

Abstract
Modulation of immune/inflammatory responses by diverse strategies including amyloid-β (Aβ) immunization, nonsteroidal anti-inflammatory drugs, and manipulation of microglial activation states has been shown to reduce Alzheimer's disease (AD)-like pathology and cognitive deficits in AD transgenic mouse models. Human umbilical cord blood cells (HUCBCs) have unique immunomodulatory potential. We wished to test whether these cells might alter AD-like pathology after infusion into the PSAPP mouse model of AD. Here, we report a marked reduction in Aβ levels/β-amyloid plaques and associated astrocytosis following multiple low-dose infusions of HUCBCs. HUCBC infusions also reduced cerebral vascular Aβ deposits in the Tg2576 AD mouse model. Interestingly, these effects were associated with suppression of the CD40–CD40L interaction, as evidenced by decreased circulating and brain soluble CD40L (sCD40L), elevated systemic immunoglobulin M (IgM) levels, attenuated CD40L-induced inflammatory responses, and reduced surface expression of CD40 on microglia. Importantly, deficiency in CD40 abolishes the effect of HUCBCs on elevated plasma Aβ levels. Moreover, microglia isolated from HUCBC-infused PSAPP mice demonstrated increased phagocytosis of Aβ. Furthermore, sera from HUCBC-infused PSAPP mice significantly increased microglial phagocytosis of the Aβ1-42 peptide while inhibiting interferon-γ-induced microglial CD40 expression. Increased microglial phagocytic activity in this scenario was inhibited by addition of recombinant CD40L protein. These data suggest that HUCBC infusion mitigates AD-like pathology by disrupting CD40L activity.
Introduction
Alzheimer disease (AD) is the most common progressive dementia and is pathologically characterized by deposition of amyloid-β peptide (Aβ) in the brain parenchyma. Aβ plaques are potent activators of both microglia and astrocytes, central nervous system (CNS)-resident immunocompetent cells that respond to cerebral amyloidosis by chronic low-level proinflammatory activation [1]. Although it was once thought that activation of microglia and astrocytes in the AD brain was an epiphenomenon and not a pathoetiological contributor to AD, more recent studies have suggested that the Aβ-mediated inflammatory cascade is an etiological perpetrator in AD. For example, therapeutic strategies aimed at manipulating this inflammatory cascade, including Aβ immunization [2–4], nonsteroidal anti-inflammatory drugs (NSAIDs) [5–9], and modulation of microglial activation [10–14], are able to reduce AD-like pathology and improve behavioral impairment in Alzheimer's transgenic mouse models and, in some cases, reduce AD pathology in humans.
Previously, we showed that the CD40–CD40 ligand (CD40L) interaction plays a critical role in Aβ-induced proinflammatory microglial activation [11]. Moreover, we have demonstrated that disruption of this signaling pathway reduces cerebral Aβ deposits in the Tg2576 mouse model of AD and improves cognitive deficits in PSAPP AD mice [12–15]. The implication of CD40–CD40L interaction in AD-associated brain inflammatory process is supported from studies demonstrating increased expression of CD40 and CD40L in and around β-amyloid plaques in AD brain [16,17]. Recently, Desideri and colleagues [18] reported that circulating soluble CD40L (sCD40L) levels are significantly increased in AD patients versus healthy elderly controls, further supporting a role for this receptor/ligand dyad in the pathogenesis of AD.
Human umbilical cord blood cells (HUCBCs) have been shown to oppose the proinflammatory T helper cell type 1 (Th1) response, as demonstrated in an animal model of stroke where HUCBC infusion promoted a strong anti-inflammatory T helper 2 (Th2) response [19]. Importantly, this effect was associated with reduced infarct volume and rescue of behavioral deficit [19]. HUCBC infusion has also shown therapeutic benefits in other neuroinflammatory conditions, including multiple sclerosis, amyotrophic lateral sclerosis, age-related macular degeneration, and Parkinson's disease [20–22]. In AD preclinical models, administration of these cells to the PSAPP mice was associated with extension of lifespan, although high doses were administered in this paradigm [23].
On the basis of these lines of evidence, we investigated whether multiple low-dose administrations of HUCBCs to AD transgenic mouse models could reduce AD-like pathology through suppression of deleterious inflammatory responses involving the CD40 pathway. To address this possibility, we infused both double-transgenic PSAPP mice and Tg2576 mice with HUCBCs and then examined cerebral parenchymal and vascular Aβ levels/β-amyloid deposits, astrocytosis, microgliosis, and CD40 pathway-associated molecules.
Materials and Methods
Animals and administration of HUCBCs
HUCBCs (95–98% mononuclear cells) were provided by Saneron CCEL Therapeutics, Inc. (Tampa, FL). Transgenic PSAPP (APPswe, PSEN1dE9) and Tg2576 mice were obtained from the Jackson Laboratory (Bar Harbor, ME) [24,25] and Taconic, Inc. (Germantown, NY) [26], respectively, and were treated intravenously (i.v.) with HUCBCs (100,000 cells/mouse) or phosphate-buffered saline (PBS) biweekly for the first 2 months and monthly for the remaining 4 months (n = 10/group, 5 males and 5 females). Mice were treated starting at 7 months of age (after appreciable Aβ deposits), and blood was collected by submandibular bleeding at 0, 2, 4, and 6 months to monitor plasma cytokines, sCD40L and Aβ levels throughout the study. We analyzed brains of these mice for Aβ deposits and gliosis at 13 months of age (when these mice manifest well-established AD-like pathology, including Aβ deposits and gliosis). We treated PSAPP mice deficient in CD40 and controls at 8 weeks of age (preliminary studies showed that we can clearly detect plasma Aβ levels at this age) with HUCBCs. Blood samples were collected by submandibular bleeding at the second month after the treatment. Animals were housed and maintained in the College of Medicine Animal Facility at the University of South Florida (USF), and all experiments were performed in compliance with protocols approved by USF Institutional Animal Care and Use Committee.
Immunohistochemistry analysis
Mice were anesthetized with isofluorane and perfused transcardially with ice-cold physiological saline containing heparin (10 U/ml). Brains were rapidly isolated and quartered using a mouse brain slicer (Muromachi Kikai, Tokyo, Japan). The first and second anterior quarters were homogenized for western blot analysis, and the third and fourth posterior quarters were used for microtome or cryostat sectioning [15]. Brains were then fixed in 4% paraformaldehyde in PBS at 4°C overnight and routinely processed in paraffin at a core facility at the Department of Pathology (USF College of Medicine). Five coronal sections from each brain (5 μm thickness) were cut with a 150-μm interval [for cingulate cortex (CC) bregma −0.10 mm to −0.82 mm; for entorhinal cortex (EC) and hippocampus (H), bregma −2.92 mm to −3.64 mm]. Sections were routinely deparaffinized and hydrated in a graded series of ethanol before pre-blocking for 30 min at ambient temperature with serum-free protein block (Dako Cytomation, Carpinteria, CA). Aβ immunohistochemical staining was performed using anti-human amyloid-β antibody (4G8) in conjunction with the VectaStain Elite ABC kit (Vector Laboratories, Burlingame, CA) coupled with diaminobenzidine substrate. Congo Red staining was done according to standard practice using 10% (wt/vol) filtered Congo Red dye cleared with alkaline alcohol. These sections were rinsed 3× for 5 min each in 70% ethanol, hydrated for 5 min in PBS, and mounted in Vectashield fluorescence mounting medium (Vector Laboratories). β-Amyloid plaques positive for 4G8 or Congo Red were detected under bright field using an Olympus BX-51 microscope. Aβ burden was determined by quantitative image analysis. Briefly, images of five 5-μm sections (150 μm apart) through each anatomic region of interest (hippocampus and neocortex) were captured and a threshold optical density was obtained that discriminated staining from background. Manual editing of each field was used to eliminate artifacts. Data are reported as percentage of immunolabeled area captured (positive pixels divided by total pixels captured). Quantitative image analysis was performed by a single examiner (T.M.) blinded to sample identities.
Immunofluorescence analysis
Double immunofluorescence for Aβ and CD40 was performed using rat anti-mouse CD40 (1:1,000; Pharmingen, Los Angeles, CA) and rabbit anti-pan Aβ (1:100; Biosource International, Inc.) with overnight incubation followed by incubation at ambient temperature with goat anti-rat immunoglobulin G (IgG) fluorescein isothiocyanate (FITC, 1:50; PharMingen) and donkey anti-rabbit Alexa Fluor555 (1:500; Invitrogen, Carlsbad, CA) for 45 min. Double immunofluorescence for Aβ and activated astrocytes was performed using a biotinylated human amyloid-β monoclonal antibody (4G8; 1:100, Signet Laboratories, Dedham, MA) and glial fibrillary acidic protein (GFAP) polyclonal antibody (1:500, DAKO). Normal rabbit, normal mouse serum (isotype control), or PBS (0.1 M, pH 7.4) were used instead of primary antibody or avidin-biotin-peroxidase complex (ABC) reagent as negative controls. Quantitative image analysis was done based on a previous method [15] with minor modifications. Images were acquired as digitized tagged-image format files to retain maximum resolution using an Olympus BX-60 microscope with an attached digital camera system (DP-70, Olympus, Tokyo, Japan), and digital images were routed into a Windows PC for quantitative analyses using SimplePCI software (Compix, Inc. Imaging Systems, Cranberry Township, PA). The cingulate cortex region was captured from the image of the cortex adjacent to the sagittal fissure, and the entorhinal cortex region was captured from the image of the cortex ventral to the entorhinal fissure. In images from cingulate and entorhinal regions, the cortical edge was not included so that the full anatomic region of interest could be captured. The hippocampal region was captured from between a portion of the CA1 subfield of the pyramidal cell layer and the lacunosum molecular layer. The anatomical locations and boundaries of the regions analyzed were based on those previously defined [27]. Images of five 5-μm sections through each anatomic region of interest were captured, and a threshold optical density was obtained that discriminated staining from background. Each anatomic region of interest was manually edited to eliminate artifacts. For “burden” analyses, data are represented as percentage of immunolabeled area captured (positive pixels) relative to the full area captured (total pixels).
