

Summary
Sex is determined in C. elegans by a chromosome-counting mechanism that tallies X chromosome dose relative to the sets of autosomes, the X:A ratio. A group of genes on X called X signal elements (XSEs) communicates X chromosome number by repressing the activity of the master sex-determination switch gene xol-1 in a dose-dependent manner. xol-1 is repressed by transcriptional and posttranscriptional mechanisms and is inactive in XX animals (hermaphrodite) but active in XO animals (male). Prior to our work, the nature of the autosomal signal and its target(s) were unknown. Here we show the signal includes discrete, trans-acting autosomal signal elements (ASEs) that counter XSEs to coordinately control both sex determination and dosage compensation. sea-1, the first autosomal signal element, encodes a T-box transcription factor that opposes XSEs by activating transcription of xol-1. Hence, xol-1 integrates both X and autosomal signals to determine sexual fate.
Introduction
Dose-sensitive signals play essential roles in cell fate decisions during development. Small changes in the concentration of key regulators are translated into dramatically different developmental fates. A prime example is sex determination. In C. elegans and D. melanogaster, a set of dose-sensitive genes on X called X signal elements (XSEs) communicates X chromosome number to determine sex (Cline and Meyer, 1996). The single dose of XSEs present in diploid XO animals specifies male fate, while the double dose in diploid XX animals specifies female (or hermaphrodite) fate. XSEs relay X chromosome dose by controlling the activity of a master sex-determination switch gene. In C. elegans, that target is xol-1 (XO lethal) (Miller et al., 1988; Rhind et al., 1995). xol-1 is repressed by the double dose of XSEs in XX animals through transcriptional and post-transcriptional mechanisms, thereby inducing hermaphrodite development, which includes activation of the dosage compensation machinery (Figure 1A; Carmi et al., 1998; Carmi and Meyer, 1999; Nicoll et al., 1997). The single dose of XSEs in XO embryos fails to repress xol-1. Active xol-1 induces male development.
Figure 1.

Identification and Characterization of the Autosomal Component of the C. elegans X:A Sex Determination Signal
(A) Genetic hierarchy regulating sex determination and dosage compensation. The ratio of X chromosomes to sets of autosomes is the primary signal that determines sexual fate and the level of X chromosome gene expression. In XX diploids (2X:2A), xol-1 is repressed, permitting high expression of sdc-2, the pivotal XX-specific trigger of dosage compensation and hermaphrodite development. In XO diploids (1X:2A), xol-1 is active and sdc-2 is repressed, permitting male development. The dosage compensation machinery is inactive in males. A group of X-linked genes called X signal elements (XSEs) communicates X chromosome dose by repressing xol-1 when they are present in two doses but not in one dose. The autosomal signal must oppose the X signal, but prior to our current work, the nature of the autosomal signal was not known. A key question was whether a group of discrete, dosesensitive autosomal signal elements (ASEs) communicates autosomal dose in a manner analogous to XSEs.
(B) Predicted outcomes from changing the dose of X and autosomal signal elements in 2X:2A and 1X:2A animals. Mutations in X signal elements cause sex-specific, dose-dependent phenotypes. XX animals with xse mutations die from the disruption of dosage compensation, and the survivors are masculinized and dumpy (Dpy). Mutations in potential autosomal signal elements (ASEs) should also cause synergistic, dose-dependent phenotypes, but ones opposite to those caused by XSE mutations. Moreover, since the sex determination signal is the ratio of X chromosomes to autosomes, a decrease in the dose of ASEs in XX animals is predicted to compensate for a decrease in the dose of XSEs by restoring the overall balance of the perceived X:A signal. Conversely, an increase in the dose ASEs in XO animals should compensate for an increase in XSEs. Furthermore, an increase in the dose of ASEs in XX animals should enhance the mutant phenotype caused by a decrease in XSEs, and a decrease in the dose of ASEs in XO animals should enhance the mutant phenotype caused by an increase in XSEs. In the figure, a decrease in the dose of either XSEs or ASEs is represented by a downward arrow, and an increase in XSE or ASE dose is represented by an upward arrow. If the change in signal element dose is masculinizing, the arrow is aqua; if the change is feminizing, the arrow is fuchsia.
(C) Genetic screen for mutations in ASE genes. The viability of hermaphrodites doubly mutant for the XSEs fox-1 sex-1 depends on yEx483[Pdpy-30::sdc-2(+)], a mitotically unstable extrachromosomal array that overexpresses sdc-2(+) constitutively from a promoter insensitive to the X:A signal. This array, referred to as yEx[sdc-2(+)] in the figure, also carries the marker myo-2::gfp, which causes the pharynx to be green. Following mutagenesis with EMS, mutations that suppress fox-1 sex-1 lethality can be recovered from hermaphrodites that acquire the ability to live in the absence of the rescuing array and therefore do not have a green pharynx. These are candidate loss-of-function mutations in ASEs.
Rather than measuring the absolute number of X chromosomes, C. elegans assesses the number of X chromosomes relative to the ploidy, the number of sets of autosomes (Madl and Herman, 1979). Although two X chromosomes dictate hermaphrodite development in a diploid animal (X:A of 1.0), they dictate male development in a tetraploid (X:A of 0.5). This counting process is remarkably discriminating. An X:A signal of 0.67 (XX triploid) reliably induces male development, and just as reliably, a signal of 0.75 (XXX tetraploid) induces hermaphrodite development.
Our goal was to discover the signals that communicate ploidy during the sex determination process. At the outset, no information was known about the nature of the autosomal signal or its target(s). A critical question was whether a set of dose-sensitive genes on autosomes relays autosomal dose in a manner analogous to that of X signal elements.
The distinguishing genetic feature of XSEs is the reciprocal, sex-specific phenotypes caused by changing their dose in XX and XO diploid animals (Figure 1B; Carmi and Meyer, 1999). Decreasing XSE dose specifically kills hermaphrodites but not males, while increasing XSE dose kills males but not hermaphrodites. Decreasing XSE dose in XX animals blocks xol-1 repression. The active xol-1 product then masculinizes XX animals and kills them by preventing the activation of dosage compensation, the vital process that equalizes levels of X chromosome products between sexes. C. elegans dosage compensation is achieved by an XX-specific protein complex that binds both X chromosomes of hermaphrodites to reduce X expression by half (Chan et al., 2004; Chuang et al., 1994; Csankovszki et al., 2004; Meyer and Casson, 1986). XX animals expressing xol-1 are unable to form an active dosage compensation complex and hence die from overexpression of X-linked genes. Conversely, increasing the dose of XSEs in XO animals represses xol-1, causing activation of dosage compensation and male lethality from underexpression of X-linked genes. Since the multiple XSEs function together to repress xol-1, altering the dose of any single XSE does not necessarily produce a visible phenotype (Carmi and Meyer, 1999). However, combinations of two or more XSE mutations cause severe synergistic sex determination and dosage compensation defects. The XSE called fox-1 encodes an RNA binding protein that controls xol-1 posttranscriptionally, and the XSE called sex-1 encodes a nuclear receptor that controls xol-1 transcriptionally. Both the transcriptional and posttranscriptional modes of regulation are required to repress xol-1 fully in XX animals.
