Organogenesis of Endocrine Pancreas
Standard treatment for diabetes mellitus is insulin and diet. Such treatment is lifelong, painful and requires frequent monitoring of blood glucose. Tight control of glucose levels is impossible without inducing hypoglycemia. Even tight glucose control does not impact on long-term complications of diabetes.30
Transplantation therapy for type 1 diabetes consists of human pancreas or islet transplantation, the latter being an experimental therapy. Transplantation can restore normal glucose tolerance and prevent complications. However, it requires immune suppression, in effect trading one disease (diabetes) for another (immune suppression).30
There are insufficient human pancreas donor organs. Only about 1,000 pancreas transplants are performed per year in the USA for its 1.3 million type 1 diabetics. The shortage of donor organs effectively precludes transplantation for 20 million type 2 diabetics.31
In that pigs are plentiful and because porcine insulin works well in humans, the pig has been suggested as a pancreas organ donor for human diabetics.32–34 The severity of humoral rejection effectively precludes their use as whole pancreas donors for non-human primates or humans. However, because they are vascularized by the host post-transplantation, isolated islets like other cell transplants are less susceptible to humoral rejection than pancreas transplants. Pig to human islet transplantation has been carried out without evidence for humoral-rejection.34 Unfortunately, even large numbers of transplanted porcine islets do not impact upon glucose control in human recipients.34 Recent experience with pig to non-human primate islet32 or neonatal islet33 transplantation shows that sustained insulin independence can be achieved, but only through the use of immunosuppressive agents that are not approved for human use or would result in an unacceptable level of morbidity in humans.
It has been known since the 1970s35,36 that glucose tolerance in alloxan—or streptozotocin (STZ)—diabetic rats (a model for type 1 diabetes) can be normalized after isotransplantation of embryonic pancreatic primordia. Exocrine pancreas does not differentiate post-transplantation of pancreatic primordia obtained early during embryogenesis (E17).35–37 Thus, transplantation of pancreatic primordia from animal embryos represents an organogenesis solution for endocrine pancreas replacement without complications that accompany co-transplantation of exocrine pancreas such as digestion of host tissues from the action of pancreatic enzymes.30
In early experiments35,36 others transplanted embryonic pancreas beneath the renal capsule of hosts, a location from which insulin was released into the systemic venous circulation. It was shown subsequently that shunting venous flow from the systemic to portal systems post-transplantation improves diabetic control.37 This makes sense because normally, the pancreas secretes insulin directly into the portal vein from which it enters the liver directly.
We transplanted whole pancreatic primordia obtained from rat embryos just after organ formation begins (E12.5) into the mesentery of STZ -diabetic rats.38,39 On E12.5 the rat pancreas is undifferentiated and dorsal and ventral components remain separate.38 By four weeks post-transplantation of whole pancreatic primordia from E 12.5 Lewis rats into the mesentery of STZ-diabetic Lewis rats, the tissue has undergone differentiation and islets of Langerhans can be delineated amidst stroma. There is no differentiation of exocrine tissue.38,39 Abnormal glucose tolerance in STZ diabetic rat hosts is normalized within two to four weeks post-isotransplantation of pancreatic primordia as is the pattern of abnormal weight gain characteristic of diabetic animals.38,39 No host immunosuppression is required for isotransplantation of E12.5 rat embryonic pancreas. However engraftment following xenotransplantation from Lewis rat embryos to C57Bl/6J mice does require that hosts be immunosuppressed.38
Subsequently we showed that glucose tolerance can be normalized in adult STZ -diabetic Lewis rats39,40 or in diabetic ZDF rats, a model for human type 2 diabetes41 following transplantation of pancreatic primordia from E28 pig embryos into the mesentery. Formerly-diabetic rats have porcine insulin, but no rat insulin detectable in circulation. Transcripts for porcine insulin are present in mesentery,39,40 liver, pancreas and mesenteric lymph nodes41 post-transplantation. Light and electron microscopy show that individual pig beta cells are engrafted in the rat tissues. Consistent with the findings of Eloy et al who described normalization of glucose post-transplantation of E15, but not E18 embryonic chick pancreas into STZ-diabetic rats42 if pig pancreatic primordia are obtained within a developmental ‘window’ prior to E35 (E28 works best), they engraft in non-immunosuppressed immunocompetent diabetic rat hosts.39–41
To examine the utility for transplantation of pig pancreatic primordia in a non-human primate model, we implanted embryonic pancreas from E28 pig embryos into the mesentery of STZ-diabetic male rhesus macaques.2 Long-term engraftment of pig β-cells within liver, pancreas and mesenteric lymph nodes post-transplantation was demonstrated by electron microscopy, positive immunehistochemistry for insulin, and positive RT-PCR and in-situ hybridization for porcine proinsulin mRNA.2 Insulin requirements were reduced in one macaque followed over 22 months post-transplantation and porcine insulin detected in plasma using sequential affinity chromatography, HPLC and mass spectrometry. Of potential importance for application of this transplantation technology to treatment of diabetes in humans and confirmatory of our previous findings in Lewis and ZDF rats, no host immunosuppression is required.2 Figure 3 shows photomicrographs originating from a mesenteric lymph node of a transplanted rhesus macaque. Sections in Figure 3A, C and E are stained with an anti-insulin antibody. Sections in Figure 3B, D and F are incubated with control serum. Sections of medullary sinus are delineated by arrows (Fig. 3A–D). Individual cells with β-cell morphology that stain positive (red) are delineated by arrowheads (Fig. 3E). No positive-staining cells are found in sections treated with control serum (Fig. 3B, D and F). Cells with morphology similar to positive cells in Figure 3E are delineated in Figure 3F (arrowheads). No insulin-positive cells are present in mesenteric lymph nodes of non-transplanted rhesus macaques.2
Figure 3.
