

Abstract
DNA mismatch repair (MMR) within human cells is hypothesized to occur primarily at the replication fork. However, experimental models measuring MMR activity at specific phases of the cell cycle and during genomic DNA synthesis are lacking. We have investigated MMR activity within the nuclear environment of HeLa cells after enriching for G1, S and G2/M phase of the cell cycle by centrifugal elutriation. This approach preserves physiologically normal MMR activity in cell populations subdivided into different phases of the cell cycle. Here we have shown that nuclear protein concentration of hMutSα and hMutLα increases as cells progress into S phase during routine cell culture. MMR activity, as measured by both in vitro and in vivo approaches, increases during S phase to the highest extent within normally growing cells. Both fidelity and activity of MMR are highest on actively replicating templates within intact cells during S phase. The MMR pathway however, is also active at lower levels at other phases of the cell cycle, and on nonreplicating templates.
Keywords: DNA mismatch repair, cell cycle, centrifugal elutriation
1. INTRODUCTION
The DNA mismatch repair (MMR) system corrects mispaired bases, small insertion or deletion loops (IDLs), and contributes to the fidelity of DNA synthesis and recombination. This DNA repair pathway is critical for genetic stability and is conserved amongst all organisms. Hereditary nonpolyposis colon cancer (HNPCC; Lynch Syndrome) has been attributed to deficient MMR within humans [1]. Bacterial MMR has been well characterized as a methyl-directed, post-replicative genomic maintenance mechanism [2; 3; 4]. MMR activity within human cells has been hypothesized to occur at the replication fork, buttressing the notion that MMR is active primarily, if not exclusively, during S phase of the cell cycle [3; 5]. An experimental model has recently demonstrated the requirement of specific DNA replication factors in addition to MutSα and MutLα for successful 3′ and/or 5′ nick-directed MMR in a reconstituted in vitro system [11]. Experimental evidence using protein extracts and intact cells also associate MMR proteins with DNA replication proteins [10; 12; 13]. However, evidence limiting MMR activities only to the replication fork during S phase has not been demonstrated. In fact, studies using intact human cells have not yet demonstrated that MMR activity is at highest frequency and fidelity during the DNA synthesis stage of the cell cycle. To date, varying results have been obtained when measuring MMR expression levels at different phases of the cell cycle [6; 7; 8; 9; 10]. Problematic issues include cell synchronization methods that commonly rely on growth restriction such as serum starvation or a chemical treatment regimen (such as lovastatin, mimosine, aphidicolin, hydroxyurea, thymidine, nocodazole) to induce a population of cells to arrest at a specific stage of the cell cycle. These synchronization methods can significantly alter physiological events within the cell, including DNA repair pathways. As well, MMR proteins are reported to participate in additional biochemical pathways that occur throughout the cell cycle, such as genome surveillance for different types of DNA damage, as well as cell cycle arrest and apoptosis following DNA damaging events [14; 15].
We have previously reported that although MMR protein levels increase within the nucleus during S and G2 cell cycle phase, the MMR pathway appears active in all phases of the cell cycle [10]. Our current studies are a more in depth investigation of MMR dynamics at specific phases of a physiologically normal cell cycle. We have employed centrifugal elutriation as a novel stress-free approach to evaluate activity and fidelity of this DNA repair pathway at specific phases of the unaltered, normal cell cycle, including the significant contribution of active DNA synthesis to the efficiency and fidelity of MMR.
2. MATERIALS AND METHODS
Cell lines and growth conditions
HeLa S3 cells (National Cell Culture Center; Minneapolis, MN) were grown in Dulbecco’s Modified Essential Media (DMEM) supplemented with 10% calf serum (CS) at 37°C in a 5% CO2 humidified atmosphere. HCT 116 cells (MMR deficient) were grown in Iscoves Modified Dulbecco’s Media (IMDM) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 humidified atmosphere.
Centrifugal elutriation optimization conditions for phase separation
HeLa cells were prepared for elutriation as previously described [10]) with the following modification. Cells collected for extraction of nuclear proteins were collected on ice. Cells collected for replating to undergo transfection or for immunofluorescence studies were collected at room temperature. HeLa cells were partitioned into enriched fraction of G1, S, and G2/M cell cycle phases using centrifugal elutriation by the following methodologies. In brief, HeLa cells were grown to a confluence of 50–60%, collected and resuspended in fresh medium at 5×106 cells/ml. Cells were then passed gently through an 18-gauge syringe for a uniform single cell suspension and 2.8 × 108 cells were loaded into a JE-5.2 counterflow elutriation rotor. Cells were equalized in the separation chamber of the elutriator in PBS containing 4.5 g/L glucose and 0.25% Calf serum for 30 minutes at constant centrifugal force and flow force (580 × g and 24 ml/min) and slowly eluted by increasing pump flow (SciLog Tandem model 1081) at 3–4 ml/min. increments. The progress of separation by elutriation is monitored by the size analyzer function (cell diameter) of the Coulter Z2 cell counter. Aliquots are removed at midpoint of each collected fraction and analyzed immediately, therefore distribution of cells is monitored in real time and can be adjusted within each elutriation (Figure 1a).
Figure 1. Flow cytometric analysis of cell cycle phases separated by a typical centrifugal elutriation.