Flow cytometric and western blot analyses of CD40 expression
For flow cytometric analysis of microglial CD40 expression, primary cultured microglial cells were plated in six-well tissue culture plates at 5 × 105 cells/well and incubated with interferon-γ (IFN-γ, 100 ng/ml) in the presence or absence of serum derived from HUCBC- or PBS-infused individual PSAPP mice. Twelve hours after incubation, microglial cells were washed with flow buffer, consisting of PBS containing 0.1% (wt/vol) sodium azide, and 2% (vol/vol) fetal calf serum (FCS), and resuspended in 250 μl of ice-cold flow buffer for fluorescence-activated cell sorting (FACS) analysis, according to methods described previously [28]. Briefly, cells were preincubated with anti-mouse CD16/CD32 monoclonal antibody (clone 2.4G2, PharMingen) for 10 min at 4°C to block nonspecific binding to Fc receptors. Cells were then centrifuged at 5,000 × g, washed three times with flow buffer, and then incubated in flow buffer with hamster anti-mouse CD40-FITC or isotype control antibody-FITC (1:100 dilution; PharMingen). After 30 min of incubation at room temperature, cells were washed twice with flow buffer, resuspended in 250 μl of flow buffer and analyzed by a FACScan™ instrument (Becton Dickinson, Franklin Lakes, NJ). A minimum of 10,000 cells were accepted for FACS analysis. Cells were gated based on morphological characteristics using CellQuest™ software (Beckton Dickinson) such that apoptotic and necrotic cells were not accepted for FACS analysis. Percentages of positive (CD40-expressing) cells were calculated as follows: for each treatment, the mean fluorescence value for the isotype-matched control antibody was subtracted from the mean fluorescence value for the CD40-specific antibody.
For western immunoblotting analysis of brain CD40 expression, mouse brain homogenates were prepared from HUCBC-and PBS-infused PSAPP mice, as previously described [15]. An aliquot corresponding to 100 μg of total protein of each sample was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred electrophoretically to immunoblotting polyvinylidene difluoride (PVDF) membranes. Nonspecific antibody binding was blocked with 5% nonfat dry milk for 1 h at room temperature in Trisbuffered saline (TBS; 20 mM Tris and 500 mM NaCl, pH 7.5). Subsequently, membranes were first hybridized with rabbit anti-CD40 antibody (1:1,500 dilution; StressGen, Victoria, Canada) for 2 h and then washed three times in TBS. Immunoblotting was by an anti-rabbit horseradish peroxidase (HRP)-conjugated IgG secondary antibody as a tracer. The luminol reagent was used to develop the blots. To demonstrate equal loading, the same membranes were then stripped with β-mercaptoethanol stripping solution (62.5 mM Tris-HCl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol) and reprobed with mouse monoclonal antibody to actin. Densitometric analysis was done as previously described [15] using a FluorS Multiimager with Quantity One™ software (BioRad, Hercules, CA).
Aβ and cytokine enyzme-linked immunosorbent assays
Mouse brains were isolated under sterile conditions on ice and placed in ice-cold lysis buffer as previously described [15]. Brains were then sonicated on ice for approximately 3 min, allowed to stand for 15 min at 4°C, and centrifuged at 15,000 rpm for 15 min. This fraction represented the detergent-soluble fraction. Detergent-insoluble Aβ1-40,42 species were further subjected to acid extraction of brain homogenates in 5 M guanidine buffer [29], followed by a 1:5 dilution in lysis buffer. Aβ1-40,42 was detected in brain homogenates prepared with lysis buffer or in plasma samples at a 1:10 or 1:5 dilution, respectively, in dilution buffer consisting of PBS + 1% bovine serum albumin (BSA) + phenylmethylsulfonyl fluoride (PMSF). Aβ1-40,42 was quantified in these samples using our own Aβ1-40,42 enzyme-linked immunosorbent assay (ELISA) kits [30] and further evaluated with commercially available Aβ1-40,42 ELISA kits (IBL-America, Minneapolis, MN) in accordance with the manufacturer's instructions, except that standards included 0.5 M guanidine buffer in some cases to facilitate Aβ aggregation. Aβ1-40,42 were represented as pg/ml of plasma and pg/mg of total protein (mean ± SD).
Cell suspensions of splenocytes from individual mice were prepared as previously described [31] and passed in 0.5-ml aliquots into 24-well plates at 3 × 106/ml. These cells were treated for 48 h with concanavalin A (ConA, 5 μg/ml). Supernatants were then collected and assayed by interleukin-10 (IL-10), tumor necrosis factor-α (TNF-α), and IL-12(p70) cytokine ELISA kits in strict accordance with the manufacturer's instructions (R&D Systems, Minneapolis, MN). The Bio-Rad protein assay was performed to measure total cellular protein from each of the cell groups under consideration just prior to quantification of cytokine release by ELISA, and cytokine secretion was expressed as pg/mg total cellular protein (mean ± SD). To verify whether stimulation of splenocytes produced any between-groups differences on cell death that might account for altered cytokine profiles, lactate dehyrogenase (LDH) release assay was carried out as described [31], and LDH was not detected in any of the wells studied. ELISAs for IgM and IgG antibodies were carried out as previously described [32]. Optical densities were determined by a microplate reader at 450 nm. The ratio of IgM to IgG was calculated using optical density values, and then the average ratio for each group was determined (mean ± SD). Brain tissue-derived (from the detergent-soluble brain homogenate fraction) and serum-derived (plasma) samples were analyzed for sCD40L (Bener MedSystems, Burlingame, CA), IL-4, IL-10, IL-2, IFN-γ, TNF-α, IL-1β, IL-12 (p70), and transforming growth factor-β (TGF-β) cytokines by Bioplex assays (Bio-Rad Laboratories) according to the manufacturer's protocol.
Microglial phagocytosis assay
Primary cultures of murine microglia were established as previously described [11,33]. For fluorometric analysis of FITC-Aβ1–42, primary murine microglia were seeded at 1 × 105 cells/well (n = 6 for each condition) in 24-well tissue-culture plates containing 0.5 ml of complete RPMI-1640 medium. These cells were treated for 60 min with “aged” Aβ1-42 conjugated with FITC (Biosource International) [33]. In the presence of FITC-Aβ1-42, microglia were then co-treated with serum (1/200, 1/400, 1/800 dilution) derived from HUCBC- or PBS-infused individual PSAPP mice in the presence or absence of CD40L protein (2 μg/ml). Microglia were rinsed three times in Aβ-free complete medium, and media were exchanged with fresh Aβ-free complete medium for 10 min both to allow for removal of nonincorporated Aβ and to promote concentration of the Aβ into phagosomes. Extracellular and cell associated FITC-Aβ were quantified using an MSF reader (SpectraMax®, Molecular Devices, Sunnyvale, CA) with an emission wavelength of 538 nm and an excitation wavelength of 485 nm. A standard curve from 0 nM to 500 nM of FITC-Aβ was run for each plate. Total cellular proteins were quantified using the Bio-Rad protein assay. The mean fluorescence values for each sample were determined by fluorometic analysis. Relative fold change values were calculated as the mean fluorescence value for each experimental sample over control. In this manner, both extracellular and cell associated FITC-Aβ were quantified. To determine the extent to which cell death might have influenced phagocytic activity in the various treatment groups, we performed LDH release assay, and no significant cell death was detected over the 3 h timeframe in any of the treatment groups (p > 0.05).
Primary culture peripheral macrophages were collected from 3-month-old wild-type mice by infusing their peritoneal cavity with ice-cold PBS following a 4-day intraperitoneal (i.p.) injection with 1 ml of 3% (wt/vol) brewer's thyoglycollate resuspended in PBS. Cells were pooled following the isolation to decrease variance. Then they were plated in culture medium (RPMI-1640; 10% FBS and antibiotics) to give 1.5 × 106 cells/well in six-well plates. Cells were incubated overnight at 37°C under 5% CO2 in a humidified incubator, and nonadherent cells were removed by washing twice with PBS at 37°C. Following the removal of nonadherent cells, the remaining cells were tested for Aβ phagocytosis as described above with the addition of 1:200, 1:400, and 1:800 dilution of sera derived from HUCBC-treated mice, sera derived from PBS-treated mice, as well as supernatants from cultured HUCBCs.
Statistical analysis
Data are presented as mean ± SD. All statistics were calculated using one-way analysis of variance (ANOVA) for multiple comparisons. A p value of <0.05 was considered significant. The statistical package for the social sciences release 10.0.5 (SPSS Inc., Chicago, Illinois) was used for all data analysis.
