

Abstract
Recent work has identified three distinct classes of viral membrane fusion proteins based on structural criteria. In addition, there are at least four distinct mechanisms by which viral fusion proteins can be triggered to undergo fusion-inducing conformational changes. Viral fusion proteins also contain different types of fusion peptides and vary in their reliance on accessory proteins. These differing features combine to yield a rich diversity of fusion proteins. Yet despite this staggering diversity, all characterized viral fusion proteins convert from a fusion-competent state (dimers or trimers, depending on the class) to a membrane-embedded homotrimeric prehairpin, and then to a trimer-of-hairpins that brings the fusion peptide, attached to the target membrane, and the transmembrane domain, attached to the viral membrane, into close proximity thereby facilitating the union of viral and target membranes. During these conformational conversions, the fusion proteins induce membranes to progress through stages of close apposition, hemifusion, and then the formation of small, and finally large, fusion pores. Clearly, highly divergent proteins have converged on the same overall strategy to mediate fusion, an essential step in the life cycle of every enveloped virus.
Keywords: trimer-of-hairpins, hemifusion, fusion pore, Class I fusion proteins, Class II fusion proteins, Class III fusion proteins
INTRODUCTION
Virus-cell fusion is the means by which all enveloped viruses, including devastating human pathogens such as human immunodeficiency virus (HIV) and Ebola virus, enter cells and initiate disease-causing cycles of replication. In all cases virus-cell fusion is executed by one or more viral surface glycoproteins, including one that is generally denoted as the fusion protein. Certain viral fusion proteins induce cell-cell fusion when expressed on the cell surface as a consequence of infection, and cell-cell fusion can contribute to viral spread, viruluence, persistence, and other untoward consequences (Corcoran et al., 2006; Cousens et al., 2007; Duelli and Lazebnik, 2007; Garg et al., 2007; Plattet et al., 2007). Because viral fusion proteins can mediate both virus-cell fusion leading to infection and pathological cell-cell fusion, they are increasingly viewed as targets for antiviral intervention.
Recent years have witnessed major breakthroughs in our understanding of the proteins and protein complexes that mediate fusion between enveloped viruses and their host cells. In particular, a wealth of information indicates that all characterized viral fusion proteins mediate membrane fusion by forming a common conformer—a trimer-of-hairpins—and by coercing membranes through a common pathway of membrane dynamics. Yet, remarkably, the fusion proteins themselves are quite diverse in sequence and structure and are activated by diverse triggering mechanisms. In this review we will highlight how diverse viral fusion proteins mediate a common pathway of membrane fusion. For other recent reviews on this topic, see (Earp et al., 2005; Harrison, 2005; Kielian and Rey, 2006; Weissenhorn et al., 2007).
The Unifying Trimer-of-Hairpins Fusion Mechanism
Membrane fusion requires bringing two separate membrane bilayers into intimate contact and then merging them into one (Figure 1A). A key intervening step is hemifusion, during which small regions of the outer contacting monolayers merge, while the inner monolayers remain intact (Figure 1Av). Resolution of the hemifusion intermediate results in merger of the inner monolayers and the creation of a small fusion pore, which can expand (Figure 1Avi). This pathway of membrane apposition to hemifusion to fusion pore formation and enlargement is also employed during cellular fusion reactions, both intracellular fusion events and cell-cell fusion reactions (Chernomordik and Kozlov, 2005; Chernomordik et al., 2006; Jahn and Scheller, 2006; Podbilewicz et al., 2006).
FIG. 1.

The common trimer-of-hairpins pathway of membrane fusion. (A) The model depicts a Class I fusion protein, but related structures (e.g., prehairpins and trimers-of-hairpins) form for Class II and III proteins, which also promote membrane merger through stages of close apposition (iv), hemifusion (v), small fusion pores (not shown), and large fusion pores (vi). See Table 2 and text for comparisons among the different classes of viral fusion proteins. The depicted Class I fusion protein is one that does not require any other viral surface proteins for fusion (e.g., influenza HA or a retroviral Env); it contains both a receptor binding subunit (labeled rb in image i) and a fusion subunit (labeled f in images i to iii). The target and viral membranes are, respectively, at the top and bottom of the images. The receptor binding subunit (rb) is not shown beyond image i as its location at the later stages is not known; in all cases studied, however, the rb subunit of this type of class I fusion protein must move out of the way, thus unclamping the fusion subunit in the metastable fusion competent state and allowing fusion to proceed. For Class I fusion proteins six helix bundles (6HBs) are seen in their bundle (v) and trimer-of-hairpins (vi) forms; the length and position of the 6HB varies for different proteins. The starting (i) and final (vi) images represent structures that are known for several viral fusion proteins; high level structural information is currently lacking on the intermediates. (B) The key features of a class I fusion protein from N- to C-terminus: a fusion peptide (FP) at or near the N-terminus, an N-heptad repeat (N-HR aka HR1 or HRA), a C-heptad repeat (C-HR aka HR2 or HRB), a transmembrane domain (TMD), and a cytoplasmic tail (squiggle). Linkers of variable lengths are indicated as straight lines. (The // between the N- and C-heptad repeats indicates that the length of these linkers varies considerably). Peptide analogs of the N-HR and C-HR helices can inhibit fusion and infection.
Virus-cell fusion is mediated by one or more surface glycoproteins of the mature virus envelope. Fusion can occur at the cell surface at neutral pH or within an endosomal compartment at low pH. For some viruses (e.g., retroviruses) only a single viral surface glycoprotein is required, whereas for others (e.g., paramyxoviruses and herpesviruses) additional viral proteins are required (Table 1). The fusion protein exists on the mature viral surface in a ‘native’ fusion-competent state, which is most often, but not always, metastable. Following triggering, the protein converts to a prehairpin intermediate, which embeds in the target membrane (Figure 1Aii) through an apolar region termed the fusion peptide. In all cases studied, the membrane-embedded prehairpin intermediate is a homotrimer of the fusion subunit, irrespective of whether the ectodomain of the native fusion protein is a dimer or a trimer (Tables 1 and 2). The next stages of fusion are generally envisioned to involve recruitment of several membrane-embedded trimeric prehairpins to the fusion site (Figure 1Aiii) (but see Yang et al., 2005), followed by a dramatic sequence of fold-back steps, during which the fusion subunit converts to a compact rod-like trimer-of-hairpins (Figure 1Aiv-vi), which is generally its most energetically stable conformation. Conversion to the final state is therefore thought to help overcome the large energy barrier to membrane merger (Chernomordik and Kozlov, 2005; Cohen and Melikyan, 2004).
TABLE 1.
Fusion proteins from different families of enveloped viruses
| Family | Proteins Needed | Fusion Protein fusion subunit | Class | Fusion pH | Fusion Peptide Location |
|---|---|---|---|---|---|
| Orthomyxoviridae | HA | HA1-S-S-HA2 | I | Low | N-terminal |
| Retroviridae | Env | SU-S-S-TM, SU/TM(a) | I | Neutral (Low)(b) | N-terminal (most) Internal (ASLV) |
| Paramyxoviridae | F, HN(c) | F2-S-S-F1 | I | Neutral(b) | N-terminal |
| Coronaviridae | S | S1/S2 | I | Neutral (Low)(b) | Internal |
| Filoviridae | GP | GP1-S-S-GP2 | I | Low(d) | Internal |
| Arenaviridae | GP, SSP | GP1/GP2/SSP | I | Low | (N-terminal)(e) |
| Togaviridae | E1/E2 | E1/E2 | II | Low | Internal |
| Flaviviridae | E(TBEV), E1/E2 (HCV) | E, E1/E2(f) | II | Low | Internal |
| Bunyaviridae | GN/GC | GN/GC(g) | II(g) | Low | Internal(g) |
| Rhabdoviridae | G | G | III | Low | Internal (bipartite) |
| Herpesviridae | gB, gD, gH/L | gB(h), gH/gL | III | Neutral (Low)(b) | Internal (bipartite) |
| Poxviridae | 8 proteins(i) | nd | NC | Neutral (Low)(b) | nd |
| Hepadnaviridae | S, L(j) | S(j) | NC | Low(j) | (N-terminal)(j) |
Information is given for 13 of the 17 families of enveloped viruses and is updated from Table 1 of (Earp et al., 2005). Viruses in parentheses represent examples. NC, not classified. See text for more details.
SU/TM, S1/S2, etc. denote that the indicated subunits are associated, but not disulfide bonded.
Some family members fuse at neutral pH, while others require low pH, in some cases in addition to receptor binding. In the case of herpesviruses, cell type differences have been seen. A need for low pH for fusion of some paramyxoviruses with cells is under investigation (see text).
Paramyxovirus receptor binding (attachment) proteins are denoted HN, H, or G depending on the virus.
Ebola virus requires low endosomal pH for entry but the only confirmed low pH requirement is for the activity of endosomal cathepsins.
Two regions of the Lassa Fever virus GP2, one at and one very close to its N-terminus, have been implicated in fusion (Klewitz et al., 2007), but more work is needed to clarify the locations of arenavirus fusion peptides.
For HCV, several regions in E1 and E2 have been implicated (Lavillette et al., 2007; Poumbourios and Drummer, 2007).
These entries are based only on a predictive analysis (Garry and Garry, 2004).
gB is central to fusion and is a Class III fusion protein, but the gH/gL complex also participates.
Vaccinia virus employs a complex of eight (Wagenaar and Moss, 2007), and perhaps additional (Kochan et al., 2007) proteins.
These are tentative assignments based in part on observations suggesting a need for proteolytic processing within S during virus entry (Glebe and Urban, 2007; Li et al., 2004; Maenz et al., 2007).
TABLE 2.
Properties of Class I, Class II, and III fusion proteins
| Property | Class I | Class II | Class III |
|---|---|---|---|
| Examples | Influenza HA, Paramyxovirus F | TBEV E, SFV E1/E2 | VSV G, HSV-1 gB |
| Type of integral membrane protein | Type I | Type I(a) | Type I |
| Requires proteolytic processing to generate fusion competent form | Yes(b) (of fusion protein) | Yes (ofaccessory protein(c)) | No(d) |
| Metastable on virion | Yes | Yes | VSV G, No; HSV-1 gB, Not known |
| Orientation with respect to viral membrane | Perpendicular (project as a spike) | Parallel (close to viral membrane) | VSV G, Perpendicular; HSV-1 gB, Not known(e) |
| Major secondary structure (of native fusion subunit) | α−helix | β-sheet | α-helix and β-sheet |
| Oligomeric structure of native fusion protein | Trimer | Dimer | VSV G, Trimer; HSV-1 gB, Not known(e),(f) |
| Location of fusion peptide in native fusion protein | Buried in subunit interface | Masked in trimer interface, at tip of extended β-strands | VSV G, Exposed, at tips of extended xtended β-strands; HSV-1 gB, Not known |
| Location of fusion peptide in primary sequence | At or near N-terminus | Internal | Internal (bipartite)(g) |
| Activated to fusogenic form by | Lo Low pH, receptor(s), or receptor followed by low pH(h) | Low pH | VSV G, Low pH; HSV-1 gB, Receptors Receptors(i) |
| Oligomeric structure of fusion-active form (membrane-embedded prehairpin and bundles) | Trimer | Trimer | Trimer(j) |
| Structure of the post-fusion form | Trimer-of-hairpins (central α-helical coiled-coil, 6HB) | Trimer-of-hairpins (mainly β-structure) | Trimer-of-hairpins (central α-helical coiled-coil and significant β-structure)(k) |
This table was updated from Table 2 of (Earp et al., 2005). See text and Table 1 for more details and additional references.
The fusion subunits of all flaviviruses except HCV have two membrane spanning domains near its C-terminii.
Proteolytic processing into two subunits is required by many class I fusion proteins (e.g., influenza HA, paramyxovirus F). For others (e.g., Ebola virus GP) processing into the two subunits occurs for the wt protein, but is not essential for infection (Neumann et al., 2007; Wool-Lewis and Bates, 1999). Some coronavirus S precursors are (e.g., MHV, Qiu et al., 2006), whereas others (e.g., SARS S) are not, proteolytically processed during biosynthesis. For these latter coronaviruses S proteins as well as for Ebola virus GP and Hendra and Nipah virus F, post synthetic cleavage by extracellular or intracellular (e.g., endosomal cathepsins) proteases may substitute (Chandran et al., 2005; Follis et al., 2006; Matsuyama et al., 2005; Pager et al., 2006; Pager and Dutch, 2005; Schornberg et al., 2006; Simmons et al., 2005).
p62 (precursor to E2) in the case of SFV; prM in the case of TBEV.
Neither VSV G nor HSV-1 gB are proteolytically processed. Bovine herpesvirus gB and human cytomegalovirus gBs are processed, but processing is not needed for cell entry (Kopp et al., 1994; Strive et al., 2002).
The pre-fusion form of gB is thought to be a trimer that projects from the virion surface.
The recently determined crystal structure of HSV-1 gB (Heldwein et al., 2006) is thought to represent its post-fusion form (Heldwein et al., 2006; Lin and Spear, 2007). The structure of the (presumed) pre-fusion trimer is not yet known.
The fusion peptides of VSV G and HSV-1 gB are comprised of two loops found at the tips of two neighboring β-strands.
Ebola GP has been suggested to use a fourth fusion trigger, a cathepsin dependent activity (Chandran et al., 2005; Kaletsky et al., 2007; Schornberg et al., 2006).
Some strains of herpesviruses require (additional) exposure to low pH in some cell types (Delboy et al., 2006).
The post-fusion forms of VSV G and HSV-1 gB are trimers. It is thought that their respective membrane embedded prehairpins are trimers.
The post-fusion form of VSV G contains a small 6HB; the apparently post fusion form of HSV-1 gB does not.
Conversion from the membrane embedded homotrimeric prehairpin to the trimer-of-hairpins may occur in steps, for example by sequential packing of the three C-terminal regions against the central N-terminal trimeric core. Formation of the fully zipped trimer-of-hairpins ectodomain may be followed by complex formation between the fusion peptide and the transmembrane domain (Armstrong et al., 2000; Tamm, 2003). During these fold-back steps, the membranes would be brought into increasingly intimate contact and could progress through (restricted) hemifusion followed by the opening of small (labile) fusion pores (not shown in Figure 1A) and finally large (robust) fusion pores that allow passage of the viral nucleocapsid into the cytoplasm to initiate replication. The exact conformers (degrees of trimer-of-hairpins formation) that bring about close membrane apposition, hemifusion, and then small and large fusion pore formation will likely vary for different fusion proteins (Melikyan et al., 2004; Melikyan et al., 2000) depending on the exact architecture of their membrane proximal ends.
High-resolution structures are available for (portions of) the trimer-of-hairpins of many viral fusion proteins and for the pre-fusion forms of several of them (Earp et al., 2005; Harrison, 2005; Kielian and Rey, 2006; Lamb and Jardetzky, 2007; Weissenhorn et al., 2007). What is remarkable is that even though the fusion proteins are structurally quite diverse, all of their fusion subunits ultimately fold back into a trimer-of-hairpins, in which three C-terminal regions pack on the outside of a central N-terminal trimeric core. The common final trimer-of-hairpins is formed irrespective of whether the fusion subunit has a prominent central α-helical coiled-coil (Class I viral fusion proteins), whether it consists largely of β-structure (Class II viral fusion proteins), or whether it displays a combination of α-helical and β-structure (Class III viral fusion proteins). When the fold-back is complete the fusion peptide and the transmembrane anchor are brought together, thereby pulling the two attached membranes (target cell and viral, respectively) together and facilitating their merger (Figure 1A).
The final common trimer-of-hairpins conformation is a viable target for therapeutic intervention. This was first shown with T20, a peptide containing part of the second (more C-terminal) heptad repeat (C-HR, a.k.a HR2) of the fusion subunit (gp41) of the HIV Env glycoprotein (Figure 1B). T20 (a.k.a Fuzeon and Enfuvirtide) was the first antifusion antiviral approved for clinical use (Kilby et al., 1998). Similar peptides inhibit fusion and infection by an array of viruses harboring Class I fusion proteins (Netter et al., 2004 and references therein), and recent studies indicate that similar strategies apply to Class II fusion proteins (Chin et al., 2007; Hrobowski et al., 2005; Liao and Kielian, 2005). For more information on anti-fusion antivirals, see (Este and Telenti, 2007; Fenouillet et al., 2007; Frey et al., 2006; Liu et al., 2007; Munch et al., 2007; Pohlmann and Reeves, 2006; Welch et al., 2007).
Despite the existence of many common features among viral fusion proteins (common pathway of membrane dynamics, common prehairpin and trimer-of-hairpins conformations), viral fusion proteins differ in several important respects (Tables 1 and 2). They vary in terms of their detailed structures, how they are triggered, and the number of different viral surface proteins involved. These and other differences combine to yield a rich diversity of fusion proteins (Figure 2). Apparently diverse proteins have converged on a common mechanism (Figure 1A) to merge lipid bilayers (Chernomordik and Kozlov, 2005; Cohen and Melikyan, 2004; Teissier and Pecheur, 2007).
FIG. 2.

Diversity of viral fusion proteins. The major differences among viral fusion proteins are their structural class (left) and mode of fusion triggering (right). Representative fusion proteins of different classes that employ different fusion triggers are: (A) influenza HA, (B) paramyxovirus F and most retroviral Env proteins, (C) α-retroviral Env proteins, (D) Ebola GP, (E) the E and E1 proteins, respectively, of TBEV and SFV, (F) VSV G, and (G) HSV gB. Some herpesviruses require low pH (likely in addition to receptor binding) for fusion in certain cell types (see text). Additional differences among viral fusion proteins include the locations and types of their fusion peptides and whether they require additional viral surface proteins (e.g., separate receptor binding proteins) for fusion.
DIVERSITY OF FUSION TRIGGERING MECHANISMS
Fusion proteins sit on the virion surface in a ‘native’ fusion-competent state, in which the fusion subunit is often referred to as being ‘clamped.’ Generation of the fusion-competent state is often achieved by a prior priming event, which in the studied cases is achieved through proteolytic cleavage of either the fusion protein precursor or an accessory protein (Table 2). For fusion to occur, the fusion-competent protein must be activated by a fusion trigger, which we define as the environmental stimulus (or stimuli) that converts the native fusion-competent protein (Figure 1Ai) to a membrane-embedded prehairpin (Figure 1Aii and iii) and then to its final trimer-of-hairpins conformation (Figure 1Avi). We therefore define fusion activation as all steps involved in converting the fusion protein from its fusion-competent to its final trimer-of-hairpins form (i.e., all steps shown in Figure 1A).
As indicated in Figure 2, there are at least four types of fusion triggers: low pH, receptor binding, a combination of receptor binding followed by low pH, and a novel, still incompletely characterized mechanism employed by filoviruses. Specific Class I proteins can be activated by each of the known triggers, while all characterized Class II proteins are activated by low pH. Among the known Class III proteins, one is activated by low pH, whereas the other requires interactions of a companion protein with specific host cell receptor(s) (and in some cases low pH as well). It was formerly thought that the fusion proteins of a given virus family use a common fusion trigger (e.g., receptor binding or low pH). It now seems apparent, however, that specific members of a family can use different triggers. For example, whereas many paramyxo-, herpes-, retro-, and some coronaviruses are activated by interactions with host cell receptors, others have been reported to require low pH (Chu et al., 2006; Delboy et al., 2006; Eifart et al., 2007; Freed and Mouland, 2006; Mothes et al., 2000; Ross et al., 2002; Schowalter et al., 2006; Seth et al., 2003).
We will now elaborate on each of the four types of fusion triggers. More detailed information on specific fusion triggers will be presented as we discuss fusion mechanisms for selected Class I, II, and III viral fusion proteins whose structures are known in both their pre- and post-fusion conformations (see below).
