

Abstract
Ascorbate (vitamin C) is a vital antioxidant molecule in the brain. However, it also has a number of other important functions, participating as a co-factor in several enzyme reactions including catecholamine synthesis, collagen production and regulation of HIF-1α. Ascorbate is transported into the brain and neurons via the Sodium-dependent Vitamin C Transporter-2 (SVCT2), which causes accumulation of ascorbate within cells against a concentration gradient. Dehydroascorbic acid, the oxidized form of ascorbate, is transported via glucose transporters of the GLUT family. Once in cells, it is rapidly reduced to ascorbate. The highest concentrations of ascorbate in the body are found in the brain and neuroendocrine tissues such as adrenal, although the brain is the most difficult organ to deplete of ascorbate. Combined with regional asymmetry in ascorbate distribution within different brain areas, these facts suggest an important role for ascorbate in the brain. Ascorbate is proposed as a neuromodulator of glutamatergic, dopaminergic, cholinergic and GABAergic transmission and related behaviors. Neurodegenerative diseases typically involve high levels of oxidative stress and thus ascorbate has been posited to have potential therapeutic roles against ischemic stroke, Alzheimer's disease, Parkinson's disease and Huntingdon's disease.
Keywords: ascorbate transport, ascorbate recycling, dehydroascorbic acid transport, SVCT1, SVCT2, glucose transporters (GLUT), brain
1. Vitamin C chemistry, cellular uptake and recycling
Vitamin C, or ascorbic acid, has many different functions in humans and other mammals. In addition to its well known role as an antioxidant, the vitamin serves as a co-factor in several important enzyme reactions, including those involved in the synthesis of catecholamines, carnitine, cholesterol, amino acids, and certain peptide hormones [1]. Of course, its best known function is to facilitate the hydroxylation of proline and lysine residues in collagen, allowing proper intracellular folding of pro-collagen for export and deposition as mature collagen. Ascorbate has also been shown to assist other prolyl and lysyl hydroxylases in the hydroxylation of hypoxia-inducible factor 1α (HIF-1α). HIF-1α is a transcription factor responsible for the cellular response to low oxygen conditions through activation of genes controlling diverse cellular pathways including glycolysis, iron transport, angiogenesis and cell survival [2]. Hydroxylation of HIF-1α targets it for destruction in the proteosome, thereby down-regulating the pathways dependent on transcriptional regulation by hypoxia.
In all of its known functions, ascorbate serves as a one-electron donor, generating the ascorbate free radical (AFR) (Fig. 1). The AFR is reduced back to ascorbate within cells by NADH- and NADPH-dependent reductases that have a high affinity for the low concentrations of the radical generated [3-5]. If the AFR accumulates significantly in areas not accessible to these enzymes, or if its concentration exceeds their capacity, two molecules of the AFR react or dismutate to form one molecule each of ascorbate and dehydroascorbic acid (DHA) [6]. The latter is the two-electron-oxidized form of ascorbate, which is very unstable, having a half-life in blood and physiologic buffers of 2-6 minutes [7,8]. DHA can also be recycled back to ascorbate by many mechanisms within cells, including direct reduction by GSH and enzymatic reduction by various thiol transferases or NADPH-dependent reductases [9,10]. Ascorbate is recycled from both its oxidized forms within cells, although cell-surface AFR reduction has also been described [11,12]. Whilst ascorbate can efflux from various types of cells [13,14], including neurons [15], because of its hydrophilic nature and negative charge at physiologic pH, it appears to do so slowly over several hours in the absence of an efflux mechanism [16]. Intracellular ascorbate concentrations in the low millimolar range (much higher than those in plasma) appear to be necessary to support its role as an antioxidant [17] and as a co-factor for dioxygenase enzymes [18,19]. Whereas multiple intracellular recycling mechanisms help to maintain intracellular high ascorbate levels, sodium- and energy-dependent transport of the vitamin is necessary to achieve a concentration gradient from blood or the interstitium into cells.
Figure 1. Ascorbate metabolism.