Results
Cerebral parenchymal and vascular β-amyloid plaques are reduced in AD transgenic mice peripherally infused with HUCBCs
Previous work in a mouse model of stroke has shown that HUCBC infusion results in significant reduction in infarct volume as well as rescue of behavioral deficits associated with decreased proinflammatory cytokine production [19]. We sought to determine whether HUCBC (95–98% mononuclear cells) infusion could impact Aβ-associated pathology in PSAPP double-transgenic mice. These animals were injected i.v. with HUCBCs (100,000 cells/mouse) beginning at 7 months of age (when β-amyloid deposits have already accumulated). At 13 months of age, mice were sacrificed and evaluated for changes in AD-like pathology. We chose to administer multiple low doses of HUCBCs because, in our pilot studies, we observed that this strategy was superior compared to a single high dose of HUCBCs on reducing cerebral amyloidosis in Tg2576 mice (data not shown). HUCBC infusion in PSAPP mice resulted in marked reduction of cerebral β-amyloid pathology as assayed by Aβ antibody (4G8) immunohistochemistry (Fig. 1A) and Congo Red histochemistry (Fig. 1C). Quantitative image analysis revealed statistically significant differences for each brain region examined (p < 0.001) between PSAPP mice infused with HUCBCs (PSAPP/HUCBC) and PSAPP mice peripherally infused with PBS (PSAPP/PBS) for both Aβ antibody (Fig. 1B) and Congo Red staining (Fig. 1D). Furthermore, ELISA analysis of brain extracts showed that levels of both detergent-soluble and -insoluble Aβ1-40,42 peptides were reduced in PSAPP mice infused with HUCBCs (by 62% and 70%, respectively; Fig. 1E). A t-test for independent samples revealed significant between-groups differences for each group examined (p < 0.001).
FIG. 1.
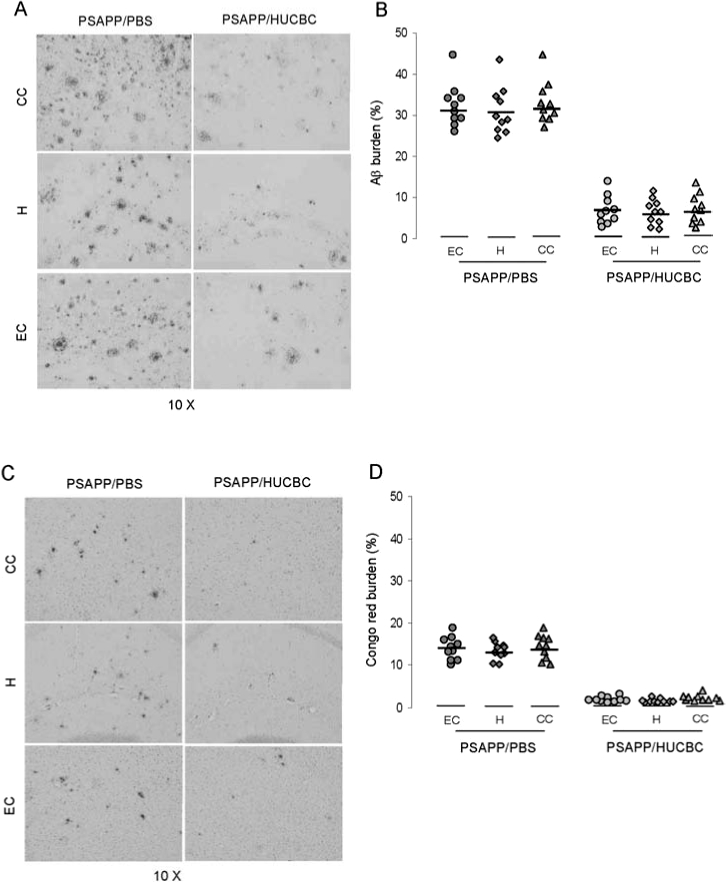

Cerebral Aβ/β-amyloid pathology is reduced in PSAPP and Tg2576 mice peripherally infused with HUCBCs. Mouse paraffin-embedded coronal brain sections from the cingulate cortex (CC), hippocampus (H), and entorhinal cortex (EC) were stained with monoclonal human Aβ antibody 4G8 (A) or Congo Red (C). Percentages (plaque area/total area) of Aβ antibody-immunoreactive deposits (B) or of Congo Red-stained deposits (D) were calculated by quantitative image analysis (mean ± SD; n = 10, 5 females and 5 males per group). (E) Aβ ELISA analysis was carried out for both levels of detergent-soluble Aβ1-40,42 (top panel) or 5 M guanidine-extracted Aβ1-40,42 (bottom panel). Data are represented as mean ± SD of Aβ1-40,42 (pg/mg protein). Mouse paraffin-embedded coronal brain sections from hippocampal regions of Tg2576 mice were stained with Congo Red (F). Positions of the hippocampal subfields CA1, CA3, and dentate gyrus (DG) are indicated (upper left panel). Arrows indicate Aβ deposit-affected vessels. (G) Percentages (% labeled area) of Congo Red-stained plaques/vessels were quantified by image analysis (mean ± SD; n = 10, 5 females and 5 males), and percentage reduction is indicated.
Given that peripheral administration of HUCBCs reduces cerebral parenchymal Aβ deposits and brain Aβ levels in PSAPP mice, we wished to investigate the impact of HUCBC infusion on cerebral amyloid angiopathy (CAA), which is characterized by Aβ deposits in the cerebral vasculature and is known to occur in 83% of AD patients [34]. For this analysis, we used the Tg2576 mouse model of AD, which is known to manifest copious Aβ deposits in cerebral vessels at 15–20 months of age [35–39]. We peripherally infused these mice with HUCBC or controls (PBS vehicle treatment or no treatment) (n = 10, 5 males and 5 females per group) at 12 months of age using the identical procedure above. Six months thereafter, these mice were sacrificed for analyses of cerebral parenchymal or vascular β-amyloid deposits by Congo Red histochemistry. As shown in Fig. 1F, Tg2576 mice receiving HUCBC treatment demonstrated reduction of both cerebral parenchymal and vascular Congo Red deposits compared with controls. Quantitative image analysis revealed statistically significant differences between Tg2576/HUCBC and Tg2576/PBS or nontreated control groups when examining total (78%), vascular (86%), or parenchymal (74%) Congo Red staining (p < 0.001; Fig. 1G). No significant difference was revealed between Tg2576/PBS and nontreated Tg2576 control mice (p > 0.05). In addition, we also analyzed cerebral Aβ levels/β-amyloid deposits by Aβ ELISA and Aβ antibody immunohistochemistry, and we obtained statistically significant results similar to those observed in HUCBC-infused PSAPP mice (p < 0.001; data not shown).
Reduced CD40+ microglia and GFAP+ astrocytes in PSAPP mice peripherally infused with HUCBCs
Previously, it has been suggested that brain inflammation resulting from activated microglia and astrocytes contributes to β-amyloid plaque formation, and we have previously shown that ligation of microglial CD40 enables activation in response to Aβ peptides [11,42,43]. To investigate whether HUCBCs could inhibit brain inflammation, we examined co-localization of β-amyloid deposits with CD40+ microglia (an in vivo microgliosis marker [17]) or reactive GFAP+ astrocytes by immunohistochemistry and western blot analyses in PSAPP mice. As shown in Fig. 2A, CD40+ microglial cells were reduced in the PSAPP/HUCBC-infused group. Quantitative image analysis revealed statistically significant reductions when comparing PSAPP/HUCBC-infused and PSAPP/PBS-infused groups for both Aβ and CD40 staining in hippocampal dentate gyrus and CA1 regions (**p < 0.001) (Fig. 2B). Western blot analysis of CD40 expression showed a statistically significant decrease in brain homogenates from HUCBC-infused PSAPP mice (p < 0.001) (Fig. 2C). Furthermore, immunohistochemistry/histochemistry and immunofluorescence analyses showed reductions in β-amyloid-associated astrocytosis in PSAPP/HUCBC mice versus PSAPP/PBS-treated mice (Fig. 2D), and morphometry revealed reductions for neocortex and hippocampus by 84% and 86%, respectively in PSAPP/HUCBC mice (p < 0.001) (Fig. 2E).
FIG. 2.