Low pH
Low pH is the sole known fusion trigger for orthomyxo-, rhabdo-, alpha-, flavi-, bunya-, and arenaviruses. These viruses enter cells by endocytosis (Marsh and Helenius, 2006; Sieczkarski and Whittaker, 2005) and fuse with early (e.g., SFV) or late (e.g., influenza virus) endosomes depending on the pH that elicits key conformational changes. The mechanisms by which three structurally well characterized fusion proteins, influenza HA (Class I), TBEV E (Class II), and VSV G (Class III) are activated by low pH will be described in detail below. In all cases low pH results in structural rearrangements that first allow repositioning of the fusion peptide so that it can bury into the target membrane (Figure 1Aii). For low pH activated Class I fusion proteins, this involves separation of the globular head domains that clamp the fusion subunit in its pre-fusion state (Godley et al., 1992; Kemble et al., 1992; Rachakonda et al., 2007). However, the degree to which the head domains must separate (White and Wilson, 1987) to allow formation of the prehairpin (Figure 1Aii), and whether further separation is required to form the trimer-of-hairpins (Figure 1Avi), is not yet clear. Many residues must be protonated to activate this group of fusion proteins (Rachakonda et al., 2007; Skehel and Wiley, 2000). These protonation events likely affect salt bridges and other local environmental features of the fusion protein, but in most cases we only have limited information on the full spectrum of residues that must be protonated. In many cases histidine residues, which can function as pH sensors, have been implicated (Chanel-Vos and Kielian, 2004; Kampmann et al., 2006; Skehel and Wiley, 2000; Stevens et al., 2004; Thoennes et al., 2007; Zavorotinskaya et al., 2004).
Attachment to host cell receptors is clearly a prerequisite for entry of all viruses into appropriate cells. For viruses whose fusion proteins are activated solely by low pH, however, binding to host cell receptors does not play an active role in fusion per se. In fact, if receptor binding is too tight, it may impede fusion (Ohuchi et al., 2002). Furthermore, low pH activated viruses (e.g., influenza, TBEV and VSV) can fuse in vitro with simple artificial liposomes composed solely of phospholipids and cholesterol. Receptor moieties can influence the overall rate of fusion (Niles and Cohen, 1993), but they are not absolutely required. Similarly, many solely low pH activated fusion proteins (e.g., influenza HA and VSV G) do not require any other viral proteins to elicit fusion (reviewed in Earp et al., 2005). In contrast, the G proteins of arenaviruses require their stable signal peptides (SSP) (Table I) (Saunders et al., 2007; Schrempf et al., 2007; York and Nunberg, 2006), and a recent study has provided evidence that the M protein can modulate the pH dependence of a flavivirus E-mediated fusion reaction (Maier et al., 2007).
Binding of Host Cell Receptors
The fusion proteins of most paramyxo-, retro-, and herpesviruses, as well as some coronaviruses, are activated by interactions with host cell receptors at neutral pH (Bossart and Broder, in press; Earp et al., 2005; Freed and Mouland, 2006; Gallo et al., 2003; Matsuyama and Taguchi, 2002; McClure et al., 1990; Zelus et al., 2003); other members of these families require (additional) exposure to low pH for virus-cell fusion (Chu et al., 2006; Delboy et al., 2006; Eifart et al., 2007; Ross et al., 2002). As described below and in Figure 3 there are, however, several different ways in which a host cell receptor(s) can activate a viral fusion protein.
FIG. 3.

Receptor activation of viral fusion proteins. Many viral fusion proteins are activated by host cell receptors, but there are many variations on how this happens (see text). Selected examples are cartooned: (A) Moloney MLV Env, (B) a paramyxovirus F according to the “association” model, (C) a paramyxovirus F according to the “dissociation” model. (D) A highly speculative model for HSV. Specific protein names are indicated under the respective proteins. The models show only the first step of fusion activation, formation of the prehairpin intermediate; subsequent steps (right arrow) lead in all cases to formation of a trimer-of-hairpins. Designations are: R, receptor; rb, receptor binding subunit; f, fusion subunit. The up arrows associated with the known fusion subunits (f) in the right hand images of all panels indicate the exposed fusion peptides, which insert into the target membrane. In (A, right panel) the receptor binding subunit is shown loosely attached (dotted line) to the fusion subunit (f). In a subsequent step, the rb dissociates (see text and Figure 4). In (D) gB is shown in a possible prehairpin conformation as is gH/gL (large upward arrow) consistent with the participation of both gB and gH/gL in fusion, but their exact roles at all stages of fusion remain to be clarified. Receptors are not shown in the right hand images, as it is not yet clear whether they remain bound at this and/or later stages.
Binding of a Single Host Cell Receptor
For some viruses an interaction with a single host cell receptor is sufficient to trigger fusion (Figures 3 and 4). This is the case for many retroviruses (Freed and Mouland, 2006; Gallo et al., 2003; McClure et al., 1990) and coronaviruses (Beniac et al., 2007; Howard et al., 2006; Matsuyama and Taguchi, 2002; Zelus et al., 2003), which all employ a single viral protein (Env or S, respectively) for fusion (Table 1). For fusion proteins that function independently of other viral proteins (Figures 3A and 4), the host cell receptor (R) can directly activate the fusion protein by engaging the receptor binding domain located within the receptor binding subunit (rb) of the fusion protein/fusion subunit(f). For paramyxo- (Figures 3B,C) and herpesviruses (Figure 3D), a single receptor indirectly activates the fusion protein by binding to a separate receptor binding glycoprotein, which relays the information of receptor binding to the fusion protein. We will now briefly describe a few examples of each scenario.
FIG. 4.

Model for receptor activation of the Moloney MLV Env protein. The figure is modeled after Figure 7D in (Wallin et al., 2004), but modified based on new information (see text). In the first image (i) the fusion subunit is depicted as being largely hidden. Following receptor (R) binding, a conformational change exposes the CXXC motif in the receptor binding subunit (rb) allowing the fusion subunit to form the prehairpin intermediate (image ii). A subsequent internal thiol disulfide exchange reaction (image iii) leads to dissociation of the receptor binding subunit, allowing the fusion subunit to fold back into a trimer-of-hairpins. At some point, the receptor binding subunit (rb) likely disengages from its receptor, but it is not yet clear when this happens. Similar models may apply to some, but not all, other retroviral Env proteins (see text).
Receptor Binding to the Receptor Binding Subunit of a Fusion Protein: The Case of Moloney Murine Leukemia Virus
The γ retrovirus Moloney murine leukemia virus (MoMLV) employs a single host cell receptor, a multi-membrane spanning cationic amino acid transporter, to enter host cells. A single viral protein called Env mediates binding and fusion. Like all retroviral Envs, MoMLV Env is composed of a receptor binding subunit (rb in Figures 3A and 4; SU in retrovirus nomenclature) associated with a fusion subunit (f in Figure 3A; TM in retrovirus nomenclature), that has all of the key features of a Class I fusion protein (Tables 1 and 2 and Figure 1B). For MoMLV Env the two subunits are covalently associated through a single disulfide bond. Available findings suggest the following model for fusion activation (Figure 4). Receptor binding to the N-terminal portion of the receptor binding subunit causes conformational changes that are transmitted through a proline rich hinge region (Lavillette et al., 1998) to the C-terminal region. The C-terminal region (of the receptor binding subunit) contains a CXXC motif found in disulfide exchange enzymes such as protein disulfide isomerase. One of the cysteines in the CXXC motif in the receptor binding subunit is linked to a cysteine in a CX6CC motif (Pinter et al., 1997) in the fusion subunit (Figure 4). Upon receptor binding, Ca++ is released (Wallin et al., 2004), the CXXC motif is exposed (Wallin et al., 2006; Wallin et al., 2005a, 2005b), and then executes a previously proposed (Opstelten et al., 1998; Pinter et al., 1997; Sanders, 2000) disulfide bond exchange reaction (Figure 4iii) that severs the covalent interaction between the receptor binding and the fusion subunit (Figure 4iv) (Li et al., 2007; Wallin et al., 2004; Wallin et al., 2006; Wallin et al., 2005; Wallin et al., 2005). The receptor binding subunit can then dissociate (Figure 4, image iv), allowing the fusion subunit to undergo a critical chain reversal reaction (Maerz et al., 2000; Maerz et al., 2001) and to fold from its prehairpin to its final trimer-of-hairpins conformation containing a six-helix bundle (Wallin et al., 2006). Recent work suggests that a cleavage event in the receptor binding domain may potentiate this process (Kumar et al., 2007).
The Env glycoproteins of most β (e.g., MPMV), all γ-(e.g., MoMLV), and all δ retroviruses (e.g., HTLV) contain the internal thiol exchange motif, CXXC, near the C-terminal end of their receptor binding subunits, and a CX6CC motif in their fusion subunits (Li et al., 2007; Sanders, 2000). Hence similar scenarios of receptor induced conformational changes in the receptor binding subunit followed by CXXC mediated thiol exchange leading to loss of the intersubunit disulfide bond, and consequent separation of the receptor binding subunit from the fusion subunit (Figure 4), may be a mechanism used by some other retroviruses. Lentiviruses including HIV (See the section on Binding to Multiple Receptors) and SIV lack the CXXC motif, as do α-retroviruses such as avian sarcoma/leukosis virus (ASLV; see the section on Avian Retroviruses), and so these latter retroviruses likely use other mechanisms to induce the requisite conformational changes in their fusion subunits. Disulfide bond reduction, facilitated by cellular reductases, has been proposed to play roles in the entry mechanisms of HIV and several other enveloped viruses (Fenouillet et al., 2007), but details of these processes are less clear.
Receptor Binding to an Independent Viral Receptor Binding Protein
For the majority of paramyxoviruses and for herpesviruses, an envelope glycoprotein spike that is separate from the fusion protein mediates binding to host cell receptors (Table 1). For these viruses, a current general model posits that engagement of the host cell receptor leads to conformational changes in the receptor binding protein that affect its interaction with the fusion protein (a complex of proteins in the case of herpesviruses), thereby initiating a cascade of conformational changes that activates the fusion protein (Bossart and Broder, in press; Lamb, 1993; Lamb and Jardetzky, 2007; Russell and Luque, 2006; Spear et al., 2006). Related models may apply to poxviruses, which employ even more proteins in their fusion complexes (Table 1) (Wagenaar and Moss, 2007). For most paramyxoviruses and herpesviruses, fusion occurs at the cell surface at neutral pH. Recently, however, some exceptions to this generality have been revealed (discussed below).
Paramyxoviruses
In the case of paramyxoviruses, host cell receptors bind to a viral receptor binding glycoprotein (also referred to as an attachment protein) and this information is somehow relayed to the fusion protein, which is then triggered to undergo dramatic conformational changes (Connolly et al., 2006; Yin et al., 2005; Yin et al., 2006) (detailed in the section on paramyxovirus F), which facilitate the membrane fusion process. The receptor binding proteins (termed H, HN, or G, depending on the virus) are homotetramers. The fusion proteins (F) are homotrimers that are proteolytically processed from a precursor, F0, to a fusion-competent metastable form in which F2, a small subunit, is disulfide bonded to F1, the larger subunit which houses the key elements of a Class I fusion protein: a fusion peptide, two heptad repeats (with a long intervening linker region), and a transmembrane domain (Figure 1B). With few exceptions, efficient fusion requires co-expression of the receptor binding and fusion proteins from the same paramyxovirus.
Presently there are two favored models for how paramyxovirus fusion proteins are activated (Bossart and Broder, in press, Morrison, 2003). In the first (Figure 3B), the receptor binding tetramer is not (tightly) associated with the fusion protein trimer on the virion surface. Following binding of the receptor, the receptor binding protein associates with the fusion protein inducing it to convert to its prehairpin and then its trimer-of-hairpins conformation (containing a prominent six-helix bundle). In the alternate model (Figure 3C), the receptor binding and fusion glycoproteins are associated on the virion, and receptor binding causes them to dissociate, thus unclamping the fusion protein. Substantial evidence indicates that the receptor binding and fusion subunits do physically associate (Bossart and Broder, in press). Support for the dissociation model (Figure 3C) has come from mutant analyses showing that if the interaction between the receptor binding and fusion proteins is too tight, fusion is impeded; this was shown for Nipah virus and for measles virus (Aguilar et al., 2006, 2007; Corey and Iorio, 2007; Plemper et al., 2002). However, mutations in the NDV HN protein have been found that inhibit fusion and weaken its association with F (Melanson and Iorio, 2004). It is clear, therefore, that the exact manner in which the interactions between paramyxovirus receptor binding and fusion proteins change (strengthen and/or weaken) at different stages of membrane fusion remains to be determined. Given the evidence that receptor binding subunits move away from the fusion subunits of influenza HA and MoMLV Env (Godley et al., 1992; Kemble et al., 1992; Wallin et al., 2004), it seems likely that at some stage of fusion the paramyxovirus receptor binding proteins must dissociate from their fusion protein partners. It should be recognized, however, that details of the fusion activation process might vary for different paramyxovirus species. This may be especially true for paramyxoviruses that use protein (e.g., measles, Hendra and Nipah) vs. carbohydrate (e.g., NDV, hPIV3, and PIV5) receptors, and for respiratory syncytial virus and metapneumonia viruses, which can replicate, albeit at reduced efficiency, in the absence of their receptor binding proteins (Bossart and Broder, in press).
Irrespective of which model (Figure 3B or 3C) or combination of models applies, there remain at least two key questions regarding how the consequences of receptor binding are relayed to the fusion protein. The first involves the nature of the fusion-promoting conformational changes in the receptor binding protein. Arguments for and against conformational changes in the head domains of the HN proteins of hPIV3, PIV5, and NDV, which all bind sialic acid containing carbohydrate structures, have been presented (Crennell et al., 2000; Lawrence et al., 2004; Porotto et al., 2007; Yuan et al., 2005; Zaitsev et al., 2004). Nonetheless, there appears to be a general consensus that some conformational change must occur in the HN tetramer (e.g., in the stalk) for fusion activation to proceed (Bossart and Broder, in press, Russell and Luque, 2006; Yuan et al., 2005). The second key question is what specific parts of the receptor binding protein interact with what specific parts of the fusion protein to elicit fusion (Lee et al., 2008). Many studies suggest that residues in the stalk domain of the receptor binding protein are important for (strain-specific) interactions with the F protein, and that residues in the stalk and head domains of the receptor binding protein are involved in fusion activation (Bossart and Broder, in press). Further, several specific regions of the F protein have been implicated in the fusion triggering process. These include small conserved regions within the N-heptad repeat (N-HR, a.k.a. HRA) and within the large (∼240 amino acid) linker between the N- and C-heptad repeats, residues near the fusion peptide including a conserved region in F2, and residues between the globular head and the stalk domain of the fusion-competent (pre-fusion) trimer (Gardner and Dutch, 2007; Gardner et al., 2007; Lamb and Jardetzky, 2007; Luque and Russell, 2007; Russell and Luque, 2006; Russell et al., 2003). As mentioned above, mechanistic details of fusion activation may vary for paramyxoviruses that employ protein vs. sialic acid-containing carbohydrate receptors. Certain paramyxoviruses have been reported to require low pH for virus-cell fusion (Seth et al., 2003; but see Bissonnette et al., 2006) and cell-cell fusion (Schowalter et al., 2006). If a low pH requirement for a productive paramyxovirus infection process is confirmed, it will be important to evaluate the specific roles of receptor binding and low pH in fusion activation.
Herpesviruses
Four (of 12) viral surface glycoproteins are essential for fusion by the α-herpesvirus, herpes simplex virus (HSV). These are: gD, the receptor binding protein, as well as gB and a heterodimer of gH/gL. gB and gH/gL are conserved in all herpesviruses and constitute their core fusion machinery (Table 1). A general model for how the HSV fusion machine is activated (reviewed in Krummenacher et al., in press; Rey, 2006; Spear et al., 2006) shares themes with models for the activation of paramyxovirus F proteins (Figures 3B and C): binding of gD to a host cell receptor induces changes in gD that somehow send a signal to and thereby activate the core fusion machinery (gB and gH/gL). Receptor-induced changes in gD are well characterized and involve loss of interactions between the N-terminal receptor binding domain and the C-terminal profusion domain (Krummenacher et al., in press; Krummenacher et al., 2005; Rey, 2006), but exactly how the unleashed pro-fusion domain of gD sparks the fusion cascade remains to be elucidated.
Substantial evidence indicates that gB, the most highly conserved protein in the fusion machine, is central to the fusion process. First, cell-cell fusion is altered when gB with mutations in its cytoplasmic tail is co-expressed with gD and gH/gL (Ruel et al., 2006; Spear et al., 2006). Second, a gB protein with a shortened cytoplasmic tail from the γ-herpesvirus, Epstein-Barr Virus, can promote fusion in the absence of other viral glycoproteins (McShane and Longnecker, 2004). Thirdly, gB contains sequences consistent with those of a bipartite fusion peptide (Backovic et al., 2007; Backovic et al., 2007; Hannah et al., 2007). Importantly, gB bears striking and unexpected structural similarity (Heldwein et al., 2006) to VSV G (Roche et al., 2006, 2007), a well characterized fusion protein that will be described later. Since VSV G and HSV gB display a unique combination of features seen in Class I and in Class II fusion proteins, they have been designated as Class III fusion proteins (Table 2). Other evidence reviewed in (Rey, 2006) supports a role for the gH/gL complex, and possible sequential interactions among gD, gB, and gH/gL have been discussed. One study reported that following receptor binding to gD, gH/gL induces hemifusion and then gB promotes full fusion (Subramanian and Geraghty, 2007). And a recent study, using bimolecular fluorescence complementation, has provided evidence that in the absence of receptor, complexes of gD and gB and of gD and gH/gL can be found on cells expressing these proteins (Figure 3D, left). Following receptor binding, and coincident with fusion, a novel complex containing gB and gH/gL was found (Atanasiu et al., 2007), as illustrated in Figure 3D (right).
Despite these advances, considerable work is needed to elucidate details of how HSV and other herpesviruses mediate fusion. Some of the key questions are: How do structurally diverse host cell receptors, which bind to diverse surfaces of gD (Spear et al., 2006), induce gD to trigger fusion? What is the sequence of interactions between gD, gB and gH/gL, and what are the interacting parts of each pair of interacting proteins? What are the exact roles of gB and gH/gL in specific steps of fusion? Some evidence indicates that the recently determined gB structure (Heldwein et al., 2006) represents its post-fusion conformation (Lin and Spear, 2007). If so, what is the structure of gB in its pre-fusion form? If there is a fusogenic complex between gB and gH/gL (Figure 3D), what does it look like? And, what are the common steps in the fusion cascade of divergent herpesviruses? The preceding discussion pertains to situations where HSV fuses with cells at neutral pH. However it has recently come to light that entry (into some cell types) of some strains of HSV and other herpesviruses requires exposure to low pH, presumably reflecting endocytic uptake (Clement et al., 2006; Delboy et al., 2006; Frampton et al., 2007; Milne et al., 2005; Nicola et al., 2003; Ryckman et al., 2006). So, is the mechanism of fusion activation altered in cases (e.g., a specific cell type) requiring the virus to pass through a low pH endocytic compartment? Bearing on this discussion, it is interesting that when HSV particles bind soluble HVEM, one of the HSV gD receptors, they can bind to protein-free liposomes, but only if the virus-receptor complexes are exposed to low pH (Whitbeck et al., 2006).
Binding to Multiple Receptors: The Case of HIV Env
The HIV fusion protein is a Class I fusion protein in which gp120, the outer receptor binding subunit, is noncovalently associated with gp41, the fusion subunit. The two subunits arise through proteolytic processing (priming) of the precursor, gp160. Like other Class I fusion proteins (Table 2), HIV Env is a metastable trimer consisting of three gp120/gp41 heterodimers. The fusion subunit, gp41, contains an N-terminal fusion peptide, an N-terminal heptad repeat region (termed N-HR or HR1), a linker region, a C-terminal heptad repeat region (termed C-HR or HR2), a tryptophan rich juxtamembrane region, a transmembrane domain, and a cytoplasmic tail (Figure 1B). The gp41 ectodomain forms a trimer-of-hairpins in its final form with a prominent six-helix bundle in which three C-HR (HR2) helices pack, in an antiparallel fashion, in the grooves created by a central triple helical coiled-coil composed of three N-HR (HR1) regions (Eckert and Kim, 2001; Gallo et al., 2003; Moore and Doms, 2003; Pohlmann and Reeves, 2006).