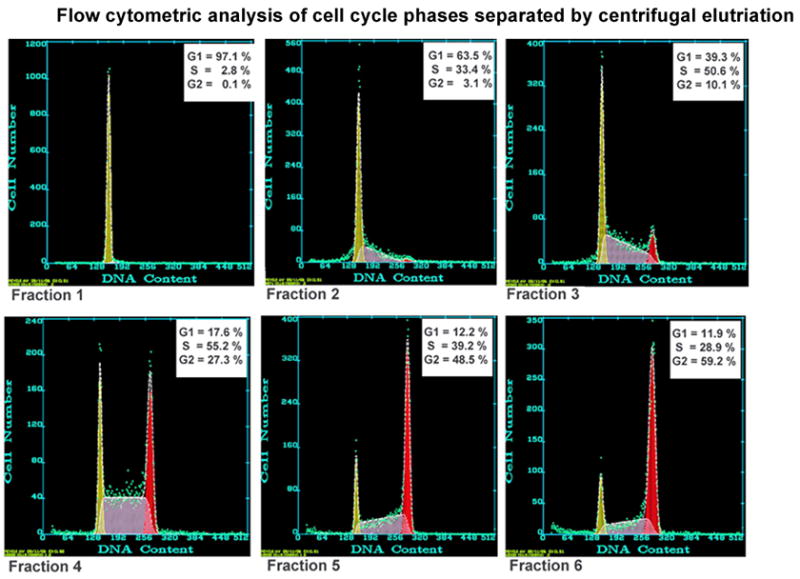
Verification of cell cycle phase distribution for each elutriation trial was accomplished by propidium iodide staining followed by detection using a Beckman/Coulter EPICS Elite flow cytometer. The resulting data was analyzed by Multicycle software (Phoenix Flow Systems) and reported as the percentage of cells in G1, S, or G2/M phase. This is a representative profile demonstrating 6 sequential fractions recovered from a single elutriation run. Enriched fractions for G1, S and G2/M were selected for subsequent experiments.
Colony survival
Control and treated cells were seeded at either 200 or 400 cells per 60 mm plate and incubated at 37°C, 5% CO2. After 7 days media was removed and plates were rinsed with PBS. Cells were fixed with 100% methanol and stained with 1% crystal violet in 20% ethanol. All colonies of ≥100 cells were counted. Triplicate plates were averaged for each experimental condition.
Nuclear protein purification and immunoblot analysis
Nuclear protein extracts were prepared as previously described [10; 16; 17]. After nuclear protein concentrations were measured in triplicate, extracts were stored at −80°C. Immunoblot analysis was conducted as previously described, after SDS protein electrophoresis and transfer to PVDF membrane using equal protein per electrophoretic lane (40 μg) [10; 18]. Specific antibodies used: MSH6 (#610919, BD Biosciences), MLH1 (#554073, BD Biosciences); PMS2 (#556415, BD Biosciences), Cyclin A (#06-138, Upstate Cell Signaling), Nucleoporin (#610832, BD Biosciences). Immunoreactive protein bands were visualized by enhanced chemiluminescence (ECL) via exposure to X-ray film. Protein bands were quantified using Kodak Image station.
Microscopic immunofluorescence
Indirect immunofluorescence was conducted to visualize the nuclear localization of MMR proteins at specific phases of the cell cycle. Laminin (Sigma) pretreated glass coverslips were placed into 60 mm plates and cells from elutriated fractions were seeded at a density of 5 × 105 cells per plate. After incubation, the attached cells were fixed and prepared for immunofluorescence staining as described previously [10; 18]. Primary antibodies used; MSH2 (#NA27, Oncogene Research Products) and MSH6 (#610919, BD Biosciences). Secondary antibody was conjugated to Alexa Fluor-546 goat anti-mouse (Molecular Probes Inc.). Emission fluorescence images were acquired with a Nikon Eclipse 800 fluorescence microscope equipped with a Senys digital camera and ImagePro software (Media Cybermetrics).
Replication-competent plasmid construction containing site-specific G:T or G:A mismatch
The p220.pbc+H/B plasmid containing the entire genomic sequence of human H-ras and a site-specific mismatch of either G:T or G:A at the codon 12 oncogenic hot spot was constructed as described previously [19; 20], using a plasmid containing the Epstein Barr Virus (EBV) origin of replication (OriP) and expressing the Epstein Barr Nuclear Antigen 1 (EBNA 1). This plasmid replicates synchronously with the mammalian cell cycle as an episome within the nucleus [21; 22].
Mismatch Repair Assays
The in vitro mismatch repair assay was performed essentially as described previously [10] using elutriated G1, S, or G2/M phase-specific HeLa nuclear extracts and the mismatch-containing p220.pbc+H/B plasmid. The in vivo MMR assay was conducted by transfecting the mismatch-containing p220.pbc+H/B plasmid into G1, S, or G2/M phase HeLa cells separated by centrifugal elutriation. For transfection, 5 × 106 cells from each elutriated population were pelleted by centrifugation and resuspended in 1 ml of complete medium and incubated at 37°C in a 5% C02 humidified atmosphere for 15 min. Cells were then repelleted and resuspended with 100 μl of Nucleofectin Solution R (Amaxa Biosystems) and 5 μg of mismatch containing plasmid. Equal aliquots of cells from each elutriated population were incubated with 5 μg of p220.pbc+H/B homoduplex plasmid to verify cell cycle phase by flow cytometric analysis. Electroporation of all cell/plasmid suspensions was conducted using the Amaxa Nucleofectin apparatus per manufacturer’s protocol (Amaxa Biosystems). The transfected cells were then transferred into 60 mm plates containing pre-warmed RPMI with 10% FBS and incubated for 2–2.5 hr at 37°C in a 5% C02 humidified atmosphere. Each plate was then rinsed 3x in PBS and cells were trypsinized, pelleted, and resuspended in 5 ml PBS. Plasmid DNA was extracted using the Wizard SV Miniprep System per manufacturer’s directions (Promega) and transformed into MMR deficient bacteria (NR9161 MutL−) for MMR analysis as previously described [10]. Purified plasmid DNA was digested with Nae I to determine correct (nick-directed), incorrect, or lack of repair of the mismatch as previously described [17; 19].
3. RESULTS
We have employed centrifugal elutriation to separate proliferating cells into discrete phases of the cell cycle to avoid cellular response to chemical treatment or serum starvation. We have also constructed a replication-competent plasmid for the investigation of both in vitro (nuclear extracts) and in vivo (whole cell transfection) repair of either a G:T mismatch that is repaired with high efficiency and fidelity, or a G:A mismatch that is repaired poorly or not at all, at the H-ras codon 12 oncogenic hot spot of mutation [10; 19; 23].