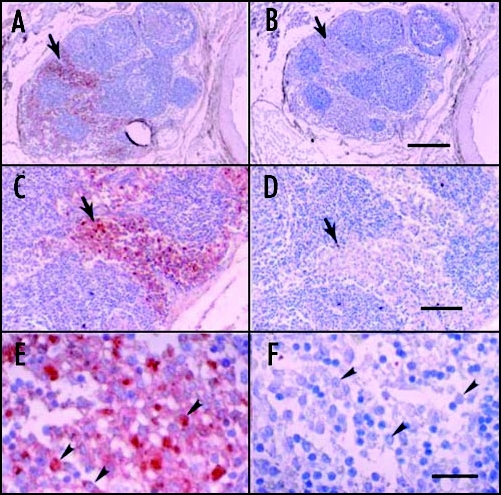
Photomicrographs of mesenteric lymph node from a STZ-diabetic rhesus macaque, 78 days post-transplantation of E28 pig pancreatic primordia. (A, C and E) are stained with an anti-insulin antibody. (B, E and F) are stained using a control serum. Sections of medullary sinus are delineated by arrows (A–D). Individual cells with beta cell morphology are delineated by arrowheads (E and F). Scale bars 120 µm (A and B); 80 µm (C and D); and 20 µm (E and F). Reproduced with permission (ref. 2).
Shown in Figure 4 are sections of mesenteric lymph node from a diabetic rhesus macaque that had been transplanted more than a year previously with E28 pig pancreatic primordia. In situ hybridization was performed using pig proinsulin antisense (Fig. 4A and C) or sense (Fig. 4B and E) probes. Cells within medullary sinuses stain (red) with the antisense, (Fig. 4A and C) but not the sense (Fig. 4B and D) probe. Intravenous glucose infusion was performed in the rhesus macaque at 600 days following the original transplantation. No rhesus macaque insulin (<0.1 ng/ml) was detected in circulation at any time. Analysis of circulating insulin at five minutes post-infusion revealed porcine insulin in circulation (Fig. 5A and B) Rhesus macaque insulin only was detected in plasma from a non-fasting non-diabetic non-transplanted rhesus macaque (Fig. 5C and D). Only porcine insulin was detected in plasma obtained from pigs.2
Figure 4.
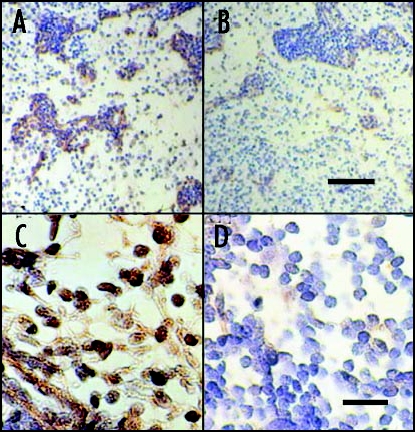
In situ hybridization was performed using pig proinsulin antisense (A and C) or sense probes (B and D) on sections of mesenteric lymph node originating from a STZ-diabetic rhesus macaque 407 days post-transplantation of E28 pig pancreatic primordia. Scale bars 80 µm (A and B) and 30 µm (C and D). Reproduced with permission (ref. 2).
Figure 5.
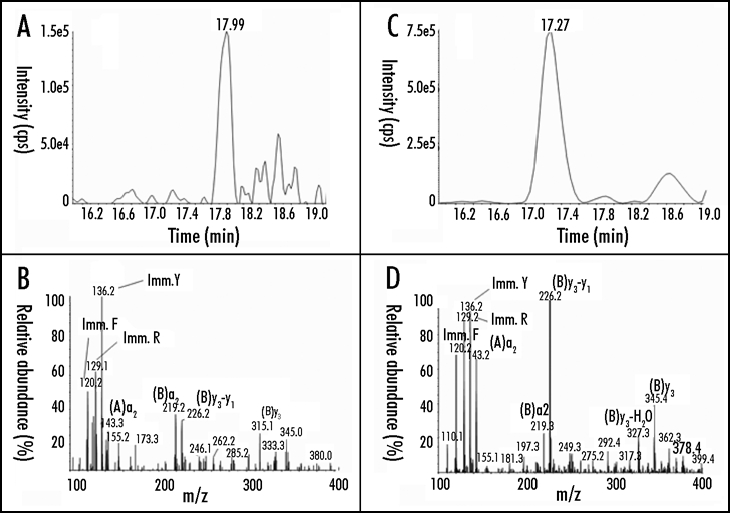
Chromatogram (A) and product ion mass spectrum (B) of porcine insulin (precursor ion [M+5H]5+ m/z 1156.3) extracted from 2 ml plasma obtained five minutes after IV glucose administration to a STZ-diabetic transplanted rhesus macaque. The retention time and diagnostic product ions derived from the five-fold charged precursor ion unambiguously identify porcine insulin. chromatogram (C) and product ion mass spectrum (D) of human/macaque insulin (precursor ion [M+5H]5+ m/z 1162.3) extracted from 1 ml of non-diabetic rhesus macaque plasma. The retention time and diagnostic product ions derived from the five-fold charged precursor ion unambiguously identify human/macaque insulin. Reproduced with permission (ref. 2).
