

Abstract
Alzheimer's disease is the most common dementia and is pathologically characterized by deposition of amyloid-β peptide (Aβ) into β-amyloid plaques, neuronal injury and low-level, chronic activation of brain immunity1. Transforming growth factor-βs (TGF-βs) are pleiotropic cytokines that have key roles in immune cell activation, inflammation and repair after injury2. We genetically interrupted TGF-β and downstream Smad2/3 signaling (TGF-β–Smad2/3) in innate immune cells by inducing expression of CD11c promoter–driven dominant-negative TGF-β receptor type II in C57BL/6 mice (CD11c-DNR)3, crossed these mice with mice overexpressing mutant human amyloid precursor protein, the Tg2576 Alzheimer's disease mouse model4, and evaluated Alzheimer's disease-like pathology. Aged double-transgenic mice showed complete mitigation of Tg2576-associated hyperactivity and partial mitigation of defective spatial working memory. Brain parenchymal and cerebrovascular β-amyloid deposits and Aβ abundance were markedly (up to 90%) attenuated in Tg2576–CD11c-DNR mice. This was associated with increased infiltration of Aβ-containing peripheral macrophages around cerebral vessels and β-amyloid plaques. In vitro, cultures of peripheral macrophages, but not microglia, from CD11c-DNR mice showed blockade of classical TGF-β–activated Smad2/3 but also showed hyperactivation of alternative bone morphogenic protein–activated Smad1/5/8 signaling and increased Aβ phagocytosis. Similar effects were noted after pharmacological inhibition of activin-like kinase-5, a type I TGF-β receptor. Taken together, our results suggest that blockade of TGF-β–Smad2/3 signaling in peripheral macrophages represents a new therapeutic target for Alzheimer's disease.
Alzheimer's disease is characterized by deposition of the 40–42–amino-acid Aβ peptide, which is proteolytically derived from amyloid precursor protein (APP), resulting in cerebral β-amyloid plaques1. Despite low-level, chronic activation of innate immunity in Alzheimer's disease5, microglia ultimately do not clear β-amyloid deposits6. TGF-βs are pleiotropic cytokines with central roles in immune suppression, immune homeostasis and repair after injury2. TGF-β1 in brain dampens microglial activation7. However, TGF-β1 overexpression promotes brain inflammation8, simultaneously accelerates brain vascular β-amyloid deposits and reduces parenchymal β-amyloid deposits9,10, and elicits neuronal Aβ secretion11.
We hypothesized that blocking innate immune TGF-β signaling would impair cerebral Aβ clearance, but, as detailed below, we found support for the converse hypothesis. We bred CD11c-DNR transgenic mice3 to Tg2576 Alzheimer's disease model mice4 and characterized the behavior of 16–17-month-old progeny (Supplementary Methods online). Tg2576 mice showed hyperactivity4 probably resulting from disinhibition associated with hippocampal or cortical injury, whereas the Tg2576–CD11c-DNR mice showed complete hyperactivity mitigation (Fig. 1a). Overall analysis of variance (ANOVA) showed significant effects of time (P < 0.001) and genotype (P < 0.05), and post-hoc comparison showed significant differences when comparing Tg2576 mice to the other groups (Fig. 1a). Similar results were observed during novel Y-maze exploration4,12 (significant effect of genotype, P < 0.01; Fig. 1b,c). Spontaneous Y-maze alternation, a measure of spatial working memory, was less frequent in Tg2576 versus wild-type4 or CD11c-DNR control mice, and Tg2576–CD11c-DNR mice had partial amelioration (significant effect trend of genotype, P = 0.07; Fig. 1d). ANOVA models revealed that gender did not confound these results (Supplementary Table 1 online).
Figure 1.

Reduced behavioral impairment in Tg2576–CD11c-DNR mice at 16–17 months of age. Four groups of littermate mice, including wild-type (WT, n = 13), CD11c-DNR (n = 6), Tg2576 (n = 11) and Tg2576–CD11c-DNR (n = 9) mice, were subjected to behavioral testing. (a) Mice were individually placed into a novel environment, and the distance traveled in cm (y axis) is represented over a 20-min time course in bins of 2.5 min each (x axis). (b) Mice were individually placed into a radially symmetric Y-maze and total number of arm entries (y axis) is shown for each genotype (x axis). (c) Number of mice (y axis) are shown grouped by bins of arm entries (10 entries per bin, x axis; Gaussian curves are shown for each mouse group) in the Y-maze. (d) Percentage alternation between Y-maze arms (y axis; chance level is shown with the dotted line) is represented for each genotype (x axis). Data are represented as group means ± s.e.m. For c, there is a rightward shift of the Tg2576 mouse group relative to WT and CD11c-DNR littermate controls (combined as they did not significantly differ) and to the Tg2576–CD11c-DNR mouse group. All post hoc statistical comparisons are versus Tg2576 mice, ***P < 0.001, **P < 0.01, *P < 0.05, †P < 0.10. No significant difference was found between WT and CD11c-DNR mouse groups (P > 0.05).