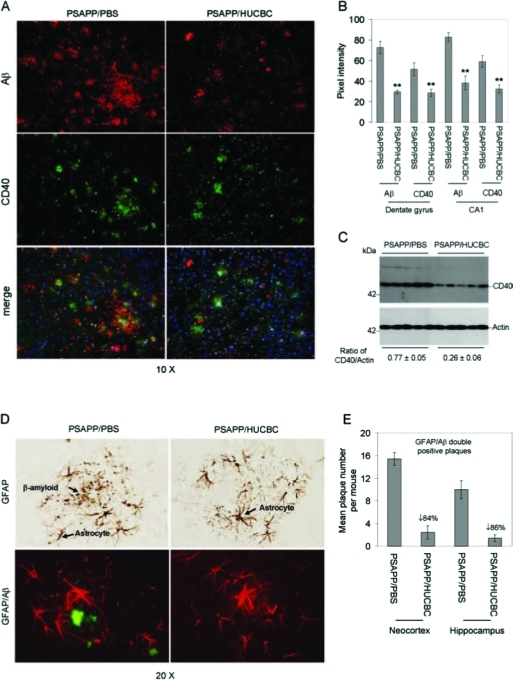
β-Amyloid-associated microgliosis and astrocytosis are reduced in HUCBC infused-PSAPP mice. (A) Immunofluorescence was performed on mouse brain coronal paraffin sections prepared from PSAPP mice infused with HUCBC or PBS. The red signal indicates Aβ+ (top panels); green indicates CD40+ (middle panels), and merged images (bottom panels) reveal co-localization of CD40 and Aβ. DAPI (blue) was used as a nuclear counterstain. (B) Immunofluorescence intensity for Aβ and CD40 was determined. (C) Western blot analysis shows reduced CD40 expression in brain homogenates from PSAPP/HUCBC versus PSAPP/PBS mice as indicated (actin was used as an internal reference control). Densitometry analysis shows the ratio of CD40 to actin as indicated below the figure. (D) Immunohistochemistry analysis shows GFAP staining (top panel), and immunofluorescence (bottom panel) reveals co-localization of GFAP (red signal) and Aβ (green signal). (E) Morphometric analysis results (mean GFAP/β-amyloid double positive plaques per mouse ± SD) are shown for the neocortex and the hippocampus of PSAPP/HUCBC versus PSAPP/PBS mice. Percent reduction of plaques double positive for GFAP and Aβ in PSAPP/HUCBC mice is indicated.
Increased plasma Aβ levels correlate with decreased CD40–CD40L interaction in HUCBC-infused PSAPP mice
Previously, we have shown that administration of neutralizing CD40L antibody to PSAPP mice results in increased levels of plasma Aβ concomitant with reduced cerebral Aβ/β-amyloid pathology, suggesting that depletion of CD40L promotes brain-to-blood clearance of Aβ [15]. It is well-known that the CD40–CD40L interaction promotes proinflammatory Th1 and opposes anti-inflammatory Th2 immune responses [44,45]. In addition, HUCBC treatment has been shown to be an immunoregulator in an animal model of stroke [19,46]. We investigated whether reduction of cerebral Aβ levels/β-amyloid deposits in HUCBC-infused PSAPP mice might result from increased brain-to-blood clearance of Aβ, and be associated with suppression of the proinflammatory CD40–CD40L interaction. We probed individual blood samples from PSAPP mice infused with HUCBCs or PBS for Aβ1-40,42 and sCD40L. ELISA revealed increased plasma Aβ1-40,42 levels in PSAPP/HUCBC mice that correlated inversely with decreased levels of plasma sCD40L in these animals (Figs. 3A–C). One-way ANOVA followed by post hoc comparison revealed significant differences between PSAPP/HUCBC-infused and PSAPP/PBS-infused mice for plasma Aβ1-40,42 levels and plasma sCD40L levels at each time point indicated (Figs. 3A–C) (**p < 0.001). It is well established that CD40–CD40L interaction on B cells is required for IgM to IgG antibody class switching. Therefore, we went on to evaluate the functional consequence of HUCBC-mediated suppression of the CD40–CD40L interaction on IgM and IgG titers in mouse blood samples obtained at the time of sacrifice. ELISA data revealed a significantly increased ratio of IgM to IgG in PSAPP/HUCBC mice when compared to control (Fig. 3D, *p < 0.05), suggesting that the CD40 signaling pathway is functionally suppressed in HUCBC-infused PSAPP mice. It has been recently reported that CD40 deficiency in APP transgenic mice confers a decrease in Aβ/β-amyloid loads [10]. Although to a lesser extent than PSAPP/CD40+/+ mice, we also found that PSAPP/CD40−/− mice do clearly manifest β-amyloid deposits (data not shown), allowing us to test whether administration of HUCBC to PSAPP/CD40−/− mice resulted in further amelioration of amyloidosis in these animals. Thus, we treated PSAPP/CD40−/− at 8 weeks of age with HUCBCs or PBS (control) and assayed circulating Aβ levels, which correlate with cerebral amyloid levels in transgenic AD mice [47]. Results indicate no further benefit of HUCBC in PSAPP/CD40−/− mice on enhanced Aβ plasma levels (Figs. 3E,F; p < 0.05), a presumed indicator of Aβ brain-to-blood efflux. These data suggest that HUCBCs mediate beneficial effects on reduction of amyloidosis via reducing CD40 pathway bioactivity, and are consistent with our previous studies showing that genetic or pharmacologic ablation of CD40–CD40L interaction mitigates AD-like pathology in transgenic mice [11,15].
FIG. 3.
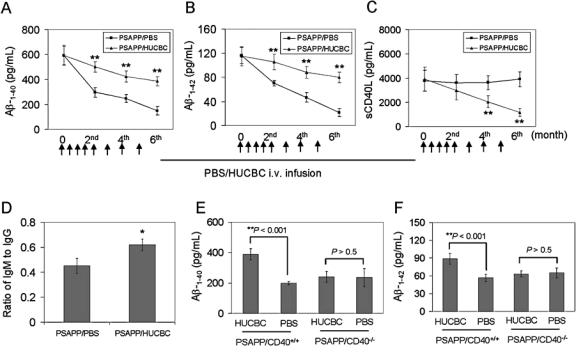
HUCBC infusion results in CD40-dependent increased plasma Aβ levels in PSAPP mice. ELISA analysis results are shown from blood (plasma) for Aβ1-40 (A), Aβ1-42 (B), sCD40L (C), and IgM/IgG (D). Data are presented as mean ± SD (n = 10) for Aβ1-40, Aβ1-42, or sCD40L (pg/ml plasma). Arrows below the panels show the time for each peripheral infusion with HUCBC or PBS. (D) Data are presented as a ratio of IgM to IgG in blood (plasma) from mice at the 6th month following the treatment. Aβ ELISA analysis for Aβ1-40 (E) and Aβ1-42 (F) in blood (plasma) derived from PSAPP/CD40+/+ or PSAPP/CD40−/− mice at the 2nd month, following the third HUCBC infusion. Data in E and F are presented as mean ± SD (n = 4, 2 males and 2 females) of Aβ1-40 or Aβ1-42 (pg/ml plasma).
We hypothesized that, if HUCBCs mediated reduced amyloidosis by reducing CD40 pathway activity, this should be associated with a shift from pro- to anti-inflammatory cytokines in HUCBC-infused PSAPP mice. Consistent with this hypothesis, we found that plasma levels of the anti-inflammatory cytokines IL-4 and IL-10 were increased in HUCBC-infused PSAPP mice (Fig. 4A, **p < 0.001). Furthermore, primary splenocytes from HUCBC-infused PSAPP mice showed reduced proinflammatory TNF-α and IL-12 (p70) and increased anti-inflammatory IL-10 secretion (Fig. 4B, **p < 0.001). We also analyzed brain cytokine levels by ELISA, and results showed statistically significant increases in anti-inflammatory TGF-β1 and IL-10 levels in PSAPP/HUCBC-infused mouse brain homogenates (Fig. 4C; **p < 0.001). Consistent with our data showing reduction in circulating sCD40L after HUCBC treatment, we also measured sCD40L in brain homogenates and found a significant decrease in PSAPP/HUCBC mice compared to control (Fig. 4D, **p < 0.001).
FIG. 4.
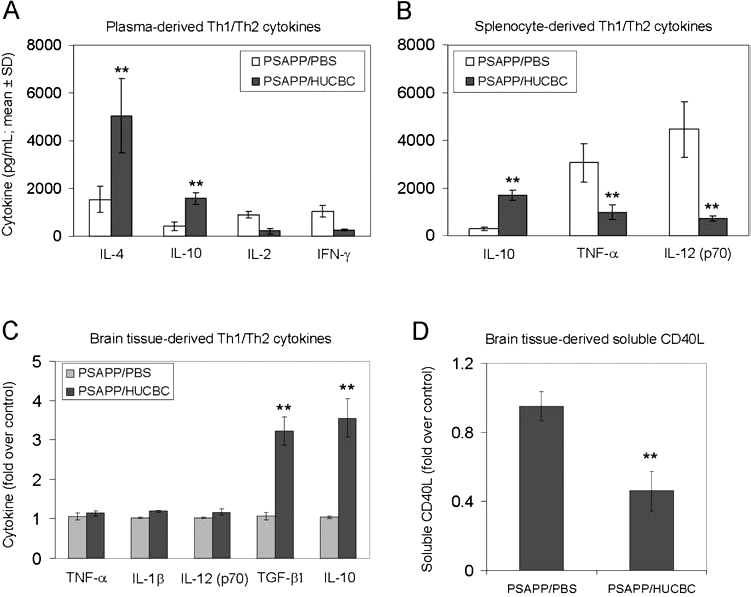
HUCBC infusion promotes anti-inflammatory/Th2 responses and decreases sCD40L in the CNS. ELISA results are shown for plasma-derived (A), splenocyte culture-derived (B), brain tissue-derived cytokines (C), and brain tissue-derived sCD40L (D). Data are presented as mean ± SD (n = 10) values of cytokines (pg/ml plasma or medium) (A and B) or fold increase of cytokines over control (untreated) mice (C and D).