Transplantation of embryonic pancreas offers theoretical advantages relative to transplantation of either pluripotent embryonic stem (ES) cells, or of fully differentiated (adult) pancreas or islets from adults or neonatal animals. Specifically: (1) Unlike ES cells, pancreatic primordia obtained at the proper time during embryogenesis differentiate along defined organ-committed lines. There is no requirement to steer differentiation and no risk of teratoma formation;43 (2) Unlike the case for insulin-secreting cells derived from ES cells glucose-sensing and insulin-releasing functions are linked following in situ differentiation of transplanted pancreatic primordia;40 (3) The growth potential of cells within embryonic pancreas is enhanced relative to those in terminally-differentiated adult pancreas or islets. It is possible to restore glucose tolerance in a diabetic rat by transplanting a single pancreatic primordium;44 (4) Only endocrine tissue differentiates following transplantation of pancreatic primordia obtained sufficiently early during embryogenesis obviating problems of host tissue digestion by exocrine pancreatic components44 and (5) The cellular immune response to transplanted embryonic pancreas obtained early during embryogenesis is attenuated relative to that directed against organs obtained at later times.45
Our finding that pig pancreatic primordia engraft long-term in non-immunosuppressed STZ diabetic rhesus macaques opens a window for their potential for their use in human diabetics. If applicable in humans, successful organogenesis of endocrine pancreas (or kidney) could provide in essence, an unlimited supply of donor organs. This would result in a paradigm shift in how the world thinks about organ replacement (Fig. 6):
there will be no need to transport organs across long distances;
transplantation can be done electively at a convenient time;
transplantation can be offered to high-risk individuals and can be repeated as needed
transplantation can be offered to patients currently not candidates including type 2 diabetics.41
Figure 6.

Advantages of organogenesis.
Questions and Answers
Dr. Melvin Blanchard, Associate Professor of Medicine, Washington University School of Medicine: Thank you, Marc, for an interesting talk. How far away are we from using pig pancreatic primordia in humans?
Dr. Hammerman: I think the observation that it is possible to transplant pig pancreatic primordia to non-human primates without an immunosuppression requirement is very important. The primordia engraft and secrete porcine insulin in the primate circulation. We can lower exogenous insulin requirements, but we have yet to normalize glucose tolerance in any transplanted primate. Until we do so it is premature to think about applying this technology in humans. Also, there is the issue of safety. We are about 2 ½ years out from our first pig-to-primate transplant.2 That animal is doing well. However, we have a long way to go and many additional transplants to perform before we can be sure that what we are doing is safe for humans.
Dr. Bob Karsh (Professor of Medicine, Washington University School of Medicine): You have told us about organogenesis of the kidney and endocrine pancreas. Is organogenesis applicable to other organs such as the liver?
Dr. Hammerman: In the 1980s two different groups attempted allotransplantation of liver and kidney from E15 rat embryos into adult rats.17,18 The renal primordia engrafted in non-immunosuppressed hosts, but the livers were rejected. Co-transplantation of E15 rat embryo liver and kidney resulted in rejection of both organs. The authors' suggested that E15 rat livers, in contrast to kidneys, contain host-derived APCs that mediate direct antigen presentation resulting in rejection of liver or of liver and kidney transplanted at the same time.18 Hagihara et al. transplanted fragments of E18–19 fetal Wistar rat liver or E90 fetal swine liver into the omentum of non-immunosuppressed Wistar rats to which a lethal dose of D-galactosamine had been administered. The isografts formed nodules of hepatocytes at 72 hours post-transplantation, but the xenografts were necrotic by that time. Both isotransplantation and xenotransplantation improved survival.46
Eventov-Friedman et al. transplanted minced embryonic pig liver in the spleen or beneath the renal capsule of nonobese diabetic (NOD) severe combined immunodeficient (SCID) mice. Both hepatocytes and bile ducts differentiated post-transplantation and porcine albumin was detected in the circulation of host mice. Transplantation of liver primordia obtained from embryos younger than E28 resulted in teratoma formation beneath the renal capsule.43
Dr. Bharath M. Reddy (Instructor in Medicine, Washington University School of Medicine): It looks like you have had more success with pancreas organogenesis than with kidney organogenesis.
