

Abstract
Iron accumulates in the brain and contributes to brain injury after intracerebral hemorrhage (ICH). The c-Jun-N-terminal kinase (JNK) signaling pathway mediates cell death after ischemic stroke, however, the involvement of JNK in ICH is not well known. This study investigated whether the JNK signaling pathway is activated by iron after ICH. Male Sprague-Dawley rats received an infusion of autologous whole blood (as a model of ICH) or ferrous iron into the right basal ganglia and control rats had an infusion of saline. Some ICH rats were treated with either deferoxamine (DFX), an iron chelator, or vehicle. Activation of JNK was measured by Western blot analysis and immunohistochemistry. Free iron in cerebrospinal fluid (CSF) and behavioral outcomes following ICH were also examined. We found that activated-JNK in the brain were increased after ICH, and an intracerebral infusion of ferrous iron also upregulated brain activated-JNK. Free iron accumulated in CSF and Systemic administration of DFX after ICH reduces free iron contents in CSF, suppresses JNK activation and improves ICH-induced neurological deficits. Our results demonstrated that the JNK signaling pathway is activated after ICH and iron may contribute to this activation. DFX reduces free iron levels and attenuates activation of JNK suggesting iron chelation may be useful therapy for ICH patients.
Keywords: cerebral hemorrhage, c-Jun-N-terminal kinase, deferoxamine, iron
1. Intoduction
c-Jun N-terminal kinase (JNK), a member of the mitogen-activated protein kinase (MAPK) superfamily, has already been widely investigated for its active actions in response to various stimuli, such as heat shock, inhibition of protein glycosylation, exposure to inflammatory cytokines, ultraviolet irradiation, and ischemia (Kyriakis et al., 2001). Activated JNK phosphorylates serine on a variety of cellular targets and, consequently, influences cell survival via transcriptional and posttranslational regulation of proteins involved in apoptosis (Manning et al., 2003). A growing number of studies have demonstrated that JNK could be activated by various forms of brain insults, such as cerebral ischemia and subarachnoid hemorrhage, and inhibited JNK activation could attenuate brain injury (Okuno et al., 2004; Gao et al., 2005; Yatsushige et al., 2005). Previous study also illuminated activation of JNK in another form of stroke, intracerebral hemorrhage (ICH), and ICH-induced neuron loss could be prevented by JNK inhibitor (Ohnishi et al., 2007). However, the involvement of JNK in ICH is still far less understood.
One potential stress that might induce JNK activation after ICH is iron. Experimental studies have demonstrated that iron overload occurs after ICH and contributes to ICH-induced brain injury (Wu et al., 2003; Hua et al., 2006; Xi et al., 2006). The major sources of iron accumulation in the brain are iron in the plasma and hemoglobin after erythrocyte lysis (Xi et al., 1998; Wu et al.,, 2003; Nakamura et al., 2005). Free iron can cause free radical formation and oxidative brain damage. The time course of free iron accumulation after ICH is unknown but we have found that deferoxamine (DFX), an iron chelator, can attenuate acute brain edema and oxidative stress following ICH (Nakamura et al., 2004).
The present study investigated whether JNK is activated in the brain after ICH and whether intracerebral injection of iron can activate JNK. The effects of DFX on JNK activation, free iron in the CSF and behavioral outcomes following ICH were also examined.
2. Materials and methods
2.1. Animal preparation and intracerebral infusion
The University of Michigan Committee on the Use and Care of Animals approved the animal protocols. A total of 147 male Sprague-Dawley rats (weighing 300–400 g; Charles River Laboratories) were used. Rats were anesthetized with pentobarbital (50 mg/kg, i.p.). The right femoral artery was catheterized for blood pressure monitoring and blood sampling. Blood was obtained from the catheter for analysis of pH, PaO2, PaCO2, hematocrit and glucose and as the source for the intracerebral blood infusion. Body temperature was maintained at 37.5 °C using a feedback-controlled heating pad. The animals were positioned in a stereotactic frame (Model 500, Kopf Instruments, Tujunga, CA, USA) and a cranial burr hole (1 mm) was drilled in the right coronal suture 4.0 mm lateral to the midline. Either 100μL autologous blood (as a model of ICH), 30μL ferrous chloride (1 mM or 10 mM, 1mM ferrous chloride is similar to the dilution of iron in the blood), or 100μL saline (as a model of control) were infused into the right basal ganglia through a 26-gauge needle at a rate of 10 μL per minute using a microinfusion pump (Harvard Apparatus Inc., Holliston, MA, USA). The coordinates were 0.2 mm anterior and 3.5 mm lateral to the bregma and a depth of 5.5 mm. After intracerebral infusion, the needle was removed, the burr hole was filled with bone wax, and the skin incision was closed with suture.