Autosomal signal elements (ASEs) would be expected to counter the action of XSEs and thereby have opposite effects: increasing ASE dose would harm hermaphrodites, while reducing ASE dose would harm males. However, the autosomal signal need not be composed of discrete autosomal signal element genes that produce trans-acting products. Instead, the autosomal signal could be an indirect reflection of chromosome number, which changes with ploidy. For example, as chromosome number increases, so does the concentration of nonspecific DNA sites that might bind XSEs with low affinity. An increase in such sites would reduce XSE binding to bona fide target sites within xol-1, thereby disrupting repression of xol-1. Alternatively, the autosomal signal might act more specifically to counter XSEs, but be composed of discrete elements that act in cis, unlike XSEs, which act in trans. For example, the autosomal signal could include high-affinity, cis-acting binding sites for XSEs, which would titrate XSEs away from their xol-1 targets, thereby activating xol-1.
Like the X signal, the autosomal signal could regulate the master sex switch gene xol-1, the target of XSEs. Alternatively, downstream genes in the sex determination and dosage compensation pathway might interpret the autosomal signal (Figure 1A). The autosomal signal might act like the X signal to coordinately control both sex determination and dosage compensation by regulating a single target gene that directs both processes, or it might act instead on more specialized targets to control the two processes independently.
Our work reveals the genetic and molecular character of the autosomal signal in C. elegans sex determination. The autosomal signal includes discrete, trans-acting ASEs that counter XSEs to coordinately control both sex determination and dosage compensation. Furthermore, sea-1 (signal element on an autosome), the first ASE identified in C. elegans, encodes a T-box transcription factor that opposes XSEs by activating transcription of xol-l, thereby inducing male development. Hence, xol-1 integrates both X and autosomal signals to determine sexual fate.
Results and Discussion
Genetic Screen for Autosomal Signal Elements
To isolate loss-of-function mutations in potential ASE genes, we designed a sensitized genetic screen that took advantage of the synergistic behavior of X chromosome signal elements (Figure 1C). Although XX animals carrying a null mutation in the XSE fox-1 have no obvious phenotype and XX animals carrying a null mutation in the XSE sex-1 have only reduced viability, all XX animals homozygous for both null mutations, fox-1(y303) and sex-1(y263), die from the failure to assemble the dosage compensation complex on X (Figures 2A and 2B; Carmi et al., 1998; Nicoll et al., 1997). We reasoned that disruption of an ASE gene, even one with a small contribution to the autosomal signal, would help restore the X:A balance and hence suppress the XX lethality, at least in part (Figure 1B).
Figure 2.
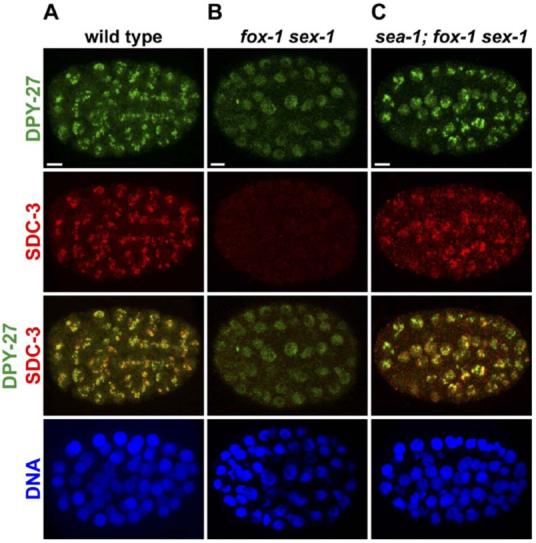
A sea-1 Mutation Permits the Dosage Compensation Complex to Assemble onto the X Chromosomes of fox-1 sex-1 XX Mutant Embryos
Confocal images of mutant or wild-type XX embryos at approximately the 100-cell stage, costained with antibodies against the dosage compensation proteins DPY-27 (green) and SDC-3 (red) and with DAPI (blue), a DNA-intercalating dye.
(A) In wild-type XX embryos, DPY-27 and SDC-3 localize specifically to X chromosomes.
(B) In fox-1 sex-1 mutant embryos, the dosage compensation complex cannot assemble on X, causing XX animals to die from overexpression of X-linked genes. In these mutants, SDC-3 is absent and DPY-27 is greatly reduced in abundance. Residual DPY-27 is diffusely distributed within nuclei.
(C) sea-1(y356) suppresses fox-1 sex-1 lethality by restoring the levels of dosage compensation proteins and permitting assembly of the complex onto hermaphrodite X chromosomes. Scale bars equal 5 μm.
For the screen, fox-1 sex-1 doubly mutant hermaphrodites were maintained as a viable strain by a transgenic array that ectopically expresses sdc-2(+) from the non-sex-specific promoter dpy-30. SDC-2 produced from yEx483[Pdpy-30::sdc-2(+)] induces assembly of the dosage compensation complex on X chromosomes independent of either the X:A signal or level of xol-1 expression (Dawes et al., 1999). The array also carries the myo-2::gfp transformation marker, which causes the pharynx to be green. The 55% of fox-1 sex-1 progeny that inherit this mitotically unstable array survive (green), while progeny that fail to inherit the array die (non-green) (Figure 1C). We sought mutations that permitted fox-1 sex-1 hermaphrodites to live without the array (non-green). These suppressor mutations are candidate ase(lf) mutations.
In principle, many classes of mutations could be recovered from this screen: loss-of-function (lf) mutations in ASEs or in xol-1 and gain-of-function (gf) mutations in XSEs. Among the suppressors isolated were one xol-1(lf) allele and the allele y356, a loss-of-function mutation shown below to disrupt an autosomal signal element.
Criteria for an Autosomal Signal Element
Autosomal signal elements would counter the X signal, allowing us to predict the consequences of changing the dose of a candidate ASE gene. Loss-of-function mutations in an ASE gene should suppress the XX lethality caused by loss-of-function mutations in XSE genes and enhance the XO lethality caused by increasing XSE dose. Conversely, increasing ASE dose should enhance the XX lethality from XSE loss-of-function mutations and suppress the XO lethality from increasing XSE dose. An ASE must act in a dose-dependent manner in the zygote, because the zygotic X:A signal determines sex. As shown below, the gene defined by the fox-1 sex-1 suppressor mutation y356 meets these criteria, and moreover, it controls sex determination as well as dosage compensation. We named the gene sea-1 (signal element on an autosome).
sea-1 Mutation Suppresses the Hermaphrodite Lethality and Masculinization Caused by Reducing XSE Dose
The mutation sea-1(y356) suppresses the XX-specific lethality caused by double mutant combinations of known XSEs, consistent with predictions for loss-of-function mutations in an ASE gene. While all fox-1 sex-1 XX animals die, 52% of the sea-1; fox-1 sex-1 XX mutants survive (Figure 3B). Suppression is recessive: all sea-1(y356)/+; fox-1(y303) sex-1(y263) XX animals die (data not shown).
Figure 3.