We also assayed spatial reference learning and memory in the Morris water maze. Tg2576 and Tg2576–CD11c-DNR mice did not differ significantly, either during visible platform (learning phase) or hidden platform (probe trials) testing, irrespective of gender (P > 0.05, data not shown). However, all mice with the Tg2576 transgene showed greater learning latencies versus Tg2576 transgene–negative mice, but no difference on the day 10 probe trial, whereas female Tg2576–positive mice showed consistent learning and memory deficits in both the learning phase and the probe trial versus female Tg2576-negative mice (Supplementary Fig. 1a–c online). A similar result has been reported showing that females drive the Tg2576 transgene spatial reference learning and memory deficit13. Thus, although the CD11c-DNR transgene completely abrogates Tg2576-associated hyperactivity, it only modestly attenuates defective spatial working memory and does not modify defective spatial reference learning and memory.
We next evaluated Aβ and β-amyloid pathology in 17–18-month-old Tg2576 mice and Tg2576–CD11c-DNR mice by four strategies (see Methods and Supplementary Methods). Tg2576 mice had typical β-amyloid burden14, which was markedly reduced in cortical areas (entorhinal cortex and cingulate cortex) and the hippocampus by 62–82% in Tg2576–CD11c-DNR mice (Fig. 2a,b; Supplementary Fig. 1d); these effects were gender independent (Supplementary Fig. 1e)15. Morphometric analysis revealed no consistent reductions in the number of small β-amyloid plaques, whereas medium- and large-sized plaque number was substantially reduced by 46–92% in Tg2576 versus Tg2576–CD11c-DNR mice (Supplementary Fig. 2a–c online), suggesting CD11c-DNR transgene–dependent reduction of β-amyloid plaque maturation.
Figure 2.
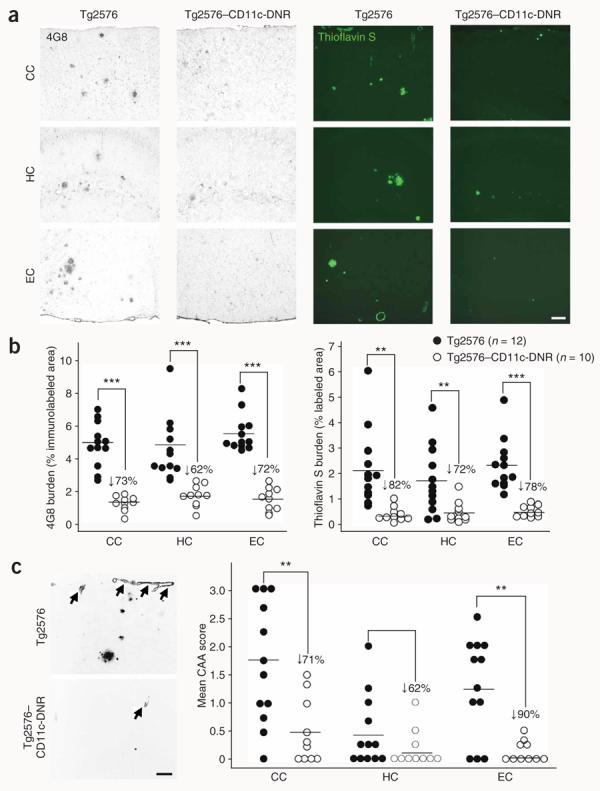
Reduced cerebral parenchymal and vascular β-amyloid deposits in Tg2576–CD11c-DNR mice at 17–18 months of age. (a) Photomicrographs from Tg2576 or Tg2576–CD11c-DNR mouse brain sections with median values by image analysis for human Aβ immunohistochemistry (antibody 4G8, bright-field photomicrographs, left) or histochemistry for thioflavin S (dark-field photomicrographs, right) are shown. CC, cingulate cortex; HC, hippocampus; EC, entorhinal cortex. (b) Photomicrographs were taken from cortical areas or hippocampus and quantitative image analysis for 4G8 (left) or thioflavin S burden (right) was conducted for Tg2576 (n = 12) and Tg2576–CD11c-DNR mice (n = 10). 4G8 or thioflavin S burden (% labeled area) is shown on the y axis, and brain region is represented on the x axis. Percentage reductions in Tg2576–CD11c-DNR versus littermate Tg2576 mice are indicated for each brain region. (c) Representative photomicrographs of thioflavin S histochemistry (inverted gray-scale, left) showing cerebral vascular β-amyloid deposits in Tg2576 or Tg2576–CD11c-DNR mice as indicated (arrows). Semiquantitative image analysis was performed (right), and severity of cerebral amyloid angiopathy (CAA score) is shown on the y axis with brain region indicated on the x axis. Scale bars in (a,c) denote 100 μm. Quantitative data are represented as group means (bars). All statistical comparisons are within brain region and between Tg2576 and Tg2576–CD11c-DNR mice, **P < 0.01 and ***P < 0.001.