The HIV fusion protein is unusual in that the Envs from most HIV strains are activated for fusion by sequential interactions of gp120 with two receptors: CD4, often referred to as the “receptor,” and a member of the chemokine family of receptors, usually CXCR4 or CCR5, generally referred to as the “coreceptor”. Although a structure is not available for a complete pre-fusion HIV (or SIV) Env trimeric ectodomain, biochemical, biophysical, and structural findings have led to a model for how the HIV fusion protein is activated (Gallo et al., 2003; Moore and Doms, 2003; Pohlmann and Reeves, 2006). The salient structural findings that contributed to the model include those for an unliganded SIV gp120 core (Chen et al., 2005), HIV-1 gp120 cores bound with (two domains of) CD4 and either Fab fragments from neutralizing antibodies or a tyrosine sulfated peptide corresponding to the N-terminal arm of CCR5 (Huang et al., 2007; Kwong et al., 1998), and structures of the core of the gp41 trimer-of-hairpins (Chan et al., 1997; Weissenhorn et al., 1997). The main features of the HIV/SIV fusion activation model are as follows: Binding of CD4 to a pocket in gp120 leads to dramatic conformational changes. The V3 loop protrudes and two sets of β-ribbons, that were previously ∼20-25 Å apart, come together to form the β-bridging sheet. These conformational changes create and position the coreceptor binding site affording it better access to a coreceptor on the target cell (Chen et al., 2005; Kwong, 2005). CD4 binding causes other changes in gp120, and importantly, changes in gp41, notably exposure of the N-heptad region (N-HR) as a triple helical coiled-coil that can bind fusion inhibitory C-HR peptide analogues such as T20 and C34 (Furuta et al., 1998; Mkrtchyan et al., 2005).
Since some isolates of HIV and many isolates of SIV are CD4-independent (reviewed in Pohlmann and Reeves, 2006), binding to the “coreceptor” can be viewed as the critical interaction for fusion activation. CD4 binding is clearly important for viral pathogenesis, as evidenced by the observation that the majority of (CD4 dependent) primary HIV-1 strains are resistant to neutralization by antibodies directed against the bridging sheet, which is occluded before CD4 binding (Chen et al., 2005; Kwong, 2005). Also, as discussed above, CD4 binding initiates prehairpin formation. However, binding of a coreceptor enhances this process (Furuta et al., 1998; He et al., 2003; Mkrtchyan et al., 2005). The exact stage at which gp41 (fully) folds into its prehairpin is still unclear, but coreceptor binding is considered a requirement for efficient fusion peptide insertion into the target membrane. Coreceptor binding is clearly required for six-helix bundle formation, which leads to fusion pore formation and enlargement (Abrahamyan et al., 2003; Finnegan et al., 2001; Gallo et al., 2001; Markosyan et al., 2003; Mkrtchyan et al., 2005; Platt et al., 2007). In the case of HIV Env, it is hypothesized that a prebundle conformation in which one C-HR region is packed in the grooves of the trimeric N-HR coiled coil (e.g., between images iv and v in Figure 1A) mediates hemifusion, and that packing of the second and then the third C-HR helices mediate, respectively, formation of initial labile fusion pores (which can close) and then enlarged (robust) fusion pores that cannot close (Abrahamyan et al., 2003).
Given the need for HIV Env from most isolates to interact sequentially with CD4 and a coreceptor, it is not surprising that factors, such as the affinities of CD4 and coreceptor for Env, as well as features of the target cell plasma membrane (e.g., densities of CD4 and coreceptors, lipid and lipid raft composition, and/or structure of the underlying cytoskeleton) contribute to the ability of CD4 and the chemokine receptor to coordinate the fusion-inducing conformational changes. The need for most HIV isolates to interact with two host cell surface proteins also likely explains, in part, why receptor-induced fusion-activation of HIV Env takes longer than low pH induced activation of known low pH activated viral fusion proteins (Gallo et al., 2003, 2006; Platt et al., 2005). The need to interact with two distinct host cell receptors also provides ripe opportunities for anti-viral intervention. Inhibitors have been identified that block CD4 or chemokine receptor binding. In fact, the FDA has recently licensed maraviroc, a CCR5 antagonist, for clinical use (reviewed in Este and Telenti, 2007; Pohlmann and Reeves, 2006).
Receptor Binding Followed by Low pH: The Case of Avian Retroviruses
Avian α-retroviruses employ a unique two-step fusion activation process. As elucidated for ASLV, their Env glycoproteins are composed of a receptor binding subunit (SU) subunit that is disulfide bonded to a fusion subunit (TM) with characteristic features of Class I fusion proteins (Figure 1 and Tables 1 and 2). When a cognate receptor binds to an α-retroviral Env at physiological temperature, it causes conformational changes in Env (Delos et al., 2005; Gilbert et al., 1995) that convert it from a metastable fusion competent state to a membrane-embedded prehairpin conformation (Damico et al., 1998; Hernandez et al., 1997; Hernandez and White, 1998; Matsuyama et al., 2004), as depicted in Figure 1A (images ii and iii). Subsequent exposure to low pH converts the prehairpin to a trimer-of-hairpins containing a six-helix bundle (Matsuyama et al., 2004; Mothes et al., 2000; Netter et al., 2004; Smith et al., 2004), thereby inducing fusion (Delos et al., 2008; Melikyan et al., 2005; Melikyan et al., 2004; Mothes et al., 2000). Two things are remarkable about the ASLV Env prehairpin intermediate. Firstly, widely divergent receptors—a low density lipoprotein related receptor and a TNF receptor family member—induce subtypes A and B Envs, respectively, to form membrane-embedded prehairpins (Damico et al., 1998; Earp et al., 2003; Hernandez et al., 1997; Melikyan et al., 2004). Secondly, the prehairpin is remarkably stable both in vitro (Brecher and White, unpublished observations) and in vivo (Narayan et al., 2003). No other viral fusion system has yet been shown to employ such a clear two-step fusion activation mechanism, with one physiological trigger (receptor) promoting formation of a long-lived membrane-embedded prehairpin and a second (low pH) promoting hairpin formation.
The need for low pH for ASLV entry was not revealed in a test of the effects of lysosomotropic agents using the standard approach of washing out the agents after four hours of infection (Gilbert et al., 1990). In retrospect, this was because of the unprecedented stability of the ASLV prehairpin intermediate (Narayan et al., 2003; Brecher and White, unpublished results). Another confounding factor was that the onset of lipid mixing (of unlabeled ASLV with liposomes) has an unusually high pH threshold (Delos et al., 2008), which likely explains the ability of ASLV labeled with fluorescent lipid probes to fuse with cells at neutral pH (Earp et al., 2003; Gilbert et al., 1990). The pH dependence of the latter process is also clearly biphasic (Delos et al., 2008), which may indicate a need for lower pH for inner leaflet lipid mixing (and content mixing) than outer leaflet mixing, consistent with significant kinetic delays seen between these events (Melikyan et al., 2005).
Similar to Env from MoMLV (a γ-retrovirus), ASLV Env has a CX6CC motif between its N- and C-heptad repeats. But, in contrast to MoMLV Env, the receptor binding subunit (SU) of ASLV Env does not have a CXXC internal thiol disulfide exchange motif. Hence it has been postulated that low pH does for ASLV Env what the internal thiol disulfide exchange motif does for MoMLV Env (Li et al., 2007), that is to move the SU head domains sufficiently away from the fusion subunit to allow the membrane-embedded prehairpin to fold-back to its final trimer-of-hairpins conformation. Key remaining issues include the identification of residues critical for the pH-induced conversion of the prehairpin to the trimer-of-hairpins conformation (Babel et al., 2007), and determining whether ASLV Env employs a chain reversal reaction similar to that proposed for other retroviral Env glycoproteins (Maerz et al., 2000, 2001).
An Apparently Novel Mechanism: The Case of Ebola Virus
The Ebola virus glycoprotein (GP) is composed of a large receptor binding subunit (GP1) disulfide-bonded to a fusion subunit, GP2, which has the canonical features of a Class I fusion protein (Figure 1B and Table 1). Its internal fusion peptide is located near its N-terminus following processing of the GP precursor at a furin cleavage site. Furin processing occurs with wild type GP, but is not essential for virus entry (Neumann et al., 2002, 2007; Wool-Lewis and Bates, 1999). Several studies indicate that low endosomal pH is required for Ebola virus entry (Takada et al., 1997; Wool-Lewis and Bates, 1998), but barring one report (Bar et al., 2006), evidence indicates that low pH is not sufficient to activate GP for fusion (Schornberg et al., 2006). Instead, Ebola virus entry requires the action of cathepsins B and L (Chandran et al., 2005; Kaletsky et al., 2007; Schornberg et al., 2006), endosomal proteases with pH optima of ∼5. A working model for Ebola GP fusion activation is shown in Figure 5. In this model, cathepsins B and L whittle Ebola GP1, the outer receptor binding subunit, from a 130 kD protein to an ∼19 kD fragment, which is derived from an N-terminal region of GP1. Remarkably, in this form the fusion subunit, GP2, is still clamped. We therefore refer to these upstream cathepsin cleavage events, albeit extensive, as priming steps. Activation of GP from its clamped 19 kD GP1 state requires a still-to-be-defined cathepsin L dependent activity (Figure 5). That activity could be limited or complete proteolysis of GP1 by cathepsin L. Alternatively, the trigger activity could involve a different endosomal protein (protease, other enzyme, other protein) that is activated by cathepsin L. In addition to the molecular identity of the fusion trigger, many other important questions remain such as the roles of the host cell receptor(s) and low pH in the final triggering step. Furthermore, thermolysin, a protease that functions at neutral pH, can substitute for the cathepsins (Schornberg et al., 2006) in priming GP (Figure 5). Hence it will be interesting to determine if in different cellular contexts, different proteolytic enzymes (both extracellular and endosomal) can prime (and trigger) GP.
FIG. 5.
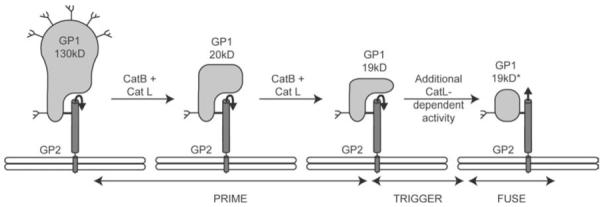
Model for priming and triggering of the Ebola virus glycoprotein. The figure is modified from Figure 4A in (Schornberg et al., 2006). GP1 is the receptor binding subunit and GP2 is the fusion subunit. Cat B and Cat L denote the endosomal cathepsins (B and L, respectively). 19 kD GP1* denotes the post fusion form of GP1. Prime denotes the cleavage of GP1 to the 19kD form. Trigger denotes the unclamping of GP2, which leads to fusion (Fuse). For clarity only one monomer of the GP trimer is shown.
Endosomal cathepsins have been implicated in the entry of several other enveloped viruses including the coronaviruses SARS and MHV, the retrovirus MoMLV, and the paramyxoviruses, Hendra and Nipah (Pager et al., 2006; Pager and Dutch, 2005). However, the exact role of cathepsins for entry of these viruses is less clear. In some of these latter cases cathepsins may make a simple proteolytic cut separating the receptor binding and fusion subunits (after virus attachment to host cells), leaving both subunits virtually intact (Li et al., 2006; Pager et al., 2006; Pager and Dutch, 2005), as opposed to the extensive cathepsin-mediated proteolysis that cleaves away a significant portion of the receptor binding subunit of the Ebola virus glycoprotein. For all of these viruses (including Ebola virus), further work is needed to clarify the roles of receptors, cathepsins, low pH, and perhaps other cellular (endosomal) factors in priming and triggering fusion.
CLASS I, II, AND III VIRAL FUSION PROTEINS
All viral fusion proteins are trimers of hairpins in their final forms, and in all cases the final structure contains a central N-terminal trimeric core surrounded by C-terminal regions that pack tightly against the outside of the central N-terminal trimeric core, thereby bringing the two critical hydrophobic elements of all viral fusion proteins—fusion peptides and transmembrane domains—into intimate functional proximity. In the ensuing text we will describe what is known about the fusion mechanisms of selected Class I, II, and III fusion proteins whose structures are known in both their pre- and post-fusion forms. In Figure 6 we point out in orange the protein segments responsible for extension (Ex) of the fusion peptides (red) towards the target membrane (i.e. for forming the prehairpin intermediate shown in Figure 1Aii and iii) and, in green, the protein segments responsible for the inversion (Inv) of the C-terminal regions (purple) so that they can pack around the central trimeric core (Figures 1 and 6; see legends). It is important to note that while high-resolution structures are available for the starting and ending states of these fusion proteins (Figure 6), the intermediates discussed and displayed in Figure 1Aii-v have been inferred from biochemical findings.
FIG. 6.
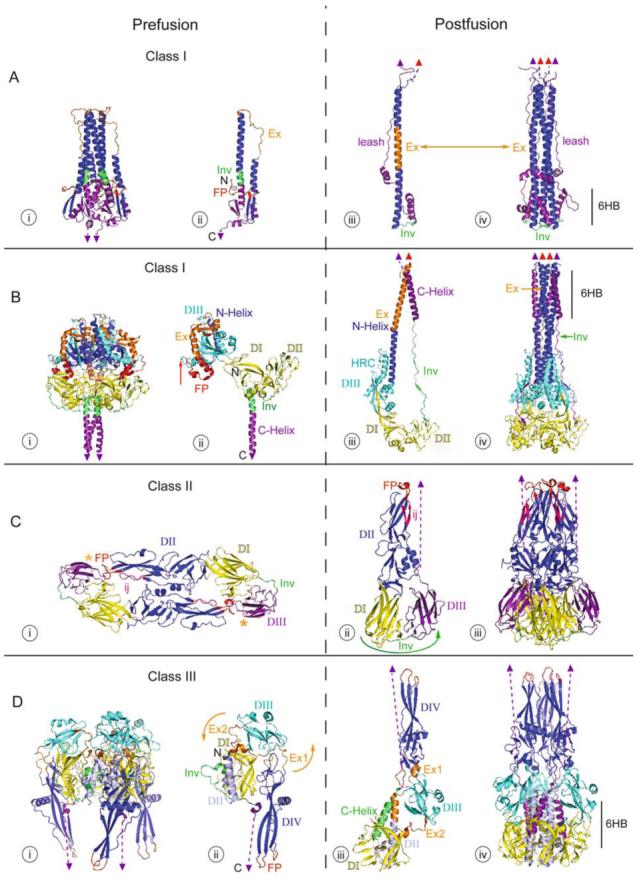
Structures of Class I, II, and III fusion proteins in their pre- and post-fusion forms. The crystal structures of the Class I fusion proteins, Influenza virus HA2 (A) and Paramyxovirus F (B), a Class II fusion protein, TBEV E (C), and a Class III fusion protein, VSV G (D), are shown. The pre-fusion states (i and ii) are on the left and the post-fusion states (iii and iv) are on the right with functional domains identified by color. Fusion peptides are in red, with the domains containing them in dark blue. C-terminal domains, which connect to the virus membrane, are in purple. C-terminal linkers, transmembrane domains, and fusion peptides not visible in the structure are represented by dashed purple lines, purple triangles, and red triangles, respectively. Regions important for the movement of the fusion peptide toward the target membrane are displayed in orange, and regions important for C-terminal inversion, that bring the fusion peptide and transmembrane domains together, are shown in green (in Ai-iii, Bi-iii, Ci-ii, and Di-iii). In the post-fusion trimer structures (iv), the orange and green are replaced by dark blue or purple domain coloring, respectively, to illustrate the similarities among all of the post-fusion forms. Other domains are represented in cyan, yellow, and blue-gray. PDB accession numbers for the structures are: 2HMG (Ai and ii); 1QU1 (Aiii and iv); 2B9B (Bi and ii); 1ZTM (Biii and iv); 1SVB (Ci); 1URZ (Cii and iii); 2J6J (Di and ii); 2CMZ (Diii and iv).
Class I Fusion Proteins
Class I fusion proteins are trimers in their pre-fusion and post-fusion states, and their final states are typified by having a central N-terminal trimeric α-helical coiled coil decorated by three “C-terminal” helices, thereby forming a 6HB. The size and position of the 6HB varies significantly among Class I proteins (see Figure 6 in Lamb and Jardetzky, 2007). Their fusion peptides are at or near the N-terminus of the fusion subunit, but their specific locations vary in the two known starting structures, those of influenza HA and a paramyxovirus F protein. For other features of Class I fusion proteins see Table 1.
Influenza HA
Influenza virus HA is the founding member of the large group of Class I fusion proteins and remains one of its best characterized. HA is produced as a precursor protein (HA0), which is cleaved into its receptor binding (HA1) and fusion (HA2) subunits, which remain disulfide linked. HA1, the receptor binding subunit, binds to sialic acid moieties on the target cell surface, allowing the virus to be internalized into the endosomal pathway. The low pH of the endosome then triggers conformational changes, which drive fusion (see above for a general description of low pH triggering) (Earp et al., 2005; Harrison, 2005; Skehel and Wiley, 2000; Weissenhorn et al., 2007).
The pre-fusion structures of HAs from several subtypes of influenza virus have been solved. Interestingly, they group into four clades based on the positioning of the globular head receptor binding domain (HA1) relative to the stem region (largely HA2), which houses the fusion activity (Russell et al., 2004). A post-fusion (acidic pH) structure has only been solved for the X:31 HA, and is shown in Figure 6A in comparison with the pre-fusion structure of its ectodomain (Bullough et al., 1994; Weis et al., 1990; Wilson et al., 1981). The receptor binding subunit, HA1, is not shown because it is not present in the post-fusion structure. The pre-fusion form (Figure 6A, i trimer and ii monomer) exists on the virion surface as a trimer with its N-terminal fusion peptide (Figure 6Ai and ii, red) protected within the trimer interface and its C-terminus extending to the transmembrane domain (purple triangle), which is inserted in the viral membrane. In the trimer three long helices form a central coiled-coil, with three shorter outer helices (the A helices, Wilson et al., 1981) packing against it. The fusion peptide is linked to the outer shorter helix by two β-strands (dark blue). Another small β-sheet (purple) links the central coiled-coil to the C-terminus.
Upon acidification, protonation of key residues in HA1 and HA2 results in movement of the receptor binding globular head domain (HA1, not shown in Figure 6A), thereby unclamping HA2, the fusion subunit. The overall structure of HA1 does not change during the process (reviewed in Skehel and Wiley, 2000). Within the fusion subunit, two major structural rearrangements, fusion peptide extension and C-terminal inversion, occur to drive fusion. Firstly, an unstructured linker (orange, labeled Ex in Figure 6Aii and iii, also referred to as the B loop in Bullough et al., 1994; Qiao et al., 1998; Wilson et al., 1981) becomes α-helical, forming one long α-helical rod in the N-terminal part of the molecule (Fig 6A, orange and dark blue in iii and dark blue in iv). The formation of this structure extends the fusion peptide (red triangle in iii and iv) more than 100 Å to the tip of the molecule, positioning it for insertion into the target membrane. This transient intermediate, the prehairpin (modeled in Figure 1A ii and iii), spans the distance between the viral and target cell membranes. Subsequently, a portion of the starting helix (green, labeled Inv in Figure 6Aii) becomes an unstructured linker (green, Inv in Figure 6Aiii and iv), allowing the C-terminal region of HA (purple) to fold back against the long N-terminal helix (dark blue in Figure 6Aiv). This movement brings the transmembrane domain (purple triangle) close to the fusion peptide (red triangle), forming a hairpin structure (iii and iv) and allowing fusion between the attached viral and target membranes.
The final, post-fusion trimer (Figure 6A iv) contains a coiled-coil superficially reminiscent of that in its pre-fusion form. Unlike those in the pre-fusion form, however, the central helices (dark blue) are longer, packed more tightly along their hydrophobic faces, and are proximal to the fusion peptide (red triangles). The outer helices (purple), which pack against the base of the central coiled-coil, are part of the C-terminal domain connected to the transmembrane domain (purple triangle). This structure, in which the outer C-helices pack against the central N-helical coiled-coil, is referred to as the six helix bundle (6HB in iv) and is a defining feature of the post-fusion structures of all Class I fusion proteins. A linker C-terminal to the 6HB connects to the transmembrane domain and has been referred to as the leash. Packing of the leash into a groove along the central coiled-coil (Chen et al., 1999) is required for fusion (Park et al., 2003), as is capping of the coiled-coil (Borrego-Diaz et al., 2003).
The pH at which these conformational changes are triggered varies for different strains of influenza virus, and many mutations have been described that alter the pH dependence of fusion (Rachakonda et al., 2007; Skehel and Wiley, 2000; Thoennes et al., 2007). Regardless of the pH at which the HA conformational change is triggered, it is generally accepted that multiple HA trimers are needed at the fusion site (Blumenthal et al., 1996; Danieli et al., 1996). Additionally, it has been hypothesized that triggered HA trimers near, but not actually at, the fusion site aid the process via a so-called “bystander effect” (Kozlov and Chernomordik, 2002).