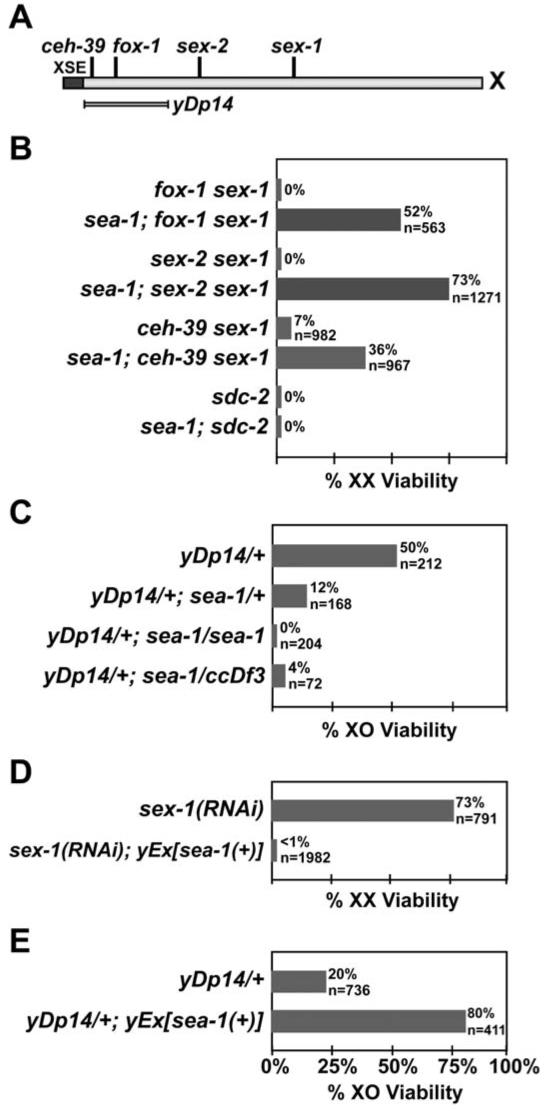
sea-1 Is an Autosomal Signal Element
(A) Genetic map of the X chromosome showing the location of XSEs (ceh-39, fox-1, sex-2, and sex-1) and yDp14, which duplicates at least two XSEs, fox-1 and ceh-39.
(B-E) Viability of XX and XO animals with different XSE and ASE configurations. The values given for n are the expected number of the genotypic class in the absence of the observed lethality; for (B) and (D), n is equal to the number of embryos laid. Only the relevant genotype is listed; the full genotype is in Experimental Procedures. (B) Suppression of hermaphrodite phenotypes by sea-1(y356). Loss of sea-1 suppresses the phenotypes caused by all known XSE mutations, including the XX-specific lethality resulting from the double mutant combinations fox-1 sex-1, ceh-39 sex-1, and sex-2 sex-1, but not the dosage compensation mutation sdc-2.
(C) Dose-dependent enhancement of male lethality by sea-1(y356). A single copy of yDp14 kills 50% of males. This lethality is enhanced by the loss of sea-1 and depends on the number of sea-1 copies present. The deficiency ccDf3 removes the sea-1 locus and behaves in a genetically similar manner to sea-1(y356), suggesting that the sea-1 mutation is null or near null.
(D) Enhancement of hermaphrodite lethality by sea-1(+) overexpression. Decreasing the function of the XSE sex-1 by RNAi decreases hermaphrodite viability; extra copies of sea-1(+) on an extrachromosomal array dramatically enhance this lethality.
(E) Suppression of male lethality by sea-1(+) overexpression. The male lethality caused by one copy of the XSE duplication yDp14 is partially suppressed by extra copies of sea-1(+).
In addition to sex-1 and fox-1, two other XSEs have been identified: ceh-39 (J. Gladden and B.J.M., unpublished) and sex-2 (Figure 3A) (C.Y. Loh and B.J.M., unpublished). While only 7% of ceh-39(y414) sex-1(y263) doubly mutant hermaphrodites survive, 36% of sea-1(y356); ceh-39(y414) sex-1(y263) triply mutant hermaphrodites survive (Figure 3B). Moreover, while all sex-2(y324) sex-1(y263) double mutants die, 73% of sea-1(y356); sex-2(y324) sex-1(y263) triply mutant hermaphrodites survive. Thus, sea-1 meets the first essential criterion for being an ASE.
sea-1(y356) suppresses the lethality of XX mutants with reduced XSE dose by restoring binding of the dosage compensation complex to X chromosomes (Figure 2C). In fox-1 sex-1 double mutants, as in wild-type XO embryos, the dosage compensation protein SDC-3 is not present, and DPY-27 is diffusely distributed throughout the nucleus rather than localized to X (Figures 2A and 2B). In sea-1; fox-1 sex-1 embryos, both DPY-27 and SDC-3 can localize to X chromosomes. However, not all SDC-3 and DPY-27 proteins are properly localized to X, consistent with the partial suppression of lethality by sea-1(y356) and the finding that live sea-1; fox-1 sex-1 animals have a mild dosage compensation defect, a dumpy (Dpy) phenotype. Together, these experiments demonstrate that sea-1 regulates dosage compensation.
Mutations in X signal elements disrupt sex determination as well as dosage compensation, causing the XX mutants to be masculinized. The rescued sea-1; fox-1 sex-1 XX animals developed as hermaphrodites rather than males, indicating that sea-1(y356) also suppressed the sex determination defects caused by XSE mutations. Thus, sea-1 acts in the known genetic hierarchy to control both sex determination and dosage compensation.
sea-1 Acts Upstream of the XX-Specific Gene that Triggers Assembly of the Dosage Compensation Complex on X
If sea-1 acts in a manner analogous to XSEs to control sex determination and dosage compensation, it will act upstream of sdc-2, the pivotal XX-specific gene that induces hermaphrodite sexual development and triggers assembly of the dosage compensation complex on X (Figure 1A; Chu et al., 2002; Dawes et al., 1999; Nusbaum and Meyer, 1989). Null alleles of sdc-2 cause masculinization and block assembly of the dosage compensation complex, causing all hermaphrodites to die. sdc-2 acts just prior to the divergence of the sex determination and dosage compensation pathways. Unlike the equally severe lethality caused by fox-1 sex-1 double mutations, sdc-2 XX lethality was not suppressed by sea-1(y356) (Figure 3B), demonstrating that sea-1 acts upstream of sdc-2 in the genetic regulatory hierarchy to control dosage compensation. Consistent with this observation, sea-1(y356) cannot suppress the lethality caused by loss-of-function mutations that disrupt members of the dosage compensation complex, such as dpy-27.
sea-1 Mutation Enhances the Male Lethality Caused by Increasing XSE Dose
The wild-type function of an ASE is to promote the male fate, and loss of ASE function should impair male development. sea-1 has this predicted property. sea-1(y356) alone does not cause lethality or any visible phenotype in males, a result expected if ASEs act cumulatively like XSEs. The cumulative nature of the X signal minimizes the impact of reducing the dose of any single element (Carmi and Meyer, 1999). However, sea-1(y356) does enhance the partial male lethality caused by the elevation of XSE dose. A single copy of yDp14, which increases the X:A ratio by duplicating fox-1 and ceh-39 (Figure 3A), kills 50% of males (Figure 3C; Carmi and Meyer, 1999). Homozygous sea-1(y356) increases yDp14/+ male lethality to 100% (Figure 3C). In this assay, the sea-1 deficiency ccDf3 behaved similarly to sea-1(y356) (Figure 3C), establishing that sea-1(y356) is a loss-of-function mutation.
sea-1 Acts in a Dose-Dependent Manner
sea-1 meets another criterion for an ASE: it acts in a dose-dependent manner. Since ASEs communicate the dose of autosomes, we expect that reducing the dose of a single ASE to one copy will shift the perceived X:A ratio enough to feminize mutants on the threshold between the male and hermaphrodite fates. Consistent with this prediction, sea-1(y356) dose dependently enhances the male lethality of yDp14/+. While 50% of yDp14/+ males and 100% of yDp14/+; sea-1(y356)/sea-1(y356) males die, only about 88% of yDp14/+; sea-1(y356)/+ males die (Figure 3C). The range of phenotypic severity caused by sea-1(y356)/sea-1(y356), sea-1(y356)/+, and +/+ in the sensitized yDp14/+ genetic background indicates that worms can distinguish among zero, one, and two doses of the wild-type sea-1.
sea-1 Acts in the Zygote
The sex of C. elegans is determined by the X:A signal of the zygote rather than the mother. Therefore, an ASE must function, at least in part, in the zygote. The experiments described previously were conducted in a manner to differentiate between the maternal and zygotic contribution of sea-1. The following summary of these experiments establishes that sea-1 acts zygotically, consistent with its role as an ASE. In this description, m represents the sea-1 maternal contribution and z the zygotic contribution.