Eighty-three percent of subjects with Alzheimer's disease present with cerebrovascular β-amyloid deposits (cerebral amyloid angiopathy (CAA))16. Tg2576 mice also develop age-dependent vascular β-amyloid deposits17, which were reduced by 62–90% in Tg2576–CD11c-DNR mice compared to Tg2576 mice (Fig. 2c). Biochemical analysis revealed Tg2576–CD11c-DNR mouse reductions in both Aβ1–40 and Aβ1–42 abundance versus Tg2576 mice ranging from 45% to 88% reduction in the detergent-soluble fraction and 45% to 53% reduction in the detergent-insoluble (but guanidine-HCl–extractable) fraction (Supplementary Fig. 2d,e, P < 0.001). Notably, although the detergent-soluble Aβ1–42/Aβ1–40 ratio was significantly (P < 0.001) reduced by 77% in Tg2576–CD11c-DNR mice, the Aβ1–42/Aβ1–40 ratio of guanidine-HCl–soluble material was not (Supplementary Fig. 2f).
A reduction in Tg2576–CD11c-DNR mouse cerebral amyloidosis could be due to attenuated APP expression, increased brain-to-blood clearance of Aβ (ref. 18), reduced amyloidogenic APP metabolism or activation of endogenous brain Aβ clearance. We probed brain homogenates from Tg2576 and Tg2576–CD11c-DNR mice for APP but did not detect differences in APP abundance between the two strains (Supplementary Fig. 2g). We also assayed blood-circulating Aβ1–40 and Aβ1–42 species abundance and did not observe differences (Supplementary Fig. 2h). To address steady-state APP metabolism, we analyzed amyloidogenic carboxyl (C)-terminal APP fragment (β-CTF, C99) and non-amyloidogenic CTF (α-CTF, C83) abundance, but again did not detect differences between the two strains (Supplementary Fig. 2i), thus turning our attention to endogenous brain Aβ clearance.
We reasoned that, if the CD11c-DNR transgene affects endogenous brain Aβ clearance, this would probably occur through modulation of brain inflammatory and immune responses. We noted substantial reductions in the number of activated glial fibrillary acidic protein (GFAP)-positive astrocytes by 44–63% in Tg2576–CD11c-DNR mice versus Tg2576 mice (Fig. 3a,b). Confocal microscopy for CD45, a leukocyte marker also expressed by activated microglia19, revealed numerous round cells in and around cerebral vessels in Tg2576–CD11c-DNR mice that were nearly absent in Tg2576 littermates (Fig. 3c). These cells had few or no processes and were also found in close vicinity to β-amyloid plaques, where they often co-localized with Aβ deposits and sometimes contained Aβ (Fig. 3c). These cells were CD11b+CD11c+ (Supplementary Fig. 3a online), stained positively for the macrophage and activated microglia marker CD68 (data not shown) and were increased in number by 66–79% in Tg2576–CD11c-DNR mice (Fig. 3d). A younger (12-month-old) cohort of mice showed 2–4 β-amyloid plaques per brain section and modest microglial and astrocytic activation that was comparable between Tg2576 and Tg2576–CD11c-DNR mice (Supplementary Fig. 3b). Of note, we did not detect round CD45+ cells in either Tg2576 or Tg2576–CD11c-DNR mice at this age (Supplementary Fig. 3b), suggesting that this relatively low level of cerebral amyloidosis does not meet a threshold limit for recruiting these cells.
Figure 3.