2.2. Experimental Groups
Rats were divided into four sets. In the first set, rats (n=3 each group) had either an intracerebral infusion of 100μL saline (control) or an infusion of 100 μL autologous blood (ICH rats) and were killed at 1, 3 and 7 days later for Western blot analysis. In the second set, rats (n=3 each group) had either an intracerebral infusion of 100μL saline (control), an infusion of 30 μL ferrous chloride (1 mM or 10 mM) or an infusion of 100 μL autologous blood (ICH rats), and then the ICH rats received either DFX treatment (100 mg/kg, i.p., 2 hours after infusion of autologous blood) or the same amount of saline (vehicle). Rat brains were sampled one day after intracerebral injection for Western blot analysis and immunohistochemistry. In the third set, rats (n=6 each group) had either an intracerebral infusion of 100μL saline (control) or an infusion of 100 μL autologous blood (ICH rats), and then the ICH rats were treated with either DFX treatment (100 mg/kg, i.p., 2 hours after infusion of autologous blood and then at 12- hour intervals for up to 7 days) or the same amount of saline (vehicle). CSF was obtained 1, 3, 7, 14 and 28 days after ICH for measurement of free iron. In the fourth set, rats (n=6 each group) had either an intracerebral infusion of 100μL saline (control) or an infusion of 100 μL autologous blood (ICH rats), and then the ICH rats were treated with either DFX treatment (100 mg/kg, i.p., 2 hours after infusion of autologous blood and then at 12- hour intervals for up to 7 days) or the same amount of saline (vehicle). All animals underwent behavioral testing and were then killed 28 days after infusion of autologous blood.
2.3. Western Blot Analysis
Rats were anesthetized and underwent intracardiac perfusion with 0.1 mol/L phosphate-buffered saline (pH 7.4). The brains were removed and a 3-mm-thick coronal brain slice was cut approximately 4mm from the frontal pole. The slice was separated into ipsilateral and contralateral basal ganglia. Western blot analysis was performed as previously described (Xi et al., 1999). Protein concentration was determined using a Bio-Rad Laboratories (Hercules, CA, USA) protein assay kit. A 50 μg portion of protein from each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a hybond-C pure nitrocellulose membrane (Amersham, Piscataway, NJ, USA). The membranes were blocked in Carnation nonfat milk and probed with primary and secondary antibodies. The primary antibody was rabbit anti-phosphorylated-JNK antibody (Cell Signaling Technology, Beverly, MA, USA; 1:1000). The secondary antibody was goat anti-rabbit IgG (Bio-Rad Laboratories, 1:200). The antigen–antibody complexes were visualized with a chemiluminescence system (Amersham) and exposed to a Kodak X-OMAT film (Rochester, NY, USA). Relative densities of bands were analyzed with the NIH Image program (Version 1.62, Bethesda, MD, USA).
2.4. Immunohistochemistry
The Immunohistochemistry method has been described previously (Hua et al., 2002). Briefly, the rats were anesthetized and subjected to intracardiac perfusion with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4). The brains were removed and kept in 4% paraformaldehyde for 12 h, then immersed in 25% sucrose for 3 to 4 days at 4°C. Brains were then placed in embedding compound and sectioned on a cryostat (18 μm thick). Immunohistochemistry staining was then performed using the avidin–biotin complex technique. The primary antibody was rabbit anti-phosphorylated-JNK antibody (Cell Signaling Technology, 1:200). The secondary antibody was biotinylated goat anti-rabbit IgG (Bio-Rad Laboratories, 1:400). Either normal rabbit IgG or the absence of primary antibody was used as negative controls.