Heterozygous sea-1(y356)/+; fox-1 sex-1 hermaphrodites from homozygous sea-1(y356); fox-1 sex-1 mothers were not viable, indicating that loss of the maternal sea-1 contribution cannot suppress fox-1 sex-1, if the zygote has a wild-type copy of sea-1 (m-z+ configuration). In contrast, sea-1(y356)/+; fox-1 sex-1; yEx483 [Pdpy-30::sdc-2(+)] hermaphrodites did produce viable non-green sea-1(y356); fox-1 sex-1 hermaphrodite self progeny, showing that loss of the zygotic sea-1(+) contribution suppresses fox-1 sex-1 mutant animals, regardless of whether the mother has a wild-type sea-1 allele (m+z-configuration). Although it was formally possible that the maternal yEx483[Pdpy-30::sdc-2(+)] array affected the phenotype of the zygote in this experiment, the sea-1(y356)/+; fox-1 sex-1; yEx483 hermaphrodites also produced +/+; fox-1 sex-1 progeny (m+z+) that died as embryos or young larvae. Therefore, maternal yEx483[Pdpy-30::sdc-2(+)] cannot rescue fox-1 sex-1 lethality in a sea-1(+) zygote and thus is unlikely to affect the phenotype of the sea-1; fox-1 sex-1 siblings. We conclude that loss of sea-1 function in the zygote is necessary and sufficient for suppression of fox-1 sex-1 mutants.
In addition, loss of sea-1 in the zygote enhances the male lethality caused by yDp14. All yDp14/+; sea-1(y356) males are dead, regardless of whether the mother is yDp14/+; sea-1(y356) (m-z-) or yDp14/+; sea-1(y356)/+(m+z-). However, not all yDp14/+; sea-1(y356)/+ males from yDp14; sea-1(y356) mothers are dead, indicating that a homozygous sea-1(y356) mutation in the mother is not sufficient to kill all male progeny. Therefore, both suppression of fox-1 sex-1 hermaphrodite lethality and enhancement of yDp14/+ male lethality are determined by the zygotic genotype of sea-1.
Increasing sea-1 Dose Has the Reciprocal Consequence of Decreasing sea-1 Dose
For sea-1 to be a bona fide ASE, increasing its dose should have the opposite effect of decreasing its dose: increasing sea-1(+) dose should enhance the XX-specific lethality caused by loss of XSE function and should reduce the XO-specific lethality caused by increasing XSE dose. Since no chromosomal duplications of the sea-1 region existed, we cloned sea-1 as described below and generated an extrachromosomal array, yEx716[sea-1(+)], which contains multiple copies sea-1(+). This sea-1(+) array enhanced the hermaphrodite lethality induced by sex-1 RNA interference (RNAi): 73% of wild-type hermaphrodites survived sex-1 RNAi but less than 1% of hermaphrodite siblings carrying yEx716[sea-1(+)] survived (Figure 3D). The converse is also true: sea-1(+) arrays rescued the male lethality caused by a single copy of the XSE duplication yDp14 (Figure 3E). In this experiment, only 20% of yDp14/+ males survived in the absence of the array, but the viability was increased to 80% in the presence of the sea-1(+) extrachromosomal array.
These genetic experiments demonstrate that sea-1 acts in a zygotic, dose-dependent manner to counter the X signal. Hence, the autosomal signal consists, at last in part, of discrete genes that control both sex determination and dosage compensation.
sea-1 Encodes a T-Box Transcription Factor
To elucidate the molecular nature of the autosomal sex signal and determine whether the signal includes trans-acting products or cis-acting sites, sea-1 was mapped using single nucleotide polymorphisms (SNPs) and then cloned by germline transformation rescue experiments (Experimental Procedures and Table S1 in the Supplemental Data available with this article online). sea-1 resides between map positions -6.33 and -5.49 on the left side of LG II, in a 270 kb region spanned by 9 cosmids, with two gaps covered by YACs. The sea-1 interval contains 92 predicted open reading frames (ORFs). Transgenic arrays containing each of the cosmids spanning the sea-1 interval were tested for their ability to rescue the enhancement of yDp14/+ male lethality by sea-1(y356)/+ and by sea-1(y356)/sea-1(y356). The F19B10 cosmid rescued these phenotypes. All rescuing activity of F19B10 was attributed to the single PCR-amplified ORF F19B10.9 (Table S1).
The wild-type product of F19B10.9 (called TBX-18 in Wormbase) is predicted to encode a 376 amino acid protein containing a C-terminal T-box DNA binding domain. Sequence analysis of F19B10.9 from the sea-1(y356) mutant provided direct evidence that we correctly determined the molecular identity of sea-1. The y356 lesion is a C to T transition 97 bases after the putative translational start site of F19B10.9, changing a glutamine codon to an ochre stop codon, which is predicted to truncate the SEA-1 protein at amino acid 33 (Figure 4A). The molecular nature of the lesion is consistent with genetic tests indicating that sea-1(y356) is a loss-of-function allele (Figure 3C).
Figure 4.
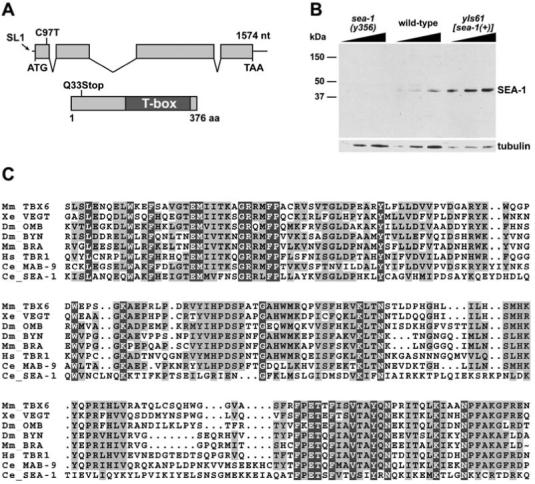
sea-1 Encodes a T-Box Protein
(A) The predicted genomic and protein structures of sea-1 are diagrammed. The site to which the SL1 trans-spliced RNA leader is attached to the sea-1 mRNA is represented on the DNA by an arrow. sea-1(y356) is a C to T transition at nucleotide 97 after the likely translational start site, replacing a glutamine codon with an ochre stop codon at position 33. SEA-1 contains a T-box DNA binding domain in the C-terminal half.
(B) Western blot of wild-type, sea-1 mutant, and sea-1(+) overexpressing embryonic extracts probed with anti-SEA-1 antibodies, with a tubulin loading control. SEA-1 protein is not detectable in sea-1(y356) mutant embryo extract but is present in wild-type extract and overexpressed in yIs61[sea-1(+)] extract. The predicted size of wild-type SEA-1 is 42.7 kDa.
(C) Alignment of SEA-1 T-box domain with T-box domains from other species. SEA-1 is significantly diverged from other T-box proteins. The SEA-1 T-box domain shares 27% identity with that of mouse BRACHYURY and frog VEGT. In contrast, the more conventional T-box domain of the C. elegans MAB-9 protein shares 52% identity with both BRACHYURY and VEGT. Similarly, the T-box domains of BRACHYURY and VEGT are 52% identical.