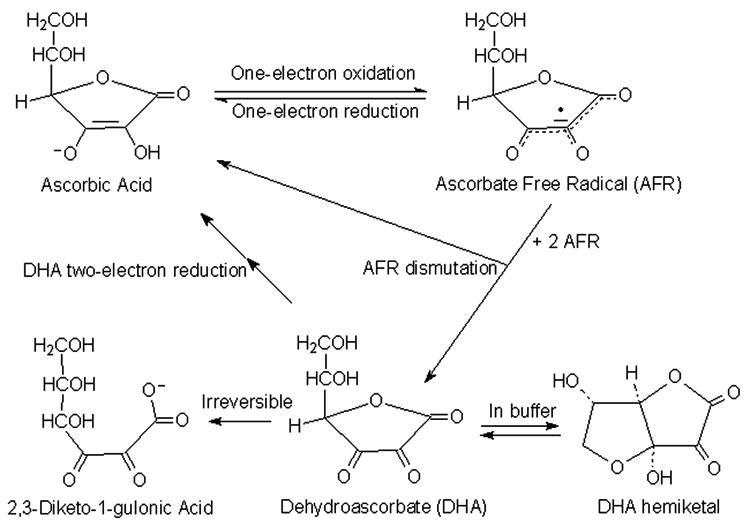
Chemical structures and reactions.
The trans-plasma membrane gradient of ascorbate is generated by specific ascorbate transporters that were first cloned in 1999 [20]. Two isoforms of these transporters are known, which although similar in amino acid sequence and structure, have different tissue distributions [21]. The Sodium-dependent Vitamin C Transporter type 1 (SVCT1, product of the SLC23A1 gene in humans; svct1 and slc23a1 in animals) resides largely in apical brush-border membranes of intestinal and renal tubular cells, where it helps to mediate absorption and re-absorption of the vitamin, respectively. Its absence in a knock-out mouse has recently been shown to impair renal ascorbate reabsorption [22]. Urinary loss of ascorbate decreases ascorbate concentrations in plasma and in most tissues, but not the brain. The SVCT2 (product of the SLC23A2 gene in humans; svct2 and slc23a2 in animals), on the other hand, is found in cells of most other tissues [23]. This transporter is especially important in brain, as discussed later. Both transporters mediate high affinity, sodium- and energy-dependent ascorbate transport into cells and as noted above are essential to establish steep concentration gradients of the vitamin across the plasma membrane [24]. The nomenclature for both isoforms has evolved since cloning of the protein in 1999 [25]. Since differentiation between human and animal isoforms is not strictly needed in this article, the term SVCT will be used.
In addition to uptake on the SVCT proteins, DHA can enter and leave cells by facilitated diffusion on the ubiquitous glucose transporters of the GLUT family (GLUTs). DHA forms a bicyclic hemiketal in solution (Fig. 1) [26,27], which is likely the species transported by the GLUTs [28,29]. Ascorbate itself is not transported on the GLUTs [30]. Once DHA has entered cells, it is rapidly reduced to ascorbate by the mechanisms noted above, and the resulting ascorbate is retained within the cells. Although such DHA uptake and reduction can at least transiently generate a trans-plasma membrane gradient of ascorbate [16], several lines of evidence suggest that it is not the major route. First, DHA concentrations in blood plasma are 2 μM or less, whereas ascorbate concentrations are 40-60 μM [31,32]. Second, glucose will compete in most cells for uptake of this low concentration of DHA on the GLUTs, as has been demonstrated in human leukocytes [33,34]. Third, and perhaps most compelling, ascorbate concentrations are low in cells lacking the SVCT. Two examples highlight this point. First, ascorbate levels are the same in erythrocytes and the plasma from which they were taken [16,35]. This is despite the observations that human erythrocytes express high numbers of GLUT1 [36] and that glucose does not compete well for DHA uptake in this cell type [37]. Key here is that failure to concentrate ascorbate in human erythrocytes corresponds to a lack SVCT proteins [38]. The second example is that knockout of the SVCT2 results in almost undetectable ascorbate concentrations in embryonic brain and pituitary [39]. These observations support the notion that the high trans-plasma membrane ascorbate concentration gradients seen in other non-epithelial cells are due to the SVCT2. Nonetheless, DHA uptake and reduction could well preserve intracellular ascorbate over short time periods in areas of high oxidant stress, where significant amounts of extracellular ascorbate are oxidized [40].