HUCBCs inhibit microglial CD40 expression and enhance in vitro phagocytosis of Aβ peptides
We and others have previously shown that microglial CD40 expression is important for CNS inflammatory responses [11,12,15–17,42,43], and IFN-γ is a strong inducer of microglial CD40 expression [48–50]. To investigate whether a soluble factor secreted following HUCBC infusion could modulate microglial expression of CD40, we treated primary microglial cells with serum from HUCBC- or PBS-infused individual PSAPP mice in the presence of IFN-γ (100 ng/ml) for 8 h. We then examined CD40 expression by FACS analysis. As shown in Fig. 5A, sera derived from HUCBC-infused PSAPP mice significantly inhibited IFN-γ-induced microglial CD40 expression compared to controls (p < 0.001). However, this effect was not directly mediated by HUCBCs or human adult mononuclear cells (HAMNCs), but was rather due to a soluble circulating factor produced by HUCBC-infused PSAPP mice (Fig. 5A).
FIG. 5.
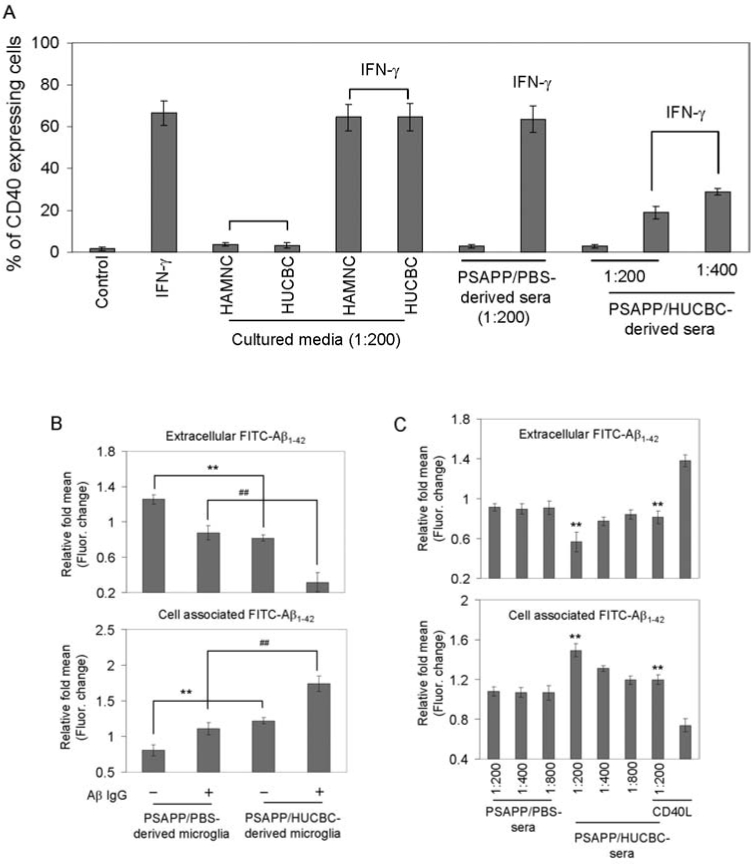
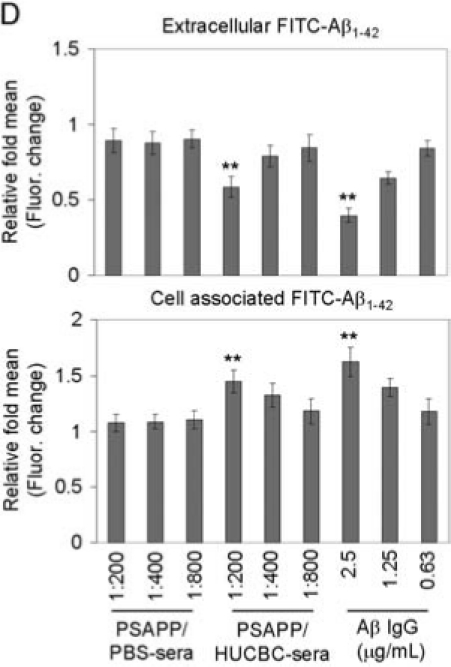
HUCBCs modulate microglial CD40 expression and promote Aβ microglial/macrophage phagocytic activity. (A) FACS analysis for CD40 expression in primary wild-type neonatal microglial cells treated with cultured medium from HUCBCs or human adult mononuclear cells (HAMNCs), or serum from individual PSAPP/HUCBC or PSAPP/PBS mice following IFN-γ challenge. Data are presented as percentage of CD40 expressing cells (mean ± SD; n = 5). (B–D) Microglial/macrophage phagocytosis assay results for extracellular and cell-associated FITC-Aβ1-42, which was detected using a fluorometer. Data are represented as the relative fold of mean ± SD fluorescence over control for each sample (n = 4 for each condition presented). Primary microglial cells from adult PSAPP/HUCBC or PSAPP/PBS mice (B), wild-type neonatal microglia (C), and primary peripheral macrophages (D) are shown.
We and others have shown that stimulation of microglial CD40 results in impaired Aβ phagocytic activity [33] and promotion of microgial neurotoxic inflammatory responses [51]. Thus, we wished to examine whether HUCBCs could enhance microglial phagocytosis of the Aβ peptide. We prepared primary cultures of adult microglia from HUCBC- and PBS-infused PSAPP mice according to previously described methods [52], and then subjected these cells to Aβ phagocytosis assay using native or Aβ antibody-opsonized fluorescent-tagged Aβ1-42 (FITC-Aβ1-42) according to our previously described methods [33]. As shown in Fig. 5B, when measuring fluorescein isothiocyanate (FITC)-tagged Aβ1-42 in cell supernatants or lysates, one-way ANOVA followed by post hoc comparison showed a significant increase in Aβ uptake by microglia derived from HUCBC-versus PBS-infused PSAPP mice (**p < 0.001). Interestingly, the presence of Aβ IgG (2.5 μg/ml) [33] significantly enhanced Aβ uptake by PSAPP/HUCBC- versus PSAPP/PBS-derived microglial cells (##p < 0.001). Given that sera from HUCBC-infused PSAPP mice suppressed IFN-γ-induced microglial CD40 expression, we wished to test if the sera could increase microglial Aβ phagocytosis. We incubated primary cultures of neonatal microglia with serum from individual PSAPP/HUCBC- versus PSAPP/PBS mice at 1:200, 1:400, and 1:800 dilutions in the presence of FITC-Aβ1-42. We found that sera at the 1:200 dilution markedly enhanced microglial phagocytosis of Aβ1-42 peptide, which was opposed by the presence of recombinant mouse CD40L protein at 2 μg/ml (Fig. 5C).
In addition, we wished to test if sera-derived from HUCBC-treated mice could increase peripheral macrophage phagocytic activity. We incubated both sera derived from HUCBCs and PBS-treated animals at 1:200, 1:400, and 1:800 dilutions with primary macrophage cells from wild-type mice in six-well tissue-culture plates in the presence of 300 nM FITC-Aβ1-42 as described above. We found that sera at the 1:200 dilution significantly enhanced macrophage phagocytosis of Aβ1-42 peptide (Fig. 5D) (**p < 0.01 with n = 4 for each treatment group presented). However these effects were not observed in cultured HUCBC media (data not shown).
Discussion
On the basis of genetic, biochemical, and post mortem evidence, Aβ peptides are key etiological contributors to AD pathogenesis [53]. In addition to parenchymal Aβ deposits, deposition of Aβ in the cerebral vasculature (known as CAA) is a pathological feature of AD, and occurs with 83% frequency in AD patients [34,54–56]. Aβ has been shown to mediate proinflammatory and neurodegenerative changes, and oligomeric forms of the peptide are neurotoxic [57]. It is well documented that brain inflammatory mechanisms mediated by reactive glia are activated in response to Aβ plaques [1,33,58,59]. Expression profiles of two such proinflammatory molecules, CD40 and CD40L, are markedly increased in and around Aβ plaques in AD patients and in mouse models of the disease [16,17], and genetic or pharmacologic blockade of the CD40–CD40L interaction reduces AD-like pathology in transgenic AD mice [15], suggesting an etiologic role of this receptor/ligand dyad in the disease [12,14]. In a recent clinical report, it was found that circulating sCD40L levels are significantly increased in AD patients [18], suggesting that peripheral as well as brain dysregulation of the CD40 pathway occurs in AD. We have previously shown that CD40 ligation promotes proinflammatory activation of microglia and reduces microglial phagocytosis of Aβ peptide in vitro [11,33], supporting a mechanistic explanation for reduced AD-like pathology after blocking the CD40–CD40L interaction [12].
HUCBCs have been shown to down-regulate the proinflammatory Th1 response in an animal model of stroke [19], and have also shown to be of therapeutic benefit in other neuroinflammatory/neurodegenerative conditions [20–22]. On the basis of this evidence, we sought to examine their putative therapeutic value in mitigating AD-like pathology in transgenic mice. After HUCBC infusion, treated mice exhibited diminished cerebral Aβ/β-amyloid pathology and down-regulation of proinflammatory responses in the brain and in the periphery. Based on the conspicuous role of the CD40–CD40L interaction in mediating brain proinflammatory response and exacerbating AD-like pathology, we investigated whether HUCBC-mediated reduction of AD-like pathology might be associated with alteration in this receptor/ligand dyad. Our results show decreased expression of microglial CD40 and reduction in both CNS and peripheral sCD40L concomitant with HUCBC-induced diminished AD-like pathology, raising the possibility that disruption of the CD40–CD40L interaction may be responsible for mitigation of AD-like pathology in this scenario. To address this hypothesis directly, we treated PSAPP mice homozygous deficient for CD40 with HUCBC and assayed circulating Aβ levels as a marker of brain-to-blood Aβ efflux, and results showed no further benefit of HUCBC in these mice.