2.5. Free iron determination
Rats were reanesthetized with pentobarbital. About 200μl–250μl cerebrospinal fluid was obtained by puncture of the cisterna magna 1, 3, 7, 14, and 28 days after ICH and stored at −80°C before determination. Free iron in CSF was measured according to the method described by Nilsson (Nilsson et al., 2002)
2.6. Behavioral Tests
All animals were tested before and after surgery and scored by investigators who were blind to both neurological and treatment conditions. Three behavioral assessments were used: corner turn, forelimb placing, and forelimb use asymmetry tests (Hua et al., 2002).
(A) Corner Turn Test
The rat was allowed to proceed into a corner, the angle of which was 30 degrees. To exit the corner, the rat could turn either to the left or the right. The direction was recorded. The test was repeated 10 to 15 times, with at least 30 seconds between each trial, and the percentage of right turns calculated. Only turns involving full rearing along either wall were included. The rats were not picked up immediately following each turn so that they did not develop an aversion for turning around. Previous study showed that the ICH rats showed a significant increase in the percentage of right (ipsilateral to the site of intracerebral infusion) turns compared with controls (Hua et al., 2002).
(B) Forelimb Placing Test
Forelimb placing was scored using a vibrissae-elicited forelimb-placing test. Independent testing of each forelimb was induced by brushing the vibrissae ipsilateral to that forelimb on the edge of a tabletop once per trial for 10 trials. Intact animals placed the forelimb quickly onto the countertop. Percent successful placing responses were determined. A previous study showed that successful responses in the forelimb contralateral to the site of injection after ICH are reduced (Hua et al., 2002).
(C) Forelimb Use Asymmetry Test
Forelimb use during explorative activity was analyzed by videotaping rats in a transparent cylinder for 3–10 min depending on the degree of activity during the trial. Behavior was quantified first by determining the occasions when the non-impaired ipsilateral (I) forelimb was used as a percentage of total number of limb use observations on the cylinder wall. Second, the occasions when the impaired forelimb contralateral (C) to the blood-injection site was used as a percentage of total number of limb use observations on the wall. Third, the occasions when both (B) forelimbs were used simultaneously as a percentage of total number of limb use observations on the wall. A single overall limb use asymmetry score was calculated as: Limb use asymmetry score = (I/(I+C+B)) − (C/(I+C+B)).
2.7. Statistical analysis
All data in this study are presented as mean ± SD. Data were analyzed with Student’s t-test, Mann-Whitney U test and analysis of variance (ANOVA), followed by Scheffe’s post hoc test. Significance levels were measured at p<0.05.
3. Results
All physiological variables including mean arterial blood pressure, blood pH, PaO2, PaCO2, hematocrit, and blood glucose level were within normal ranges (mean arterial blood pressure, 80 to 120 mmHg; blood pH, 7.40 to 7.50; PaO2, 80 to 120 mmHg; PaCO2, 35 to 45 mmHg; hematocrit, 35% to 45%; blood glucose level, 80 to 130 mg/dl).
3.1. JNK was activated in the ipsilateral basal ganglia after infusion of autologous blood
There was marked JNK activation in the ipsilateral basal ganglia after infusion of autologous blood as shown by Western blot analysis. Thus, one day after infusion of autologous blood, the 46 kDa and the 54 kDa forms of JNK increased significantly compared with control (46 kDa: 5282 ± 318 vs. 434 ± 247 pixels; 54 kDa: 2971 ± 816 vs. 112 ± 101 pixels; p<0.001). The time course of JNK activation is shown in Figure 1. Activated JNK increased markedly one day after infusion of autologous blood and remained at high levels at least for 7 days (Figure 1). Intracerebral infusion of ferrous iron also activated JNK at 24 hours (Figure 2). Activated-JNK positive cells were detected by immunohistochemistry in the ipsilateral basal ganglia after ferrous iron (Figures 3A and 3B) or autologous blood infusion (Figure 3C), while the immunoreactivity of phosphorylated-JNK was weak in the control models (Figures 3D).
Figure 1.

Time course of the levels of phosphorylated JNK in the ipsilateral basal ganglia after intracerebral infusion of autologous blood. (A) Western blots showing phosphorylated-JNK at 1 day (lanes 1–3), 3 days (lanes 4–6) and 7 days after intracerebral infusion of autologous blood. Equal amounts of protein (50 μg) were used for each sample. (B) Quantification of Western blot analysis. Values are expressed as means ± SD; there are three rats in each group. # p< 0.05 and * p<0.01 vs. day 7.