Separation of proliferating populations of HeLa cells into phases of the cell cycle has repeatedly yielded substantially enriched fractions, within our hands, of G1 (~90%), S (~50–70%), and G2/M (~55–60%) from a total of 6–7 subdivided centrifugal elutriation fractions collected from each run (Figure 1; Supplement Figures 1, 2). This method of enrichment for cell cycle phase is consistently equal to, or improved, over other methods [7; 8; 9]. To ensure that the elutriation protocol does not cause membrane damage, a trypan blue exclusion assay was conducted for pre-eluted cells and all fractions of eluted populations over each elution trial. Trypan blue was consistently excluded in > 95% of each cell population (results not shown), indicating that cell membranes were equally intact before and after elutriation. In order to verify that centrifugal elutriation did not decrease cell survival or growth dynamics overall, colony survival assays were conducted with cell populations collected before elutriation (proliferating) and after elutriation at different stages of the cell cycle. In addition, colony survival of cells using serum starvation for arrest of cells in G1 phase was determined simultaneously to compare elutriation with this common method of cell cycle synchronization [6; 23]. Figure 2 demonstrates that cell fractions placed back into culture after varying times of centrifugal elutriation have no statistical difference in subsequent survival of colonies compared to control cells (non-elutriated). However, cells that have been serum starved (0.5% calf serum) for 60 hours to induce G1 arrest undergo a significant decrease in subsequent colony viability when compared to control cells grown in complete media. Additionally, HeLa cells within G1 phase replated into complete media immediately after centrifugal elutriation show no apparent disruption in cell growth, as the cells rapidly exit G1 during the succeeding 8 hours in culture (Table 1). Contrary to this, we have previously found that virtually all serum starved cells remain in G1 for up to 12 hours [23], suggesting highly altered cell cycle physiology and greatly decreased biochemical activity. Collectively these results demonstrate cell survival and cell cycle progression after centrifugal elutriation similar to that of non-elutriated cells. Therefore, elutriated cells that have been replated appear to grow without cytotoxic consequences for subsequent MMR studies at discrete phases of the cell cycle.
Figure 2. Colony survival of serum starved versus elutriated HeLa cells.
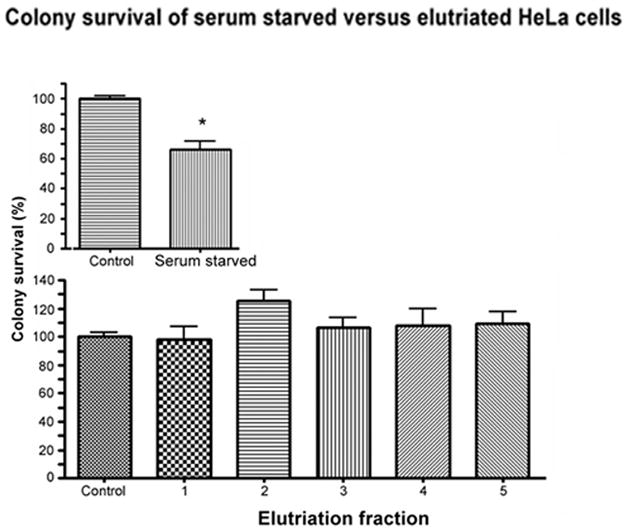
Cells were serum starved in 0.5% calf serum for 60 hr to induce G1 arrest, or were maintained on 5% calf serum (control). After treatment, cells were trypsinized and an equal number of cells were replated in complete media in triplicate (200 or 400 cells per plate). After elutriation, an equal number of cells from each fraction were replated as above. All plates were fixed and stained for colony counting after 7 days of incubation. The number of colonies from each condition was normalized to percent of control colonies (100%). Error bars represent standard error. Asterick indicates significantly decreased viability from control (P < 0.05).
Table 1.
G1 cell cycle progression after elutriation*
| 0 hr | 2 hr | 4 hr | 8 hr | |
|---|---|---|---|---|
| G1 | 83 | 78 | 36 | 9 |
| S | 14 | 16 | 37 | 72 |
| G2/M | 3 | 6 | 27 | 19 |
G1 phase of the cell cycle was enriched by elutriation and HeLa cells from this fraction were replated in complete media. Flow cytometry was used to monitor progression of cells through the cell cycle for up to 8 hours. After 2 hours cells begin to attach and flatten to the culture plate similar to that observed of non-elutriated cells. Cells remained in synchrony for up to 8 hours.
Concentration of MMR Proteins within the Nucleus Correlates with Phase of Cell Cycle
MMR protein concentrations within the nucleus of elutriated cells were compared by immunoblot analysis (Figure 3a). Equal protein concentrations of nuclear extracts from each elutriated fraction were immunoblotted for hMSH6, hMLH1 and hPMS2. All MMR proteins display a pattern of increasing expression within the nucleus from G1 through S phase and remain relatively high into G2 phase, similar to previous findings [10]. Cyclin A expression, which peaks in mid S phase [24], is consistent with flow cytometric data of DNA content and further indicates that normal cell cycle regulation has not been altered, whereas nucleoporin protein loading control indicates equal amount of nuclear protein within each lane. Eluted cells from G1 and S fractions were also replated onto coverslips and incubated for an additional 8 hours. Relative synchrony of these two phases was maintained, as indicated by flow cytometry, as cells progressed from G1 into S phase (58%) or from S into G1 phase (79%). Microscopic immunofluorescence of hMHS2 and hMSH6 within these cell populations undergoing 8 hours of incubation after elutriation (Figure 3b) verify the immunoblot analysis of elutriated cell populations collected immediately after elutriation. MMR proteins are increased within the nucleus of S phase as compared to G1 during physiological progression of the cell cycle. We were unable to perform comparable microscopic immunofluorescence of 0 hr cells as 2–4 hrs are required for cells to adhere sufficiently to the coverslip. Also, after 8 hrs of incubation, the smallest G2/M phase population was no longer sufficiently enriched for microscopic immunofluorescence. Although it is possible to maintain G2/M enriched cells (59.5%) for shorter incubation times after elutriation (2 hrs), these cells are not sufficiently adherent for comparative microscopic immunofluorescence (data not shown).