Dr. Hammerman: That's one way to interpret our results. However, it's probably too early to judge because it hard to know what the evenutal application for either technology will be. As an example of an alternative way to use the renal organogenesis technology, Yokoo et al. implanted labeled human mesenchymal stem cells (hMSC) into the nephrogenic site of E11.5 rat embryos before the metanephric kidneys had formed. Following 48 hours of whole embryo culture, the metanephric primordia had formed in vitro and contained hMSC. Metanephroi were dissected out and transplanted into the omentum of uninephrectomized rats. Transplants enlarged over two weeks in non-immunosuppressed rat hosts, became vascularized by host vessels and contained hMSC-derived cells that were morphologically identical to resident renal cells. These findings suggest that transplanted renal primordia might be useful as a scaffold to generate a ‘humanized’ kidney.11
Dr. Robert E. Schwartz (Resident in Medicine, Barnes-Jewish Hospital, Washington University School of Medicine): Have you seen any evidence for transmission of porcine endogenous retrovirus (PERV) from embryonic pig to non-human primates? Is this a concern?
Dr. Hammerman: It certainly is a concern. Several years ago Paradis et al. showed that despite persistent microchimerism (donor cells in human recipients of living pig tissue), PERV infection was not detected in human recipients.47 That is good news. Rood and Cooper have pointed out that transplantation of porcine islets into humans is being carried out all over the world. One hopes that mechanisms for informed consent and oversight are in place.48
Dr. Rashmi S. Mullur (Instructor in Medicine, Washington University School of Medicine): How do the pig pancreatic cells get into lymph nodes?
Dr. Hammerman: Good question. I can only speculate. In contrast to islet formation that occurs within stroma following transplantation of rat pancreatic primordia into diabetic rats or mice,38,39 individual endocrine cells engraft in tissues following transplantation of E28 pig pancreatic primordia into the mesentery of diabetic rats39–41 and also following transplantation of E28 pig pancreatic primordia into the mesentery of STZ-diabetic rhesus macaques.2 During normal pancreatic organogenesis, individual endocrine cells first migrate away from primitive ducts prior to coalescing into islets. Migration and coalescence are guided by a number of cell and tissue adhesion molecules. We speculated that the failure of individual pig endocrine cells to coalesce into islets post-transplantation of E28 pig pancreatic primordia into rats, may result from the absence of adhesion molecules in rat interstitium that are recognized by pig endocrine cells.40 Therefore following transplantation of pancreatic primordia from pig-to-rat, the endocrine cells cannot re-aggregate to form islets. In contrast, rat endocrine cells recognize rat or mouse adhesion molecules and islets are formed albeit in the absence of exocrine tissue.38
One explanation for our ability to transplant E28 pig pancreatic primordia without immunosuppression into rats or rhesus macaques may be that the absence of both exocrine tissue and islets results in a pattern of antigen expression that is not recognized as foreign by the host.38 An alternative explanation is host tolerance on the basis of chimerism as proposed by Abraham et al. to explain successful xenoengraftment of human pancreatic islet-derived progenitor cells in multiple tissues of non-immunosuppressed immunocompetent mice.49 Another is ‘T cell paralysis’50 on the basis host exposure to antigen in the form of cells that express class II major histocompatibility complex II (MHC II) in the absence of a co-stimulatory signal. Of course, such explanations would require that our pig beta cells express MHC II or the swine equivalent, swine leukocyte antigen (SLA) II. On first consideration, this might seem a bit far-fetched. However, under some circumstances pig β-cells do express SLA II.51,52
Neural cell adhesion molecule (NCAM) is one of the regulars for endocrine cell aggregation during islet development.53 Crnic et al. showed that loss of NCAM function causes the formation of lymph node metastasis in a transgenic model of pancreatic β-cell carcinogenesis. Metastases were facilitated by upregulated pancreatic lymphangiogenesis possibly induced by the disaggregation of endocrine cells.54 Possibly, the failure of endocrine cells to aggregate following transplantation of E28 pig pancreatic primordia in the mesentery of rats or rhesus macaques induces a comparable lymphagiogenesis that permits migration of beta cells to regional lymph nodes. A state of chimerism results and tolerance is induced on the basis of SLA II expression on beta cells in the absence of co-stimulatory molecules.2 It may be that diabetic patients will be the ultimate beneficiaries of this rather fortuitous series of events that might render hosts tolerant post-transplantation of E28 pig pancreatic primordia.
Dr. Karsh: You have told us about what happens after transplantation of pancreas from E28 embryos. Have you tried transplanting pancreas from older embryos?