2. Vitamin C accumulation and maintenance in the CNS: Role of SVCT2
Vitamin C may be more than simply a “micronutrient” in the central nervous system (CNS), since it is present in millimolar concentrations in neuron-rich areas. There are two novel aspects of how ascorbate enters the CNS that distinguish its uptake from that seen in other organ systems (Fig. 2). First, although ascorbate transport across the blood-brain barrier occurs [41,42], it is very slow [41], and second, the ability to maintain a steep ascorbate concentration gradient from blood to neuronal cells is generated by a two step mechanism: first into the cerebrospinal fluid (CSF), and then into the brain cells (Fig. 2).
Figure 2. Ascorbate uptake and metabolism in the CNS.
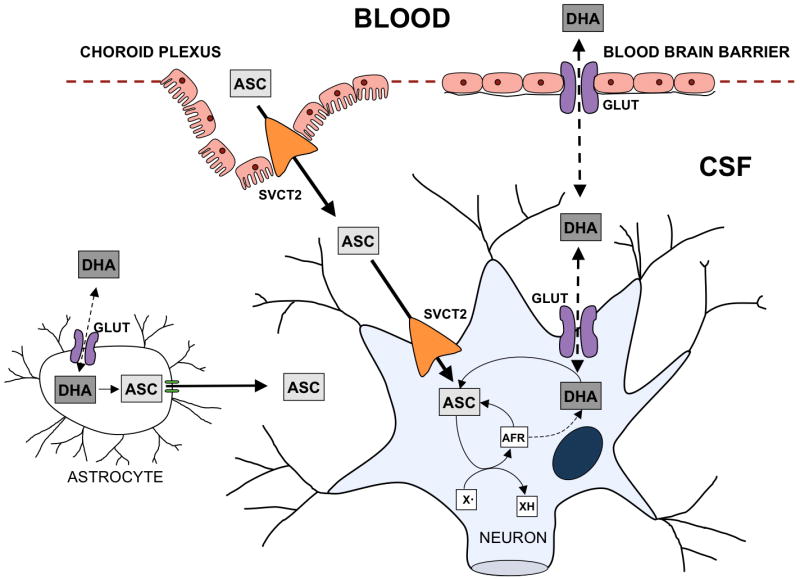
ASC, ascorbate; AFR, ascorbate free radical; DHA, dehydroascorbic acid; CSF, cerebrospinal fluid; X• oxidizing free radical species. Ascorbate enters the CSF either directly through the choroid plexus via the SVCT2 or possibly as DHA via GLUTs across the blood brain-barrier. Similarly ascorbate enters the neuron through the SVCT2 or as DHA via the GLUTs. AFR generated by X• dismutates to form DHA and ascorbate. Both the AFR and DHA are recycled back to ascorbate by cellular metabolism. Glial cells obtain ascorbate from the recycling of DHA that enters via GLUTs while neurons also have the SVCT2 for direct acquisition of ascorbate.
2.1 Routes of ascorbate entry into the CNS
The major route by which ascorbate enters the CNS involves transport from plasma to the CSF across the epithelium of the choroid plexus (Fig. 2) [43-45]. SVCT2 mRNA has been demonstrated by in situ hybridization in the choroid plexus epithelium [46,47], where it mediates high affinity and sodium-dependent ascorbate transport across the basolateral membrane into the cells [48,49]. Intracellular ascorbate then exits the cells into the CSF across the apical membrane of the choroid plexus ependymal cells [50,51], although it is not known how this occurs. This trans-cellular transport generates a 4-fold plasma-to-CSF ascorbate gradient in rats [45,52], resulting in CSF concentrations of about 200-400 μM [53,54], compared to plasma concentrations of 60 μM or less (Fig. 3). In humans CSF ascorbate concentrations tend to be slightly lower at 160 μM [55,56], but still show a pronounced gradient from plasma values of 40-60 μM. The SVCT2 has also been demonstrated in specialized α and β1 ependymal cells termed tanycytes that line the ventricular walls in the area of the hypothalamus [57]. These modified glial cells showed high affinity, sodium-, and energy-dependent ascorbate uptake in culture. However, they were not thought to move ascorbate from the CSF into the brain interstitium [58].