Here, we demonstrate that infusion of the HUCBC mononuclear fraction into PSAPP and Tg2576 mice results in reduced levels of both soluble and insoluble brain Aβ1-40,42 concomitant with increased plasma Aβ1-40,42 levels. Past studies have suggested brain-to-blood clearance mechanisms that selectively remove Aβ from the brain, potentially reducing Aβ levels in normal as well as AD patient brains [47,60–63]. Experiments in rat models demonstrating clearance of Aβ1-40 peptide from the brain via the blood–brain barrier (BBB) support this notion [62–64]. Vascular endothelial cells, which are important BBB constituents, express CD40 [14,28,49,65], and we now show that sCD40L is reduced in blood plasma from HUCBC-treated PSAPP mice, raising the possibility that interruption of CD40–CD40L interaction at the level of cerebrovascular endothelial cells may promote brain-to-blood clearance of Aβ. Furthermore, reduced circulating sCD40L levels in HUCBC-treated PSAPP mice raises the possibility that inflammatory cytokines produced by the CD40–CD40L interaction on endothelial cells are reduced. This idea is consistent with our finding of a shift toward anti-inflammatory cytokines in the CNS after HUCBC infusion. Interestingly, we also demonstrate that CAA, which is present in 83% of AD patients [34], is reduced by 68% after HUCBC treatment in Tg2576 AD mice. This result shows that reduction in parenchymal Aβ does not come at the cost of increased vascular Aβ deposits, unlike a model in which TGF-β1 overexpression reduces parenchymal plaques but increases vascular Aβ deposits [66,67].
In vitro HUCBC studies have shown that these cells secrete soluble factors that have beneficial effects [46,68]. For example, supernatants from cultured HUCBCs promote survival of NT2 neural cells and peripheral blood mononuclear cells cultured under conditions designed to induce cell stress and limit protein synthesis [20]. Additionally, HUCBCs have been shown to produce a number of neurotrophic factors and cytokines that modulate inflammatory responses, including nerve growth factor, colony stimulating factor-1, thrombopoietin, and IL-11 [19,69,70]. Previous reports have shown that HUCBC entry into the brain is not required to promote neuroprotection [71], and that recovery following brain injury is mediated through peripheral responses [72]. We did not detect infiltration of HUCBCs into brain parenchyma, either at 4 h after HUCBC administration or at the time of mouse sacrifice (data not shown), making it unlikely that these cells were directly involved in ameliorating cerebral amyloidosis. Therefore, we hypothesized that a soluble factor produced after HUCBC infusion in the periphery was responsible for reduced AD-like pathology and inflammatory response. To test this, we: (1) measured cytokines in blood plasma, spleen, and brains from HUCBC- or PBS-treated PSAPP mice, (2) evaluated the impact of sera from these treated mice on IFN-γ-induced microglial CD40 expression, and (3) assayed Aβ phagocytosis in vitro in neonatal microglia treated with sera from HUCBC/PSAPP or PBS/PSAPP mice and in adult microglial cultures derived from these mice. Results generally show a shift from proinflammatory Th1-type cytokines toward anti-inflammatory Th2 cytokines in tissues from HUCBC-treated PSAPP mice. Furthermore, sera from HUCBC-treated mice are able to reduce microglial CD40 expression and enhance Aβ phagocytosis by these cells. Finally, adult microglia from HUCBC-treated PSAPP mice have increased capacity to phagocytose Aβ.
When taken together, the above results suggest that, in addition to promoting brain-to-blood Aβ efflux, HUCBC infusion promotes production of a peripheral anti-inflammatory soluble factor that is likely able to cross the BBB and affect microglial Aβ clearance. Previous reports have show that soluble factors, including heat-shock proteins and pro-inflammatory cytokines, are capable of modulating Aβ phagocytosis by microglia [73,74], and our previous work has shown that microglial CD40–CD40L interaction retards Aβ phagocytosis/clearance [33]. Nonsaturable BBB transport mechanisms have been described for a number of cytokines including TNF-α (which is transported via TNF receptors) and IL-1, and other soluble factors such as leukemia inhibitory factor, chemoattractant-1, and epithelial growth factor [75].
Thus, it remains possible that soluble factors produced by the host in response to HUCBC treatment gain access to the brain via the BBB and encounter microglia. Ultimately, we propose that infused HUCBCs exert their effect on reducing cerebral amyloidosis by causing the host to secrete a soluble factor that acts by reducing sCD40L–CD40 interaction on microglia, which then promotes microglial clearance of Aβ. This mechanism is supported by our observations of: (1) reduced brain levels of sCD40L in HUCBC-infused PSAPP mice, (2) reduced CD40 expression on microglia cultured in the presence of HUCBC-infused PSAPP mouse sera, (3) increased Aβ phagocytosis/removal by microglia cultured in the presence of HUCBC-infused PSAPP mouse sera or cultured from adult PSAPP/HUCBC mice, and (4) our previous observations that microglial CD40 ligation shifts these cells away from a Aβ phagocytic phenotype and toward a proinflammatory response [33]. Future studies designed to identify this soluble factor are warranted and may yield additional pharmacotherapeutic target(s). Additionally, our observation of no further therapeutic benefit of HUCBCs when administered to PSAPP/CD40−/− mice establishes a CD40 pathway-dependent mechanism for HUCBC therapeutic benefit on reduction of cerebral amyloidosis. These results dovetail with our previous studies showing that the CD40–CD40L interaction mitigates AD-like pathology in transgenic mice [11,15].
Recently, it was shown that peripheral macrophages are able to infiltrate the brain and limit cerebral amyloidosis in AD mice after irradiation, suggesting that hematogenously derived macrophages are efficient at phagocytosing and clearing Aβ deposits [76]. However, earlier studies have shown that brain-resident microglia are also able to phagocytose/clear Aβ [77–79]. We did not detect the presence of brain-infiltrating macrophages in the current experimental paradigm. Specifically, we stained for CD40 (a marker for both macrophages and microglia), and noted the presence of process-bearing cells that morphologically resembled microglia in and around Aβ plaques (see Fig. 2A). Also, we did not observe vascular “cuffing” that would suggest the presence of infiltrating macrophages that are frequently observed in other CNS inflammatory conditions such as experimental autoimmune encephalomyelitis [80]. Furthermore, our results provide evidence that both primary culture microglia and macrophages posses the ability for enhanced Aβ phagocytosis following in vitro stimulation with sera derived from HUCBC-treated animals. This, too, is consistent with peripheral immunomodulation of the CD40–CD40L interaction by HUCBC treatment. Additionally, given the difficulties inherent to discriminating macrophages from microglia, and the ability of peripheral macrophages to engraft into the CNS and take up a microglial phenotype after brain injury [81], it remains possible that peripheral macrophages may contribute to reduced cerebral amyloidosis after HUCBC treatment.
In this report, we have shown that HUCBC infusion ameliorates AD-like pathology, including reductions in: (1) cerebral Aβ levels/β-amyloid pathology, (2) CAA, and (3) brain inflammation including CD40+-activated microglia and GFAP+-activated astrocytes. These effects of HUCBCs were associated with increased brain-to-blood efflux of Aβ and a shift from proinflammatory Th1 to anti-inflammatory Th2 cytokines both in the brain and in the periphery, similar to what we observed after Aβ immunization [31,82,83]. In addition, HUCBC infusion of PSAPP mice reduces both CNS and circulating sCD40L levels, and sera from these mice is able to promote microglial Aβ phagocytosis. When taken together, our results provide the basis for a novel immunomodulatory strategy for AD using HUCBCs.
Acknowledgments
This work was supported by the Johnnie B. Byrd, Sr. Alzheimer's Center & Research Institute (J.T.), the National Institutes of Health/National Institute on Aging (NIH/NIA, R41AG031586, J.T.), the NIH/National Institute of Neurological Disorders and Stroke (NINDS, R01NS048335, J.T.); Cryo-Cell International, Inc. and Saneron CCEL Therapeutic, Inc. T.T. is supported by an Alzheimer Association grant and an NIH/NIA “Pathway to Independence” award (K99 AG029726). We thank Dr. Huntington Potter and Dr. Alison Willing for helpful advice.
Disclosure
P.R.S. is a cofounder and J.T. is a consultant for Saneron CCEL Therapeutics, Inc. and are inventors on a patent application submitted by USF. P.R.S. was not involved in any data acquisition and analysis.