Dr. Hammerman: We have transplanted E28,2,40–41 E2939 or E3540 pig pancreatic primordia into non-immunosuppressed immunocompetent rats39–41 or non-human primates.2 Our experience in rats is that E35 primordia are rejected. However, either E2840–41 or E29 primordia39 engraft in rats and E28 work best in terms of regularly normalizing glucose tolerance in STZ-diabetic rats. Given the absence of an immunosuppression requirement for engraftment of E28 pig pancreatic primordia in rats, we went directly to testing their efficacy in diabetic non-human primates.2
Eventov Friedman et al. performed studies in which E21–E100 pig pancreatic primordia were transplanted under the kidney capsule of immunodeficient NOD-SCID mice. They found optimal growth potential as reflected by the highest levels of porcine insulin in circulation from tissue harvested in the E42–E56 period. Differentiation of both endocrine and exocrine tissue was observed post-transplantation beneath the kidney capsule of the NOD-SCID mice.43 Subsequently Eventov-Friedman et al. showed that E42 pig pancreatic primordia transplanted into NOD-SCID mice can restore normoglycemia in diabetic animals. Using assays for both direct and indirect T cell rejection responses to the xenogeneic tissue, they showed that E42 pig pancreatic tissue, in comparison to E56 or later embryonic tissues, exhibits markedly reduced immunogenicity. Fully immunocompetent diabetic mice grafted with the E42 pig pancreatic tissue and treated with an immunosuppression protocol consisting of CTLA4-Ig and anti-CD40 ligand attained normal blood glucose levels following transplantation of E42 primordia, eliminating the need for insulin.45
Dr. Karsh: Was there teratoma formation following transplantation of embryonic pancreas?
Dr. Hammerman: In contrast to what they reported after transplantation of embryonic pig liver obtained prior to E28 in NOD-SCID mice, Eventov-Friedman found no teratoma formation following transplantation of pig pancreas obtained from embryos at any age.43 We have never detected teratomas following transplantation of embryonic rat or pig pancreas. Our transplanted rats have a normal rat life span of about two years and outlive non-transplanted STZ-diabetic animals.
Acknowledgements
Supported by George M. O'Brien Center DK079333 and grant 1-2005-110 from JDRF.
Abbreviations
- APC
antigen presenting cell
- E
embryonic day
- ES
embryonic stem cell
- GFR
glomerular filtration rate
- MHC
major histocompatibility complex
- NOD-SCID
nonobese diabetic severe combined immunodeficient
- PAH
P-aminohippurate
- SLA
swine leukocyte antigen
- STZ
streptozotocin
- UV
urine volume
Note
Edited transcripts of research conferences sponsored by Organogenesis and the Washington University George M. O′Brien Center for Kidney Disease Research (P30 DK079333) are published in Organogenesis. These conferences cover organogenesis in all multicellular organisms including research into tissue engineering, artificial organs and organ substitutes and are participated in by faculty at Washington University School of Medicine, St. Louis Missouri, USA.
Footnotes
Previously published online as an Organogenesis E-publication: http://www.landesbioscience.com/journals/organogenesis/article/5382
References
- 1.Hammerman MR. Windows of opportunity for organogenesis. Transplant Immunology. 2005;5:1–8. doi: 10.1016/j.trim.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 2.Rogers SA, Chen F, Talcott MR, Faulkner C, Thomas JM, Thevis M, Hammerman MR. Long-term engraftment following transplantation of pig pancreatic primordia into non-immunosuppressed diabetic rhesus macaques. Xenotransplantation. 2007;14:591–602. doi: 10.1111/j.1399-3089.2007.00429.x. [DOI] [PubMed] [Google Scholar]
- 3.Hammerman MR. Treatment for end-stage renal disease: An organogenesis/tissue engineering odyssey. Transpl Immunol. 2004;12:211–218. doi: 10.1016/j.trim.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Ibrahim Z, Busch J, Awwad M, Wagner R, Wells K, Cooper DKC. Selected physiologic compatibilities and incompatibilities between human and porcine organ systems. Xenotransplantation. 2006;13:488–499. doi: 10.1111/j.1399-3089.2006.00346.x. [DOI] [PubMed] [Google Scholar]
- 5.Cozzi E, Bhatti F, Schmoekel M, Chavez G, Smith KG, Zaidi A, Bradley JR, Thiru S, Goddard M, Vial C, Ostlie D, Wallwork J, White D, Friend PJ. Long-term survival of nonhuman primates receiving life-supporting transgenic porcine kidney xenografts. Transplantation. 2000;70:15–21. [PubMed] [Google Scholar]