The T-box is a site-specific DNA binding domain present in a large group of proteins involved in diverse aspects of metazoan developmental regulation, including the specification of primary germ layer identity, limb development, and cell fate during organogenesis (Kispert and Hermann, 1993; Showell et al., 2004). Many T-box transcription factors have dose-sensitive phenotypes, like the ASE T-box protein SEA-1. For example, the founding member Brachyury (T) causes a shortened tail phenotype in heterozygous mice and a more severe phenotype in homozygous mice, including lethality accompanied by a missing posterior notochord and mesoderm (Chesley, 1935; Dobrovolskaia-Zavadskaia, 1927; Gluecksohn-Schoenheimer, 1944).
SEA-1 Accumulates in Nuclei of Young Embryos
Consistent with the molecular identity of SEA-1 as a T-box transcription factor, SEA-1 accumulates in nuclei of young embryos, as indicated by immunofluorescence experiments with anti-SEA-1 antibodies (Figure 5A). Antibodies raised to an N-terminal peptide of SEA-1 recognized a 43 kDa band in Western blots of wild-type embryonic extract, in agreement with the predicted size of SEA-1 (Figure 4B). This band is more abundant in extract made from embryos carrying the chromosomally integrated yIs61[sea-1(+)] array, which has multiple copies of sea-1(+), and is missing from sea-1(y356) mutant embryonic extract, confirming the specificity of the antibody, the overexpression of SEA-1 from the yIs61[sea-1(+)] array, and the loss of SEA-1 in the mutant. Consistent with the Western blot, SEA-1 accumulates to higher levels in overexpressing yIs61[sea-1(+)] embryos and is absent from sea-1(y356) mutant embryos (Figures 5B and 5C).
Figure 5.

sea-1 Activates xol-1 Transcription
(A-C) Confocal images of wild-type, sea-1 mutant, and sea-1(+) overexpressing embryos at approximately the 20- to 30-cell stage, costained with antibodies against SEA-1 (green) and SDC-3 (red) and with DAPI (blue), a DNA-intercalating dye.
(A) SEA-1 accumulates in nuclei of 20- to 100-cell wild-type embryos, the correct time to regulate xol-1. At the 30-cell stage, the dosage compensation protein SDC-3 is present in nuclei but has not yet localized to the X chromosomes.
(B) SEA-1 protein is not detectable in sea-1(y356) mutant embryos; SDC-3 is unaffected.
(C) sea-1 is overexpressed in yIs61[sea-1(+)] embryos.
(D) sea-1 acts upstream of xol-1. Multiple copies of sea-1 on the integrated array yIs61 enhanced the weak XX-specific lethality caused by sdc-2(RNAi). The enhancement of lethality was suppressed by a xol-1 mutation. n is the total number of embryos counted. Percent viability is the number of adults relative to the number of embryos laid.
(E) Model for sea-1 action. Our cumulative data establish sea-1 to be an ASE that acts upstream in the sex determination hierarchy to activate xol-1 transcription.
(F) DIC images of gravid wild-type XX hermaphrodites, him-8 XX hermaphrodites, and SEA-1-overexpressing XX hermaphrodites carrying yIs33[Pxol-l::lacZ], a transcriptional fusion between the xol-1 promoter and lacZ coding sequence, stained for β-galactosidase activity (Nicoll et al., 1997). Expression of this reporter mimics endogenous xol-1 expression: β-galactosidase is produced in XO embryos but not XX embryos. The him-8 mutation elevates X chromosome nondisjunction, resulting in ∼37% XO progeny compared to 0.2% XO progeny from wild-type XX hermaphrodites. In 58% of yIs33[Pxol-l::lacZ]; him-8(-) hermaphrodites examined, at least one embryo showed dark staining, indicating expression of Pxol-1::lacZ at high level (n = 53 adults containing 590 embryos). These darkly staining embryos are XO. The vast majority of all other embryos (XX) showed no β-galactosidase staining. By contrast, in 87% of gravid yIs33[Pxol-l::lacZ]; him-8(+) XX hermaphrodites examined, more than 75% of the embryos showed either light or no β-galactosidase staining (n = 69 adults containing 1260 embryos). None of the yIs33[Pxol-l::lacZ] hermaphrodites had any darkly staining embryos. Overexpression of sea-1 by the integrated yIs61[sea-1(+)] array was sufficient to partially activate Pxol-1::lacZ expression in XX embryos. About 58% of yIs61[sea-1(+)]; yIs33[Pxol-l::lacZ] gravid hermaphrodites contained at least one darkly staining XX embryo, and 25% of the other XX embryos showed medium to dark staining (n = 62 adults containing 917 embryos). Scale bars equal 5 μm.
Early expression of signal elements is key to their function as regulators of the sex determination and dosage compensation decision. Accumulation of SEA-1 becomes detectable by immunofluorescence in very young embryos, before the 30-cell stage, and is no longer detectable by the 100-cell stage. This temporal pattern of SEA-1 accumulation is similar to that of XSEs, which also accumulate prior to the 30-cell stage and become greatly diminished in older embryos (Carmi et al., 1998; Nicoll et al., 1997). Proper expression of xol-1, the direct target of the XSEs, is required from the 30-cell stage through gastrulation: functional XOL-1 is necessary for XO viability and sufficient for XX lethality during this interval in development (Rhind et al., 1995). XOL-1 prevents accumulation of SDC-2, which triggers the assembly of the dosage compensation complex on the X chromosomes of XX embryos by approximately the 40-cell stage (Dawes et al., 1999). Since SEA-1 accumulation precedes both the formation of the dosage compensation complex and the expression of xol-1, SEA-1 is present at the right time and place to regulate the choice of sexual fate, potentially by regulating xol-1.
sea-1 Is an Activator of xol-1 Transcription
The steps in the genetic pathway controlled by the autosomal signal were not known. Theoretically, an ASE could repress one or more XSE; however, SEA-1 is unlikely to repress any of the known XSEs since sea-1(y356) strongly suppresses all XSE null mutant combinations. Like the XSEs, SEA-1 could regulate xol-1 expression directly or indirectly. Alternately, it could act in parallel with xol-1 to regulate sdc-2. Distinguishing among these possibilities is critical to understanding how the autosomal signal regulates sex determination and dosage compensation.
With few exceptions, T-box transcription factors are activators of gene expression, although very few direct targets of T-box genes are known (Tada and Smith, 2001). We first sought sea-1 targets using the consensus in vitro binding site, the palindrome TTTCACACCT AGGTGTGAAA (Kispert and Hermann, 1993), for the founding T-box family member Brachyury to conduct BLAST searches of promoters and entire cosmids of sex determination and dosage compensation genes. Although most tested T-box proteins bind this sequence in vitro, sequence variants are found in the upstream regulatory regions of their targets, suggesting that in vivo conditions favor binding to variant sites (Tada and Smith, 2001). The BLAST searches identified no consensus binding sites in genes of the sex determination and dosage compensation pathway. The T-box DNA binding domain in SEA-1 is quite diverged relative to other T-box proteins (Figure 4C), suggesting that the SEA-1 DNA binding site has diverged sufficiently to be unrecognizable by a simple BLAST search.