Figure 2.

The phosphorylated-JNK levels in the ipsilateral basal ganglia 24 hours after intracerebral infusion of ferrous iron. (A) Western blots showing phosphorylated-JNK levels after intracerebral infusion of 10 mM ferrous iron (lanes 1–3), 1 mM ferrous iron (lanes 4–6) and control operation (lanes 7–9). Equal amounts of protein (50 μg) were used for each sample. (B) Quantification of Western blot analysis. Values are expressed as means ± SD. # p <0.05 and * p<0.01 compared with the control group.
Figure 3.
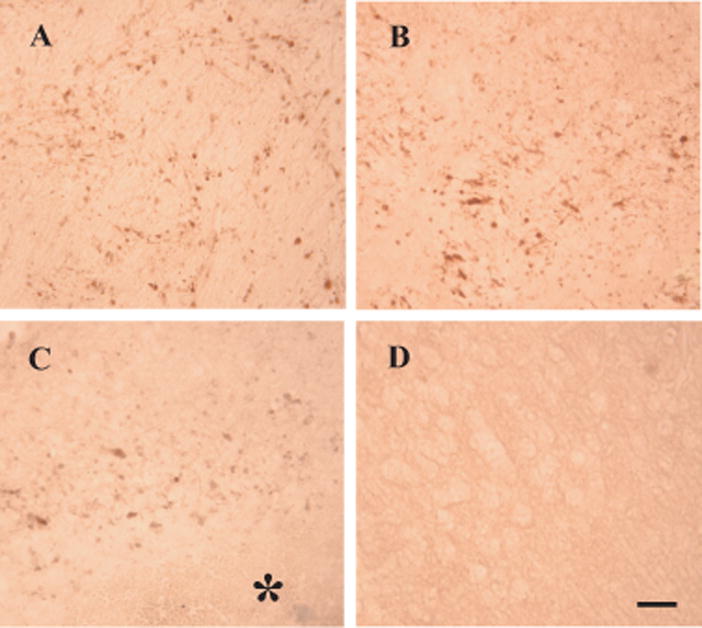
Phosphorylated-JNK immunoreactivity in the basal ganglia at 24 hours after intracerebral infusion. Phosphorylated-JNK immunoreactivity was detected in the ipsilateral basal ganglia after intracerebral infusion of 10 mM ferrous iron (A), 1 mM ferrous iron (B). Phosphorylated-JNK immunoreactivity was also detected in the ipsilateral basal ganglia after intracerebral infusion of autologous blood (C) and control operation (D). A sterisk indicates the hematoma. Scale bar = 50 μm.
3.2. DFX suppressed the upregulation of activated-JNK after infusion of autologous blood
DFX suppressed the upregulation of activated-JNK in the ipsilateral basal ganglia 24 hours after infusion of autologous blood. Western blotting showed that activated JNK levels are lower in the DFX-treated group compared with those in the vehicle-treated group (Figure 4).
Figure 4.

The effect of deferoxamine on the levels of phosphorylated JNK 1 day after intracerebral infusion of autologous blood. (A) Western blots showing phosphorylated-JNK levels in the ipsilateral basal ganglia after intracerebral infusion of autologous blood treated with vehicle (ICH+Vehicle, lanes 1–3), DFX (ICH+DFX, lanes 4–6) and Control group (lanes 7–9). Equal amounts of protein (50 μg) were used for each sample. (B) Quantification of Western blot analysis. Values are expressed as means ± SD. # p <0.05 vs. ICH+Vehicle group and * p<0.01 vs. Control group.
3.3. Free iron accumulated in CSF after infusion of autologous blood and deferoxamine reduces free iron contents in CSF
The time course of free iron levels in CSF after infusion of autologous blood is shown in Figure 5. Free iron levels in the CSF were very low in the control rat (1.1 ± 0.4 μmol/L). After infusion of autologous blood, free iron levels in CSF were increased at the first day (8.5 ± 1.3 μmol/L), peaked at the third day (14.2 ± 5.0 μmol/L) and still remained at high levels for at least 28 days (6.2 ± 1.1 μmol/L). DFX treatment reduced free iron levels in CSF at all time points (Figure 5).