- 6.Yamada K, Yazawa K, Shimizu A, Iwanaga T, Hisashi Y, Nuhn M, O'Malley P, Nobori S, Vagefi PA, Patience C, Fishman J, Cooper DKC, Hawley RJ, Greenstein J, Schuurman HJ, Awwad M, Sykes M, Sachs DH. Marked prolongation of porcine renal xenograft survival in baboons through the use of alpha 1,3 galactosyltransferase donors and the cotransplantation of vascularized thymic tissue. Nat Med. 2005;11:32–34. doi: 10.1038/nm1172. [DOI] [PubMed] [Google Scholar]
- 7.Wolf AS, Palmer SJ, Snow ML, Fine LG. Creation of a functioning mammalian chimeric kidney. Kidney Int. 1990;38:991–997. doi: 10.1038/ki.1990.303. [DOI] [PubMed] [Google Scholar]
- 8.Rogers SA, Lowell JA, Hammerman NA, Hammerman MR. Transplant developing metanephroi into adult rats. Kidney Int. 1998;54:27–37. doi: 10.1046/j.1523-1755.1998.00971.x. [DOI] [PubMed] [Google Scholar]
- 9.Marshall D, Bottomley M, Symonds K, Brenchley PEC, Bravery CA. Transplantation of metanephroi to sites within the abdominal cavity. Transplant Proc. 2005;37:194–197. doi: 10.1016/j.transproceed.2004.12.283. [DOI] [PubMed] [Google Scholar]
- 10.Dekel B, Burakova T, Arditti FD, Reich-Zeliger S, Milstein O, Aviel-Ronen S, Rechavi G, Friedman N, Kaminski N, Passwell JH, Reisner Y. Human and porcine early kidney precursors as a new source for transplantation. Nat Med. 2003;9:53–60. doi: 10.1038/nm812. [DOI] [PubMed] [Google Scholar]
- 11.Yokoo T, Fukui A, Ohashi T, Miyazaki Y, Utsunomiya Y, Kawamura T, Hosoiya T, Okabe M, Kobayashi E. Xenobiotic kidney organogenesis from human mesencymal stem cells using a growing rodent embryo. J Am Soc Nephrol. 2006;17:1026–1034. doi: 10.1681/ASN.2005101043. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong SR, Campbell GR, Campbell JH, Little MH. Establishment of metanephros transplantation in mice highlights contributions by both nephrectomy and pregnancy to developmental progression. Exp Nephrol. 2005;101:155–164. doi: 10.1159/000087939. [DOI] [PubMed] [Google Scholar]
- 13.Marshall D, Dilworth MR, Clancy M, Bravery CA, Ashton N. Increasing renal mass improves survival in anephric rats following metanephros transplantation. Exp Physiol. 2007;92:263–271. doi: 10.1113/expphysiol.2006.036319. [DOI] [PubMed] [Google Scholar]
- 14.Hammerman MR. Transplantation of developing kidneys. Transplantation Reviews. 2002;16:62–71. [Google Scholar]
- 15.Rogers SA, Hammerman MR. Prolongation of life in anephric rats following de novo renal organogenesis. Organogenesis. 2004;1:22–25. doi: 10.4161/org.1.1.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammerman MR. Transplantation of renal primordia: Renal organogenesis. Pediatric Nephrology. 2007;22:1991–1998. doi: 10.1007/s00467-007-0554-7. [DOI] [PubMed] [Google Scholar]
- 17.Foglia RP, LaQuaglia M, Statter MB, Donahoe PK. Fetal allograft survival in immunocompetent recipients is age dependent and organ specific. Ann Surg. 1986;204:402–410. doi: 10.1097/00000658-198610000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Velasco A, Hegre OD. Decreased immunogenicity of fetal kidneys: the role of passenger leukocytes. J Pediatric Surg. 1989;24:59–63. doi: 10.1016/s0022-3468(89)80303-3. [DOI] [PubMed] [Google Scholar]
- 19.Rogers SA, Liapis H, Hammerman MR. Transplantation of metanephroi across the major histocompatibility complex in rats. Am J Physiol. 2001;280:R132–R136. doi: 10.1152/ajpregu.2001.280.1.R132. [DOI] [PubMed] [Google Scholar]
- 20.Statter M, Fahrner KJ, Barksdale EM, Parks DE, Flavell RA, Donahoe PK. Correlation of fetal kidney and testis congenic graft survival with reduced major histocompatibility complex burden. Transplantation. 1989;47:651–660. doi: 10.1097/00007890-198904000-00017. [DOI] [PubMed] [Google Scholar]
- 21.Dekel B, Marcus H, Herzel BH, Bocher WO, Passwell JH, Reisner Y. In vivo modulation of the allogeneic immune response by human fetal kidneys: The role of cytokines, chemokines, and cytolytic effector molecules. Transplantation. 2000;69:1470–1478. doi: 10.1097/00007890-200004150-00044. [DOI] [PubMed] [Google Scholar]
- 22.Dekel B, Amariglio F, Kaminski N, Schwartz A, Goshen E, Arditti FD, Tsarfaty I, Passwell JH, Reisner R, Rechavi G. Engraftment and differentiation of human renal anlagen into functional mature nephrons after transplantation into mice is accompanied by a profile of gene expression similar to normal human kidney. J Am Soc Nephrol. 2002;13:977–990. doi: 10.1681/ASN.V134977. [DOI] [PubMed] [Google Scholar]