Paramyxovirus F
Fusion of paramyxoviruses with target cells is mediated by the fusion protein, F. Interaction between the viral receptor binding protein and the host cell receptor changes the association between the viral receptor binding and fusion proteins, thereby triggering fusion. Unlike other Class I fusion proteins, the receptor binding function is carried out by a separate receptor binding protein (HN in HPIV3 and SV5, now referred to as PIV5; see above). Like influenza HA, F is produced in an uncleaved form, F0, which must be cleaved in order for F to function in fusion. This cleavage produces F2 and F1, which remain disulfide linked. The fusion peptide, which was previously internal in the F0 sequence, is now N-terminal in the F1 sequence and competent to mediate fusion (Bossart and Broder, in press; Lamb and Jardetzky, 2007).
The crystal structures of the ectodomains of PIV5 (SV5) F (pre-fusion) and hPIV3 (post-fusion) are shown in Figure 6B (Yin et al., 2005, 2006). The F pre-fusion structure (Figure 6B i and ii) exists as a trimer resembling a ball on a stick with the fusion peptide (red) “wedged” between domains DII and DIII of its own and another subunit. The F protein consists of three domains. Domains I and II (both colored yellow in Figure 6B) consist primarily of β-sheet and linker structures, and form a relatively stable structure around which the more dramatic conformational changes occur during fusion. Since the pre-fusion structure is uncleaved (F0), the N-terminus (N) shown in DI represents the start of the F2 segment. The furin cleavage site, representing the start of the F1 fragment (red arrow in Bii) is exposed on the surface of the trimer. Domain III (cyan) consists of both α-helical and β-sheet structure. Four short helices, two beta strands, and the linkers between them in the pre-fusion structure (orange and dark blue in Figure 6Biii) become the N-helix in the post fusion structure (dark blue in iv). In the pre-fusion structure (Figure 6Bi and ii), the C-helix (purple; also referred to as HRB) extends from a linker (green, labeled Inv in ii) and connects to the transmembrane domain (purple triangle). Interactions between the C-helices (purple) and between DI and DII (both yellow) of different subunits help stabilize the pre-fusion trimer.
Upon triggering by its cognate receptor binding protein (Figure 3B and 3C), the paramyxovirus F protein undergoes conformational changes broadly analogous to those of Influenza HA, but quite different in detail. First, it is hypothesized that interactions between the trimeric C-helices (purple) are broken. This helix dissociation is believed to occur early in the conformational change since fusion can be inhibited at an early stage by N-helix derived peptides (which bind the C-helix) but not by C-helix derived peptides (Joshi et al., 1998; Russell et al., 2001). Once the C-helices of the pre-fusion trimer have dissociated, a region consisting of small α-helices, linkers, and a sheet consisting of two β-strands (orange, labeled Ex in ii and iii) becomes entirely α-helical, extending the fusion peptide toward the target membrane by building on a short preexisting α-helix (dark blue in ii and iii). This newly structured extension region (Ex) becomes part of the long N-terminal helix. This long new helix is thought to be stabilized by the formation of an additional helix (HRC, not to be confused with the C-helix) in Domain III (Cyan). As a result a prehairpin intermediate spanning the distance between the target and viral membranes is formed, as described above for influenza HA and as modeled in Figure 1A ii and iii. Following extension of the fusion peptide, an α-helical region (green, Inv) between Domain II and the C-Helix (purple, also referred to as HRB) unwinds, extending the length of the linker between Domain II and the C-Helix. This extended linker (Inv, green in ii and iii) allows the C-Helix (HRB) to fold back and pack in an inverted orientation and pack in an anti-parallel fashion against the elongated N-Helix (dark blue in Figure 6Biv) thereby facilitating fusion between the viral and target cell membranes (Russell et al., 2001).
The post-fusion F trimer looks very different from its prefusion form. It forms a long trimeric coiled-coil, with the fusion peptides (red triangles) inserted in the membrane as seen for influenza HA. Unlike influenza HA, however, two domains (DI and DII, yellow) lie below the central coiled-coil in the post-fusion structure, and the 6HB is located at the membrane proximal end of F, as opposed to HA, where the 6HB is located at the membrane distal end, followed by a long C-terminal leash. The membrane distal end of the F coiled-coil is decorated by an additional set of outer helices (HRC, cyan), part of which superficially resembles the C-helix in the HA 6HB (Figure 6Aiv), but which is formed from a different region of the protein.
A comparison of the structural transitions in influenza HA and paramyxovirus F illustrates many of the commonalities shared by Class I fusion proteins (Table 1). They exist as metastable trimers with the fusion peptide protected until an external trigger (receptor binding or low pH) releases a clamp allowing extension (Ex) of an N-terminal helix and insertion of the fusion peptide into the target cell membrane. Once the N-helical coiled-coil has formed, the C-helix inverts by means of a flexible linker (Inv), and packs into the grooves of the central N-terminal coiled-coil, thus forming a 6HB. This event plus packing of other C-terminal regions brings the viral membrane into proximity with the target cell membrane, and allows fusion (Bossart and Broder, in press; Lamb and Jardetzky, 2007).
Class II Fusion Proteins
Class II fusion proteins consist primarily of β-sheet structure with internal fusion peptides formed as loops at the tips of β-strands. They are associated with a chaperone protein (p62 for SFV E1 and prM for TBEV E), which is cleaved during or soon after viral assembly (Table 2). Following maturation, the fusion protein ectodomains exist as anti-parallel dimers that lie low along the virion surface, with each stem at a threefold axis of symmetry on the virion. Once triggered, Class II fusion proteins realign as trimers that project from the viral membrane at threefold axes. In addition to TBEV E and SFV E1 (discussed below), the pre-fusion and post-fusion structures of the Dengue and West Nile Virus E proteins have also been solved, and are similar in most respects to TBEV E. For further information, see Kielian (2006) and Kielian and Rey (2006).
TBEV E
The crystal structures of pre-fusion and post-fusion forms of the TBEV E protein are shown in Figure 6C (Bressanelli et al., 2004; Rey et al., 1995). The pre-fusion E protein is an antiparallel homodimer (i, view from above). It consists of three domains, consisting almost entirely of β-sheet structure. It is primarily contacts between the subunits in Domain II (dark blue) that maintain the homodimer. This domain also contains the fusion peptide loop (FP, red) and the ij loop (ij, pink), which play critical roles in target membrane binding. The fusion loop peptide is not exposed in the native structure, being masked by nearby residues in Domain I (yellow) and Domain III (purple) of the opposite subunit.
Upon exposure to low pH, the E protein undergoes a great deal of movement, but few conformational changes within individual domains. First, the dimer contacts between the subunits are broken. This is followed by rotation of almost the entire protein around the C-terminal stem region, which is not visible in the crystal structure (an orange * denotes where the stem would connect with the ectodomain in the pre-fusion structure). The ectodomain then moves toward the target membrane into which the fusion loop peptide inserts. Since each C-terminal stem sits on a threefold axis on the viral surface (Ferlenghi et al., 2001) these rearrangements create a membrane-embedded homotrimer (Kielian, 2006), a state analogous to the homotrimeric prehairpin of Class I fusion proteins. Domain III (purple) is connected to Domain I (yellow) by a flexible linker (Inv, green). During the final stage of fusion, this linker allows Domain III to foldback at the “side” of Domain I to a position 33 Å from its pre-fusion location (motion represented by green arrow in ii), bringing the C-terminal stem region (dashed purple line; connected to the viral transmembrane domain) and the tip of the structure containing the fusion loop peptide (inserted into the target membrane) together, thus driving fusion. Note that i represents a top view and ii and iii side views of the E1 structure.
SFV E1/E2
Both the TBEV E and SFV E1 ectodomains start as antiparallel dimers in their pre-fusion states, but differ in important respects. For example, TBEV E has receptor binding activity and it forms an antiparallel homodimer that shields the fusion peptide of its partner E subunit; in comparison, for SFV, receptor binding and fusion loop peptide shielding are carried out by a companion protein, E2 (the cleavage product of p62), which is found in association with E1 as a heterodimer (Lescar et al., 2001; Mancini et al., 2000; Wu et al., 2007). Nonetheless, upon acidification, both TBEV E and SFV E1 form similar post-fusion trimer-of-hairpin structures reminiscent of those of Class I fusion proteins. The TBEV E and SFV E1 post-fusion trimers differ in the relative positions of their fusion loops. Whereas the TBEV E post-fusion trimer has a “closed” conformation, in which the three fusion loops are in close contact (at the membrane proximal end of the protein), the SFV E1 post fusion trimer has an “open” conformation, in which the three fusion loops are separated by 45 Å (Bressanelli et al., 2004; Gibbons et al., 2004). The reasons for and significance of this difference are not yet clear. When triggered in the presence of membranes, SFV E1 has been observed to form ring structures containing five or six trimers suggesting cooperativity between trimers during the fusion process (Gibbons et al., 2004; Kielian and Rey, 2006). Similar rings (with six trimers) have been observed for TBEV E (Stiasny et al., 2004). It has been hypothesized that these rings may form volcano-like structures that may influence membrane curvature during fusion.
Class III Fusion Proteins
As discussed above, recent structural work on VSV G (Roche et al., 2006, 2007) and HSV-1 gB (Heldwein et al., 2006) has identified a third class of fusion proteins that shares features with both Class I and Class II fusion proteins. Like Class I proteins, they are trimers in their pre-fusion forms and they contain a central α-helical coiled-coil. However, their fusion domains more closely resemble those of Class II proteins in that their fusion loops are found at the tips of extended β-strands (Table 2).
VSV G
The crystal structures of the neutral and low pH forms of the VSV G ectodomain are shown in Figure 6D (neutral i and ii, low pH iii and iv). In agreement with previous biochemical work, the pre-fusion form of VSV G is a trimer, but the trimer interface is small. In contrast to those in the pre-fusion conformations of all other fusion proteins known to date, the fusion loops (red) are located on the outside of the structure, not protected at an interface.
Upon acidification, a series of conformational changes occur in VSV G that reposition the fusion loops (red) into the vicinity of the target membrane. A second series of conformational changes then bend the protein back, reorienting the C-terminal portion anti-parallel to the N-terminal segment, thereby bringing the viral and target membranes together. These conformational changes, reminiscent of the changes proposed for HA in their consequences, are modeled to happen as follows: In the first step, conformational changes occur in two regions, Ex1 and Ex2 (orange in i, ii, and iii). Each of these regions consists of two pieces, one of which is an unstructured linker, the other of which has helical structure (Roche et al., 2007). During the conformational change the unstructured linker of Ex1 becomes helical and the helical residues become unstructured, resulting in movement of the fusion domain (DIV) approximately 90°. The motion of DIV is completed by changes in Ex2, in which linkers between DII (blue-gray in ii) and DIII (cyan), become helical, extending each of the two DII helices (blue-gray and orange in iii). The result is the rotation of both DIII and DIV such that the fusion loops (red) are now in the vicinity of the target membrane. Finally, inversion of the C-terminal stem is accomplished by additional structural rearrangements in DomainII. In particular, an unstructured loop (Inv, green in ii) becomes an α-helix, which we refer to in Figure 6D as the “C-helix” (green in iii), that is oriented antiparallel to the core structure. Consequently, the “C-helix” packs against the now elongated helix of Domain II (blue-gray and orange in iii), bringing the C-terminus and viral membrane into proximity with the target membrane, thereby facilitating fusion. The reader is referred to the supplemental movie in (Roche et al., 2007) for an animation of how these conformational changes are thought to occur.
VSV G bears some similarities with both Class I and Class II fusion proteins. For example, Domain IV, the fusion domain (dark blue), is similar in structure to the fusion domain of Class II fusion proteins. Like the Class II fusion domains, those of VSV G do not change significantly in conformation during fusion, but move as a whole and pack as trimers with their fusion loops inserted in the target membrane. Additionally, in the post-fusion form, the inner helix of Domain II (blue-gray in iv) is reminiscent of the membrane distal end of the N-helix in the post fusion forms of both of the Class I fusion proteins shown in Figure 6. The “C-helix” of VSV G (green in iii and purple in iv) packs against the central trimeric coiled-coil (blue-gray in iv), forming a small 6HB. Unlike Class I fusion proteins, however, the N-terminal end of the N-helix in the VSV G post-fusion trimer is located a significant distance from the fusion loop. On the C-terminal end of the “C-helix” (Figure 6D, iii and iv), a long linker (not present in the structure; dashed purple line in Figure 6Diii and iv) extends from the membrane distal end of the molecule to the target membrane.
A potentially unique feature of VSV G and other rhabdovirus G proteins is that their low pH-induced conformational changes are reversible. The pre-fusion and post-fusion states are in thermodynamic equilibrium, with the equilibrium shifted towards the post fusion state at low pH (Gaudin, 2000). This is different from the majority of other viral fusion proteins, which are metastable and irreversibly inactivated (lose the capacity to mediate fusion with a subsequently presented target membrane) if triggered in the absence of a target membrane. It is not yet known whether conformational changes in HSV-1 gB, the other known Class III fusion protein, are reversible.
MEMBRANE INTERACTING AND JUXTAMEMBRANE REGIONS
The above sections describe the general characteristics of viral fusion proteins, their known structural features in their native and post-fusion conformations, and the presumed intermediates along the fusion pathway. In addition, the various known triggering mechanisms have been presented. Below, we describe the roles of regions of the protein often not present in the available crystal structures of the fusion proteins, namely the fusion peptides, transmembrane domains, juxtamembrane ectodomains, and cytoplasmic regions, all of which can have direct effects on either the target or viral membrane during fusion. Most of our understanding of the roles of these sequences comes from mutagenesis and domain swapping experiments. In a few cases structures of peptides corresponding to these sequences have been elucidated. This is particularly true for fusion peptides, the hydrophobic sequences that interact directly with target membranes. Consistent with the themes described above for the structure and function of fusion protein ectodomain sequences, the membrane associated and juxtamembrane regions share many common features but differ in some particulars as discussed below.
Fusion Peptides and Fusion Loops
Some, but not all, fusion peptides can be preliminarily identified by sequence analysis as regions of intermediate hydrophobicity with predicted membrane binding potential. Support for the identity of a fusion peptide comes from mutating select residues within the candidate sequence within the context of the full length fusion protein and demonstrating decreased fusion activity with otherwise wild-type functions (e.g., protein expression processing, and virion incorporation). The ability of synthetic candidate fusion peptides corresponding to wild type, but not mutant, fusion peptide sequences to bind artificial membranes (liposomes or planar bilayers) and induce fusion provides additional evidence for fusion peptide identity and function. Confirmation that these peptide sequences actually interact with the target membrane in the context of the triggered full-length fusion protein has come from identifying sequences that can be labeled by cross-linking probes embedded in the target membrane.
Fusion peptides come in three forms: N-terminal, internal single loops, and internal bipartite loops. Most Class I fusion proteins have N-terminal fusion peptides that are generated by proteolytic processing of a precursor protein. Processing of the α-retrovirus and filovirus (Class I) fusion proteins leaves the fusion peptide in an internal location, but near the N-terminus of the fusion subunit. The fusion peptides of Class II fusion proteins are located at an internal position at the tip of a loop formed by antiparallel β-strands, and they are stabilized by disulfide bonds. A second loop, also formed by antiparallel β-strands and stabilized by disulfide bonds, appears to control a requirement for cholesterol in the target membrane (at least for SFV and SIN), but may not interact directly with the membrane. Class III fusion proteins have bipartite internal fusion peptides consisting of two rigid loops formed by three antiparallel β-strands that form a sheet and an additional strand. The two fusion peptide tips are equally positioned for interaction with target membranes. All fusion peptides appear to interact exclusively with the outer leaflet of the target membranes, at least during the initial interaction stage. With the exception of that of VSV G, hydrophobic fusion peptides are found at subunit interfaces in the native structure (Table 2) where their hydrophobic surfaces are shielded by surrounding protein (Figure 6).
N-terminal Fusion Peptides
The native-state ectodomain structures of two glycoproteins with N-terminal fusion peptides have been solved: those for the influenza virus HA and for the SV5 (PIV5) F (Figure 6) (Wilson et al., 1981; Yin et al., 2006). In each case, the fusion peptide resides at a subunit interface in an extended conformation. The HA fusion peptide is located near the viral membrane, packed between HA2 helices of its own subunit and an adjacent one, with the N- and C-terminal strands of HA1 helping to clamp the fusion peptide in place (Wilson et al., 1981). In contrast, the SV5 F fusion peptide lies in a groove between domains II and III of the head region of F (Yin et al., 2006). Each of these fusion peptides relocates to the end of an extended α-helix in the post-fusion conformation (Figure 6).
Our knowledge of how N-terminal fusion peptides interact with target membranes comes primarily from studies using synthetic peptides and model membranes. N-terminal fusion peptide sequences are characterized by having structural plasticity. In general, these peptides tend to exist as random coils in solution and as α-helix, β-sheet, or a combination of these structures upon interaction with target membranes. The relevance of these peptide structures to respective segments in the full-length fusion proteins as they interact with membranes has been debated. An oblique angle of insertion is, however, generally thought to be important (reviewed in Durell et al., 1997; Martin and Ruysschaert, 2000).
Recent structural analyses of the HA and HIV fusion peptides, two well characterized fusion peptides (for example, Reichert et al., 2007 and references therein), in micelles have provided our best window on the membrane embedded conformations of N-terminal fusion peptides. The structure of the wild-type HA fusion peptide in DPC micelles reveals a kinked structure in which the charged residues of the fusion peptide reside at the membrane-water interface and both the N- and C-terminal regions penetrate the outer leaflet of the bilayer as α-helices (Han et al., 2001). The inner surface of the kink contains the hydrophobic residues, consistent with their location in the bilayer, and a glycine ridge is seen on the outer surface of the N-terminal helix (Han et al., 2001) (Figure 7A). Subsequent structural analyses of two mutant fusion peptides that, in the context of full-length HA, are compromised in fusion (Qiao et al., 1999), revealed that they either failed to form the kinked structure or penetrated the target membrane less deeply (Li et al., 2005). A rigid kink, held in place by hydrophobic interactions between aromatic residues (F9, I10 and W14) on the interior of the kink is required for optimal fusion (Lai et al., 2006; Lai and Tamm, 2007). Whether this structure remains in the final-post fusion conformation or collapses to interact with the transmembrane domain as has been proposed (Armstrong et al., 2000; Park et al., 2003; Tamm, 2003) remains to be seen.
FIG. 7.

Structures of the HA and HIV fusion peptides in DPC micelles. (A) The HA fusion peptide, residues 1-20 of HA2, sits in the membrane as a kinked structure with E11, N12, and E15 at the membrane interface and the majority of the hydrophobic residues on the interior of the kink (PDB entry 1IBN). F9, I10, and W14 are on the interior at the apex of the kink where they stabilize the bent structure (Lai and Tamm, 2007). The glycine ridge, formed by residues G1, G4, and G8, is visible along the N-terminal helix. (B) The HIV fusion peptide exists as an extended helix from residues I4 to A15 in DPC micelles (PDB entry 2PJV). Both the N- and C-terminal portions of the protein are disordered in the structure. ATR-FTIR studies have shown that the HIV fusion peptide inserts into bilayers at an angle (Martin and Ruysschaert, 2000), but its exact orientation and depth of penetration await further study.
Not all N-terminal fusion peptides mediate fusion via a kinked structure; the HIV fusion peptide, which is unstructured in solution, adopts an extended α-helical structure in either SDS or DPC micelles (Figure 7B) (Gabrys and Weliky, 2007; Jaroniec et al., 2005; Li and Tamm, 2007). Residues 4-14 are α-helical in DPC micelles (Gabrys and Weliky, 2007; Li and Tamm, 2007). In SDS micelles, the helical character of the peptide extends to Gly20 (Jaroniec et al., 2005). A slight break in the helix at Gly10 was observed in the majority of structures in DPC micelles but was a minor structure in SDS micelles (Li and Tamm, 2007). No evidence of the type of kink observed for the HA fusion peptide was observed in any of these studies. Although it has been reported that the physiologically relevant fusogenic structure of the HIV fusion peptide is β-sheet (Nieva et al., 1994; Peisajovich et al., 2002; Zheng et al., 2006), a recent study suggested that, at least during the early stages of fusion, α-helical structure is more important (Li and Tamm, 2007), as indicated in earlier studies (Martin et al., 1993; Rafalski et al., 1990). Another recent study using HIV fusion peptides tagged with bulky side-groups that either allow or disfavor β-sheet formation did not reveal any differences in the fusion activity between these two types of peptides (Reichert et al., 2007) concluded that the structural plasticity of these fusion peptides was their most important feature (Reichert et al., 2007). A major difference between these studies was the use of liposomes containing cholesterol and sphingomyelin in the latter, but not the former, case. The presence of these lipids in target membranes has been reported to favor the β-strand conformation of the HIV fusion peptide (Buzon and Cladera, 2006; Zheng et al., 2006). Perhaps both α-helical and β-sheet structures are important at different stages of the HIV fusion process.