Figure 5.

Bar graph showing the time course of free iron levels in CSF after intracerebral infusion of autologous blood and the effect of deferoxamine treatment on the free iron levels in CSF after intracerebral infusion of autologous blood. Values are expressed as means ± SD. * p<0.01 between ICH+Vehicle group and the Control group, # p <0.05 between DFX-treated group and the vehicle-treated group.
3.4. DFX treatment ameliorates neurological deficits after infusion of autologous blood
The neurological deficits induced by intracerebral infusion of autologous blood were also reduced by DFX treatment. Corner turn scores were improved at all time points in the DFX-treated group compared with the vehicle-treated group (e.g., day 7: 69.6 ± 20.0% versus 83.9 ± 16.8%, p < 0.05). Forelimb-placing scores were also improved in the DFX-treated group compared with the vehicle-treated group (e.g., day 3: 73.3 ± 30.5% versus 46.7 ± 33.6%, p < 0.05). There was also an improvement in forelimb-use asymmetry deficits associated with DFX therapy (e.g., day 1: 26.8 ± 17.2% versus 47.2 ± 17.4%, p < 0.01) (Figure 6).
Figure 6.

Bar graphs showing the effects of deferoxamine on neurological deficits following intracerebral infusion of autologous blood. Corner turn test (A), forelimb placing test (B) and forelimb using asymmetry test (C) were examined after ICH. Values are expressed as means ± SD. * p<0.05 and ** p<0.01 between ICH+Vehicle group and Control group; # p <0.05 and ## p<0.01 between ICH+Vehicle group and ICH+DFX group.
4. Discussion
The present study found that: 1) JNK is activated in the brain after infusion of autologous blood. 2) An intracerebral infusion of ferrous iron activates JNK. 3) Free iron accumulated in CSF and systemic administration of DFX after intracerebral infusion of autologous blood reduces free iron contents in CSF, suppresses JNK activation and improves neurological deficits induced by intracerebral infusion of autologous blood.
Increasing evidence suggests that JNK signaling pathway is activated by stress and cytokines, and play important roles in mediating cell death and differentiation (Martin et al., 1996). Activated JNK phosphorylates the substrates including nuclear substrates such as c-Jun and cytosol substrates including Bcl-2 then leading to neuronal death, and blocking the JNK signaling pathway inhibits neuronal cell death and attenuates brain injury after brain ischemia/reperfusion(Maroney et al., 1998; Okuno et al., 2004; Gao et al., 2005; Yatsushige et al., 2005). Previous study also demonstrated activation of JNK in another form of stroke, ICH, and ICH-induced neuron loss could also be prevented by JNK inhibitor (Ohnishi et al., 2007). However, the involvement of JNK in ICH is still not well understood. In our current study, JNK was activated in the first day after intracerebral infusion of autologous blood and remained at high levels for seven days. Our previous and current results have shown that iron accumulates in the brain after infusion of autologous blood. Therefore, we examined whether intracerebral infusion of ferrous iron activates JNK. We found that there was a marked increase in JNK activation after iron infusion. DFX could partially block JNK activation indicating that iron does play a role in JNK activation after intracerebral hemorrhage. However, the block was incomplete suggesting that other factors also result in JNK activation.
It is well-known that thrombin formation and iron are two major factors causing brain injury after intracerebral hemorrhage (Xi et al., 2003; Xi et al., 2006; Hua et al., 2007). The role of thrombin in JNK activation after intracerebral hemorrhage needs further studies, although thrombin can activate JNK (Waetzig et al., 2005). In addition, iron and thrombin can interact to cause brain damage (Nakamura et al., 2005). Nakamura et al. (2005) found that intracerebral co-administration of holo-transferrin (holo-Tf), an iron-loaded transferrin, with thrombin causes brain edema, oxidative damage and DNA fragmentation. Thus, the presence of holo-Tf and thrombin during the formation of an intracerebral hematoma may participate in ICH-induced brain injury and JNK activation.