References
- 1.Benzing WC. Wujek JR. Ward EK. Shaffer D. Ashe KH. Younkin SG. Brunden KR. Evidence for glialmediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20:581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 2.Bard F. Cannon C. Barbour R. Burke RL. Games D. Grajeda H. Guido T. Hu K. Huang J. Johnson-Wood K. Khan K. Kholodenko D. Lee M. Lieberburg I. Motter R. Nguyen M. Soriano F. Vasquez N. Weiss K. Welch B. Seubert P. Schenk D. Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 3.Nicoll JA. Wilkinson D. Holmes C. Steart P. Markham H. Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nature Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 4.Schenk D. Barbour R. Dunn W. Gordon G. Grajeda H. Guido T. Hu K. Huang J. Johnson-Wood K. Khan K. Kholodenko D. Lee M. Liao Z. Lieberburg I. Motter R. Mutter L. Soriano F. Shopp G. Vasquez N. Vandevert C. Walker S. Wogulis M. Yednock T. Games D. Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 5.Lim GP. Chu T. Yang F. Beech W. Frautschy SA. Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim GP. Yang F. Chu T. Chen P. Beech W. Teter B. Tran T. Ubeda O. Ashe KH. Frautschy SA. Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim GP. Yang F. Chu T. Gahtan E. Ubeda O. Beech W. Overmier JB. Hsiao-Ashec K. Frautschy SA. Cole GM. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001;22:983–991. doi: 10.1016/s0197-4580(01)00299-8. [DOI] [PubMed] [Google Scholar]
- 8.Szekely CA. Thorne JE. Zandi PP. Ek M. Messias E. Breitner JC. Goodman SN. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004;23:159–169. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- 9.in t’ Veld BA. Ruitenberg A. Hofman A. Launer LJ. van Duijn CM. Stijnen T. Breteler MM. Stricker BH. Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- 10.Laporte V. Ait-Ghezala G. Volmar CH. Mullan M. CD40 deficiency mitigates Alzheimer's disease pathology in transgenic mouse models. J Neuroinflammation. 2006;3:3. doi: 10.1186/1742-2094-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan J. Town T. Paris D. Mori T. Suo Z. Crawford F. Mattson MP. Flavell RA. Mullan M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- 12.Tan J. Town T. Mullan M. CD40-CD40L interaction in Alzheimer's disease. Curr Opin Pharmacol. 2002;2:445–451. doi: 10.1016/s1471-4892(02)00180-7. [DOI] [PubMed] [Google Scholar]
- 13.Todd Roach J. Volmar CH. Dwivedi S. Town T. Crescentini R. Crawford F. Tan J. Mullan M. Behavioral effects of CD40-CD40L pathway disruption in aged PSAPP mice. Brain Res. 2004;1015:161–168. doi: 10.1016/j.brainres.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Town T. Tan J. Mullan M. CD40 signaling and Alzheimer's disease pathogenesis. Neurochem Int. 2001;39:371–380. doi: 10.1016/s0197-0186(01)00044-4. [DOI] [PubMed] [Google Scholar]
- 15.Tan J. Town T. Crawford F. Mori T. DelleDonne A. Crescentini R. Obregon D. Flavell RA. Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nature Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- 16.Calingasan NY. Erdely HA. Altar CA. Identification of CD40 ligand in Alzheimer's disease and in animal models of Alzheimer's disease and brain injury. Neurobiol Aging. 2002;23:31–39. doi: 10.1016/s0197-4580(01)00246-9. [DOI] [PubMed] [Google Scholar]
- 17.Togo T. Akiyama H. Kondo H. Ikeda K. Kato M. Iseki E. Kosaka K. Expression of CD40 in the brain of Alzheimer's disease and other neurological diseases. Brain Res. 2000;885:117–121. doi: 10.1016/s0006-8993(00)02984-x. [DOI] [PubMed] [Google Scholar]
- 18.Desideri G. Cipollone F. Necozione S. Marini C. Lechiara MC. Taglieri G. Zuliani G. Fellin R. Mezzetti A. di Orio F. Ferri C. Enhanced soluble CD40 ligand and Alzheimer's disease: Evidence of a possible pathogenetic role. Neurobiol Aging. 2006;29:348–356. doi: 10.1016/j.neurobiolaging.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 19.Vendrame M. Cassady J. Newcomb J. Butler T. Pennypacker KR. Zigova T. Sanberg CD. Sanberg PR. Willing AE. Infusion of human umbilical cord blood cells in a rat model of stroke dose-dependently rescues behavioral deficits and reduces infarct volume. Stroke. 2004;35:2390–2395. doi: 10.1161/01.STR.0000141681.06735.9b. [DOI] [PubMed] [Google Scholar]
- 20.El-Badri NS. Hakki A. Saporta S. Liang X. Madhusodanan S. Willing AE. Sanberg CD. Sanberg PR. Cord blood mesenchymal stem cells: Potential use in neurological disorders. Stem Cells Dev. 2006;15:497–506. doi: 10.1089/scd.2006.15.497. [DOI] [PubMed] [Google Scholar]
- 21.Garbuzova-Davis S. Gografe SJ. Sanberg CD. Willing AE. Saporta S. Cameron DF. Desjarlais T. Daily J. Kuzmin-Nichols N. Chamizo W. Klasko SK. Sanberg PR. Maternal transplantation of human umbilical cord blood cells provides prenatal therapy in Sanfilippo type B mouse model. FASEB J. 2006;20:485–487. doi: 10.1096/fj.05-4684fje. [DOI] [PubMed] [Google Scholar]
- 22.Henning RJ. Burgos JD. Ondrovic L. Sanberg P. Balis J. Morgan MB. Human umbilical cord blood progenitor cells are attracted to infarcted myocardium and significantly reduce myocardial infarction size. Cell Transplant. 2006;15:647–658. doi: 10.3727/000000006783981611. [DOI] [PubMed] [Google Scholar]
- 23.Ende N. Chen R. Ende-Harris D. Human umbilical cord blood cells ameliorate Alzheimer's disease in transgenic mice. J Med. 2001;32:241–247. [PubMed] [Google Scholar]
- 24.Garcia-Alloza M. Robbins EM. Zhang-Nunes SX. Purcell SM. Betensky RA. Raju S. Prada C. Greenberg SM. Bacskai BJ. Frosch MP. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 25.Jankowsky JL. Slunt HH. Ratovitski T. Jenkins NA. Copeland NG. Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 26.Hsiao K. Chapman P. Nilsen S. Eckman C. Harigaya Y. Younkin S. Yang F. Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 27.Mori T. Town T. Tan J. Yada N. Horikoshi Y. Yamamoto J. Shimoda T. Kamanaka Y. Tateishi N. Asano T. Arundic Acid ameliorates cerebral amyloidosis and gliosis in Alzheimer transgenic mice. J Pharmacol Exp Ther. 2006;318:571–578. doi: 10.1124/jpet.106.105171. [DOI] [PubMed] [Google Scholar]
- 28.Tan J. Town T. Suo Z. Wu Y. Song S. Kundtz A. Kroeger J. Humphrey J. Crawford F. Mullan M. Induction of CD40 on human endothelial cells by Alzheimer's beta-amyloid peptides. Brain Res Bull. 1999;50:143–148. doi: 10.1016/s0361-9230(99)00122-7. [DOI] [PubMed] [Google Scholar]
- 29.Johnson-Wood K. Lee M. Motter R. Hu K. Gordon G. Barbour R. Khan K. Gordon M. Tan H. Games D. Lieberburg I. Schenk D. Seubert P. McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rezai-Zadeh K. Shytle D. Sun N. Mori T. Hou H. Jeanniton D. Ehrhart J. Townsend K. Zeng J. Morgan D. Hardy J. Town T. Tan J. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci. 2005;25:8807–8814. doi: 10.1523/JNEUROSCI.1521-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Town T. Vendrame M. Patel A. Poetter D. DelleDonne A. Mori T. Smeed R. Crawford F. Klein T. Tan J. Mullan M. Reduced Th1 and enhanced Th2 immunity after immunization with Alzheimer's beta-amyloid(1-42) J Neuroimmunol. 2002;132:49–59. doi: 10.1016/s0165-5728(02)00307-7. [DOI] [PubMed] [Google Scholar]
- 32.Nikolic WV. Bai Y. Obregon D. Hou H. Mori T. Zeng J. Ehrhart J. Shytle RD. Giunta B. Morgan D. Town T. Tan J. Transcutaneous beta-amyloid immunization reduces cerebral beta-amyloid deposits without T cell infiltration and microhemorrhage. Proc Natl Acad Sci USA. 2007;104:2507–2512. doi: 10.1073/pnas.0609377104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend KP. Town T. Mori T. Lue LF. Shytle D. Sanberg PR. Morgan D. Fernandez F. Flavell RA. Tan J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol. 2005;35:901–910. doi: 10.1002/eji.200425585. [DOI] [PubMed] [Google Scholar]
- 34.Ellis RJ. Olichney JM. Thal LJ. Mirra SS. Morris JC. Beekly D. Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- 35.Christie R. Yamada M. Moskowitz M. Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol. 2001;158:1065–1071. doi: 10.1016/S0002-9440(10)64053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedlich AL. Lee JY. van Groen T. Cherny RA. Volitakis I. Cole TB. Palmiter RD. Koh JY. Bush AI. Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer's disease. J Neurosci. 2004;24:3453–3459. doi: 10.1523/JNEUROSCI.0297-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J. Onstead L. Randle S. Price R. Smithson L. Zwizinski C. Dickson DW. Golde T. McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L. Cao D. Garber DW. Kim H. Fukuchi K. Association of aortic atherosclerosis with cerebral beta-amyloidosis and learning deficits in a mouse model of Alzheimer's disease. Am J Pathol. 2003;163:2155–2164. doi: 10.1016/s0002-9440(10)63572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robbins EM. Betensky RA. Domnitz SB. Purcell SM. Garcia-Alloza M. Greenberg C. Rebeck GW. Hyman BT. Greenberg SM. Frosch MP. Bacskai BJ. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006;26:365–371. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frackowiak J. Wisniewski HM. Wegiel J. Merz GS. Iqbal K. Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol (Berl) 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 41.Potter H. Wefes IM. Nilsson LN. The inflammation-induced pathological chaperones ACT and apo-E are necessary catalysts of Alzheimer amyloid formation. Neurobiol Aging. 2001;22:923–930. doi: 10.1016/s0197-4580(01)00308-6. [DOI] [PubMed] [Google Scholar]