Internal Fusion Peptides/Loops
α-Retrovirus and Filovirus Fusion Peptides
Although they are found near the N-terminus, the fusion peptides of α-retroviruses and filoviruses are internal to their fusion subunits. They are flanked by two Cys residues that are thought to be linked by a disulfide bond, and they contain two Pro residues near their centers. Mutagenesis studies have shown that the second of these Pro residues and the flanking pair of Cys residues are important for fusion (Delos et al., 2000; Delos and White, 2000; Ito et al., 1999; Jeffers et al., 2002). Recent work has revealed that the Cys residues flanking the fusion peptide of ASLV EnV are important for the lipid mixing stage of fusion (Delos et al., 2008). Apparently the Cys residues are needed either to maintain an appropriate and stable membrane disrupting structure during the fold-back stage of fusion and/or to allow folding of the fusion protein into a sufficiently compact hairpin structure to allow merging of the membranes.
For the α-retroviruses, separation of the SU (receptor binding) and TM (fusion) subunits by cleavage at a furin cleavage site is required for fusion and infection (Perez and Hunter, 1987). Similar cleavage of the filovirus glycoproteins occurs (during the virion assembly/maturation part of the viral life cycle), but is not essential for infection (Neumann et al., 2002; Neumann et al., 2007; Wool-Lewis and Bates, 1999). Given the role of cathepsins in Ebola virus entry (Chandran et al., 2005; Kaletsky et al., 2007; Schornberg et al., 2006) it is possible that endosomal proteases can substitute for furin, as appears to be the case for Hendra and Nipah virus F proteins (Pager et al., 2006; Pager and Dutch, 2005). Alternatively, the Ebola virus fusion peptide may not need to be near the N-terminus. The latter idea is suggested by the observation that the putative furin cleavage site of the Marburg virus glycoprotein is further upstream of its fusion peptide than those of ASLV Env and Ebola GP.
Peptides corresponding to the central, membrane-interacting portions of both the ASLV and the Ebola virus fusion peptides have been studied (Adam et al., 2004; Cheng et al., 2004; Freitas et al., 2007; Gomara et al., 2004; Ruiz-Arguello et al., 1998). The solution NMR structure of the ASLV fusion peptide (residues 16-43) revealed a type II β turn about the central Pro, consistent with an earlier prediction (Delos et al., 2000). The remainder of the peptide was less structured. In SDS micelles, the peptide acquired a more α-helical character, particularly in the N-terminal region, with the central Pro at the micelle-water interface. The peptide inserted into membranes at an angle and more deeply at low pH (Cheng et al., 2004), consistent with the need for low pH to induce fusion (Delos et al., 2008; Melikyan et al., 2005; Melikyan et al., 2004; Mothes et al., 2000). Interestingly, the ASLV fusion peptide induces more lipid mixing than comparable concentrations of the HA fusion peptide, and displays a tendency to self-associate into a variety of oligomeric states (Cheng et al., 2004). The synthetic fusion peptide of the Ebola virus glycoprotein has been studied by several investigators (Adam et al., 2004; Freitas et al., 2007; Gomara et al., 2004; Ruiz-Arguello et al., 1998). Although differing in detail, each of these structures is consistent with the Ebola virus fusion peptide adopting some α-helical character in membranes with the critical Pro at the membrane-water interface.
Internal Fusion Peptides/Loops of Class II Fusion Proteins
Class II fusion peptides are found at the ends of a pair of anti-parallel β-strands, and are stabilized by disulfide bonds. A second loop (the “ij loop”), found at the tip of another set of anti-parallel β-strands and stabilized by disulfide bonds, appears to assist in membrane fusion, but does not extend as far as the fusion loop and may not interact directly with the target membrane. Unlike the fusion peptides of Class I fusion proteins, whose hallmark appears to be structural plasticity, the highly disulfide-stabilized fusion loops of Class II fusion proteins are not likely to change structure during the fusion process.
The fusion proteins of the alphaviruses SFV and SIN have been shown to have a specific requirement for cholesterol in the target membrane for fusion (reviewed in Kielian, 2006) and this requirement has been mapped to the ij loop of the SFV E1 protein (Gibbons et al., 2004). Cholesterol enhances fusion mediated by TBEV E and HCV E1/E2, but is not absolutely required (Corver et al., 2000; Stiasny et al., 2003; Teissier and Pecheur, 2007). SFV has an additional stereospecific requirement for sphingomyelin (Moesby et al., 1995). The cholesterol and sphingomyelin requirements do not, however, indicate a requirement for lipid rafts per se as analogs of these lipids that are refractory for fusion still form lipid rafts (Ahn et al., 2002) and, conversely, sphingomyelins that do not form lipid rafts still support fusion (Waarts et al., 2002).
Bipartate Internal Fusion Peptides of Class III Fusion Peptides
The first indications that the rhabdoviruses might harbor bipartite fusion peptides came from mutagenesis studies that identified two independent regions of the G proteins of VSV and VHSV that were important for cell-cell fusion (Rocha et al., 2004; Shokralla et al., 1999). However no direct lipid binding studies have been performed to corroborate these sequences as fusion peptides. The recently published structures of the pre- and post-fusion forms of VSV G and subsequent mutagenesis studies now confirm these predictions (Roche et al., 2006, 2007; Sun et al., 2008). By virtue of the similarity of the structure of HSV-1 gB (Heldwein et al., 2006) to that of VSV G (Roche et al., 2006, 2007), gB is now also classified as a Class III fusion protein, consistent with mutagenesis studies identifying bipartite fusion peptide activity (Backovic et al., 2007; Hannah et al., 2007). The fusion peptides of VSV G and HSV-1 gB are found at the tips of two loops formed by a three-stranded β-sheet and an unstructured strand. These fusion loops are stabilized by disulfide bonds similar to the fusion loops of Class II fusion proteins. In contrast to the ij loop of Class II proteins, both of the Class III loops are positioned such that they both could interact with the target membrane.
Although no structure of a bipartite fusion peptide in the presence of membranes is yet available, given the combined β-sheet and disulfide bond stabilization of the fusion peptide loops it seems unlikely that the structure of a bipartite (ClassIII) fusion peptide will be significantly altered in the presence of membranes. It will be interesting, but challenging, to see what the fusion loops of Class II and III fusion proteins look like in membranes.
Transmembrane Domains (TMDs)
Studies on the relevance of the transmembrane domains (TMDs) of viral fusion proteins are often complicated by the fact that changes within the TMD can affect the folding, trimerization, surface expression, and/or virion incorporation of the fusion protein. Nevertheless, it is clear that the TMD plays an important role in fusion. For example, replacement of the TMD of the X:31 HA and other fusion proteins with a GPI anchor blocks fusion at the hemifusion or small pore stage (Kemble et al., 1994; Markosyan et al., 2000; Melikyan et al., 1995; Tong and Compans, 2000). Further studies have indicated minimal length requirements for individual TMDs (Armstrong et al., 2000; Lin et al., 2003; Melikyan et al., 2000; West et al., 2001). In the case of X:31 HA, deletion of 12 residues from the TMD (Δ12HA) generated a hemifusion mutant reminiscent of GPI-HA. Full fusion was restored by the addition of a single Arg residue to the C-terminus of Δ12HA. The added Arg most likely allowed the shortened TMD to span the bilayer such that its guanido group could interact with phosphate head groups of the inner viral bilayer (Armstrong et al., 2000). Interestingly, many retroviral glycoproteins contain a conserved basic residue within the TMD that is important for fusion (Owens et al., 1994; Pietschmann et al., 2000; West et al., 2001). This residue is predicted to “snorkel” to the membrane interface and interact with phosphate head groups (West et al., 2001). Collectively, these studies suggest that the TMD must span the viral membrane bilayer and must be firmly anchored at the inner leaflet surface to support full fusion.
A comprehensive understanding of the TMD sequences needed to support fusion remains elusive. In some cases, specific TMD sequences do not appear to be critical for fusion. In these cases, domain swapping studies have shown that chimeras containing the TMDs from other viral fusion proteins and even those of nonfusogenic cell membrane proteins still support fusion (Kozerski et al., 2000). However, for reasons that are not yet clear, not all TMD chimeras are functional. In other cases, a need for specific residues within the TMD has been reported (Cleverley and Lenard, 1998; Dennison et al., 2002; Harman et al., 2002; Melikyan et al., 2000; Owens et al., 1994; Pietschmann et al., 2000; Taylor and Sanders, 1999). A possible role for conserved basic residues within certain retroviral Env TMDs has been described above. In several other cases, a requirement for helix disrupting residues (e.g., Pro or Gly) in the middle of a TMD has been observed (Cleverley and Lenard, 1998; Dennison et al., 2002; Harman et al., 2002; Melikyan et al., 2000; Taylor and Sanders, 1999). These residues have been proposed to be important for structural plasticity of the TMD during the fusion process. An alternative possibility is that specific residues are required to accommodate interactions with the fusion peptide during late stages of fusion.
Membrane Proximal Regions
Membrane Proximal Ectodomain Regions (MPERs)
Many Class I viral fusion proteins have unstructured regions between their fusion peptides and their N-terminal heptad repeat helix, and between their C-terminal heptad repeat helix and their TMD, that are important for fusion. The sequence between the C-terminal helix and the TMD is termed the membrane proximal ectodomain region (MPER). A clear role for this region of a fusion protein was first elucidated by showing that mutations of aromatic residues, which are found frequently in MPERs, abrogated HIV Env mediated fusion (Salzwedel et al., 1999). The mutations affected a late stage in fusion: conversion from a small to a large fusion pore (Munoz-Barroso et al., 1999). Using the “hydrophobic-at-interface” algorithm (White et al., 1998), MPER sequences have been predicted to partition into membrane interfaces. In keeping with this prediction, synthetic peptides corresponding to the HIV Env MPER partitioned into membrane interfaces (Schibli et al., 2001; Suarez et al., 2000) whereupon they formed α-helical structures and oligomerized (Schibli et al., 2001; Suarez et al., 2000; Sun et al., 2008). Fusion-defective mutant peptides bound membranes, but did not oligomerize. The importance of the HIV Env MPER is further highlighted by the finding that several neutralizing antibodies bind the HIV MPER (Lorizate et al., 2006; Ofek et al., 2004; Sanchez-Martinez et al., 2006) and by the realization that this is a major site of action of the HIV peptide inhibitor, T20 (Kliger et al., 2001). A current issue is the disposition of the MPER sequence in native lentivirus Env proteins, whether they are oligomeric and perpendicular to the viral membrane (Zanetti et al., 2006) or whether they splay as a tripod (Zhu et al., 2006). The latter possibility seems accordant with the need for membrane association of MPER sequences for reactivity with neutralizing antibodies (Dimitrov et al., 2007; Ofek et al., 2004; Sanchez-Martinez et al., 2006). Another question revolves around possible interactions between the MPER and the fusion peptide region (Bellamy-McIntyre et al., 2007; Lorizate et al., 2006).
The importance of MPERs of other viral fusion proteins has also been established. For example, mutations of MPER residues in FIV Env (Giannecchini et al., 2004), HPIV2 F (Tong et al., 2001) and SARS S (Howard et al., 2008) inhibit fusion. Peptides corresponding to the MPERs of the Ebola virus, FIV and SARS fusion proteins have been shown to require their aromatic residues for membrane destabilization (Giannecchini et al., 2004; Saez-Cirion et al., 2003; Sainz et al., 2005). The above proteins are all Class I fusion proteins, although similar requirements for aromatic residues in the VSV G and HSV-1 gH MPERs for fusion suggest that the effect is not limited to Class I fusion proteins (Galdiero et al., 2007; Jeetendra et al., 2003; Jeetendra et al., 2002). Interestingly an inverse correlation between the hydrophobicity of the MPER and that of the fusion loops of several herpes virus gB proteins has led to the hypothesis that hydrophobicity in MPER sequences may help compensate for less hydrophobic fusion peptides (Backovic et al., 2007).
Cytoplasmic Tails (CTs)
The cytoplasmic tails (CTs) of viral fusion proteins have a variety of functions that differ both among and within virus families. Therefore, no broad generalizations about these intraviral sequences can be made. Furthermore, results are often difficult to interpret because alterations in the CT can affect stability, surface expression, membrane domain localization, and virion incorporation of the fusion protein. Nevertheless, the CTs of many fusion proteins have direct effects on fusion.
The CT can influence the structure of the ectodomain (referred to as “inside-out” signaling) and thereby affect fusion (Aguilar et al., 2007; Earp et al., 2005; Kalia et al., 2005; Taylor and Sanders, 2003; Waning et al., 2002). A number of viruses have long CTs that must be proteolytically cleaved (by a viral protease) for fusion. CT cleavage may alter the ectodomain conformation in a manner that affects trimer stability (Plemper et al., 2002; Taylor and Sanders, 2003; Waning et al., 2002) or interaction with a companion protein (Aguilar et al., 2007; Murakami et al., 2004; Wyma et al., 2004), thereby lowering the energy barrier for fusion activation (Aguilar et al., 2007; Kalia et al., 2005; Plemper et al., 2002; Waning et al., 2002). In support of the latter hypothesis, cell-cell fusion was faster with an HIV Env that lacked a CT and this correlated with faster 6HB formation (Abrahamyan et al., 2005). Similarly, truncations (and mutations) in the CT of HSV gB correlated with altered syncytia formation (Ruel et al., 2006).
AFM studies have shown that immature HIV particles are stiffer than mature particles and that this difference is primarily mediated by the gp41 CT (Kol et al., 2007). Several possible reasons for this are suggested in the literature. The HIV CT appears to interact with the capsid protein gag, and gag cleavage during maturation is required for gp41-mediated fusion, suggesting that gp41-gag interactions may serve to prevent premature fusion activation for HIV (Wyma et al., 2004). The CT of the HIV-1 Env has three sequences that are predicted to form amphipathic α-helices. Two of these domains have been suggested to interact with membranes (Bultmann et al., 2001; Chen et al., 2001; Kliger et al., 1997; Wyss et al., 2005). Differences in the rate of fusion for HIV-1 and HIV-2 Envs have been correlated with differences in the rate of CD4-induced opening of the coreceptor binding site, which in turn, has been attributed to differences in the predicted CT helices of these two Envs (Gallo et al., 2006). Interestingly, the inhibition of HIV fusion by AME, a cholesterol-binding compound, has been mapped to the gp41 CT, and a genetic screen for resistance yielded a gp41 with a new viral protease site in the CT (Waheed et al., 2007). Perhaps the gp41 CT interacts specifically with cholesterol in the viral membrane through one or more of the α-helices described above, and this interaction helps modulate the fusion reaction.
There have been a number of reports of a role for membrane proximal regions of cytoplasmic tails in late stages of fusion similar to those for MPERs. For example, many viral fusion proteins have Cys residues in the membrane proximal regions of their cytoplasmic tails that become palmitoylated. Palmitoylation primarily affects localization of the fusion protein to specific plasma membrane domains and its incorporation into virus particles. However, palmitoylation has been shown to be important for the fusion function of SARS S (Petit et al., 2007) and some, but not all, subtypes of influenza HA (Chen et al., 2005; Jin et al., 1996; Sakai et al., 2002; Steinhauer et al., 1991; Wagner et al., 2005). Palmytoylation is apparently not needed for VSV G (Whitt and Rose, 1991), MLV Env (Yang and Compans, 1996), HIV gp41 (Chan et al., 2005), or for SIN E1 or E2 (Smit et al., 2001). Deletion of either the entire CT or the palmitoylation sites for HA (subtype H3) resulted in a decrease in the rate of transfer of large dyes, but not small dyes, and abolished “flickering,” the transient opening and closing of small pores (Melikyan et al., 1997). Similar results were obtained for the influenza B HA (Ujike et al., 2004) and the PIV5 F protein (Dutch and Lamb, 2001). The transition from small to large fusion pores is the most energy demanding step of fusion (Cohen and Melikyan, 2004); interaction of the membrane proximal region of the cytoplasmic tail with the inner viral membrane may help destabilize and/or dehydrate the viral membrane to minimize the energy required for pore enlargement, a role also proposed for MPER sequences.
SUMMARY
In conclusion, we hope that we have given the reader an appreciation for how highly diverse fusion proteins mediate a common pathway of membrane fusion. Although they employ similar general protein structures—a native fusion competent, often metastable, state, a membrane embedded trimeric prehairpin, and a final, lowest energy, trimer-of-hairpins conformation, and although they mediate fusion through a common set of steps—close membrane apposition, hemifusion, small fusion pores, and large fusion pores—the specific proteins that mediate fusion are highly divergent. They differ in sequence and detailed architecture, in how they are triggered for fusion, in their need for accessory proteins, in the type of fusion peptide they deploy, and in other features. It is fascinating that these diverse viral fusion proteins have apparently converged on a common solution to the membrane fusion problem.
Supplementary Material
ACKNOWLEDGMENTS
Our research was supported by NIH grants AI22470, R21AI055925, and U54 AI57168. We thank Christopher Broder, Robert Doms, Rebecca Dutch, Roselyn Eisenberg, Yves Gaudin, Henrik Garoff, Katja Heldwein, Margaret Kielian, and Lukas Tamm for helpful comments, and David Shultis for help with Figure 6.