- 23.Dekel B, Burakova T, Ben-Hur H, Hadar M, Oren R, Laufer J, Reisner Y. Engraftment of human kidney tissue in rat radiation chimera: II Human fetal kidneys display reduced immunogenicity to adoptively transferred human peripheral blood mononuclear cells and exhibit rapid growth and development. Transplantation. 1997;64:1550–1558. doi: 10.1097/00007890-199712150-00008. [DOI] [PubMed] [Google Scholar]
- 24.Rogers SA, Hammerman MR. Transplantation of rat metanephroi into mice. Am J Physiol. 2001;280:R1865–R1869. doi: 10.1152/ajpregu.2001.280.6.R1865. [DOI] [PubMed] [Google Scholar]
- 25.Takeda S, Rogers SA, Hammerman MR. Differential origin for endothelial and mesangial cells after transplantation of pig fetal renal primordia into rat. Transpl Immunol. 2006;15:211–215. doi: 10.1016/j.trim.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Rogers SA, Talcott M, Hammerman MR. Transplantation of pig renal anlagen. ASAIO J. 2003;49:48–52. doi: 10.1097/00002480-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Rogers SA, Liapis H, Hammerman MR. Normalization of glucose post-transplantation of pig pancreatic anlagen into non-immunosuppressed diabetic rats depends on obtaining anlagen prior to embryonic day 35. Transpl Immunol. 2005;14:67–75. doi: 10.1016/j.trim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Sorenson CM, Rogers SA, Korsmeyer SJ, Hammerman MR. Fulminant metanephric apoptosis and abnormal kidney development in bcl-2-deficient mice. Am J Physiol. 1995;268:F73–F81. doi: 10.1152/ajprenal.1995.268.1.F73. [DOI] [PubMed] [Google Scholar]
- 29.Hammerman MR. Regulation of cell survival during renal development. Pediatr Nephrol. 1998;12:596–602. doi: 10.1007/s004670050513. [DOI] [PubMed] [Google Scholar]
- 30.Bottino R, Trucco M. Multifaceted therapeutic approaches for a multigenic disease. Diabetes. 2005;54:S79–S86. doi: 10.2337/diabetes.54.suppl_2.s79. [DOI] [PubMed] [Google Scholar]
- 31.Danovitch GM, Cohen DJ, Weir MR, Stock RG, Bennett WM, Christiansen LL, Sung RS. Current status of kidney and pancreas transplantation in the United States 1994–2003. Am J Transplant. 2005;5:904–915. doi: 10.1111/j.1600-6135.2005.00835.x. [DOI] [PubMed] [Google Scholar]
- 32.Hering B, Wijkstrom M, Graham M, Hardstedt M, Aasheim TC, Jie T, Ansite JD, Nakano M, Cheng J, Li W, Moran K, Christians U, Finnegan C, Mills CD, Sutherland DE, Bansal-Pakala P, Murtaugh MP, Kirchhof N, Schuurman HJ. Prolonged diabetes reversal after intraportal xenotransplantation of wild-type porcine islets in immunosuppressed nonhuman primates. Nat Med. 2006;12:301–303. doi: 10.1038/nm1369. [DOI] [PubMed] [Google Scholar]
- 33.Cardona K, Korbutt GS, Milas Z, Lyon J, Cano J, Jiang W, Bello-Lahorn H, Hacquoil B, Strobert E, Gangappa S, Weber CJ, Pearson TC, Rajotte RV, Larsen CP. Long-term survival of neonatal porcine islets in non-human primates by targeting costimulation pathways. Nat Med. 2006;12:304–306. doi: 10.1038/nm1375. [DOI] [PubMed] [Google Scholar]
- 34.Groth CG, Korsgren O, Tibell A, Tollemar J, Moller E, Bolinder J, Ostman J, Reinholt FR, Hellerstrom C, Andersson A. Transplantation of porcine fetal pancreas to diabetic patients. J Lancet. 1994;344:1402–1404. doi: 10.1016/s0140-6736(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 35.Brown J, Molnar IG, Clark W, Mullen Y. Control of experimental diabetes mellitus in rats by transplantation of fetal pancreases. Science. 1974;184:1377–1379. doi: 10.1126/science.184.4144.1377. [DOI] [PubMed] [Google Scholar]
- 36.Hegre OD, Leonard RJ, Erlandsen SL, McEvoy RC, Parsons RP, Lazarow A. Transplantation of islet tissue in the rat. Acta Endocrinol. 1976;205:257–278. [PubMed] [Google Scholar]
- 37.Brown J, Mullen YS, Clark WR, Molnar G, Heinsinger D. Importance of hepatic portal circulation for insulin action in streptozotocin-diabetic rats transplanted with fetal pancreases. J Clin Invest. 1979;64:1688–1694. doi: 10.1172/JCI109631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogers SA, Liapis H, Hammerman MR. Intraperitoneal transplantation of pancreatic anlagen. ASAIO J. 2003;49:527–532. doi: 10.1097/01.mat.0000084174.33319.7f. [DOI] [PubMed] [Google Scholar]
- 39.Rogers SA, Chen F, Talcott M, Hammerman MR. Islet cell engraftment and control of diabetes in rats following transplantation of pig pancreatic anlagen. Am J Physiol. 2004;286:E502–E509. doi: 10.1152/ajpendo.00445.2003. [DOI] [PubMed] [Google Scholar]