The term “free iron” refers to iron bound in such a way that it retains its ability to catalyse the formation of reactive oxygen species, and it is therefore a highly relevant parameter to measure during periods of oxidative stress (Otterbein et al., 2003). The sources of increased free iron after intracerebral hemorrhage are either the clot itself or circulating blood in the presence of a disrupted blood-brain barrier. Before erythrocyte lysis and hemoglobin breakdown, free iron may come mainly from serum of the clot or through a disrupted blood-brain barrier or both. After erythrocyte lysis, hemoglobin released from erythrocytes results in increased extracellular heme; heme is then transported into neurons and glia and metabolized by hemeoxygenase generating free iron (Wagner et al., 2003). Previous studies have shown iron-positive cells (Perls’ staining), that appear to be neurons, located around the clot within 24 hours after intracerebral infusion of autologous blood and DFX reduces acute brain edema when given 6 hours after intracerebral infusion of autologous blood (Wu et al., 2003; Nakamura et al., 2004).
Oxidative brain injury and apoptotic cell death occurs in brain after intracerebral infusion of autologous blood (Nakamura et al., 2005; Xi et al., 2006). Iron-induced brain damage may result from oxidative stress (Wu et al., 2002) and free iron can stimulate the formation of free radicals leading to neuronal damage. Although the role of JNK in apoptosis following intracerebral hemorrhage is not well known, it has been reported that inhibition of JNK activation reduces apoptotic neuronal cell death after intracerebral hemorrhage (Ohnishi et al., 2007).
Systemic administration of DFX reduced neurological deficits induced by intracerebral infusion of autologous blood. Earlier we have reported that systemic use of DFX attenuates brain edema in rats after intracerebral infusion of autologous whole blood or hemoglobin (Huang et al., 2002; Nakamura et al., 2004). These results suggest that iron chelation with DFX may be useful therapy for patients with ICH. DFX is a drug approved by the FDA for the treatment of acute iron intoxication and for chronic iron overload due to transfusion-dependent anemias. DFX can penetrate the blood-brain barrier rapidly and reach a high concentration in the brain after systemic administration (Palmer et al., 1994). DFX chelates iron by forming a stable complex that prevents iron from entering into further chemical reactions. In addition, DFX reduces hemoglobin-induced brain Na+/K+ ATPase inhibition and neuronal toxicity (Sadrzadeh et al., 1987; Regan et al., 1993; Guo et al., 2001). Favorable effects of iron chelator therapy have also been reported in various cerebral ischemia models (Hurn et al., 1995; Liachenko et al., 2003). Furthermore, DFX can serve as a direct free radical scavenger (Hurn et al., 1995; Liachenko et al., 2003) and can induce delayed tolerance to cerebral ischemia (Prass et al., 2002).
In summary, JNK activation occurs in the brain after intracerebral infusion of autologous blood and ferrous iron. Systemic administration of DFX after infusion of autologous blood reduces free iron levels in CSF, suppresses activation of JNK and improves functional outcome. The mechanisms of iron-induced JNK activation and its role in ICH-induced brain injury need to be investigated further. These results suggest that iron chelation with DFX may be useful therapy for intracerebral hemorrhage.
Acknowledgments
This study was supported by grants NS-017760, NS-039866, NS-047245 and NS-052510 from the National Institutes of Health (NIH) and 0755717Z from American Heart Association (AHA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH and AHA.