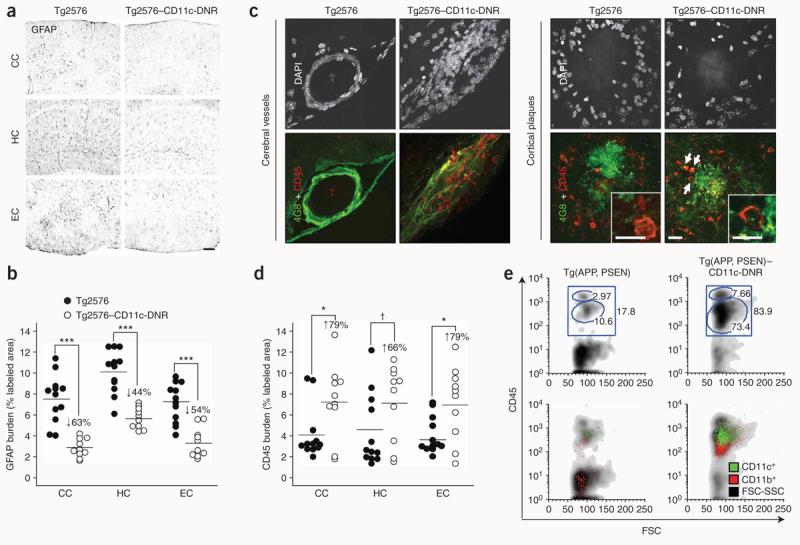
The CD11c-DNR transgene reduces astrocytosis but increases infiltrating macrophages in Alzheimer's disease mouse models. (a,b) Photomicrographs from Tg2576 (n = 12) and Tg2576–CD11c-DNR (n = 10) mouse brain sections (a), with median values by image analysis for GFAP immunohistochemistry (b); GFAP burden (% labeled area) is shown on the y axis and brain region is represented on the x axis. (c) Confocal micrographs of Tg2576 or Tg2576–CD11c-DNR brain sections (left, cerebrovessels; right, entorhinal cortex β-amyloid plaques) immunolabeled for human Aβ and mouse CD45 and counterstained with DAPI. Colocalization of Aβ with CD45+ cells in Tg2576–CD11c-DNR mice is denoted by arrows, and some of these cells contain Aβ deposits (high-magnification single optical section insets). (d) Quantitative image analysis for CD45 burden. All statistical comparisons are within brain region and between Tg2576 and Tg2576–CD11c-DNR mice, ***P < 0.001, *P < 0.05, †P < 0.10. (e) Double-transgenic Tg(APP,PSEN) mice were crossed with CD11c-DNR mice (designated Tg(APP,PSEN)–CD11c-DNR), and five mouse brains per group were pooled for FACS analysis with fluorescently tagged antibodies to CD45, CD11b and CD11c, as indicated. Log-fluorescence intensity for CD45 is indicated on the y axis, and forward scatter (FSC, a measure of cell size) is indicated on the x axis. Percentages of cells within each gate are indicated in top plots, and overlays of CD11b and CD11c are shown in bottom plots. Scale bars denote 100 μm (a) and 20 μm (c; 10 μm for insets).
To better characterize these cells, we crossed an accelerated Alzheimer's disease mouse model bearing both mutant APP and presenilin-1 transgenes (designated Tg(APP,PSEN)20) with CD11c-DNR mice and performed FACS analysis of brains from aged progeny. CD45+CD11b+CD11c+ cell numbers were greatly increased in Tg(APP,PSEN)–CD11c-DNR mouse brains (Fig. 3e and Supplementary Table 2 online, P < 0.01). Similar to the cells from Tg2576–CD11c-DNR mice, these cells most closely resemble brain-infiltrating macrophages that became ‘sensitized’ by the CD11c-DNR transgene to enter Alzheimer's disease-like brains. Two functionally distinct subpopulations of monocytes exist—the proinflammatory (Ly-6C+) and anti-inflammatory (Ly-6C−) subsets21. We noted that a majority (>90–95%) of CD45+ cells in Tg(APP,PSEN)–CD11c-DNR mice were Ly-6C− (Supplementary Fig. 4a online). Brain infiltration by these cells seems to occur in response to increasing cerebral amyloidosis, as they are not detected in brains of CD11c-DNR mice (data not shown) or Tg2576–CD11c-DNR mice at a younger age (12 months, Supplementary Fig. 3b) and do not accumulate in the periphery of Tg(APP,PSEN)–CD11c-DNR mice (Supplementary Fig. 4b).
We did not detect CD11c expression by microglia in Tg2576 mice (Supplementary Fig. 3a). Additionally, CD11c-DNR transgene mRNA levels trended toward an increase in Tg2576–CD11c-DNR versus CD11c-DNR mouse brains (quantitative real-time PCR unitless ratio of CD11c-DNR/hypoxanthine phosphoribosyltransferase 1 (Hprt1) ± s.e.m.: 11.8 ± 3.7 versus 6.6 ± 0.9; P = 0.10), and we detected CD11c-DNR mRNA in peripheral macrophages but not microglia (data not shown). To determine whether TGF-β signaling was reduced in peripheral macrophages or microglia, we challenged primary peripheral macrophage cultures from wild-type and CD11c-DNR mice with a dose range of TGF-β1 and with immunostimulatory lipopolysaccharide (LPS). Wild-type microglia and macrophages showed phosphorylated Smad2/3 (ref. 2) after TGF-β1 challenge, and whereas CD11c-DNR microglia also responded to challenge, CD11c-DNR macrophages were nonresponsive (Fig. 4a). In the relative absence of TGF-β–activated Smad2/3 signaling, we detected constitutively increased phosphorylation of the parallel bone morphogenic protein–activated Smad1/5/8–p21–activated protein kinase pathway2 in CD11c-DNR macrophages (Fig. 4b). Exogenous TGF-β1 and LPS further augmented Smad1/5/8 phosphorylation; however, CD11c-DNR macrophages did not show increased extracellular signal–related kinase-1/2 phosphorylation in response to general LPS activation (Fig. 4b).
Figure 4.