REFERENCES
- Abrahamyan L, Markosyan R, Moore J, Cohen F, Melikyan G. Human immunodeficiency virus type 1 Env with an intersubunit disulfide bond engages coreceptors but requires bond reduction after engagement to induce fusion. J Virol. 2003;77:5829–5836. doi: 10.1128/JVI.77.10.5829-5836.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamyan L, Mkrtchyan S, Binley J, Lu M, Melikyan G, Cohen F. The cytoplasmic tail slows the folding of human immunodeficiency virus type 1 Env from a late prebundle configuration into the six-helix bundle. J Virol. 2005;79:106–115. doi: 10.1128/JVI.79.1.106-115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam B, Lins L, Stroobant V, Thomas A, Brasseur R. Distribution of hydrophobic residues is crucial for the fusogenic properties of the Ebola virus GP2 fusion peptide. J Virol. 2004;78:2131–2136. doi: 10.1128/JVI.78.4.2131-2136.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar HC, Matreyek KA, Choi DY, Filone CM, Young S, Lee B. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signaling. J Virol. 2007;81:4520–4532. doi: 10.1128/JVI.02205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar HC, Matreyek KA, Filone CM, Hashimi ST, Levroney EL, Negrete OA, Bertolotti-Ciarlet A, Choi DY, McHardy I, Fulcher JA, Su SV, Wolf MC, Kohatsu L, Baum LG, Lee B. N-Glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J Virol. 2006;80:4878–4889. doi: 10.1128/JVI.80.10.4878-4889.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn A, Gibbons D, Kielian M. The fusion peptide of Semliki Forest virus associates with sterol-rich membrane domains. J Virol. 2002;76:3267–3275. doi: 10.1128/JVI.76.7.3267-3275.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong R, Kushnir A, White J. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J Cell Biol. 2000;151:425–437. doi: 10.1083/jcb.151.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A. 2007;104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babel AR, Bruce J, Young JA. The hr1 and fusion peptide regions of the subgroup B avian sarcoma and leukosis virus envelope glycoprotein influence low pH-dependent membrane fusion. PLoS ONE. 2007;2:e171. doi: 10.1371/journal.pone.0000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backovic M, Jardetzky TS, Longnecker R. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein B are critical for fusion activity. J Virol. 2007;81:9596–9600. doi: 10.1128/JVI.00758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backovic M, Leser G, Lamb R, Longnecker R, Jardetzky T. Characterization of EBV gB indicates properties of both class I and class II viral fusion proteins. Virology. 2007;368:102–113. doi: 10.1016/j.virol.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar S, Takada A, Kawaoka Y, Alizon M. Detection of cell-cell fusion mediated by ebola virus glycoproteins. J Virol. 2006;80:2815–2822. doi: 10.1128/JVI.80.6.2815-2822.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy-McIntyre A, Lay C, Bäar S, Maerz A, Talbo G, Drummer H, Poumbourios P. Functional links between the fusion peptide-proximal polar segment and membrane-proximal region of human immunodeficiency virus gp41 in distinct phases of membrane fusion. J Biol Chem. 2007;282:23104–23116. doi: 10.1074/jbc.M703485200. [DOI] [PubMed] [Google Scholar]
- Beniac D, Devarennes S, Andonov A, He R, Booth T. Conformational Reorganization of the SARS coronavirus spike following receptor binding: Implications for membrane fusion. PLoS ONE. 2007;2:e1082. doi: 10.1371/journal.pone.0001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonnette ML, Connolly SA, Young DF, Randall RE, Paterson RG, Lamb RA. Analysis of the pH requirement for membrane fusion of different isolates of the paramyxovirus parainfluenza virus 5. J Virol. 2006;80:3071–3077. doi: 10.1128/JVI.80.6.3071-3077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal R, Sarkar DP, Durell S, Howard DE, Morris SJ. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrego-Diaz E, Peeples ME, Markosyan RM, Melikyan GB, Cohen FS. Completion of trimeric hairpin formation of influenza virus hemagglutinin promotes fusion pore opening and enlargement. Virology. 2003;316:234–244. doi: 10.1016/j.virol.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Bossart KN, Broder CC. Paramyxovirus entry. In: Pohlmann S, Simmons G, editors. Viral Entry into Host Cells. Landes Bioscience; Austin, TX: 2008. in press. [Google Scholar]
- Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. Embo J. 2004;23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Bultmann A, Muranyi W, Seed B, Haas J. Identification of two sequences in the cytoplasmic tail of the human immunodeficiency virus type 1 envelope glycoprotein that inhibit cell surface expression. J Virol. 2001;75:5263–5276. doi: 10.1128/JVI.75.11.5263-5276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzon V, Cladera J. Effect of cholesterol on the interaction of the HIV GP41 fusion peptide with model membranes. Importance of the membrane dipole potential. Biochemistry. 2006;45:15768–15775. doi: 10.1021/bi060622i. [DOI] [PubMed] [Google Scholar]
- Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- Chan WE, Lin HH, Chen SS. Wild-type-like viral replication potential of human immunodeficiency virus type 1 envelope mutants lacking palmitoylation signals. J Virol. 2005;79:8374–8387. doi: 10.1128/JVI.79.13.8374-8387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Sullivan N, Felbor U, Whelan S, Cunningham J. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanel-Vos C, Kielian M. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J Virol. 2004;78:13543–13552. doi: 10.1128/JVI.78.24.13543-13552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Vogan E, Gong H, Skehel J, Wiley D, Harrison S. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- Chen BJ, Takeda M, Lamb RA. Influenza virus hemagglutinin (H3 subtype) requires palmitoylation of its cytoplasmic tail for assembly: M1 proteins of two subtypes differ in their ability to support assembly. J Virol. 2005;79:13673–13684. doi: 10.1128/JVI.79.21.13673-13684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Skehel JJ, Wiley DC. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci USA. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SS, Lee SF, Wang CT. Cellular membrane-binding ability of the C-terminal cytoplasmic domain of human immunodeficiency virus type 1 envelope transmembrane protein gp41. J Virol. 2001;75:9925–9938. doi: 10.1128/JVI.75.20.9925-9938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SF, Wu CW, Kantchev EA, Chang DK. Structure and membrane interaction of the internal fusion peptide of avian sarcoma leukosis virus. Eur J Biochem. 2004;271:4725–4736. doi: 10.1111/j.1432-1033.2004.04436.x. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM. Membrane hemifusion: crossing a chasm in two leaps. Cell. 2005;123:375–382. doi: 10.1016/j.cell.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Zimmerberg J, Kozlov MM. Membranes of the world unite α. J Cell Biol. 2006;175:201–207. doi: 10.1083/jcb.200607083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JF, Chu JJ, Ng ML. The envelope glycoprotein domain III of dengue virus serotypes 1 and 2 inhibit virus entry. Microbes Infect. 2007;9:1–6. doi: 10.1016/j.micinf.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Chu VC, McElroy LJ, Chu V, Bauman BE, Whittaker GR. The avian coronavirus infectious bronchitis virus undergoes direct low-pH-dependent fusion activation during entry into host cells. J Virol. 2006;80:3180–3188. doi: 10.1128/JVI.80.7.3180-3188.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleverley DZ, Lenard J. The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc Natl Acad Sci USA. 1998;95:3425–3430. doi: 10.1073/pnas.95.7.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen FS, Melikyan GB. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol. 2004;199:1–14. doi: 10.1007/s00232-004-0669-8. [DOI] [PubMed] [Google Scholar]
- Connolly S, Leser G, Yin H, Jardetzky T, Lamb R. Refolding of a paramyxovirus F protein from pre-fusion to post-fusion conformations observed by liposome binding and electron microscopy. Proc Natl Acad Sci USA. 2006;103:17903–17908. doi: 10.1073/pnas.0608678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran J, Salsman J, de Antueno R, Touhami A, Jericho M, Clancy E, Duncan R. The p14 fusion-associated small transmembrane (FAST) protein effects membrane fusion from a subset of membrane microdomains. J Biol Chem. 2006;281:31778–31789. doi: 10.1074/jbc.M602566200. [DOI] [PubMed] [Google Scholar]
- Corey EA, Iorio RM. Mutations in the stalk of the measles virus hemagglutinin protein decrease fusion but do not interfere with virus-specific interaction with the homologous fusion protein. J Virol. 2007;81:9900–9910. doi: 10.1128/JVI.00909-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corver J, Ortiz A, Allison SL, Schalich J, Heinz FX, Wilschut J. Membrane fusion activity of tick-borne encephalitis virus and recombinant subviral particles in a liposomal model system. Virology. 2000;269:37–46. doi: 10.1006/viro.1999.0172. [DOI] [PubMed] [Google Scholar]
- Cousens C, Maeda N, Murgia C, Dagleish M, Palmarini M, Fan H. In vivo tumorigenesis by Jaagsiekte sheep retrovirus (JSRV) requires Y590 in Env TM, but not full-length orfX open reading frame. Virology. 2007;367:413–421. doi: 10.1016/j.virol.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crennell S, Takimoto T, Portner A, Taylor G. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat Struct Biol. 2000;7:1068–1074. doi: 10.1038/81002. [DOI] [PubMed] [Google Scholar]
- Damico RL, Crane J, Bates P. Receptor-triggered membrane association of a model retroviral glycoprotein. Proc Natl Acad Sci USA. 1998;95:2580–2585. doi: 10.1073/pnas.95.5.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delboy MG, Patterson JL, Hollander AM, Nicola AV. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol J. 2006;3:105. doi: 10.1186/1743-422X-3-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delos SE, Brecher MB, Chen Z, Melder DC, Federspiel MJ, White JM. Cysteines flanking the internal fusion peptide enable the avian sarcoma/leukisus virus glycoprotein to mediate the lipid mixing stage of fusion with high efficiency. J. Virol. 2008;82:3131–3134. doi: 10.1128/JVI.02266-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delos SE, Gilbert JM, White JM. The central proline of an internal viral fusion peptide serves two important roles. J Virol. 2000;74:1686–1693. doi: 10.1128/jvi.74.4.1686-1693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delos SE, Godby JA, White JM. Receptor-induced conformational changes in the SU subunit of the avian sarcoma/leukosis virus a envelope protein: Implications for fusion activation. J Virol. 2005;79:3488–3499. doi: 10.1128/JVI.79.6.3488-3499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delos SE, White JM. Critical role for the cysteines flanking the internal fusion peptide of avian sarcoma/leukosis virus envelope glycoprotein. J Virol. 2000;74:9738–9741. doi: 10.1128/jvi.74.20.9738-9741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennison SM, Greenfield N, Lenard J, Lentz BR. VSV transmembrane domain (TMD) peptide promotes PEG-mediated fusion of liposomes in a conformationally sensitive fashion. Biochemistry. 2002;41:14925–14934. doi: 10.1021/bi0203233. [DOI] [PubMed] [Google Scholar]
- Dimitrov AS, Jacobs A, Finnegan CM, Stiegler G, Katinger H, Blumenthal R. Exposure of the membrane-proximal external region of HIV-1 gp41 in the course of HIV-1 envelope glycoprotein-mediated fusion. Biochemistry. 2007;46:1398–1401. doi: 10.1021/bi062245f. [DOI] [PubMed] [Google Scholar]
- Duelli D, Lazebnik Y. Cell-to-cell fusion as a link between viruses and cancer. Nat Rev Cancer. 2007;7:968–976. doi: 10.1038/nrc2272. [DOI] [PubMed] [Google Scholar]
- Durell SR, Martin I, Ruysschaert JM, Shai Y, Blumenthal R. What studies of fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion (review) Mol Membr Biol. 1997;14:97–112. doi: 10.3109/09687689709048170. [DOI] [PubMed] [Google Scholar]
- Dutch RE, Lamb RA. Deletion of the cytoplasmic tail of the fusion protein of the paramyxovirus simian virus 5 affects fusion pore enlargement. J Virol. 2001;75:5363–5369. doi: 10.1128/JVI.75.11.5363-5369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earp LJ, Delos SE, Netter RC, Bates P, White JM. The avian retrovirus avian sarcoma/leukosis virus subtype A reaches the lipid mixing stage of fusion at neutral pH. J Virol. 2003;77:3058–3066. doi: 10.1128/JVI.77.5.3058-3066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earp LJ, Delos SE, Park HE, White JM. The many mechanisms of viral membrane fusion proteins. Curr Top Microbiol Immunol. 2005;285:25–66. doi: 10.1007/3-540-26764-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- Eifart P, Ludwig K, Bottcher C, de Haan CA, Rottier PJ, Korte T, Herrmann A. The role of endocytosis and low pH in cell entry of the murine hepatitis virus MHV-A59. J Virol. 2007;81:10758–10768. doi: 10.1128/JVI.00725-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Este JA, Telenti A. HIV entry inhibitors. Lancet. 2007;370:81–88. doi: 10.1016/S0140-6736(07)61052-6. [DOI] [PubMed] [Google Scholar]
- Fenouillet E, Barbouche R, Jones IM. Cell entry by enveloped viruses: redox considerations for HIV and SARS-coronavirus. Antioxid Redox Signal. 2007;9:1009–1034. doi: 10.1089/ars.2007.1639. [DOI] [PubMed] [Google Scholar]
- Ferlenghi I, Clarke M, Ruttan T, Allison SL, Schalich J, Heinz FX, Harrison SC, Rey FA, Fuller SD. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol Cell. 2001;7:593–602. doi: 10.1016/s1097-2765(01)00206-4. [DOI] [PubMed] [Google Scholar]
- Finnegan C, Berg W, Lewis G, DeVico A. Antigenic properties of the human immunodeficiency virus envelope during cell-cell fusion. J Virol. 2001;75:11096–11105. doi: 10.1128/JVI.75.22.11096-11105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follis K, York J, Nunberg J. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell-cell fusion but does not affect virion entry. Virology. 2006;350:358–369. doi: 10.1016/j.virol.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton AR, Jr., Stolz DB, Uchida H, Goins WF, Cohen JB, Glorioso JC. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J Virol. 2007;81:10879–10889. doi: 10.1128/JVI.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Mouland AJ. The cell biology of HIV-1 and other retroviruses. Retrovirology. 2006;3:77. doi: 10.1186/1742-4690-3-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas MS, Gaspar LP, Lorenzoni M, Almeida FC, Tinoco LW, Almeida MS, Maia LF, Degreve L, Valente AP, Silva JL. Structure of the Ebola fusion peptide in a membrane-mimetic environment and the interaction with lipid rafts. J Biol Chem. 2007;282:27306–27314. doi: 10.1074/jbc.M611864200. [DOI] [PubMed] [Google Scholar]
- Frey G, Rits-Volloch S, Zhang XQ, Schooley RT, Chen B, Harrison SC. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc Natl Acad Sci USA. 2006;103:13938–13943. doi: 10.1073/pnas.0601036103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta R, Wild C, Weng Y, Weiss C. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol. 1998;5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- Gabrys CM, Weliky DP. Chemical shift assignment and structural plasticity of a HIV fusion peptide derivative in dodecylphosphocholine micelles. Biochim Biophys Acta. 2007;1768:3225–3234. doi: 10.1016/j.bbamem.2007.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdiero S, Falanga A, Vitiello M, D’Isanto M, Collins C, Orrei V, Browne H, Pedone C, Galdiero M. Evidence for a role of the membrane-proximal region of herpes simplex virus Type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem. 2007;8:885–895. doi: 10.1002/cbic.200700044. [DOI] [PubMed] [Google Scholar]
- Gallo S, Finnegan C, Viard M, Raviv Y, Dimitrov A, Rawat S, Puri A, Durell S, Blumenthal R. The HIV Env-mediated fusion reaction. Biochim Biophys Acta. 2003;1614:36–50. doi: 10.1016/s0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- Gallo S, Puri A, Blumenthal R. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry. 2001;40:2231–12236. doi: 10.1021/bi0155596. [DOI] [PubMed] [Google Scholar]
- Gallo S, Reeves J, Garg H, Foley B, Doms R, Blumenthal R. Kinetic studies of HIV-1 and HIV-2 envelope glycoprotein-mediated fusion. Retrovirology. 2006;3:90. doi: 10.1186/1742-4690-3-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner A, Dutch R. A conserved region in the F(2) subunit of paramyxovirus fusion proteins is involved in fusion regulation. J Virol. 2007;81:8303–8314. doi: 10.1128/JVI.00366-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner A, Martin K, Dutch R. A conserved region between the heptad repeats of paramyxovirus fusion proteins is critical for proper F protein folding. Biochemistry. 2007;46:5094–5105. doi: 10.1021/bi6025648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg H, Joshi A, Freed E, Blumenthal R. Site-specific mutations in HIV-1 gp41 reveal a correlation between HIV-1-mediated bystander apoptosis and fusion/hemifusion. J Biol Chem. 2007;282:16899–16906. doi: 10.1074/jbc.M701701200. [DOI] [PubMed] [Google Scholar]
- Garry C, Garry R. Proteomics computational analyses suggest that the carboxyl terminal glycoproteins of Bunyaviruses are class II viral fusion protein (beta-penetrenes) Theor Biol Medi Model. 2004;1:10. doi: 10.1186/1742-4682-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin Y. Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell Biochem. 2000;34:379–408. doi: 10.1007/0-306-46824-7_10. [DOI] [PubMed] [Google Scholar]
- Giannecchini S, Bonci F, Pistello M, Matteucci D, Sichi O, Rovero P, Bendinelli M. The membrane-proximal tryptophan-rich region in the transmembrane glycoprotein ectodomain of feline immunodeficiency virus is important for cell entry. Virology. 2004;320:156–166. doi: 10.1016/j.virol.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Gibbons D, Ahn A, Liao M, Hammar L, Cheng R, Kielian M. Multistep regulation of membrane insertion of the fusion peptide of Semliki Forest virus. J Virol. 2004;78:3312–3318. doi: 10.1128/JVI.78.7.3312-3318.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons D, Vaney M, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey F. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 2004;427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- Gilbert JM, Hernandez LD, Balliet JW, Bates P, White JM. Receptor-induced conformational changes in the subgroup A avian leukosis and sarcoma virus envelope glycoprotein. J Virol. 1995;69:7410–7415. doi: 10.1128/jvi.69.12.7410-7415.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JM, Mason D, White JM. Fusion of Rous sarcoma virus with host cells does not require exposure to low pH. J Virol. 1990;64:5106–5113. doi: 10.1128/jvi.64.10.5106-5113.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glebe D, Urban S. Viral and cellular determinants involved in hepadnaviral entry. World J Gastroenterol. 2007;13:22–38. doi: 10.3748/wjg.v13.i1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godley L, Pfeifer J, Steinhauer D, Ely B, Shaw G, Kaufmann R, Suchanek E, Pabo C, Skehel JJ, Wiley DC, Wharton S. Introduction of intersubunit disulfide bonds in the membranedistal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell. 1992;68:635–645. doi: 10.1016/0092-8674(92)90140-8. [DOI] [PubMed] [Google Scholar]
- Gomara MJ, Mora P, Mingarro I, Nieva JL. Roles of a conserved proline in the internal fusion peptide of Ebola glycoprotein. FEBS Lett. 2004;569:261–266. doi: 10.1016/j.febslet.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat Struct Biol. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J Virol. 2007;81:4858–4865. doi: 10.1128/JVI.02755-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman A, Browne H, Minson T. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J Virol. 2002;76:10708–10716. doi: 10.1128/JVI.76.21.10708-10716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SC. Mechanism of membrane fusion by viral envelope proteins. Adv Virus Res. 2005;64:231–261. doi: 10.1016/S0065-3527(05)64007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Vassell R, Zaitseva M, Nguyen N, Yang Z, Weng Y, Weiss C. Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J Virol. 2003;77:1666–1671. doi: 10.1128/JVI.77.3.1666-1671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- Hernandez L, Peters R, Delos S, Young J, Agard D, White J. Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J Cell Biol. 1997;139:1455–1464. doi: 10.1083/jcb.139.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, White J. Mutational analysis of the candidate internal fusion peptide of the avian leukosis and sarcoma virus subgroup A envelope glycoprotein. J Virol. 1998;72:3259–3267. doi: 10.1128/jvi.72.4.3259-3267.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MW, Travanty EA, Jeffers SA, Smith MK, Wennier ST, Thackray LB, Holmes KV. Aromatic amino acids in the juxtamembrane domain of severe acute respiratory syndrome coronavirus spike glycoprotein are important for receptor-dependent virus entry and cell-cell fusion. J Virol. 2008;82:2883–2894. doi: 10.1128/JVI.01805-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MW, Tripet B, Jobling MG, Holmes RK, Holmes KV, Hodges RS. Dissection of the fusion machine of SARS-coronavirus. Adv Exp Med Biol. 2006;581:319–322. doi: 10.1007/978-0-387-33012-9_56. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrobowski YM, Garry RF, Michael SF. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol J. 2005;2:49. doi: 10.1186/1743-422X-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lam SN, Acharya P, Tang M, Xiang SH, Hussan SS, Stanfield RL, Robinson J, Sodroski J, Wilson IA, Wyatt R, Bewley CA, Kwong PD. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Watanabe S, Sanchez A, Whitt MA, Kawaoka Y. Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J Virol. 1999;73:8907–8912. doi: 10.1128/jvi.73.10.8907-8912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs-engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Jaroniec CP, Kaufman JD, Stahl SJ, Viard M, Blumenthal R, Wingfield PT, Bax A. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005;44:16167–16180. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- Jeetendra E, Ghosh K, Odell D, Li J, Ghosh HP, Whitt MA. The membrane-proximal region of vesicular stomatitis virus glycoprotein G ectodomain is critical for fusion and virus infectivity. J Virol. 2003;77:12807–12818. doi: 10.1128/JVI.77.23.12807-12818.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeetendra E, Robison CS, Albritton LM, Whitt MA. The membrane-proximal domain of vesicular stomatitis virus G protein functions as a membrane fusion potentiator and can induce hemifusion. J Virol. 2002;76:12300–12311. doi: 10.1128/JVI.76.23.12300-12311.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers SA, Sanders DA, Sanchez A. Covalent modifications of the ebola virus glycoprotein. J Virol. 2002;76:12463–12472. doi: 10.1128/JVI.76.24.12463-12472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Subbarao K, Bagai S, Leser GP, Murphy BR, Lamb RA. Palmitylation of the influenza virus hemagglutinin (H3) is not essential for virus assembly or infectivity. J Virol. 1996;70:1406–1414. doi: 10.1128/jvi.70.3.1406-1414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SB, Dutch RE, Lamb RA. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology. 1998;248:20–34. doi: 10.1006/viro.1998.9242. [DOI] [PubMed] [Google Scholar]