Taking a candidate gene approach, we asked whether SEA-1 regulates xol-1 in vivo. Using a genetic approach, we showed that sea-1 acts upstream of xol-1 (Figures 5D and 5E). Multiple copies of sea-1 on the integrated array yIs61 enhanced the weak XX-specific lethality caused by sdc-2(RNAi). The enhancement of lethality was suppressed by a xol-1 mutation (Figure 5D). While 65% of sdc-2(RNAi) XX embryos are viable, only 8% of yIs61; sdc-2(RNAi) XX embryos are viable. A xol-1 mutation restores viability to 74%, indicating that sea-1 functions upstream of xol-1 (Figure 5E).
xol-1 is regulated at both transcriptional and post-transcriptional levels (Carmi and Meyer, 1999; Nicoll et al., 1997). To assess whether SEA-1 controls xol-1 at the level of transcription, we assayed the effect of an increase in sea-1 dose on the transcription of a lacZ reporter controlled by the xol-1 promoter. Expression of the integrated transgenic reporter yIs33[Pxol-1::lacZ] mimics expression of the endogenous xol-1 gene. β-galactosidase is expressed at high levels in XO but not XX embryos. If an ASE regulates xol-1, by definition it must be an activator of xol-1 expression. We found that the increased dose of sea-1 provided by the integrated array yIs61[sea-1(+)] activates expression of yIs33 [Pxol-1::lacZ] in hermaphrodite embryos (Figure 5F). Moreover, quantitative RT-PCR analysis of xol-1 transcript levels in yIs61[sea-1(+)] XX animals compared to wild-type XX animals revealed a 2.1-fold increase (SEM of 0.18) in transcript levels in the SEA-1-overexpressing strain. We have yet to find direct evidence of SEA-1 binding to the xol-1 promoter, but the latter two experiments conclusively demonstrate that xol-1 is the target (direct or indirect) of SEA-1 and that SEA-1 activates xol-1 transcription.
Conclusions
In summary, sea-1 meets all the criteria for an autosomal element of the C. elegans sex determination signal. Remarkably, ASEs are highly analogous to XSEs. Loss of sea-1 suppresses the XX-specific masculinization and lethality caused by reducing XSE dose and enhances the XO-specific lethality caused by increasing XSE dose. Increasing the dose of sea-1(+) has the reciprocal phenotype: it enhances the hermaphrodite sex determination and dosage compensation defects caused by reducing XSE dose and suppresses the male lethality caused by increasing XSE dose. Moreover, sea-1 acts in a dose-dependent, zygotic manner. sea-1 is thus the first identified component of the C. elegans autosomal sex signal. sea-1 encodes a T-box transcription factor, defining a role for this key class of cell fate regulators in sex determination. SEA-1 activates xol-1 transcription, either directly or indirectly, establishing that xol-1 acts as the master regulator to integrate both X chromosome and autosomal signals to determine sexual fate in C. elegans.
Drosophila melanogaster is the only other species with a well-characterized X:A sex-determination signal, although it uses a completely distinct set of genes and molecular mechanisms to assess this signal (Cline and Meyer, 1996). In flies, four XSEs activate transcription of the single feminizing target gene Sex-lethal (Sxl). Despite extensive genetic screens, only one significant ASE has been found. The ASE called deadpan (dpn) represses transcription of Sxl, but repression by dpn is not robust (Barbash and Cline, 1995; Younger-Shepherd et al., 1992). Unlike sea-1, deadpan plays essential roles in other developmental processes, as do all fly, but not worm, XSE genes. If the fly X:A denominator is composed of other discrete loci, each is likely to have a much weaker effect than dpn. In contrast, our recent experiments have revealed two other distinct C. elegans ASE genes, each at least similar in strength to sea-1. The ASEs act cumulatively to control sex determination and dosage compensation (P. Nix, M.M.J., J.R.P., and B.J.M., unpublished). Thus, unlike in flies, the C. elegans autosomal sex signal is composed of a small set of discrete autosomal signal element genes that oppose X signal element genes to control the choice of sexual fate and the activation of dosage compensation. Future experiments will determine whether ASEs, like XSEs, function through diverse molecular mechanisms and will reveal the molecular strategy by which ASEs counter XSEs to determine sexual fate. Analysis of sea-1 has already provided important answers to the long-standing enigma of how small changes in the concentration of dose-sensitive regulators can be translated into different developmental fates.
Experimental Procedures
Mutagenesis
Progeny from EMS-mutagenized (Brenner, 1974) L4 fox-1 sex-1; yEx483[dpy-30::sdc-2(+), myo-2::gfp] hermaphrodites were grown semiclonally at 15°C to reduce the number of nonmutant escapers. F1, F2, and F3 progeny, representing 4500 mutagenized haploid genomes, were screened for live non-green hermaphrodites. Of 19 strong suppressors of fox-1 sex-1 lethality, 7 were not X-linked. Two of these alleles, y356 and y379, mapped to discrete autosomal loci and were analyzed further.
Hermaphrodite Viability
Hermaphrodite viability was assessed by counting embryos laid by gravid hermaphrodites over a 4-8 hr period and then counting viable adults 3-6 days later. Percent viability was calculated as the (number of adults)/(number of embryos).
The strains fox-1(y303) sex-1(y263), sex-2(y324) sex-1(y263), and ceh-39(y414) sex-1(y263) are inviable. These homozygous double mutants were therefore maintained with the rescuing array yEx483 [Pdpy-30::sdc-2(+), myo-2::gfp], or the doubly mutant X chromosome was balanced by the szT1 chromosome. szT1 was identified by its dominant Him and recessive lethal phenotypes. To assess ceh-39(y414) sex-1(y263); yEx483 viability, progeny from rare nongreen escaper adults were scored. Viability counts for the lethal null sdc-2(y74) allele were performed with the heterozygous strains sdc-2(y74)/szT1 and sea-1(y356); sdc-2(y74)/szT1.
Male Lethality
The effect of changing sea-1 dose on male viability was assessed in the context of the X duplication yDp14, which has an extra copy of fox-1 and at least one other XSE (Figure 3A). One copy of yDp14 kills 50% of males, as determined from the relative number of +/+; him-8; unc-2 (Unc) and yDp14/+; him-8; unc-2 (non-Unc) male self progeny from yDp14/+; him-8(e1489); unc-2(e55) hermaphrodites. Out of 552 adult progeny counted, 212 yDp14/+ males were expected, but only 107 observed.
To determine whether reduction of sea-1 dose enhances the male lethality of one copy of yDp14, progeny of yDp14/+; sea-1(y356); him-8(e1489); unc-2(e55) hermaphrodites were examined. Out of 723 adult progeny counted, 204 yDp14/+; sea-1; him-8; unc-2 (non-Unc) males were expected but none found. To learn whether this enhancement was dose sensitive, broods of yDp14/+; mIn1 mIs14/sea-1; him-8; unc-2 hermaphrodites (1129 total adult progeny counted) were compared with broods of yDp14/+; mIn1 mIs14/+; him-8; unc-2 control hermaphrodites (781 total adult progeny counted). mIn1 mIs14 is a chromosome II balancer marked with a recessive dpy-10 allele and the dominant myo-2::gfp marker. Relative to the control broods, only 12% of the expected yDp14/+; mIn1 mIs14/sea-1; him-8; unc-2 (non-Unc, green, non-Dpy) males survived (20 observed, 168 expected).
To assess whether sea-1(y356) has a phenotype equivalent to the deletion ccDf3, yDp14; sea-1; unc-2 hermaphrodites were crossed with ccDf3/mIn1 mIs14 males (4135 total adult progeny counted) and with control +/mIn1 mIs14 males (1323 total adult progeny counted). 4% of the expected yDp14/+; sea-1/ccDf3; unc-2 (non-Unc, non-green) males survived (3 observed, 72 expected), relative to the number of yDp14/+; sea-1/+; unc-2 (non-Unc, non-green) male progeny from the parallel control cross. These three viable males were very sick, small, and slow-growing; therefore, it is unlikely that these rare escapers truly indicate a less severe phenotype for sea-1(y356)/ccDf3 than for sea-1(y356)/sea-1(y356).