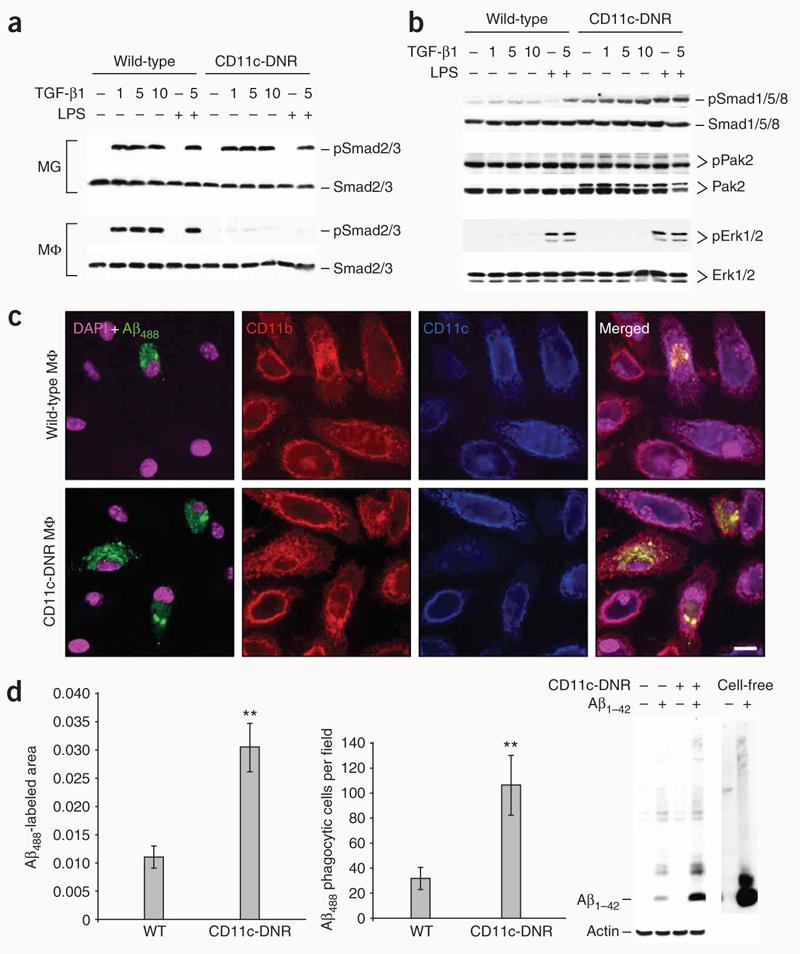
TGF-b1 shifts CD11c-DNR macrophages from canonical to alternate Smad signaling and increases Aβ phagocytosis in vitro. (a) Primary microglia (MG) or macrophages (MΦ) from wild-type or CD11c-DNR mice went untreated or were treated for 30 min with a dose range of recombinant TGF-β1 (1, 5 or 10 ng/ml as indicated) with or without 50 ng/ml of LPS. Cell lysates were western blotted for phosphorylated (p) and total Smad2/3 proteins as an indicator of canonical TGF-β-activated signaling. (b) Primary macrophages were treated as above and western blotted for phosphorylated and total Smad1/5/8 or p21-activated kinase 2 (Pak2; both activated in the alternate Smad signaling pathway), or extracellular signal-regulated kinase (Erk) 1/2. (c) Primary macrophages were pulsed for 4 h with 2 μg/ml of preaggregated Aβ488 and chased for 15 min before analysis by confocal microscopy with antibodies to CD11b or CD11c (merged images are shown on the right). (d) Quantification of confocal images (n = 3 randomly-selected fields per group) was performed, and Aβ488− labeled area is shown on the left. Numbers of Aβ488 phagocytic cells per field are shown in the middle graph. Data are represented as group means ± s.d. Cell lysates were prepared from macrophages treated in parallel with 2 μg/ml of unlabeled human synthetic Aβ1–42, and 2 ng of the peptide (cell-free) was western blotted side-by-side with antibody 6E10 (right). Data shown in (a–d) are representative of three to four independent experiments in which similar results were obtained.
We pulse-chased peripheral macrophages from wild-type and CD11c-DNR mice with fluorescently tagged Aβ1–42 (Aβ488) to test for Aβ phagocytosis. Notably, quantitative confocal microscopy analyses revealed approximately threefold increased Aβ488 phagocytosis in CD11c-DNR macrophages as compared to wild-type macrophages, which was not altered by the presence of the fluorescent Aβ1–42 tag (Fig. 4c,d). Both monomeric and oligomeric species of Aβ1–42 could be detected in greater quantities in CD11c-DNR macrophages than in wild-type cells (Fig. 4d), suggesting that CD11c-DNR macrophages engulf Aβ species irrespective of aggregation status.