- Kaletsky R, Simmons G, Bates P. Proteolysis of the Ebola glycoproteins enhances virus binding and infectivity. J Virol. 2007;81:13378–13384. doi: 10.1128/JVI.01170-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Gupta P, Montelaro RC. Antibody neutralization escape mediated by point mutations in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41. J Virol. 2005;79:2097–2107. doi: 10.1128/JVI.79.4.2097-2107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann T, Mueller DS, Mark AE, Young PR, Kobe B. The Role of histidine residues in low-pH-mediated viral membrane fusion. Structure. 2006;14:1481–1487. doi: 10.1016/j.str.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Kemble GW, Bodian DL, Rose J, Wilson IA, White JM. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J Virol. 1992;66:4940–4950. doi: 10.1128/jvi.66.8.4940-4950.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble GW, Danieli T, White JM. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- Kielian M. Class II virus membrane fusion proteins. Virology. 2006;344:38–47. doi: 10.1016/j.virol.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Kielian M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol. 2006;4:67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- Klewitz C, Klenk H, ter Meulen J. Amino acids from both N-terminal hydrophobic regions of the Lassa virus envelope glycoprotein GP-2 are critical for pH-dependent membrane fusion and infectivity. J Gen Virol. 2007;88:2320–2328. doi: 10.1099/vir.0.82950-0. [DOI] [PubMed] [Google Scholar]
- Kliger Y, Aharoni A, Rapaport D, Jones P, Blumenthal R, Shai Y. Fusion peptides derived from the HIV type 1 glycoprotein 41 associate within phospholipid membranes and inhibit cell-cell Fusion. Structure-function study. J Biol Chem. 1997;272:13496–13505. doi: 10.1074/jbc.272.21.13496. [DOI] [PubMed] [Google Scholar]
- Kliger Y, Gallo SA, Peisajovich SG, Munoz-Barroso I, Avkin S, Blumenthal R, Shai Y. Mode of action of an antiviral peptide from HIV-1. Inhibition at a post-lipid mixing stage. J Biol Chem. 2001;276:1391–1397. doi: 10.1074/jbc.M004113200. [DOI] [PubMed] [Google Scholar]
- Kochan G, Escors D, González J, Casasnovas J, Esteban M. Membrane cell fusion activity of the vaccinia virus A17-A27 protein complex. Cell Microbiol. 2007;10:149–164. doi: 10.1111/j.1462-5822.2007.01026.x. [DOI] [PubMed] [Google Scholar]
- Kol N, Shi Y, Tsvitov M, Barlam D, Shneck RZ, Kay MS, Rousso I. A stiffness switch in human immunodeficiency virus. Biophys J. 2007;92:1777–1783. doi: 10.1529/biophysj.106.093914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp A, Blewett E, Misra V, Mettenleiter T. Proteolytic cleavage of bovine herpesvirus 1 (BHV-1) glycoprotein gB is not necessary for its function in BHV-1 or pseudorabies virus. J Virol. 1994;68:1667–1674. doi: 10.1128/jvi.68.3.1667-1674.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozerski C, Ponimaskin E, Schroth-Diez B, Schmidt MF, Herrmann A. Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J Virol. 2000;74:7529–7537. doi: 10.1128/jvi.74.16.7529-7537.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov MM, Chernomordik LV. The protein coat in membrane fusion: lessons from fission. Traffic. 2002;3:256–267. doi: 10.1034/j.1600-0854.2002.030403.x. [DOI] [PubMed] [Google Scholar]
- Krummenacher C, Carfi A, Eisenberg RJ, Cohen GH. Entry of herpesviruses into cells: the enigma variations. In: Pohlmann S, Simmons G, editors. Viral Entry into Host Cells. Landes Bioscience; Austin, TX: 2008. in press. [Google Scholar]
- Krummenacher C, Supekar V, Whitbeck J, Lazear E, Connolly S, Eisenberg R, Cohen G, Wiley D, Carfí A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Nachagari D, Fields C, Franks J, Albritton LM. Host cell cathepsins potentiate Moloney murine leukemia virus infection. J Virol. 2007;81:10506–10514. doi: 10.1128/JVI.02853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong P. Human immunodeficiency virus: refolding the envelope. Nature. 2005;433:815–816. doi: 10.1038/433815a. [DOI] [PubMed] [Google Scholar]
- Kwong P, Wyatt R, Robinson J, Sweet R, Sodroski J, Hendrickson W. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AL, Park H, White JM, Tamm LK. Fusion peptide of influenza hemagglutinin requires a fixed angle boomerang structure for activity. J Biol Chem. 2006;281:5760–5770. doi: 10.1074/jbc.M512280200. [DOI] [PubMed] [Google Scholar]
- Lai AL, Tamm LK. Locking the kink in the influenza hemagglutinin fusion domain structure. J Biol Chem. 2007;282:23946–23956. doi: 10.1074/jbc.M704008200. [DOI] [PubMed] [Google Scholar]
- Lamb R. Paramyxovirus fusion: a hypothesis for changes. Virology. 1993;197:1–11. doi: 10.1006/viro.1993.1561. [DOI] [PubMed] [Google Scholar]
- Lamb RA, Jardetzky TS. Structural basis of viral invasion: lessons from paramyxovirus F. Curr Opin Struct Biol. 2007;17:427–436. doi: 10.1016/j.sbi.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillette D, Maurice M, Roche C, Russell SJ, Sitbon M, Cosset FL. A proline-rich motif downstream of the receptor binding domain modulates conformation and fusogenicity of murine retroviral envelopes. J Virol. 1998;72:9955–9965. doi: 10.1128/jvi.72.12.9955-9965.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillette D, Pécheur E, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, Cosset F. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol. 2007;81:8752–8765. doi: 10.1128/JVI.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Borg N, Streltsov V, Pilling P, Epa V, Varghese J, McKimm-Breschkin J, Colman P. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus typeIII. J Mol Biol. 2004;335:1343–1357. doi: 10.1016/j.jmb.2003.11.032. [DOI] [PubMed] [Google Scholar]
- Lee JK, Prussia A, Paal T, White LK, Snyder JP, Plemper RK. Functional interaction between paramyxovirus fusion and attachment proteins. J Biol Chem. 2008 doi: 10.1074/jbc.M801018200. Epub, accession # 18426797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Wengler G, Rey FA. The Fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105:137–148. doi: 10.1016/s0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- Li F, Berardi M, Li W, Farzan M, Dormitzer P, Harrison S. Conformational states of the severe acute respiratory syndrome coronavirus spike protein ectodomain. J Virol. 2006;80:6794–6800. doi: 10.1128/JVI.02744-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tong S, Lee HB, Perdigoto AL, Spangenberg HC, Wands JR. Glycine decarboxylase mediates a postbinding step in duck hepatitis B virus infection. J Virol. 2004;78:1873–1881. doi: 10.1128/JVI.78.4.1873-1881.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Zhang S, Kronqvist M, Ekström M, Wallin M, Garoff H. The conserved His8 of the Moloney murine leukemia virus Env SU subunit directs the activity of the SU-TM disulphide bond isomerase. Virology. 2007;361:149–160. doi: 10.1016/j.virol.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Li Y, Han X, Lai AL, Bushweller JH, Cafiso DS, Tamm LK. Membrane structures of the hemifusion-inducing fusion peptide mutant G1S and the fusion-blocking mutant G1V of influenza virus hemagglutinin suggest a mechanism for pore opening in membrane fusion. J Virol. 2005;79:12065–12076. doi: 10.1128/JVI.79.18.12065-12076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tamm LK. Structure and plasticity of the human immunodeficiency virus gp41 fusion domain in lipid micelles and bilayers. Biophys J. 2007;93:876–885. doi: 10.1529/biophysj.106.102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Kielian M. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus membrane fusion. J Cell Biol. 2005;171:111–120. doi: 10.1083/jcb.200507075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin E, Spear P. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc Natl Acad Sci USA. 2007;104:13140–13145. doi: 10.1073/pnas.0705926104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Derdeyn CA, Blumenthal R, West J, Hunter E. Progressive truncations C terminal to the membrane-spanning domain of simian immunodeficiency virus Env reduce fusogenicity and increase concentration dependence of Env for fusion. J Virol. 2003;77:7067–7077. doi: 10.1128/JVI.77.12.7067-7077.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wu S, Jiang S. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr Pharm Des. 2007;13:143–162. doi: 10.2174/138161207779313722. [DOI] [PubMed] [Google Scholar]
- Lorizate M, Gomara MJ, de la Torre BG, Andreu D, Nieva JL. Membrane-transferring sequences of the HIV-1 Gp41 ectodomain assemble into an immunogenic complex. J Mol Biol. 2006;360:45–55. doi: 10.1016/j.jmb.2006.04.056. [DOI] [PubMed] [Google Scholar]
- Luque L, Russell C. Spring-loaded heptad repeat residues regulate the expression and activation of paramyxovirus fusion protein. J Virol. 2007;81:3130–3141. doi: 10.1128/JVI.02464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenz C, Chang S, Iwanski A, Bruns M. Entry of duck hepatitis B virus into primary duck liver and kidney cells after discovery of a fusogenic region within the large surface protein. J Virol. 2007;81:5014–5023. doi: 10.1128/JVI.02290-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerz A, Center R, Kemp B, Kobe B, Poumbourios P. Functional implications of the human T-lymphotropic virus type 1 transmembrane glycoprotein helical hairpin structure. J Virol. 2000;74:6614–6621. doi: 10.1128/jvi.74.14.6614-6621.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerz A, Drummer H, Wilson K, Poumbourios P. Functional analysis of the disulfide-bonded loop/chain reversal region of human immunodeficiency virus type 1 gp41 reveals a critical role in gp120-gp41 association. J Virol. 2001;75:6635–6644. doi: 10.1128/JVI.75.14.6635-6644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier C, Delagrave S, Zhang Z, Brown N, Monath T, Pugachev K, Guirakhoo F. A single M protein mutation affects the acid inactivation threshold and growth kinetics of a chimeric flavivirus. Virology. 2007;362:468–474. doi: 10.1016/j.virol.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Mancini EJ, Clarke M, Gowen BE, Rutten T, Fuller SD. Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki Forest virus. Mol Cell. 2000;5:255–266. doi: 10.1016/s1097-2765(00)80421-9. [DOI] [PubMed] [Google Scholar]
- Markosyan RM, Cohen FS, Melikyan GB. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol Biol Cell. 2003;14:926–938. doi: 10.1091/mbc.E02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan RM, Cohen FS, Melikyan GB. The lipidanchored ectodomain of influenza virus hemagglutinin (GPI-HA) Is capable of Inducing nonenlarging fusion pores. Mol. Biol. Cell. 2000;11:1143–1152. doi: 10.1091/mbc.11.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Defrise-Quertain F, Decroly E, Vandenbranden M, Brasseur R, Ruysschaert JM. Orientation and structure of the NH2-terminal HIV-1 gp41 peptide in fused and aggregated liposomes. Biochim Biophys Acta. 1993;1145:124–133. doi: 10.1016/0005-2736(93)90389-h. [DOI] [PubMed] [Google Scholar]
- Martin I, Ruysschaert JM. Common properties of fusion peptides from diverse systems. Biosci Rep. 2000;20:483–500. doi: 10.1023/A:1010454803579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Delos SE, White JM. Sequential roles of receptor binding and low pH in forming prehairpin and hairpin conformations of a retroviral envelope glycoprotein. J Virol. 2004;78:8201–8209. doi: 10.1128/JVI.78.15.8201-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Taguchi F. Receptor-induced conformational changes of murine coronavirus spike protein. J Virol. 2002;76:11819–11826. doi: 10.1128/JVI.76.23.11819-11826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci USA. 2005;102:12543–12547. doi: 10.1073/pnas.0503203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure MO, Sommerfelt MA, Marsh M, Weiss RA. The pH independence of mammalian retrovirus infection. J Gen Virol. 1990;71:767–773. doi: 10.1099/0022-1317-71-4-767. [DOI] [PubMed] [Google Scholar]
- McShane M, Longnecker R. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc Natl Acad Sci USA. 2004;101:17474–17479. doi: 10.1073/pnas.0404535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melanson VR, Iorio RM. Amino acid substitutions in the F-specific domain in the stalk of the newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J Virol. 2004;78:13053–13061. doi: 10.1128/JVI.78.23.13053-13061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan G, Barnard R, Abrahamyan L, Mothes W, Young J. Imaging individual retroviral fusion events: from hemifusion to pore formation and growth. Proc Natl Acad Sci USA. 2005;102:8728–8733. doi: 10.1073/pnas.0501864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan G, Barnard R, Markosyan R, Young J, Cohen F. Low pH is required for avian sarcoma and leukosis virus Env-induced hemifusion and fusion pore formation but not for pore growth. J Virol. 2004;78:3753–3762. doi: 10.1128/JVI.78.7.3753-3762.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan G, Markosyan R, Hemmati H, Delmedico M, Lambert D, Cohen F. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151:413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Jin H, Lamb RA, Cohen FS. The role of the cytoplasmic tail region of influenza virus hemagglutinin in formation and growth of fusion pores. Virology. 1997;235:118–128. doi: 10.1006/viro.1997.8686. [DOI] [PubMed] [Google Scholar]
- Melikyan GB, Markosyan RM, Brener SA, Rozenberg Y, Cohen FS. Role of the cytoplasmic tail of ecotropic moloney murine leukemia virus Env protein in fusion pore formation. J Virol. 2000;74:447–455. doi: 10.1128/jvi.74.1.447-455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Markosyan RM, Roth MG, Cohen FS. A point mutation in the transmembrane domain of the hemagglutinin of influenza virus stabilizes a hemifusion intermediate that can transit to fusion. Mol Biol Cell. 2000;11:3765–3775. doi: 10.1091/mbc.11.11.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, White JM, Cohen FS. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J Cell Biol. 1995;131:679–691. doi: 10.1083/jcb.131.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol. 2005;79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mkrtchyan S, Markosyan R, Eadon M, Moore J, Melikyan G, Cohen F. Ternary complex formation of human immunodeficiency virus type 1 Env, CD4, and chemokine receptor captured as an intermediate of membrane fusion. J Virol. 2005;79:11161–11169. doi: 10.1128/JVI.79.17.11161-11169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moesby L, Corver J, Erukulla RK, Bittman R, Wilschut J. Sphingolipids activate membrane fusion of Semliki Forest virus in a stereospecific manner. Biochemistry. 1995;34:10319–10324. doi: 10.1021/bi00033a001. [DOI] [PubMed] [Google Scholar]
- Moore J, Doms R. The entry of entry inhibitors: a fusion of science and medicine. Proc Natl Acad Sci USA. 2003;100:10598–10602. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison T. Structure and function of a paramyxovirus fusion protein. Biochim Biophys Acta. 2003;1614:73–84. doi: 10.1016/s0005-2736(03)00164-0. [DOI] [PubMed] [Google Scholar]
- Mothes W, Boerger AL, Narayan S, Cunningham JM, Young JA. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell. 2000;103:679–689. doi: 10.1016/s0092-8674(00)00170-7. [DOI] [PubMed] [Google Scholar]
- Munch J, Standker L, Adermann K, Schulz A, Schindler M, Chinnadurai R, Pohlmann S, Chaipan C, Biet T, Peters T, Meyer B, Wilhelm D, Lu H, Jing W, Jiang S, Forssmann WG, Kirchhoff F. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell. 2007;129:263–275. doi: 10.1016/j.cell.2007.02.042. [DOI] [PubMed] [Google Scholar]
- Munoz-Barroso I, Salzwedel K, Hunter E, Blumenthal R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J Virol. 1999;73:6089–6092. doi: 10.1128/jvi.73.7.6089-6092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Ablan S, Freed EO, Tanaka Y. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol. 2004;78:1026–1031. doi: 10.1128/JVI.78.2.1026-1031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan S, Barnard RJ, Young JA. Two retroviral entry pathways distinguished by lipid raft association of the viral receptor and differences in viral infectivity. J Virol. 2003;77:1977–1983. doi: 10.1128/JVI.77.3.1977-1983.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netter R, Amberg S, Balliet J, Biscone M, Vermeulen A, Earp L, White J, Bates P. Heptad repeat 2-based peptides inhibit avian sarcoma and leukosis virus subgroup a infection and identify a fusion intermediate. J Virol. 2004;78:13430–13439. doi: 10.1128/JVI.78.24.13430-13439.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G, Feldmann H, Watanabe S, Lukashevich I, Kawaoka Y. Reverse genetics demonstrates that proteolytic processing of the Ebola virus glycoprotein is not essential for replication in cell culture. J Virol. 2002;76:406–410. doi: 10.1128/JVI.76.1.406-410.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G, Geisbert T, Ebihara H, Geisbert J, Daddario-DiCaprio K, Feldmann H, Kawaoka Y. Proteolytic processing of the Ebola virus glycoprotein is not critical for Ebola virus replication in nonhuman primates. J Virol. 2007;81:2995–2998. doi: 10.1128/JVI.02486-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, McEvoy AM, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol. 2003;77:5324–53032. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieva JL, Nir S, Muga A, Goni FM, Wilschut J. Interaction of the HIV-1 fusion peptide with phospholipid vesicles: different structural requirements for fusion and leakage. Biochemistry. 1994;33:3201–3209. doi: 10.1021/bi00177a009. [DOI] [PubMed] [Google Scholar]
- Niles WD, Cohen FS. Single event recording shows that docking onto receptor alters the kinetics of membrane fusion mediated by influenza hemagglutinin. Biophys J. 1993;65:171–176. doi: 10.1016/S0006-3495(93)81049-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchi M, Ohuchi R, Sakai T, Matsumoto A. Tight binding of influenza virus hemagglutinin to its receptor interferes with fusion pore dilation. J Virol. 2002;76:12405–12413. doi: 10.1128/JVI.76.24.12405-12413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opstelten DJ, Wallin M, Garoff H. Moloney murine leukemia virus envelope protein subunits, gp70 and Pr15E, form a stable disulfide-linked complex. J Virol. 1998;72:6537–6545. doi: 10.1128/jvi.72.8.6537-6545.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens RJ, Burke C, Rose JK. Mutations in the membrane-spanning domain of the human immunodeficiency virus envelope glycoprotein that affect fusion activity. J Virol. 1994;68:570–574. doi: 10.1128/jvi.68.1.570-574.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pager CT, Craft WW, Jr., Patch J, Dutch RE. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology. 2006;346:251–257. doi: 10.1016/j.virol.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pager CT, Dutch RE. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J Virol. 2005;79:12714–12720. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HE, Gruenke JA, White JM. Leash in the groove mechanism of membrane fusion. Nat Struct Biol. 2003;10:1048–1053. doi: 10.1038/nsb1012. [DOI] [PubMed] [Google Scholar]
- Peisajovich SG, Epand RF, Epand RM, Shai Y. Sendai virus N-terminal fusion peptide consists of two similar repeats, both of which contribute to membrane fusion. Eur J Biochem. 2002;269:4342–4350. doi: 10.1046/j.1432-1033.2002.03132.x. [DOI] [PubMed] [Google Scholar]