Figure 3.
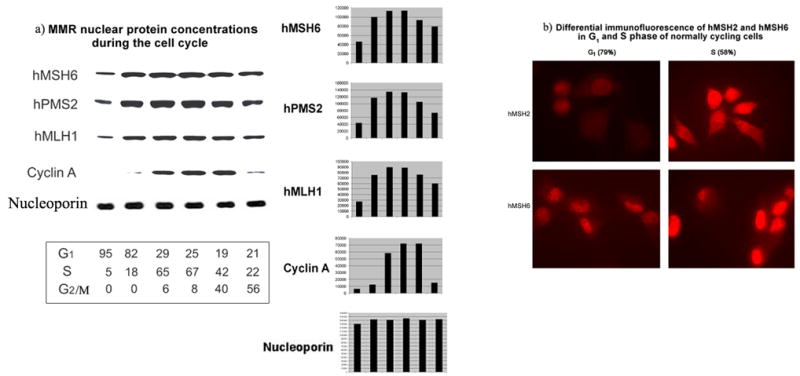
a) HeLa MMR nuclear protein concentrations during the cell cycle. Proliferating HeLa populations were separated into cell cycle phases by centrifugal elutriation and each fraction was used to prepare nuclear lysates. Equal amounts of nuclear protein from each fraction were separated by electrophoresis and subjected to immunoblot analysis (see materials and methods). Antibody to cyclin A was used to monitor phase of cell cycle in each fraction (peaks in mid S phase [10; 24]). Antibody to nucleoporin was used as a nuclear protein loading control. Flow cytometry was used to monitor phase of cell cycle in each fraction. Distribution of the cell cycle phase for each fraction is listed under each immunoblot lane. Bar graphs were generated by densitometry measurements of each band. b) Differential immunofluorescence of hMSH2 and hMSH6 in G1 and S phase of normally cycling cells. Proliferating HeLa cells were subjected to centrifugal elutriation for separation of cell cycle phases. S and G1 collected fractions were plated onto coverslips, incubated for a further 8 hours for progression into G1 or S phase respectively, and subjected to immunofluorescence staining for hMSH2 or hMSH6. Each photograph is an equally exposed representative microscopic field from each coverslip.
MMR efficiency and fidelity increases with DNA replicative activity
We have previously reported that MMR nuclear protein concentrations are highest in S and G2/M phases, although mismatch recognition and repair activity decreases in G2/M as compared to S phase [10]. These MMR studies, similar to other MMR assays, were accomplished using nuclear extracts and a plasmid that does not replicate in mammalian cells [25; 26; 27; 28]. Therefore increased MMR activity within S phase can be demonstrated even within nonreplicating DNA using an in vitro assay.
We have now investigated MMR activity in nuclear extracts and within intact cells from discrete phases of the cell cycle. For these studies, we use a plasmid (p220.pbc+H/B) containing sequences required for mammalian replication (EBV oriP and EBNA 1). The current in vitro investigations were accomplished by constructing a G:T or G:A mismatch located at the H-ras codon 12 site that was subsequently ligated into the replication-competent plasmid, as previously described [19; 20]. The mismatch-containing plasmid was then incubated with HeLa nuclear extracts prepared from elutriated populations of cells enriched for G1, S, or G2/M portion of the cell cycle. Figure 4 demonstrates the results of the in vitro MMR experiments. Similar to previous results, the G:T mismatch appears to be repaired more efficiently overall (total repair above background) than G:A at this mutagenic hot spot, except for virtually equal low repair during G1. The observed increase in repair of G:T over that of G:A is not statistically significant however. Both mismatches are repaired with highest efficiency (Total repair) and fidelity (Correct repair) within S phase nuclear extracts, with the exception of G:A correct repair in G1 phase extracts (94%) as compared with G:T correct repair in S phase extracts (92%) having no statistical difference. Interestingly, lowest overall MMR is of G:A within nuclear extracts from G2/M phase. Significantly, highest efficiency of total repair for both mismatches is observed only during S phase using the current replicating plasmid. Fidelity of correct repair within the replicating plasmid is much higher than previous results using a plasmid that was unable to replicate in mammalian cells in the same in vitro assay (p < 0.001; except for G:A mismatch within G2/M phase nuclear extracts with 80% correct repair previously and only 50% correct repair currently) (Table 2 and [10]). Both plasmid constructs have demonstrated similar and consistent efficiencies of background repair by transformation into both MMR competent (DH5α) and incompetent (NR9161) E. coli within several different investigations [10; 17; 20].
Figure 4. In vitro MMR of a replication-competent plasmid.

In vitro MMR assays were performed as described (materials and methods). The p220.pbc+H/B plasmid contains a mammalian origin of replication and a site-specific G:T or G:A mismatch. Nuclear extracts were purified from elutriated fractions of HeLa cells enriched for each phase of the cell cycle. Total repair frequency above the MMR negative E. coli (NR9161) background was calculated and the percent of correct (nick-directed repair) versus incorrect (opposite nicked strand) MMR was determined from total repair frequency [27]. MMR negative control E. coli (NR9161) were used to determine background MMR frequency, and MMR positive E. coli (DH5α were included in each experiment. The bar graph below the table is a graphical representation of the data within the table. The frequency above background of total repair for each mismatch at each phase of the cell cycle (black bars), and fractions of correct (nick-directed MMR; grey bars) and incorrect (opposite nicked strand; white bars) mismatch repair within each total repair frequency, were plotted separately as indicated.
Table 2.