To further validate results from a genetic approach, we used two activin-like kinase 5 (ALK5, a key TGF-β receptor I that pairs with TGF-β receptor II for signaling) inhibitors: SB-505124 and SB-431542 (ref. 11). Treatment of peripherally isolated macrophages with exogenous TGF-β1 increased phosphorylation of both Smad2/3 and Smad1/5/8, and this effect on Smad2/3 phosphorylation was blocked by ALK5 inhibition in a dose-dependent manner (Supplementary Fig. 5a online). Of note, ALK5 inhibitor treatment alone increased the ratio of phospho-Smad1/5/8 to phospho-Smad2/3, which was associated with increased Aβ phagocytosis in a dose-dependent fashion (Supplementary Fig. 5a–c). Thus, both genetic and pharmacologic means of TGF-β signaling inhibition promote increased macrophage Aβ phagocytosis.
Exogenous addition of TGF-β1 to microglia promotes increased Aβ uptake10, raising an apparent discrepancy with our findings. We were able to reproduce these results in both wild-type microglia (data not shown) and wild-type macrophages. Notably, we show that either the CD11c-DNR transgene or ALK5 inhibitors promote blockade of TGF-β–activated Smad2/3 phosphorylation, but they promote increased phosphorylation of alternate Smad1/5/8 signaling pathway molecules in macrophages, which is further inducible by exogenous TGF-β1 (Fig. 4a,b and Supplementary Fig. 5a). Thus, activation of the alternate Smad1/5/8 signaling cascade in response to TGF-β may resolve this apparent discrepancy. Specifically, it seems that the act of blocking Smad2/3 signaling results in promotion of Smad1/5/8 signaling, which is associated with increased macrophage Aβ phagocytosis.
Although Tg2576–CD11c-DNR mice had substantial reductions in detergent-soluble and guanidine HCl–soluble Aβ1–42 and detergent-soluble Aβ1–42/Aβ1–40 ratio, guanidine HCl–soluble Aβ1–42/Aβ1–40 ratio was apparently unaltered. When comparing male and female Tg2576 mice, we observed that females had greater behavioral impairment than males (Supplementary Fig. 1a–c). Female Tg2576 mice had increased detergent-soluble and guanidine HCl–soluble Aβ1–42 and increases in both detergent-soluble and guanidine HCl–soluble Aβ1–42/Aβ1–40 ratio (Supplementary Fig. 5d,e). Insoluble Aβ is probably primarily responsible for Tg2576-associated Morris water maze impairment22, and our data suggest that the ratio of more aggregated Aβ1–42/Aβ1–40 is particularly crucial for this behavioral phenotype.
Our results imply that reduction of TGF-β signaling in peripheral macrophages promotes increased brain infiltration of blood-derived macrophages and Aβ clearance in Alzheimer's disease mice. But is infiltration associated with a proinflammatory response, as was observed after active Aβ vaccination in subjects with Alzheimer's disease23? We found that a panel of proinflammatory cytokines in brain homogenates from Tg2576 and Tg2576–CD11c-DNR mice were either unchanged between groups or significantly lower in Tg2576–CD11c-DNR mice (data not shown). Furthermore, we observed reduced levels of proinflammatory tumor necrosis factor-α mRNA in Tg2576 and Tg2576–CD11c-DNR mice compared to wild-type mice, and increased levels of anti-inflammatory interleukin-10 mRNA in Tg2576–CD11c-DNR brains compared to wild-type mice (Supplementary Fig. 5f), suggesting that infiltrating CD11c-DNR macrophages are shifted to an anti-inflammatory phenotype endorsing Aβ phagocytosis24.
Does infiltration require blockade of TGF-β signaling, or do TGF-β signaling–sufficient peripheral macrophages routinely enter Alzheimer's disease mouse brains and limit cerebral amyloidosis? Reports suggest that brain infiltration of blood-derived macrophages occurs to a limited extent. For example, blood-derived monocytes have been found near cerebral vessels and β-amyloid plaques25, and ablation of these cells increases cerebral amyloidosis26,27. These effects seem specific to Alzheimer's disease mice, as recent approaches did not show similar results after CNS injury unless the mice were irradiated28,29. Future studies designed to establish chimeric mice without using irradiation will be useful to directly evaluate brain entry of peripheral monocytes and macrophages. Nonetheless, our results dovetail with findings that peripheral, blood-derived macrophages can clear cerebral Aβ, as blocking TGF-β signaling on peripheral macrophages leads to en masse brain infiltration and beneficial cerebral Aβ clearance. Notably, TGF-β signaling inhibitors may either be beneficial or deleterious depending on where the inhibition is targeted (that is, directly in the CNS11 or in the periphery). Yet, we suggest that inhibition of TGF-β signaling on peripheral monocytes and macrophages may represent an advantageous anti-amyloid therapeutic approach for Alzheimer's disease.