- Perez LG, Hunter E. Mutations within the proteolytic cleavage site of the Rous sarcoma virus glycoprotein that block processing to gp85 and gp37. J Virol. 1987;61:1609–1614. doi: 10.1128/jvi.61.5.1609-1614.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit CM, Chouljenko VN, Iyer A, Colgrove R, Farzan M, Knipe DM, Kousoulas KG. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology. 2007;360:264–274. doi: 10.1016/j.virol.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann T, Zentgraf H, Rethwilm A, Lindemann D. An evolutionarily conserved positively charged amino acid in the putative membrane-spanning domain of the foamy virus envelope protein controls fusion activity. J Virol. 2000;74:4474–4482. doi: 10.1128/jvi.74.10.4474-4482.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter A, Kopelman R, Li Z, Kayman SC, Sanders DA. Localization of the labile disulfide bond between SU and TM of the murine leukemia virus envelope protein complex to a highly conserved CWLC motif in SU that resembles the activesite sequence of thiol-disulfide exchange enzymes. J Virol. 1997;71:8073–8077. doi: 10.1128/jvi.71.10.8073-8077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Durnin JP, Kabat D. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J Virol. 2005;79:4347–4356. doi: 10.1128/JVI.79.7.4347-4356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Durnin JP, Shinde U, Kabat D. An allosteric rheostat in HIV-1 gp120 reduces CCR5 stoichiometry required for membrane fusion and overcomes diverse entry limitations. J Mol Biol. 2007;374:64–79. doi: 10.1016/j.jmb.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattet P, Cherpillod P, Wiener D, Zipperle L, Vandevelde M, Wittek R, Zurbriggen A. Signal peptide and helical bundle domains of virulent canine distemper virus fusion protein restrict fusogenicity. J Virol. 2007;81:11413–11425. doi: 10.1128/JVI.01287-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper R, Hammond A, Gerlier D, Fielding A, Cattaneo R. Strength of envelope protein interaction modulates cytopathicity of measles virus. J Virol. 2002;76:5051–5061. doi: 10.1128/JVI.76.10.5051-5061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbilewicz B, Leikina E, Sapir A, Valansi C, Suissa M, Shemer G, Chernomordik LV. The C. elegans developmental fusogen EFF-1 mediates homotypic fusion in heterologous cells and in vivo. Dev Cell. 2006;11:471–481. doi: 10.1016/j.devcel.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Pohlmann S, Reeves JD. Cellular entry of HIV: Evaluation of therapeutic targets. Curr Pharm Des. 2006;12:1963–1973. doi: 10.2174/138161206777442155. [DOI] [PubMed] [Google Scholar]
- Porotto M, Fornabaio M, Kellogg G, Moscona A. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J Virol. 2007;81:3216–3228. doi: 10.1128/JVI.02617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poumbourios P, Drummer H. Recent advances in our understanding of receptor binding, viral fusion and cell entry of hepatitis C virus: new targets for the design of antiviral agents. Antivir Chem Chemother. 2007;18:169–189. doi: 10.1177/095632020701800402. [DOI] [PubMed] [Google Scholar]
- Qiao H, Armstrong RT, Melikyan GB, Cohen FS, White JM. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol Biol Cell. 1999;10:2759–2769. doi: 10.1091/mbc.10.8.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao H, Pelletier S, Hoffman L, Hacker J, Armstrong RT, White JM. Specific single or double proline substitutions in the “spring-loaded” coiled-coil region of the influenza hemagglutinin impair or abolish membrane fusion activity. J Cell Biol. 1998;141:1335–1347. doi: 10.1083/jcb.141.6.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Hingley ST, Simmons G, Yu C, Das Sarma J, Bates P, Weiss SR. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J Virol. 2006;80:5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachakonda PS, Veit M, Korte T, Ludwig K, Bottcher C, Huang Q, Schmidt MF, Herrmann A. The relevance of salt bridges for the stability of the influenza virus hemagglutinin. Faseb J. 2007;21:995–1002. doi: 10.1096/fj.06-7052hyp. [DOI] [PubMed] [Google Scholar]
- Rafalski M, Lear JD, DeGrado WF. Phospholipid interactions of synthetic peptides representing the N-terminus of HIV gp41. Biochemistry. 1990;29:7917–7922. doi: 10.1021/bi00486a020. [DOI] [PubMed] [Google Scholar]
- Reichert J, Grasnick D, Afonin S, Buerck J, Wadhwani P, Ulrich AS. A critical evaluation of the conformational requirements of fusogenic peptides in membranes. Eur Biophys J. 2007;36:405–413. doi: 10.1007/s00249-006-0106-2. [DOI] [PubMed] [Google Scholar]
- Rey F. Molecular gymnastics at the herpesvirus surface. EMBO Rep. 2006;7:1000–1005. doi: 10.1038/sj.embor.7400807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- Rocha A, Ruiz S, Tafalla C, Coll JM. Conformation- and fusion-defective mutations in the hypothetical phospholipid-binding and fusion peptides of viral hemorrhagic septicemia salmonid rhabdovirus protein G. J Virol. 2004;78:9115–9122. doi: 10.1128/JVI.78.17.9115-9122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- Roche S, Rey FA, Gaudin Y, Bressanelli S. Structure of the pre-fusion form of the vesicular stomatitis virus glycoprotein G. Science. 2007;315:843–848. doi: 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- Ross SR, Schofield JJ, Farr CJ, Bucan M. Mouse transferrin receptor 1 is the cell entry receptor for mouse mammary tumor virus. Proc Natl Acad Sci USA. 2002;99:12386–12390. doi: 10.1073/pnas.192360099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruel N, Zago A, Spear PG. Alanine substitution of conserved residues in the cytoplasmic tail of herpes simplex virus gB can enhance or abolish cell fusion activity and viral entry. Virology. 2006;346:229–237. doi: 10.1016/j.virol.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Ruiz-Arguello MB, Goni FM, Pereira FB, Nieva JL. Phosphatidylinositol-dependent membrane fusion induced by a putative fusogenic sequence of Ebola virus. J Virol. 1998;72:1775–1781. doi: 10.1128/jvi.72.3.1775-1781.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell C, Luque L. The structural basis of paramyxovirus invasion. Trends Microbiol. 2006;14:243–246. doi: 10.1016/j.tim.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. Embo J. 2001;20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J Cell Biol. 2003;163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RJ, Gamblin SJ, Haire LF, Stevens DJ, Xiao B, Ha Y, Skehel JJ. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology. 2004;325:287–296. doi: 10.1016/j.virol.2004.04.040. [DOI] [PubMed] [Google Scholar]
- Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol. 2006;80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Cirion A, Gomara MJ, Agirre A, Nieva JL. Pre-transmembrane sequence of Ebola glycoprotein. Interfacial hydrophobicity distribution and interaction with membranes. FEBS Lett. 2003;533:47–53. doi: 10.1016/s0014-5793(02)03747-x. [DOI] [PubMed] [Google Scholar]
- Sainz B, Jr., Rausch JM, Gallaher WR, Garry RF, Wimley WC. The aromatic domain of the coronavirus class I viral fusion protein induces membrane permeabilization: putative role during viral entry. Biochemistry. 2005;44:947–958. doi: 10.1021/bi048515g. [DOI] [PubMed] [Google Scholar]
- Sakai T, Ohuchi R, Ohuchi M. Fatty acids on the A/USSR/77 influenza virus hemagglutinin facilitate the transition from hemifusion to fusion pore formation. J Virol. 2002;76:4603–4611. doi: 10.1128/JVI.76.9.4603-4611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzwedel K, West JT, Hunter E. A conserved tryptophanrich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Envmediated fusion and virus infectivity. J Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Martinez S, Lorizate M, Hermann K, Kunert R, Basanez G, Nieva JL. Specific phospholipid recognition by human immunodeficiency virus type-1 neutralizing anti-gp41 2F5 antibody. FEBS Lett. 2006;580:2395–2399. doi: 10.1016/j.febslet.2006.03.067. [DOI] [PubMed] [Google Scholar]
- Sanders D. Sulfhydryl involvement in fusion mechanisms. In: Fuller HA, editor. Fusion of Biological Membranes and Related Problems. Vol. 34. Kluwer Academic/Plenum Publishers; New York: 2000. [Google Scholar]
- Saunders AA, Ting JP, Meisner J, Neuman BW, Perez M, de la Torre JC, Buchmeier MJ. Mapping the landscape of the lymphocytic choriomeningitis virus stable signal peptide reveals novel functional domains. J Virol. 2007;81:5649–5657. doi: 10.1128/JVI.02759-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibli DJ, Montelaro RC, Vogel HJ. The membrane-proximal tryptophan-rich region of the HIV glycoprotein, gp41, forms a well-defined helix in dodecylphosphocholine micelles. Biochemistry. 2001;40:9570–9578. doi: 10.1021/bi010640u. [DOI] [PubMed] [Google Scholar]
- Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schowalter RM, Smith SE, Dutch RE. Characterization of human metapneumovirus F protein-promoted membrane fusion: critical roles for proteolytic processing and low pH. J Virol. 2006;80:10931–10941. doi: 10.1128/JVI.01287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrempf S, Froeschke M, Giroglou T, von Laer D, Dobberstein B. Signal Peptide requirements for lymphocytic choriomeningitis virus glycoprotein C maturation and virus infectivity. J Virol. 2007;81:12515–12524. doi: 10.1128/JVI.01481-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth S, Vincent A, Compans R. Activation of fusion by the SER virus F protein: a low-pH-dependent paramyxovirus entry process. J Virol. 2003;77:6520–6527. doi: 10.1128/JVI.77.11.6520-6527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokralla S, Chernish R, Ghosh HP. Effects of double-site mutations of vesicular stomatitis virus glycoprotein G on membrane fusion activity. Virology. 1999;256:119–129. doi: 10.1006/viro.1999.9606. [DOI] [PubMed] [Google Scholar]
- Sieczkarski SB, Whittaker GR. Viral entry. Curr Topics Microbiol Immunol. 2005;285:1–23. doi: 10.1007/3-540-26764-6_1. [DOI] [PubMed] [Google Scholar]
- Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- Smit JM, Bittman R, Wilschut J. Deacylation of the transmembrane domains of Sindbis virus envelope glycoproteins E1 and E2 does not affect low-pH-induced viral membrane fusion activity. FEBS Lett. 2001;498:57–61. doi: 10.1016/s0014-5793(01)02495-4. [DOI] [PubMed] [Google Scholar]
- Smith JG, Mothes W, Blacklow SC, Cunningham JM. The mature avian leukosis virus subgroup A envelope glycoprotein is metastable, and refolding induced by the synergistic effects of receptor binding and low pH is coupled to infection. J Virol. 2004;78:1403–1410. doi: 10.1128/JVI.78.3.1403-1410.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear P, Manoj S, Yoon M, Jogger C, Zago A, Myscofski D. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology. 2006;344:17–24. doi: 10.1016/j.virol.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Steinhauer DA, Wharton SA, Wiley DC, Skehel JJ. Deacylation of the hemagglutinin of influenza A/Aichi/2/68 has no effect on membrane fusion properties. Virology. 1991;184:445–448. doi: 10.1016/0042-6822(91)90867-b. [DOI] [PubMed] [Google Scholar]
- Stevens J, Corper AL, Basler CF, Taubenferger JK, Palese P, Wilson IA. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- Stiasny K, Bressanelli S, Lepault J, Rey FA, Heinz FX. Characterization of a membrane-associated trimeric low-pH-induced Form of the class II viral fusion protein E from tick-borne encephalitis virus and its crystallization. J Virol. 2004;78:3178–3183. doi: 10.1128/JVI.78.6.3178-3183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiasny K, Heinz F. Flavivirus membrane fusion. J Gen Virol. 2006;87:2755–2766. doi: 10.1099/vir.0.82210-0. [DOI] [PubMed] [Google Scholar]
- Stiasny K, Koessl C, Heinz FX. Involvement of lipids in different steps of the flavivirus fusion mechanism. J Virol. 2003;77:7856–7862. doi: 10.1128/JVI.77.14.7856-7862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strive T, Borst E, Messerle M, Radsak K. Proteolytic processing of human cytomegalovirus glycoprotein B is dispensable for viral growth in culture. J Virol. 2002;76:1252–1264. doi: 10.1128/JVI.76.3.1252-1264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez T, Nir S, Goni FM, Saez-Cirion A, Nieva JL. The pre-transmembrane region of the human immunodeficiency virus type-1 glycoprotein: a novel fusogenic sequence. FEBS Lett. 2000;477:145–149. doi: 10.1016/s0014-5793(00)01785-3. [DOI] [PubMed] [Google Scholar]
- Subramanian R, Geraghty R. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci USA. 2007;104:2903–2908. doi: 10.1073/pnas.0608374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Belouzard S, Whittaker GR. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J Biol Chem. 2008;283:6418–6427. doi: 10.1074/jbc.M708955200. [DOI] [PubMed] [Google Scholar]
- Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. A system for functional analysis of Ebola virus glycoprotein. PNAS. 1997;94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm LK. Hypothesis: spring-loaded boomerang mechanism of influenza hemagglutinin-mediated membrane fusion. Biochim Biophys Acta. 2003;1614:14–23. doi: 10.1016/s0005-2736(03)00159-7. [DOI] [PubMed] [Google Scholar]
- Taylor GM, Sanders DA. Structural criteria for regulation of membrane fusion and virion incorporation by the murine leukemia virus TM cytoplasmic domain. Virology. 2003;312:295–305. doi: 10.1016/s0042-6822(03)00297-6. [DOI] [PubMed] [Google Scholar]
- Taylor GM, Sanders DA. The role of the membrane-spanning domain sequence in glycoprotein-mediated membrane fusion. Mol Biol Cell. 1999;10:2803–2815. doi: 10.1091/mbc.10.9.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier E, Pecheur EI. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur Biophys J. 2007;36:887–899. doi: 10.1007/s00249-007-0201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoennes S, Li ZN, Lee BJ, Langley WA, Skehel JJ, Russell RJ, Steinhauer DA. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology. 2007;370:403–414. doi: 10.1016/j.virol.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S, Compans RW. Oligomerization, secretion, and biological function of an anchor-free parainfluenza virus type 2 (PI2) fusion protein. Virology. 2000;270:368–376. doi: 10.1006/viro.2000.0286. [DOI] [PubMed] [Google Scholar]
- Tong S, Yi F, Martin A, Yao Q, Li M, Compans RW. Three membrane-proximal amino acids in the human parainfluenza type 2 (HPIV 2) F protein are critical for fusogenic activity. Virology. 2001;280:52–61. doi: 10.1006/viro.2000.0755. [DOI] [PubMed] [Google Scholar]
- Ujike M, Nakajima K, Nobusawa E. Influence of acylation sites of influenza B virus hemagglutinin on fusion pore formation and dilation. J Virol. 2004;78:11536–1143. doi: 10.1128/JVI.78.21.11536-11543.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waarts B, Bittman R, Wilschut J. Sphingolipid and cholesterol dependence of alphavirus membrane fusion. Lack of correlation with lipid raft formation in target liposomes. J Biol Chem. 2002;277:38141–38147. doi: 10.1074/jbc.M206998200. [DOI] [PubMed] [Google Scholar]
- Wagenaar T, Moss B. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J Virol. 2007;81:6286–6293. doi: 10.1128/JVI.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Herwig A, Azzouz N, Klenk HD. Acylation-mediated membrane anchoring of avian influenza virus hemagglutinin is essential for fusion pore formation and virus infectivity. J Virol. 2005;79:6449–6458. doi: 10.1128/JVI.79.10.6449-6458.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed AA, Ablan SD, Roser JD, Sowder RC, Schaffner CP, Chertova E, Freed EO. HIV-1 escape from the entry-inhibiting effects of a cholesterol-binding compound via cleavage of gp41 by the viral protease. Proc Natl Acad Sci USA. 2007;104:8467–8471. doi: 10.1073/pnas.0701443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin M, Ekström M, Garoff H. Isomerization of the intersubunit disulphide-bond in Env controls retrovirus fusion. EMBO J. 2004;23:54–65. doi: 10.1038/sj.emboj.7600012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin M, Ekström M, Garoff H. Receptor-triggered but alkylation-arrested env of murine leukemia virus reveals the transmembrane subunit in a prehairpin conformation. J Virol. 2006;80:9921–9925. doi: 10.1128/JVI.00380-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin M, Ekström M, Garoff H. The fusion-controlling disulfide bond isomerase in retrovirus Env is triggered by protein destabilization. J Virol. 2005a;79:1678–1685. doi: 10.1128/JVI.79.3.1678-1685.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin M, Löving R, Ekström M, Li K, Garoff H. Kinetic analyses of the surface-transmembrane disulfide bond isomerization-controlled fusion activation pathway in Moloney murine leukemia virus. J Virol. 2005b;79:13856–13864. doi: 10.1128/JVI.79.22.13856-13864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning DL, Schmitt AP, Leser GP, Lamb RA. Roles for the cytoplasmic tails of the fusion and hemagglutinin-neuraminidase proteins in budding of the paramyxovirus simian virus 5. J Virol. 2002;76:9284–9297. doi: 10.1128/JVI.76.18.9284-9297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis WI, Brunger AT, Skehel JJ, Wiley DC. Refinement of the influenza virus hemagglutinin by simulated annealing. J Mol Biol. 1990;212:737–761. doi: 10.1016/0022-2836(90)90234-D. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W, Hinz A, Gaudin Y. Virus membrane fusion. FEBS Lett. 2007;581:2150–2155. doi: 10.1016/j.febslet.2007.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch B, Vandemark A, Heroux A, Hill C, Kay M. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JT, Johnston PB, Dubay SR, Hunter E. Mutations within the putative membrane-spanning domain of the simian immunodeficiency virus transmembrane glycoprotein define the minimal requirements for fusion, incorporation, and infectivity. J Virol. 2001;75:9601–9612. doi: 10.1128/JVI.75.20.9601-9612.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitbeck J, Zuo Y, Milne R, Cohen G, Eisenberg R. Stable association of herpes simplex virus with target membranes is triggered by low pH in the presence of the gD receptor, HVEM. J Virol. 2006;80:3773–3780. doi: 10.1128/JVI.80.8.3773-3780.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM, Wilson IA. Anti-peptide antibodies detect steps in a protein conformational change: low-pH activation of the influenza virus hemagglutinin. J Cell Biol. 1987;105:2887–2896. doi: 10.1083/jcb.105.6.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SH, Wimley WC, Ladokhin AS, Hristova K. Protein folding in membranes: determining energetics of peptidebilayer interactions. Methods Enzymol. 1998;295:62–87. doi: 10.1016/s0076-6879(98)95035-2. [DOI] [PubMed] [Google Scholar]
- Whitt MA, Rose JK. Fatty acid acylation is not required for membrane fusion activity or glycoprotein assembly into VSV virions. Virology. 1991;185:875–878. doi: 10.1016/0042-6822(91)90563-q. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Wool-Lewis R, Bates P. Endoproteolytic processing of the ebola virus envelope glycoprotein: cleavage is not required for function. J Virol. 1999;73:1419–1426. doi: 10.1128/jvi.73.2.1419-1426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool-Lewis RJ, Bates P. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J Virol. 1998;72:3155–3160. doi: 10.1128/jvi.72.4.3155-3160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SR, Haag L, Hammar L, Wu B, Garoff H, Xing L, Murata K, Cheng RH. The dynamic envelope of a fusion class II virus. pre-fusion stages of semliki forest virus revealed by electron cryomicroscopy. J Biol Chem. 2007;282:6752–6762. doi: 10.1074/jbc.M609125200. [DOI] [PubMed] [Google Scholar]
- Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol. 2004;78:3429–3435. doi: 10.1128/JVI.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss S, Dimitrov AS, Baribaud F, Edwards TG, Blumenthal R, Hoxie JA. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J Virol. 2005;79:12231–12241. doi: 10.1128/JVI.79.19.12231-12241.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Compans RW. Palmitoylation of the murine leukemia virus envelope glycoprotein transmembrane subunits. Virology. 1996;221:87–97. doi: 10.1006/viro.1996.0355. [DOI] [PubMed] [Google Scholar]
- Yang X, Kurteva S, Ren X, Lee S, Sodroski J. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J Virol. 2005;79:12132–13147. doi: 10.1128/JVI.79.19.12132-12147.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci USA. 2005;102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, pre-fusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York J, Nunberg J. Role of the stable signal peptide of Junín arenavirus envelope glycoprotein in pH-dependent membrane fusion. J Virol. 2006;80:7775–7780. doi: 10.1128/JVI.00642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Thompson T, Wurzburg B, Paterson R, Lamb R, Jardetzky T. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure. 2005;13:803–815. doi: 10.1016/j.str.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Zaitsev V, von Itzstein M, Groves D, Kiefel M, Takimoto T, Portner A, Taylor G. Second sialic acid binding site in Newcastle disease virus hemagglutinin-neuraminidase: implications for fusion. J Virol. 2004;78:3733–3741. doi: 10.1128/JVI.78.7.3733-3741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti G, Briggs JA, Grunewald K, Sattentau QJ, Fuller SD. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2006;2:e83. doi: 10.1371/journal.ppat.0020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavorotinskaya T, Qian Z, Franks J, Albritton LM. A point mutation in the binding subunit of a retroviral envelope protein arrests virus entry at hemifusion. J Virol. 2004;78:473–481. doi: 10.1128/JVI.78.1.473-481.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelus BD, Schickli JH, Blau DM, Weiss SR, Holmes KV. Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37 degrees C either by soluble murine CEACAM1 receptors or by pH 8. J Virol. 2003;77:830–840. doi: 10.1128/JVI.77.2.830-840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Yang R, Bodner ML, Weliky DP. Conformational flexibility and strand arrangements of the membrane-associated HIV fusion peptide trimer probed by solid-state NMR spectroscopy. Biochemistry. 2006;45:12960–12975. doi: 10.1021/bi0615902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Liu J, Bess J, Jr., Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.