- 42.Tan J. Town T. Paris D. Placzek A. Parker T. Crawford F. Yu H. Humphrey J. Mullan M. Activation of microglial cells by the CD40 pathway: relevance to multiple sclerosis. J Neuroimmunol. 1999;97:77–85. doi: 10.1016/s0165-5728(99)00053-3. [DOI] [PubMed] [Google Scholar]
- 43.Tan J. Town T. Saxe M. Paris D. Wu Y. Mullan M. Ligation of microglial CD40 results in p44/42 mitogen-activated protein kinase-dependent TNF-alpha production that is opposed by TGF-beta 1 and IL-10. J Immunol. 1999;163:6614–6621. [PubMed] [Google Scholar]
- 44.Grewal IS. Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 45.Mackey MF. Barth RJ., Jr. Noelle RJ. The role of CD40/CD154 interactions in the priming, differentiation, and effector function of helper and cytotoxic T cells. J Leukoc Biol. 1998;63:418–428. doi: 10.1002/jlb.63.4.418. [DOI] [PubMed] [Google Scholar]
- 46.Newman MB. Willing AE. Manresa JJ. Sanberg CD. Sanberg PR. Cytokines produced by cultured human umbilical cord blood (HUCB) cells: implications for brain repair. Exp Neurol. 2006;199:201–208. doi: 10.1016/j.expneurol.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 47.DeMattos RB. Bales KR. Cummins DJ. Paul SM. Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- 48.Carson MJ. Reilly CR. Sutcliffe JG. Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 49.Chen K. Huang J. Gong W. Zhang L. Yu P. Wang JM. CD40/CD40L dyad in the inflammatory and immune responses in the central nervous system. Cell Mol Immunol. 2006;3:163–169. [PubMed] [Google Scholar]
- 50.Tan J. Town T. Mullan M. CD45 inhibits CD40L-induced microglial activation via negative regulation of the Src/p44/42 MAPK pathway. J Biol Chem. 2000;275:37224–37231. doi: 10.1074/jbc.M002006200. [DOI] [PubMed] [Google Scholar]
- 51.Ponomarev Shriver LP. Dittel BN. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J Immunol. 2006;176:1402–1410. doi: 10.4049/jimmunol.176.3.1402. [DOI] [PubMed] [Google Scholar]
- 52.Lue LF. Walker DG. Modeling Alzheimer's disease immune therapy mechanisms: interactions of human postmortem microglia with antibody-opsonized amyloid beta peptide. J Neurosci Res. 2002;70:599–610. doi: 10.1002/jnr.10422. [DOI] [PubMed] [Google Scholar]
- 53.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 54.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 55.Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 56.Vinters HV. Farag ES. Amyloidosis of cerebral arteries. Adv Neurol. 2003;92:105–112. [PubMed] [Google Scholar]
- 57.Malinin NL. Wright S. Seubert P. Schenk D. Griswold-Prenner I. Amyloid-beta neurotoxicity is mediated by FISH adapter protein and ADAM12 metallo-protease activity. Proc Natl Acad Sci USA. 2005;102:3058–3063. doi: 10.1073/pnas.0408237102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eikelenboom P. van Gool WA. Neuroinflammatory perspectives on the two faces of Alzheimer's disease. J Neural Transm. 2004;111:281–294. doi: 10.1007/s00702-003-0055-1. [DOI] [PubMed] [Google Scholar]
- 59.Rozemuller AJ. van Gool WA. Eikelenboom P. The neuroinflammatory response in plaques and amyloid angiopathy in Alzheimer's disease: therapeutic implications. Curr Drug Targets CNS Neurol Disord. 2005;4:223–233. doi: 10.2174/1568007054038229. [DOI] [PubMed] [Google Scholar]
- 60.Crossgrove JS. Li GJ. Zheng W. The choroid plexus removes beta-amyloid from brain cerebrospinal fluid. Exp Biol Med (Maywood) 2005;230:771–776. doi: 10.1177/153537020523001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeMattos RB. Bales KR. Parsadanian M. O'Dell MA. Foss EM. Paul SM. Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002;81:229–236. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- 62.Shibata M. Yamada S. Kumar SR. Calero M. Bading J. Frangione B. Holtzman DM. Miller CA. Strickland DK. Ghiso J. Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shiiki T. Ohtsuki S. Kurihara A. Naganuma H. Nishimura K. Tachikawa M. Hosoya K. Terasaki T. Brain insulin impairs amyloid-beta(1-40) clearance from the brain. J Neurosci. 2004;24:9632–9637. doi: 10.1523/JNEUROSCI.2236-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Terasaki T. Ohtsuki S. Brain-to-blood transporters for endogenous substrates and xenobiotics at the blood-brain barrier: an overview of biology and methodology. NeuroRx. 2005;2:63–72. doi: 10.1602/neurorx.2.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suo Z. Tan J. Placzek A. Crawford F. Fang C. Mullan M. Alzheimer's beta-amyloid peptides induce inflammatory cascade in human vascular cells: the roles of cytokines and CD40. Brain Res. 1998;807:110–117. doi: 10.1016/s0006-8993(98)00780-x. [DOI] [PubMed] [Google Scholar]
- 66.Wyss-Coray T. Borrow P. Brooker MJ. Mucke L. Astroglial overproduction of TGF-beta 1 enhances inflammatory central nervous system disease in transgenic mice. J Neuroimmunol. 1997;77:45–50. doi: 10.1016/s0165-5728(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 67.Wyss-Coray T. Lin C. Yan F. Yu GQ. Rohde M. McConlogue L. Masliah E. Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nature Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 68.Vendrame M. Gemma C. de Mesquita D. Collier L. Bickford PC. Sanberg CD. Sanberg PR. Pennypacker KR. Willing AE. Anti-inflammatory effects of human cord blood cells in a rat model of stroke. Stem Cells Dev. 2005;14:595–604. doi: 10.1089/scd.2005.14.595. [DOI] [PubMed] [Google Scholar]
- 69.McGowan E. Sanders S. Iwatsubo T. Takeuchi A. Saido T. Zehr C. Yu X. Uljon S. Wang R. Mann D. Dickson D. Duff K. Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol Dis. 1999;6:231–244. doi: 10.1006/nbdi.1999.0243. [DOI] [PubMed] [Google Scholar]
- 70.Suen Y. Lee SM. Schreurs J. Knoppel E. Cairo MS. Decreased macrophage colony-stimulating factor mRNA expression from activated cord versus adult mononuclear cells: altered posttranscriptional stability. Blood. 1994;84:4269–4277. [PubMed] [Google Scholar]
- 71.Borlongan CV. Hadman M. Sanberg CD. Sanberg PR. Central nervous system entry of peripherally injected umbilical cord blood cells is not required for neuroprotection in stroke. Stroke. 2004;35:2385–2389. doi: 10.1161/01.STR.0000141680.49960.d7. [DOI] [PubMed] [Google Scholar]
- 72.Sanberg PR. Willing AE. Garbuzova-Davis S. Saporta S. Liu G. Sanberg CD. Bickford PC. Klasko SK. El-Badri NS. Umbilical cord blood-derived stem cells and brain repair. Ann NY Acad Sci. 2005;1049:67–83. doi: 10.1196/annals.1334.008. [DOI] [PubMed] [Google Scholar]
- 73.Kakimura J. Kitamura Y. Takata K. Umeki M. Suzuki S. Shibagaki K. Taniguchi T. Nomura Y. Gebicke-Haerter PJ. Smith MA. Perry G. Shimohama S. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16:601–603. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- 74.Koenigsknecht-Talboo J. Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quan N. Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21:727–735. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 76.Simard AR. Soulet D. Gowing G. Julien JP. Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 77.Paresce DM. Ghosh RN. Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 78.Paresce DM. Chung H. Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid beta-protein by microglial cells. J Biol Chem. 1997;272:29390–29397. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- 79.Chung H. Brazil MI. Soe TT. Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- 80.Imrich H. Harzer K. On the role of peripheral macrophages during active experimental allergic encephalomyelitis (EAE) J Neural Transm. 2001;108:379–395. doi: 10.1007/s007020170060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Priller J. Flugel A. Wehner T. Boentert M. Haas CA. Prinz M. Fernandez-Klett F. Prass K. Bechmann I. de Boer BA. Frotscher M. Kreutzberg GW. Persons DA. Dirnagl U. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nature Med. 2001;7:1356–1361. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- 82.Town T. Nikolic V. Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Town T. Tan J. Flavell RA. Mullan M. T-cells in Alzheimer's disease. Neuromolecular Med. 2005;7:255–264. doi: 10.1385/NMM:7:3:255. [DOI] [PubMed] [Google Scholar]