Comparison of efficiency (total repair) and fidelity (correct repair) of MMR at the same DNA site in different conditions*
| % total repair (efficiency | ||||||
|---|---|---|---|---|---|---|
| A | G:T | G:A | G:T | G:A | G:T | G:A |
| G1 | 40 | 26 | 30 | 31 | 37 | 35 |
| S | 45 | 33 | 55 | 48 | 82 | 59 |
| G2/M | 41 | 23 | 37 | 13 | 72 | 40 |
| % correct repair (fidelity) | ||||||
| B | G:T | G:A | G:T | G:A | G:T | G:A |
| G1 | 53 | 30 | 88 | 94 | 94 | 100 |
| S | 70 | 40 | 97 | 92 | 96 | 100 |
| G2/M | 59 | 80 | 73 | 50 | 91 | 81 |
| Nuclear extract; nonreplicating plasmid [10] | Nuclear extract; replicating plasmid (Fig. 4) | Intact cell; replicating plasmid (Fig. 5) | ||||
Total repair and correct repair values only were extracted from each experimental model to condense into Table 2 for comparison purposes.
We then used an in vivo approach to determine if active DNA replication within an intact cellular environment would contribute to increased MMR activity. The same mismatched plasmid construct as above was transfected into G1, S, or G2/M-enriched HeLa cells immediately after elutriation and cells were incubated for 2 hours (Figure 5a). Almost all in vivo repair events for both G:T and G:A were improved as compared to in vitro conditions (p < 0.05; compare Figures 4a and 5a). However, lowest MMR activity within G1 phase was not significantly different from current or previous in vitro conditions (Table 2 and [10]). This further indicates that components of DNA replication, lacking in G1 phase, do play a substantial role in MMR activities. Also, similar to previous results, the G:T mismatch has been repaired significantly better than G:A in both S and G2/M phases of the cell cycle (p < 0.001) in regard to total repair efficiency above background. Fidelity of correct repair of both G:T and G:A is also consistently above 90% in all phases of the cell cycle, with the exception of G:A in G2/M phase (81%), similar to previous results of 80% (Figure 5a, Table 2, and [10]). Flow cytometric analysis demonstrates that all transfected cell populations, although actively moving through the cell cycle, were still enriched for each selected phase after 2 hours of incubation (Figure 5b).
Figure 5.
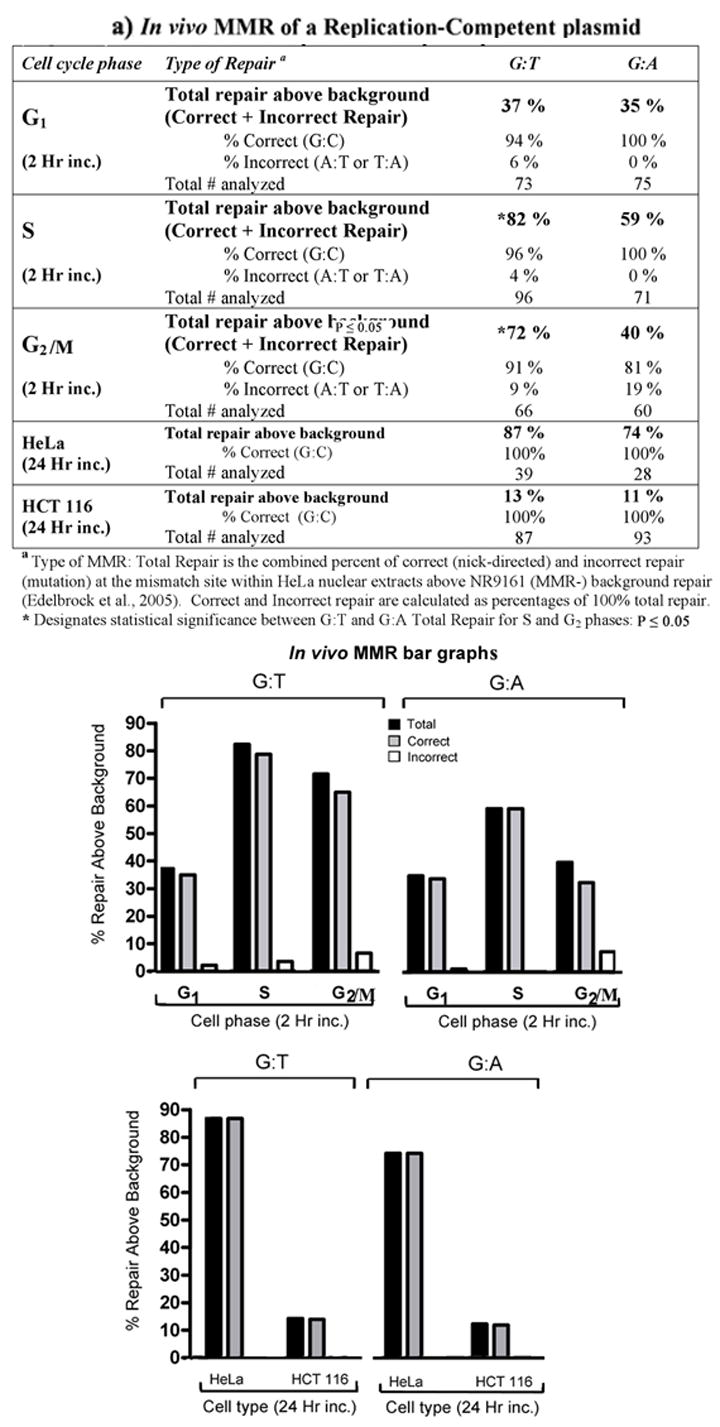

a) In vivo MMR of replication-competent plasmid. In vivo MMR assays were performed as described (materials and methods). The p220.pbc+H/B) plasmid contains a mammalian origin of replication and a site-specific G:T or G:A mismatch. HeLa cells enriched for each phase of the cell cycle by elutriation and transfected with the mismatch-containing plasmid were incubated for 2 hr. Total repair frequency above the MMR negative E. coli (NR9161) background was calculated and the percent of correct (nick-directed repair) versus incorrect (opposite nicked strand) MMR was determined from total repair frequency [27]. Negative MMR (proliferating HCT116) and positive MMR (proliferating HeLa) controls were incubated for 24 hr before plasmid was extracted. The bar graphs below the table is a graphical representation of the data within the table. The frequency above background of total repair for each mismatch at each phase of the cell cycle (black bars), and fractions of correct (nick-directed MMR; grey bars) and incorrect (opposite nicked strand; white bars) mismatch repair within each total repair frequency, were plotted separately as indicated. b) HeLa cell cycle progression during 2 hr incubation. Elutriated HeLa cell populations enriched for each phase of the cell cycle (bolded) were immediately transfected with the replication-competent plasmid and replated as described (materials and methods). Each fraction was subjected to flow cytometry immediately after transfection, as well as 2 hours after transfection. c) HeLa cell cycle progression during 24 hr incubation. Elutriated HeLa cells enriched for G1 phase of the cell cycle were immediately transfected with the replication-competent plasmid and replated as described (materials and methods). Each fraction was subjected to flow cytometry immediately after transfection, as well as 24 hours after transfection.