Abbreviations
- ATPase
adenosine triphosphatase
- CSF
cerebrospinal fluid
- DFX
deferoxamine
- ECL
enhanced chemiluminescence
- FDA
food and drug administration
- holo-Tf
holo-transferrin
- ICH
intracerebral hemorrhage
- IgG
immunoglobulin G
- i.p
intraperitoneal injection
- JNK
c-Jun-N-terminal kinase
- kDa
kilodalton
- PaCO2
partial pressure of carbon dioxide in artery
- PaO2
partial pressure of oxygen in artery
- pH
power of hydrogen
- PBS
phosphate-buffered saline
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
References
- Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, Luo Y, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- Guo Y, Regan RF. Delayed therapy of hemoglobin neurotoxicity. Academic Emergency Medicine. 2001;8:510. [Google Scholar]
- Hua Y, Keep R, Hoff J, Xi G. Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke. 2007;38:759–762. doi: 10.1161/01.STR.0000247868.97078.10. [DOI] [PubMed] [Google Scholar]
- Hua Y, Nakamura T, Keep R, Wu J, Schallert T, Hoff J, Xi G. Long-term effects of experimental intracerebral hemorrhage: the role of iron. J Neurosurg. 2006;104:305–312. doi: 10.3171/jns.2006.104.2.305. [DOI] [PubMed] [Google Scholar]
- Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- Hua Y, Xi G, Keep RF, Wu J, Jiang Y, Hoff JT. Plasminogen activator inhibitor-1 induction after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. 2002;22:55–61. doi: 10.1097/00004647-200201000-00007. [DOI] [PubMed] [Google Scholar]
- Huang F, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Koehler RC, Blizzard KK, Traystman RJ. Deferoxamine reduces early metabolic failure associated with severe cerebral ischemic acidosis in dogs. Stroke. 1995;26:688–694. doi: 10.1161/01.str.26.4.688. discussion 694–695. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Liachenko S, Tang P, Xu Y. Deferoxamine improves early postresuscitation reperfusion after prolonged cardiac arrest in rats. Journal of Cerebral Blood Flow & Metabolism. 2003;23:574–581. doi: 10.1097/01.WCB.0000057742.00152.3F. [DOI] [PubMed] [Google Scholar]
- Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Jr, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JH, Mohit AA, Miller CA. Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Brain Res Mol Brain Res. 1996;35:47–57. doi: 10.1016/0169-328x(95)00181-q. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Keep R, Hua Y, Schallert T, Hoff J, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100:672–678. doi: 10.3171/jns.2004.100.4.0672. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G. Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res. 2005;1039:30–36. doi: 10.1016/j.brainres.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Xi G, Park JW, Hua Y, Hoff JT, Keep RF. Holo-transferrin and thrombin can interact to cause brain damage. Stroke. 2005;36:348–352. doi: 10.1161/01.STR.0000153044.60858.1b. [DOI] [PubMed] [Google Scholar]
- Nilsson UA, Bassen M, Savman K, Kjellmer I. A simple and rapid method for the determination of “free” iron in biological fluids. Free Radic Res. 2002;36:677–684. doi: 10.1080/10715760290029128. [DOI] [PubMed] [Google Scholar]
- Ohnishi M, Katsuki H, Fujimoto S, Takagi M, Kume T, Akaike A. Involvement of thrombin and mitogen-activated protein kinase pathways in hemorrhagic brain injury. Exp Neurol. 2007;206:43–52. doi: 10.1016/j.expneurol.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- Palmer C, Roberts RL, Bero C. Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke. 1994;25:1039–1045. doi: 10.1161/01.str.25.5.1039. [DOI] [PubMed] [Google Scholar]
- Prass K, Ruscher K, Karsch M, Isaev N, Megow D, Priller J, Scharff A, Dirnagl U, Meisel A. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. Journal of Cerebral Blood Flow & Metabolism. 2002;22:520–525. doi: 10.1097/00004647-200205000-00003. [DOI] [PubMed] [Google Scholar]
- Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- Sadrzadeh SM, Anderson DK, Panter SS, Hallaway PE, Eaton JW. Hemoglobin potentiates central nervous system damage. J Clin Invest. 1987;79:662–664. doi: 10.1172/JCI112865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, Goetz M, Lucius R, Herdegen T, Hanisch UK. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia. 2005;50:235–246. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: Role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Research. 2002;953:45–52. doi: 10.1016/s0006-8993(02)03268-7. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep R, Hoff J. Mechanisms of brain injury after intracerebral hemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hua Y, Xiang JM, Hoff JT. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke. 1999;30:1247–1255. doi: 10.1161/01.str.30.6.1247. [DOI] [PubMed] [Google Scholar]
- Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? Journal of Neurochemistry. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]
- Yatsushige H, Yamaguchi M, Zhou C, Calvert JW, Zhang JH. Role of c-Jun N-terminal kinase in cerebral vasospasm after experimental subarachnoid hemorrhage. Stroke. 2005;36:1538–1543. doi: 10.1161/01.STR.0000170713.22011.c8. [DOI] [PubMed] [Google Scholar]