METHODS
Mice
We obtained Tg2576 mice from Taconic and maintained them as heterozygotes on a hybrid C57BL/6 × SJL background by intercrossing breeding pairs4. We maintained CD11c-DNR mice3 as heterozygotes on a C57BL/6 background and crossed them with Tg2576 mice to yield four genotypes of littermates, which we studied between 16 and 18 months of age: wild-type (n = 14, four males and ten females), CD11c-DNR (6 males), Tg2576 (n = 12, five males and 7 females) and Tg2576–CD11c-DNR (n = 10, seven males and three females). We also analyzed a younger (12-month-old) cohort of these mice (during initial deposition of β-amyloid4), including Tg2576 (n = 5, three females and two males) and Tg2576–CD11c-DNR (n = 5, three females and two males) genotypes. We obtained an accelerated doubly-transgenic mouse model of Alzheimer's disease (Tg(APPswe,PSEN1dE9)85 (ref. 20), designated Tg(APP,PSEN) in this report) from the Jackson Laboratory, and we maintained these mice as heterozygotes on a hybrid C57BL/6 × C3H background by intercrossing breeding pairs. CD11c-DNR mice were also bred to Tg(APP,PSEN) mice, and both Tg(APP,PSEN) mice (n = 5, three males and two females) and Tg(APP,PSEN)–CD11c-DNR mice (n = 5, three males and two females) were analyzed at 15 months of age. We housed all mice in a 12-h light and dark cycle at Yale University in The Anylan Center Animal Housing Facility, and the Yale University Institutional Animal Care and Use Committee approved all experiments, which we conducted in accordance with Yale Animal Resources Center guidelines.
Tissue handling
We killed mice with isofluorane and transcardially perfused them with ice-cold PBS. We rapidly isolated and quartered brains as previously described30 using a mouse brain slicer (World Precision Instruments). We weighed and then snap-froze anterior quarters and randomly assigned one of these to electric homogenization (Tekmar Tissuemizer) in cell lysis buffer (containing 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% vol/vol Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4 1 μg/ml leupeptin and 1 mM PMSF) for protein isolation. We centrifuged protein homogenates at 13,000g for 30 min at 4 °C, removed the supernatant (detergent-soluble fraction), treated the remaining pellet with 5 M guanidine HCl and solubilized it by occasional mixing on ice for 30 min (guanidine HCl–soluble fraction). We immersion-fixed posterior quarters in 4% (vol/vol) paraformaldehyde for 48 h at 4 °C, randomly assigned one posterior quarter for cryoprotection in a graded series of sucrose diluted in PBS (10% to 20% to 30%, each incubation step at 4 °C overnight) and embedded cerebral pieces in optimal cutting temperature compound (OCT, Tissue-Tek, Sakura) for cryosectioning.
Immunohistochemistry and morphometry
For immunohistochemistry (Aβ, GFAP and CD45 staining) and thioflavin S histochemistry, we cryosectioned four 10-μm coronal brain sections per mouse (spaced 50 μm apart) using a Leica model CM1850 freezing microtome (Leica), applied brain sections to Superfrost Plus Gold slides (Fisher Scientific) and allowed slides to air-dry for 5 min at 25 °C. We performed immunohistochemical staining with the VectaStain Elite ABC kit (Vector Laboratories) in accordance with the manufacturer's instructions with 3′-3′ diaminobenzadine as a chromogen (Sigma-Aldrich). We performed histochemistry with thioflavin S (a fluorescent dye that binds to the β-pleated sheet conformation present in mature β-amyloid plaques) by diluting 1% (w/vol) of practical-grade thioflavin S (Sigma-Aldrich) in 70% vol/vol ethanol. We filtered the solution and then used it to incubate slides for 10 min at 25 °C followed by three rinses for 5 min each in 70% ethanol and a final rinse in PBS. We then air-dried slides in the dark, mounted them in fluorescent mounting media containing DAPI (Prolong Gold; Invitrogen–Molecular Probes) and viewed them with an automated Olympus BX-61 microscope equipped for bright and dark fields.
For confocal microscopy analyses, we cryosectioned 25-μm coronal brain sections (spaced 50 μm apart), applied PAP-pen (Invitrogen) and preblocked in serum-free protein block (Dako) for 30 min at 25 °C. We then diluted primary antibody in serum-free protein block and incubated slides overnight at 4 °C. After three rinses for 5 min each in PBS, we incubated slides for 1 h at 25 °C with appropriate Alexa Fluor 488–, Alexa Fluor 594– or Alexa Fluor 647–conjugated secondary antibodies (Invitrogen-Molecular Probes). After an additional three rinses for 5 min each with PBS at 25 °C, we air-dried slides in the dark and finally mounted them with Prolong Gold containing DAPI (Invitrogen-Molecular Probes). We imaged fluorophores in separate channels with a Zeiss 510 META laser-scanning confocal microscope (Carl Zeiss Microimaging) and generated projections from three-dimensional–rendered optical sections. We variously used the antibodies to the following proteins for immunohistochemistry: cow GFAP (1:1,000; Dako), human Aβ (clone 4G8, 1:250; Covance Research Products), mouse CD45 or CD11b (1:200; Serotec), mouse CD11c (1:50; Thermo Fisher Scientific–Pierce Biotechnology) and mouse Ly-6C (conjugated with biotin, 1:100; BD Biosciences–Pharmingen).