Additionally, we transfected the mismatched plasmid into elutriated HeLa cell fractions that were enriched for G1 phase and incubated these cells for 24 hours to complete at least one cell cycle. Flow cytometric analysis demonstrates that over 90% of the cells were in G1 at the time of transfection and were asynchronous at the time of plasmid recovery 24 hours later, as expected (Figure 5c). MMR within these transfected cells was at the highest efficiency of all assay conditions, and was 100% correct (Figure 5a). Therefore, increased transfection efficiency observed during S phase (results not shown) does not appear to affect the frequency or fidelity of MMR within the cell, but transition through the cell cycle, and presumably S phase, is important. Proliferating HCT 116 human colon cancer cells, lacking hMLH1 protein expression and therefore deficient for MMR, were also transfected with the mismatch-containing plasmids for a 24 hour incubation period as a negative MMR control (Figure 5a). These cells demonstrated less than 15% total MMR above background, similar to previous in vitro results with this cell line [17].
4. DISCUSSION
Centrifugal elutriation is considered to be the gold standard of cell separation techniques as it putatively involves little or no physiological stress [29; 30]. Here we have demonstrated that centrifugal elutriation is an excellent method for separation of cell cycle-enriched populations (G1, S and G2/M) for physiologically relevant MMR studies, as evidenced by several viability measurements. We have verified that MMR nuclear protein concentration and MMR activity are not induced nor decreased by the stress of elutriation, but rather are dependent on physiological cell cycle events. The levels of MMR proteins within the nucleus alter as cells progress through the cell cycle, low G1 levels rising as the cell enters S phase. Our current results collectively indicate that although MMR activity is present at all phases of the cell cycle, this repair pathway is at highest efficiency and is most accurate within the cell during DNA replication.
To date, characterization of MMR events has been investigated by the use of plasmids containing a nick or gap in the strand to be repaired within an in vitro system [10; 26; 27; 32; 33; 34]. While information from this approach has been extremely useful for the elucidation of several important aspects of this DNA repair pathway, plasmid replication does not occur. Thus, the contribution of DNA replication to MMR within mammalian cells has not been measured despite that MMR is thought to occur at the replication fork [35]. Therefore, to investigate if MMR activity is increased during DNA replication, we compared MMR activity in a plasmid system capable of generating a replication fork. Plasmids containing the EBV oriP and expressing EBNA 1 protein have been shown to replicate synchronously during S phase in the form of a stable episome [22; 36]. Our current results indicate that MMR is much more efficient when the plasmid design incorporates a mammalian origin of replication in comparison to previous results using a plasmid lacking this sequence [10]. In vitro analyses by others have demonstrated that MMR activity also require DNA replication components PCNA, RFC, RPA, and EXO1 [11; 37; 38]. We have previously demonstrated co-localization of MutSα during S phase to replication factories that also contain PCNA and newly synthesized DNA [10; 12]. We hypothesize that these replication factors also required for MMR are more accessible when the mispairs are located on replicating sequences.
Overall, our previous MMR model, a frequently employed protocol that consists of a nonreplicating plasmid incubated with nuclear extract, resulted in poorest efficiency of total repair and lowest fidelity of correct repair within S phase, as compared to current results using a replicatable plasmid within nuclear extracts or intact cells (Table 2A and 2B). This indicates that factors present during S phase and replication competent DNA both contribute to enhanced MMR during S phase of the cell cycle. Total repair efficiency (Table 2A) remained uniformally low in G1 phase, even within intact cells (δ40%). Total repair efficiency also remained relatively low in G2/M phase for both mismatches, except for repair of G:T, which increased to 72% within the replicating plasmid in intact cells, as compared to G:A repair, which increased to only 40%. These results may indicate an intriguing mechanism within the cell cycle for why G:A is repaired so poorly overall at this oncogenic site in mammalian cells [10; 19; 23]. We have previously reported that although MMR nuclear protein levels remain similarly high in S and G2 phase, MMR binding activity in G2 returns to the lower activity observed in G1 phase. We have suggested this may indicate posttranslational alterations of MMR proteins [10], and we are currently investigating posttranslational phosphorylation effects of hMSH6, the only MMR protein that undergoes measurable posttranslational phosphorylation (unpublished results). The fidelity of correct repair of both mismatches is similarly increased overall within the replicatable plasmid, except for the intriguing differences in regard to G:A repair within G2/M (Table 2B). Fidelity of correct repair of G:A in G2/M phase (80%) is twice that of S phase within the nonreplicating plasmid in nuclear extracts, but decreased by almost 50% of S phase repair using the replicating plasmid in nuclear extracts. The fidelity of correct repair of G:A in G2/M then returns to over 80% within intact cells using the same replicating plasmid. Perhaps because the total repair efficiency is so low within the nuclear extracts for the G:A mismatch (23% and 13%), that the correct repair fidelity of G:A to G:C is not as accurate as the other values. Or perhaps there is some inhibiting condition or specific binding molecules within nuclear extracts for a G:A mismatch within replicatable sequences that is not inhibitory within the intact cell. Conversely, the nuclear environment within intact cells is likely to be more conducive to enhanced “cross-talk” between different repair pathways, such as MMR and BER. As well, nuclear extracts are a mixture of all cells within the elutriated population, and you cannot differentiate G2 from mitotic cells by elutriation or flow cytometry. Transfection of the plasmid into this population however, may be more selective for the G2 phase, which would be more likely to have sufficient MMR activity as compared to mitotic phase cells.