- 40.Rogers SA, Liapis H, Hammerman MR. Normalization of glucose post-transplantation of pig pancreatic anlagen into non-immunosuppressed diabetic rats depends on obtaining anlagen prior to embryonic day 35. Transpl Immunol. 2005;14:67–75. doi: 10.1016/j.trim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Rogers SA, Chen F, Talcott M, Liapis H, Hammerman MR. Glucose tolerance normalization following transplantation of pig pancreatic primordia into non-immunosuppressed diabetic ZDF rats. Transpl Immunol. 2006;16:176–184. doi: 10.1016/j.trim.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Eloy R, Haffen K, Kedinger M, Grenier JF. Chick embryo pancreatic transplants reverse experimental diabetes of rats. J Clin Invest. 1979;64:361–373. doi: 10.1172/JCI109470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eventov-Friedman S, Katchman H, Shezen E, Arnivich A, Tchorsh D, Dekel B, Freud E, Reisner Y. Embryonic pig liver, pancreas and lung as a source for transplantation: Optimal organogenesis without teratoma depends on distinct time windows. Proc Natl Acad Sci. 2005;102:2928–2933. doi: 10.1073/pnas.0500177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown J, Heninger D, Kuret J, Mullen YS. Islet cells grow after transplantation of fetal pancreas and control of diabetes. Diabetes. 1981;30:9–13. doi: 10.2337/diab.30.1.9. [DOI] [PubMed] [Google Scholar]
- 45.Eventov-Friedman S, Tchorsh D, Katchman H, Shezan E, Arnovich A, Hecht G, Dekel B, Rechavi G, Blazar B, Feine I, Tal O, Freud E, Reisner Y. Embryonic pig pancreatic tissue transplantation for the treatment of diabetes. PLoS Med. 2006;7:1165–1177. doi: 10.1371/journal.pmed.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hagihara M, Shimura T, Takebe K, Munkhbat B, Tsuji K. Effects of iso and xeno fetal liver fragments transplantation on acute and chronic liver failure in rats. Cell Transplant. 1994;3:283–290. doi: 10.1177/096368979400300404. [DOI] [PubMed] [Google Scholar]
- 47.Paradis K, Langford G, Long Z, Hemline W, Sandstorm P, Switzer WM, Chapman LE, Lackey C, Onions D. The XEN 111 study group, Otto E, Search for cross-species transmission of porcine endogenous retrovirus in patients treated with living pig tissue. Science. 1999;285:1236–1241. doi: 10.1126/science.285.5431.1236. [DOI] [PubMed] [Google Scholar]
- 48.Rood PPM, Cooper DKC. Islet xenotransplantation: Are we really ready for clinical trials? Am J Transplantation. 2006;6:1269–1274. doi: 10.1111/j.1600-6143.2006.01336.x. [DOI] [PubMed] [Google Scholar]
- 49.Abraham EJ, Kodama S, Lin JC, Ubeda M, Faustman DL, Habener JF. Human pancreatic islet-derived progenitor cell engraftment in immunocompetent mice. Am J Pathology. 2004;164:817–830. doi: 10.1016/S0002-9440(10)63170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Markmann J, Lo D, Naji A, Palmiter RD, Brinster D, Heber-Katz E. Antigen presenting function of class II MHC expressing pancreatric beta cells. Nature. 1988;336:476–479. doi: 10.1038/336476a0. [DOI] [PubMed] [Google Scholar]
- 50.Burkley LC, Lo D, Flavell RA. Tolerance in transgenic mice expressing major histocompatibility molecules extrathymically on pancreatic cells. Science. 1990;248:1364–1368. doi: 10.1126/science.1694042. [DOI] [PubMed] [Google Scholar]
- 51.Murray AG, Nelson RC, Rayat GR, Elliott JF, Korbutt GS. Neonatal porcine islet cells induce human CD4+ but not CD 8+ lymphocyte proliferation and resist cell-mediated cytolytic injury in vitro. Diabetes. 1999;48:1713–1719. doi: 10.2337/diabetes.48.9.1713. [DOI] [PubMed] [Google Scholar]
- 52.Edamura K, Nasu K, Nishimura R, Ogawa H, Sasaki N, Ohgawara H. Effect of long-term culture on the expression of antigens and adhesion molecule in single porcine pancreatic endocrine cells. Xenotransplantation. 2005;12:327–332. doi: 10.1111/j.1399-3089.2005.00232.x. [DOI] [PubMed] [Google Scholar]
- 53.Cirulli V, Bettens D, Rutihauser U, Halban PA, Orci L, Roullier DG. Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet type segregation. J Cell Science. 1994;107:1429–1436. doi: 10.1242/jcs.107.6.1429. [DOI] [PubMed] [Google Scholar]
- 54.Crnic I, Strittmatter K, Cavalerro U, Kopfstein L, Jussila L, Alitaloi K, Christorori G. Loss of neural cell adhesion molecule induces tumor metastasis by upregulating lymphangiogenesis. Cancer Res. 2004;64:8630–8638. doi: 10.1158/0008-5472.CAN-04-2523. [DOI] [PubMed] [Google Scholar]