To measure the effect of sea-1 overexpression on male viability, yEx716[sea-1(+)] males were crossed to yDp14; unc-2 hermaphrodites (1626 total adult progeny), and the number of viable yDp14/+; unc-2 (non-green) males (142 observed, 736 expected) and yDp14/+; unc-2; yEx716[sea-1(+)] (green) males (325 observed, 411 expected) was counted. The expected number of each class was determined by crossing yEx716[sea-1(+)] males with N2 hermaphrodites to measure the transmission frequency of the array.
RNA Interference
RNAi against sex-1 and sdc-2 was performed as described (Kamath et al., 2000), except bacteria were grown in the presence of 100 μg/ml ampicillin and 12.5 μg/ml tetracycline. Of 4981 F1 embryos laid by yEx716[sea-1(+)]; sex-1(RNAi) hermaphrodites, less than 1% of the expected array-bearing progeny (4 observed, 1982 expected) survived, based on the transmission frequency of the array in the absence of sex-1 RNAi.
SNP Mapping
sea-1(y356) was mapped using Snip-SNPs (single nucleotide polymorphisms that alter a restriction enzyme site) between CB4856, the Hawaiian isolate of C. elegans (Koch et al., 2000), and N2, the parental strain for sea-1. sea-1; fox-1 sex-1 hermaphrodites were crossed with homozygous CB4856 males; the heterozygous male cross progeny were crossed to sea-1; fox-1 sex-1 hermaphrodites. Cross progeny hermaphrodites homozygous for sea-1 and fox-1 sex-1 may have acquired Hawaiian SNP alleles by recombination.
Multiple Snip-SNP loci were genotyped from a single recombinant worm using a bulk worm lysate generated from the self-progeny of that recombinant hermaphrodite (Wicks et al., 2001). 1 μL of lysate served as the template for a 25 μl PCR reaction (0.4 μM each primer, 0.4 mM dNTPs, 1 U Roche Expand Long polymerase mix, 1X Expand Long buffer #1). The entire reaction was digested for 4 hr in a final volume of 30 μl (5-10 U appropriate restriction enzyme), half of which was run on a 2% NuSieve agarose gel.
Linkage of sea-1 to chromosome II was demonstrated by genotyping 17 recombinant hermaphrodites for 15 SNPs spanning the 5 autosomes. The recombinants were cross progeny from mating y356/CB4856 II; fox-1 sex-1 X males with y356 II; fox-1 sex-1 X hermaphrodites. y356 was unlinked to any of the SNPs on chromosomes I, III, IV, or V. 2/17 recombinants acquired Hawaiian alleles at map positions LGII:0.12 and LGII:13.4 but retained the N2 allele at LGII:-9.2. 1/17 acquired the Hawaiian allele at -9.2 but retained N2 alleles at 0.12 and 13.4. This recombination data places y356 on the left side of chromosome II between -9.2 and 0.12.
The location of sea-1 was further narrowed to a 0.8 cM interval by genotyping 166 additional recombinants at 15 additional SNP loci between map positions -9.2 and 0.12. One recombinant acquired Hawaiian alleles at map position -6.33 and all SNPs to its left but not at -6.2 or any other SNPs to the right. Thus, y356 is to the right of map position -6.33. Another recombinant acquired Hawaiian alleles at -5.49 and all to its right. Therefore, y356 is to the left of map position -5.49. Thus, y356 is in the interval bounded on the left by -6.33 on YAC Y49F6b and on the right by -5.49 on cosmid T24E12.
Transgene Arrays
To express sdc-2(+) independently of the sex signal, the array yEx483 was made by coinjecting pTY975 Pdpy-30::sdc-2 (50 ng/μl), pRF4 rol-6(su1006) (100 ng/μl), and pPD118.33 myo-2::gfp (50 ng/μl) (from A. Fire) into wild-type worms. To make cosmid lines to test rescue of sea-1(y356), individual cosmids (10-20 ng/μl) were coinjected with pPD118.33 myo-2::gfp (30 ng/μl) into sea-1(y356) hermaphrodites. yEx716[sea-1(+)] was made from a PCR product of the genomic region corresponding to ORF F19B10.9. The primers used (JRP73: TAGTCATGACGTTCTTCCTGAAACTT and JRP74: GTGACGGATGGAGAATAGTTGTATAT) amplify the entire region between the two neighboring ORFs, F19B10.8 and F19B10.2. sea-1(y356) hermaphrodites were coinjected with this PCR product (2 ng/μl), pPD118.33 myo-2::gfp (30 ng/μl), and a mixture (100 ng/μl) of other cosmid and PCR product DNA that had been shown not to rescue sea-1. yIs61[sea-1(+)] is an autosomal integrant of yEx71-6[sea-1(+)], following UV irradiation (Frank et al., 2003). yIs33 is a Pxol-1::lacZ reporter containing 2.8 kb of xol-1 upstream regulatory sequence fused to the lacZ ORF and the 3′ UTR from unc-54 to stabilize the lacZ transcript (Nicoll et al., 1997).
Quantification of xol-1 Transcript Levels
Quantitative RT-PCR analysis (Van Gilst et al., 2005) of xol-1 transcript levels was performed on three independent cDNA preparations made from each of wild-type, him-8, and yIs61[sea-1(+)] embryos. xol-1 transcript levels were normalized to nhr-64 transcript levels.
SEA-1 Antibody
Rabbit anti-SEA-1 antibodies were raised (Covance, Inc.) and affinity-purified against an N-terminal 21 amino acid peptide plus CG linker, NQKYPQSAEEYHKVLCNYFIT (synthesized by David King). The C terminus of this peptide is located 2 amino acids N-terminal to the ochre stop codon introduced by the sea-1(y356) mutation.
Immunoblot Analysis
To prepare C. elegans embryonic extract, gravid hermaphrodites were washed off plates with M9 and treated with hypochlorite to obtain embryos. Embryos were boiled in an equal volume of SDS-PAGE buffer for 10 min and spun for 1 min at 16,000 × g. The supernatant was fractionated through a 10% SDS-PAGE gel. The Western blot was performed with rabbit anti-SEA-1, mouse antitubulin (iCN), horseradish peroxidase-conjugated donkey anti-rabbit, (Jackson ImmnunoResearch Labs), and horseradish peroxidase-conjugated donkey anti-mouse (Jackson ImmnunoResearch Labs) antibodies.
Immunofluorescence Microscopy
sea-1, wild-type, and yIs61[sea-1(+)] embryos were prepared as described (Severson et al., 2000) but were fixed for 10 min in room temperature ethanol, and the slides were incubated with antibodies overnight at 4°C. The following antibodies were used: rabbit anti-SEA-1, rat anti-SDC-3 (P. McDonel, B.J.M.), FITC-conjugated goat anti-rabbit (Jackson ImmnunoResearch Labs), and Cy3-conjugated donkey anti-rat (Jackson ImmnunoResearch Labs). At least 500 embryos of each genotype were scored.
fox-1 sex-1, wild-type, and sea-1; fox-1 sex-1 embryos were prepared as described (Chuang et al., 1994), except 2% paraformaldehyde was used in the fixative and the embryos incubated in secondary antibodies overnight at room temperature. The following antibodies were used: rabbit anti-DPY-27 (Chuang et al., 1994), rat anti-SDC-3 (P. McDonel, B.J.M.), FITC-conjugated goat-anti-rabbit (Jackson ImmnunoResearch Labs), and Cy-3-conjugated donkey anti-rat, diluted (Jackson ImmnunoResearch Labs). At least 2000 embryos of each genotype were scored.