Transfection of replication-competent plasmids containing a site-specific mismatch into cells during S phase of the cell cycle results in highest fidelity of nick-directed MMR, regardless of the type of mismatch. Conversely, MMR efficiency remains at a level similar to in vitro conditions when transfected into cells enriched for G1 phase. However, if the incubation period is extended sufficiently for the cell to complete a full cycle, including traversal through at least one cell cycle, MMR activity is measured at highest efficiency and is 100% nick-directed.
In conclusion, our results provide direct evidence that the MMR pathway functions at increased efficiency and fidelity of repair during DNA synthesis within the normal physiological cycling of the cell. Previous results indicate that the concentrated location of hMutSα to replication factories likely contributes significantly to this increased MMR activity, as well as overall increased levels of nuclear MMR proteins [10]. However, the MMR pathway can also function, albeit to a lesser extent, at other phases of the cell cycle, including G1 phase, and is likely required for repair of mismatches occurring by mechanisms other than replication errors.
Supplementary Material
Aliquots are collected at midpoint from each fraction of cells and are counted and analyzed for diameter (size) during each centrifugal elutriation by the size analyzer function of the Coulter Z2 cell counter and channelizer. Three distinct size distributions correspond to G1 (13–16 μM), S (15–18 μM) and G2/M (18–21 μM) phases of the cell cycle. The analysis of the distribution of cells by diameter is monitored in real time and elutriation collection can be adjusted as necessary.
Depicted is flow cytometry data from several different elutriation trials (elutriation trial 1 is also depicted in Figure 1). Each fraction of the cell population was collected by incrementally increasing pump flow rate. Collected fractions from each trial are reported as the percentage of cells within G1, S and G2/M phase, as determined by flow cytometry. Usually 6–7 fractions were collected within each elutriation trial, either on ice or at room temperature, as described in materials and methods. Separation of cells into different cell cycle phases by elutriation within proliferating populations of HeLa cells has consistently yielded enriched fractions of G1 (>90 %), S (~50–70 %), and G2/M (~55–60 %) within our hands. Therefore, because of multiple elutriation trials required for all of the experiments described in this paper, we use the terms G1, S, or G2/M “fraction” or “enriched” to indicate that cells used within the described experiment fall within the above ranges for each phase of the cell cycle, unless otherwise noted. These levels of cell cycle phase enrichment ranges are either comparable to, or higher, than other methods [7; 8; 9].
Acknowledgments
This work was supported by National Institutes of Health grants CA84412 (K.J.W.) and CA106575 (K.J.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marra G, Boland CR. Hereditary nonpolyposis colorectal cancer: the syndrome, the genes, and the historical perspectives. J Natl Cancer Inst. 1995;87:1114–1125. doi: 10.1093/jnci/87.15.1114. [DOI] [PubMed] [Google Scholar]
- 2.Iyer RR, Pluciennik A, Burdett V, Modrich P. DNA Mismatch Repair: Functions and Mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 3.Kunkel TA, Erie DA. DNA Mismatch Repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 4.Modrich P. Mechanisms and biological effects of mismatch repair. Annu Rev Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- 5.Jiricny J. The multifaceted mismatch-repair system. Nature Reviews Molecular Cell Biology. 2006;7:335–347. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 6.Iwanaga R, Komori H, Ohtani K. Differential regulation of expression of the mammalian DNA repair genes by growth stimulation. Oncogene. 2004:1–10. doi: 10.1038/sj.onc.1207976. [DOI] [PubMed] [Google Scholar]
- 7.Klingler H, Hemmerle C, Bannwart F, Haider R, Cattaruzza MS, Marra G. Expression of the hMSH6 mismatch-repair protein in colon cancer and Hela cells. Swiss Med Wkly. 2002;132:57–63. doi: 10.4414/smw.2002.09855. [DOI] [PubMed] [Google Scholar]
- 8.Marra G, Chang CL, Laghi LA, Chauhan DP, Young D, Boland CR. Expression of human MutS homolog 2 (hMSH2) protein in resting and proliferating cells. Oncogene. 1996;13:2189–2196. [PubMed] [Google Scholar]
- 9.Meyers M, Theodosiou M, Acharya S, Odegaard E, Wilson T, Lewis JE, Davis TW, Wilson-Van Patten C, Fishel R, Boothman DA. Cell cycle regulation of the human DNA mismatch repair genes hMSH2, hMLH1, and hPMS2. Cancer Res. 1997;57:206–208. [PubMed] [Google Scholar]
- 10.Schroering AG, Edelbrock MA, Richards TJ, Williams KJ. The cell cycle and DNA mismatch repair. Exp Cell Res. 2007;313:292–304. doi: 10.1016/j.yexcr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human Mismatch Repair: Reconstitution of a nick-directed bidirectional reaction. J Biol Chem. 2005;280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleczkowska HE, Marra G, Lettieri T, Jiricny J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes & Develop. 2001;15:724–736. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masih PJ, Kunnev D, Melendy T. Mismatch Repair proteins are recruited to replicating DNA through interaction with Proliferating Cell Nuclear Antigen (PCNA) Nucleic Acids Res. 2008;36:67–75. doi: 10.1093/nar/gkm943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, Umar A, Kucherlapati R, Lipkin M, Kunkel TA, Edelmann W. An Msh2 Point Mutation Uncouples DNA Mismatch Repair and Apoptosis. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes & Develop. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 16.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edelbrock M, He H, Schroering AG, Fernstrom M, Bathala S, Williams KJ. Recognition and binding of mismatch repair proteins at an oncogenic hot spot. BMC Molecular Biology. 2005;6(6):10. doi: 10.1186/1471-2199-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroering AG, Williams KJ. Rapid induction of chromatin-associated DNA mismatch repair proteins after MNNG treatment. DNA Repair. 2008;7:951–969. doi: 10.1016/j.dnarep.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arcangeli L, Simonetti J, Pongratz C, Williams KJ. Site- and strand-specific mismatch repair of human H-ras genomic DNA in a mammalian cell line. Carcinogenesis. 1997;18:1311–1318. doi: 10.1093/carcin/18.7.1311. [DOI] [PubMed] [Google Scholar]