Image analysis
We acquired images of brain sections stained with antibodies to 4G8, GFAP or CD45 or with thioflavin S using an automated Olympus BX-61 microscope with an attached Magnafire CCD camera system and Scion Image for Windows software, release alpha 4.0.3.2 (Scion) as previously described30. We captured images of four 10-μm sections through each anatomic region of interest, and we obtained a threshold optical density that best discriminated staining from background. For β-amyloid, GFAP, CD45 and thioflavin S burden analyses, data are reported as the percentage of labeled area captured (positive pixels) divided by the full area captured (total pixels). We determined CAA scores according to previous methods10. For β-amyloid plaque (4G8 or thioflavin S staining) morphometric analyses30, we calculated maximum diameters of plaques (small, <25 μm; medium, 25–50 μm; or large, >50 μm) by quantitative image analyses, and an examiner blind to sample identities (T.T. or T.M.) totaled numbers of plaques falling into each diameter category. For semiquantitative immunofluorescence analysis of CD45+CD11b+ macrophages in individual progeny from Tg(APP,PSEN)–CD11c-DNR matings, we blindly scored four brain sections per mouse using a five-point semiquantitative scale.
Statistical analyses
In instances of single comparisons of the means, we used Levene's test for equality of the variance followed by t-test for independent samples to assess significance, except for semi-quantitative histology (that is, ‘CAA Score’ and ‘CD45+CD11b+ infiltrating macrophages’ data), where we used the Mann-Whitney U-test. In instances of multiple means comparisons, we used ANOVA, followed by post hoc comparison by Fisher's LSD (for behavioral data) or Bonferroni's method (for all other analyses). For all analyses, we set alpha levels at 0.05 (SPSS for Windows, release 15.0, SPSS Inc.). An examiner blinded to sample identities performed all analyses, and the code was not broken until analyses were completed.
Supplementary Material
ACKNOWLEDGMENTS
We thank F. Manzo for help with preparing this manuscript and A. Ferrandino and S. Sojitra for expert technical assistance. We are grateful to Y. Wan, P. Rakic and J.J. Breunig for helpful discussion and to P. Rakic and J.J. Breunig for assistance with confocal microscopy. We also thank N. Ruddle for providing the brain FACS protocol. This work was supported by an Alzheimer's Association Grant (to R.A.F. and T.T.), and a US National Institutes of Health Pathway to Independence award (1K99AG029726 and 4R00AG029726 to T.T.). R.A.F. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 3.Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat. Immunol. 2005;6:600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 4.Hsiao K, et al. Correlative memory deficits, Aβ elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 5.Akiyama H, et al. Inflammation and Alzheimer's disease. Neurobiol. Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wisniewski HM, Wegiel J, Wang KC, Kujawa M, Lach B. Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can. J. Neurol. Sci. 1989;16:535–542. doi: 10.1017/s0317167100029887. [DOI] [PubMed] [Google Scholar]
- 7.Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-β1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- 8.Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E. Chronic overproduction of transforming growth factor-β1 by astrocytes promotes Alzheimer's disease–like microvascular degeneration in transgenic mice. Am. J. Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wyss-Coray T, et al. Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer's disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- 10.Wyss-Coray T, et al. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 11.Tesseur I, et al. Deficiency in neuronal TGF-β signaling promotes neurodegeneration and Alzheimer's pathology. J. Clin. Invest. 2006;116:3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holcomb L, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 13.King DL, et al. Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer's disease. Behav. Brain Res. 1999;103:145–162. doi: 10.1016/s0166-4328(99)00037-6. [DOI] [PubMed] [Google Scholar]
- 14.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Callahan MJ, et al. Augmented senile plaque load in aged female β-amyloid precursor protein–transgenic mice. Am. J. Pathol. 2001;158:1173–1177. doi: 10.1016/s0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellis RJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- 17.Robbins EM, et al. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J. Neurosci. 2006;26:365–371. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- 19.Lemere CA, et al. Nasal Aβ treatment induces anti-Aβ antibody production and decreases cerebral amyloid burden in PD-APP mice. Ann. NY Acad. Sci. 2000;920:328–331. doi: 10.1111/j.1749-6632.2000.tb06943.x. [DOI] [PubMed] [Google Scholar]
- 20.Jankowsky JL, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 21.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 22.Westerman MA, et al. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicoll JA, et al. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat. Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 24.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J. Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stalder AK, et al. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J. Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow–derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 27.El Khoury J, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 28.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 29.Mildner A, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 30.Tan J, et al. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat. Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.