All embryos were mounted in VectaShield (Vector Laboratories, Inc) containing 1 μg/ml diamidinophenylindole (DAPI). All fluorescent images were obtained on a Leica TCS NT confocal microscope.
Supplementary Material
Acknowledgments
We are grateful to E. Ralston for advice and technical assistance. We thank P. Nix for performing the quantitative RT-PCR analysis of xol-1 transcript levels. We also thank T. Cline, P. Nix, and A. Severson for insightful comments on the manuscript. Some nematode strains were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). J.R.P. was supported by a Howard Hughes Medical Institute predoctoral fellowship. M.M.J. was supported by NIH predoctoral training grant 2T32GM007127. B.J.M. is an investigator of the Howard Hughes Medical Institute. This work was supported by NIH grant R37GM30702 to B.J.M.
References
- Barbash DA, Cline TW. Genetic and molecular analysis of the autosomal component of the primary sex determination signal of Drosophila melanogaster. Genetics. 1995;141:1451–1471. doi: 10.1093/genetics/141.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi I, Meyer BJ. The primary sex determination signal of Caenorhabditis elegans. Genetics. 1999;152:999–1015. doi: 10.1093/genetics/152.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi I, Kopczynski JB, Meyer BJ. The nuclear hormone receptor SEX-1 is an X-chromosome signal that determines nematode sex. Nature. 1998;396:168–173. doi: 10.1038/24164. [DOI] [PubMed] [Google Scholar]
- Chan RC, Severson AF, Meyer BJ. Condensin restructures chromosomes in preparation for meiotic divisions. J. Cell Biol. 2004;167:613–625. doi: 10.1083/jcb.200408061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesley P. Develoment of the short-tailed mutant in the house mouse. J. Exp. Zool. 1935;70:429–459. [Google Scholar]
- Chu DS, Dawes HE, Lieb JD, Chan RC, Kuo AF, Meyer BJ. A molecular link between gene-specific and chromosome-wide transcriptional repression. Genes Dev. 2002;16:796–805. doi: 10.1101/gad.972702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang PT, Albertson DG, Meyer BJ. DPY-27:a chromosome condensation protein homolog that regulates C. elegans dosage compensation through association with the X chromosome. Cell. 1994;79:459–474. doi: 10.1016/0092-8674(94)90255-0. [DOI] [PubMed] [Google Scholar]
- Cline TW, Meyer BJ. Vive la difference: males vs females in flies vs worms. Annu. Rev. Genet. 1996;30:637–702. doi: 10.1146/annurev.genet.30.1.637. [DOI] [PubMed] [Google Scholar]
- Csankovszki G, McDonel P, Meyer BJ. Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science. 2004;303:1182–1185. doi: 10.1126/science.1092938. [DOI] [PubMed] [Google Scholar]
- Dawes HE, Berlin DS, Lapidus DM, Nusbaum C, Davis TL, Meyer BJ. Dosage compensation proteins targeted to X chromosomes by a determinant of hermaphrodite fate. Science. 1999;284:1800–1804. doi: 10.1126/science.284.5421.1800. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia-Zavadskaia N. Sur la mortification spontanee de la queue chez la souris nouveau-nee se sur l’existence d’un caactere heriditaire non-viable. Crit. Rev. Soc. Biol. 1927;97:114–116. [Google Scholar]
- Frank CA, Baum PD, Garriga G. HLH-14 is a C. elegans achaete-scute protein that promotes neurogenesis through asymmetric cell division. Development. 2003;130:6507–6518. doi: 10.1242/dev.00894. [DOI] [PubMed] [Google Scholar]
- Gluecksohn-Schoenheimer S. The development of normal and homozygous brachy (T/T) mouse embryos in the extraembryonic coelem of the chick. Proc. Natl. Acad. Sci. USA. 1944;30:134–140. doi: 10.1073/pnas.30.6.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2000;2:0002.0001–0002.0010. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispert A, Hermann BG. The Brachyury gene encodes a novel DNA binding protein. EMBO J. 1993;12:4898–4899. doi: 10.1002/j.1460-2075.1993.tb06179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch R, van Luenen HG, van der Horst M, Thijssen KL, Plasterk RH. Single nucleotide polymorphisms in wild isolates of Caenorhabditis elegans. Genome Res. 2000;10:1690–1696. doi: 10.1101/gr.gr-1471r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madl JE, Herman RK. Polyploids and sex determination in Caenorhabditis elegans. Genetics. 1979;93:393–402. doi: 10.1093/genetics/93.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BJ, Casson LP. Caenorhabditis elegans compensates for the difference in X chromosome dosage between the sexes by regulating transcript levels. Cell. 1986;47:871–881. doi: 10.1016/0092-8674(86)90802-0. [DOI] [PubMed] [Google Scholar]
- Miller LM, Plenefisch JD, Casson LP, Meyer BJ. xol-1: a gene that controls the male modes of both sex determination and X chromosome dosage compensation in C. elegans. Cell. 1988;55:167–183. doi: 10.1016/0092-8674(88)90019-0. [DOI] [PubMed] [Google Scholar]
- Nicoll M, Akerib CC, Meyer BJ. X-chromosomecounting mechanisms that determine nematode sex. Nature. 1997;388:200–204. doi: 10.1038/40669. [DOI] [PubMed] [Google Scholar]
- Nusbaum C, Meyer BJ. The Caenorhabditis elegans gene sdc-2 controls sex determination and dosage compensation in XX animals. Genetics. 1989;122:589–593. doi: 10.1093/genetics/122.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind NR, Miller LM, Kopczynski JB, Meyer BJ. xol-1 acts as an early switch in the C. elegans male/hermaphrodite decision. Cell. 1995;80:71–82. doi: 10.1016/0092-8674(95)90452-2. [DOI] [PubMed] [Google Scholar]
- Severson AF, Hamill DR, Carter JC, Schumacher J, Bowerman B. The aurora-related kinase AIR-2 recruits ZEN-4/CeMKLP1 to the mototic spindle at metaphase and is required for cytokinesis. Curr. Biol. 2000;10:1162–1171. doi: 10.1016/s0960-9822(00)00715-6. [DOI] [PubMed] [Google Scholar]
- Showell C, Binder O, Conlon FL. T-box genes in early embryogenesis. Dev. Dyn. 2004;229:201–218. doi: 10.1002/dvdy.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Smith JC. T-targets: clues to understanding the functions of T-box proteins. Dev. Growth Differ. 2001;43:1–11. doi: 10.1046/j.1440-169x.2001.00556.x. [DOI] [PubMed] [Google Scholar]
- Van Gilst MR, Hadjivassiliou H, Jolly A, Yamamoto KR. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 2005;3:e53. doi: 10.1371/journal.pbio.0030053. 10.1371/journal.pbio.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 2001;28:160–164. doi: 10.1038/88878. [DOI] [PubMed] [Google Scholar]
- Younger-Shepherd S, Vaessin H, Bier E, Jan LY, Jan YN. deadpan, an essential lpan-neural gene encoding an HLH protein, acts as a denominator in Drosophila sex determination. Cell. 1992;70:911–922. doi: 10.1016/0092-8674(92)90242-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.