- 20.Arcangeli L, Williams KJ. Mammalian assay for site-specific DNA damage processing using the human H-ras proto-oncogene. Nucleic Acids Res. 1995;23:2269–2276. doi: 10.1093/nar/23.12.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yates J, Warren N, Sugden B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature. 1985;313:812–815. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]
- 22.Yates JL, Guan N. Eptsein-Barr Virus-Derived Plasmids Replicate Only Once per Cell Cycle and Are Not Amplified after Entry into Cells. Journal of Virology. 1991;65:483–488. doi: 10.1128/jvi.65.1.483-488.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matton N, Simonetti J, Williams KJ. Inefficient in vivo repair of mismatches at an oncogenic hotspot correlated with lack of binding by mismatch repair proteins and with phase of the cell cycle. Carcinogenesis. 1999;20:1417–1424. doi: 10.1093/carcin/20.8.1417. [DOI] [PubMed] [Google Scholar]
- 24.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. Garland Science; New York: 2002. [Google Scholar]
- 25.Holmes JJ, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. PNAS. 1990;87:5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iams K, Larson ED, Drummond JT. DNA Template Requirements for Human Mismatch Repair in Vitro. J Biol Chem. 2002;277:30805–30814. doi: 10.1074/jbc.M200846200. [DOI] [PubMed] [Google Scholar]
- 27.Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J Biol Chem. 1991;266:3744–3751. [PubMed] [Google Scholar]
- 28.Wang H, Hays JB. Mismatch Repair in Human Nuclear Extracts: Time courses and ATP requirements for kinetically distinguishable steps leading to tightly controlled 3′ and 5′ and aphidicolin-sensitive 3′ to 5′ mispair-provoked excision. J Biol Chem. 2002;277:26143–26148. doi: 10.1074/jbc.M200358200. [DOI] [PubMed] [Google Scholar]
- 29.Bauer J. Advances in cell separation: Recent developments in counterflow centrifugal elutriation and continuous flow cell separation. Journal of Chromatography B. 1999;722:55–69. doi: 10.1016/s0378-4347(98)00308-9. [DOI] [PubMed] [Google Scholar]
- 30.Davis PK, Ho A, Dowdy SF. Biological methods for cell-cycle synchronization of mammalian cells. BioTechniques. 2001;30:1322–1331. doi: 10.2144/01306rv01. [DOI] [PubMed] [Google Scholar]
- 31.Christmann M, Kaina B. Nuclear Translocation of Mismatch Repair Proteins MSH2 and MSH6 as a Response of Cells to Alkylating Agents. J Biol Chem. 2000;275:36256–36262. doi: 10.1074/jbc.M005377200. [DOI] [PubMed] [Google Scholar]
- 32.Cooper DL, Lahue RS, Modrich P. Methyl-directed mismatch repair is bidirectional. J Biol Chem. 1993;268:11823–11829. [PubMed] [Google Scholar]
- 33.Tomer G, Buermeyer AB, Nguyen MM, Liskay RM. Contribution of human Mlh1 and Pms2 ATPase activities to DNA mismatch repair. J Biol Chem. 2002;277:21801–21809. doi: 10.1074/jbc.M111342200. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Hays JB. Mismatch Repair in Human Nuclear Extracts: Effects of internal DNA-hairpin structures between mismatches and excision-initiation nicks on mismatch correction and mismatch-provoked excision. J Biol Chem. 2003;278:28686–28693. doi: 10.1074/jbc.M302844200. [DOI] [PubMed] [Google Scholar]
- 35.Surtees J, Alani E. Replication factors license exonuclease I in mismatch repair. Mol Cell. 2004;15:164–166. doi: 10.1016/j.molcel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 36.James MR, Stary A, Daya-Grosjean L, Drougard C, Sarasin A. Comparative study of Epstein-Barr virus and SV40-based shuttle-expression vectors in human repair-deficient cells. Mutation Research. 1989;202:169–185. doi: 10.1016/0165-1110(89)90023-7. [DOI] [PubMed] [Google Scholar]
- 37.Dzantiev L, Constantin N, Genschel J, Lyer RR, Burgers PM, Modrich P. A defined human system that supports bidirectional mismatch-provoked excision. Molecular Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tompkinson AE, Gu L, Li GM. Reconstitution of 5′-Directed Human Mismatch Repair in a Purified System. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Aliquots are collected at midpoint from each fraction of cells and are counted and analyzed for diameter (size) during each centrifugal elutriation by the size analyzer function of the Coulter Z2 cell counter and channelizer. Three distinct size distributions correspond to G1 (13–16 μM), S (15–18 μM) and G2/M (18–21 μM) phases of the cell cycle. The analysis of the distribution of cells by diameter is monitored in real time and elutriation collection can be adjusted as necessary.
Depicted is flow cytometry data from several different elutriation trials (elutriation trial 1 is also depicted in Figure 1). Each fraction of the cell population was collected by incrementally increasing pump flow rate. Collected fractions from each trial are reported as the percentage of cells within G1, S and G2/M phase, as determined by flow cytometry. Usually 6–7 fractions were collected within each elutriation trial, either on ice or at room temperature, as described in materials and methods. Separation of cells into different cell cycle phases by elutriation within proliferating populations of HeLa cells has consistently yielded enriched fractions of G1 (>90 %), S (~50–70 %), and G2/M (~55–60 %) within our hands. Therefore, because of multiple elutriation trials required for all of the experiments described in this paper, we use the terms G1, S, or G2/M “fraction” or “enriched” to indicate that cells used within the described experiment fall within the above ranges for each phase of the cell cycle, unless otherwise noted. These levels of cell cycle phase enrichment ranges are either comparable to, or higher, than other methods [7; 8; 9].