Figure 3. Ascorbate contents of plasma and various vertebrate organs.
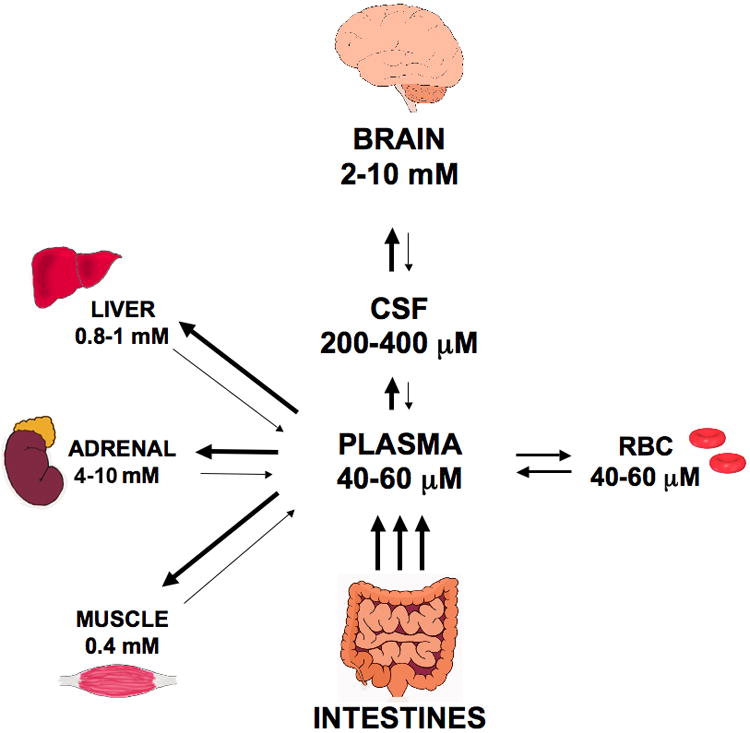
Although movement of ascorbate across the blood-brain barrier would seem a logical route of entry into the CNS, two features of the barrier prevent such movement. First, the junctions between endothelial cells that form the blood-brain barrier are very tight relative to other capillary beds [59]. Second, in contrast to most large vessel endothelial cells that have been studied, brain capillary endothelial cells do not express the SVCT2 protein in vivo [60,61]. This has been demonstrated as lack of SVCT2 mRNA by in situ hybridization [62], absent SVCT2 immunostaining of endothelium in both brain slices [63] and fragmented vessels [61], and by an inability to detect the SVCT2 in short-term cultures of brain capillary endothelium by immunoblotting [61]. Concordance of lack of the SVCT2 in brain capillary endothelium and lack of ascorbate transport across the blood-brain barrier endothelium provides support for the notion that the SVCT2 mediates ascorbate movement out of the blood in most other tissues. Such a mechanism would involve uptake of ascorbate from blood on the SVCT2, transit of ascorbate across the endothelial cell, and exit from the basolateral side of the cell by some as yet unknown mechanism. It remains to be seen whether ascorbate might also transit across less tightly apposed endothelia by paracellular movement between the cells.
In contrast to ascorbate, if DHA is present in blood in significant quantities, it can enter the CNS more rapidly than ascorbate. The proposed route of entry involves uptake of DHA on GLUT1 in the blood-brain barrier endothelium [64], failure to undergo reduction to ascorbate by intracellular GSH and enzymes, and exit from the cells on GLUT1 on the basolateral side of the cells [30,41,65]. DHA can then diffuse to neurons or glial cells where it can again be taken up, reduced to ascorbate, and retained by the cells. Ascorbate generated by DHA reduction within endothelial cells might also somehow exit the cells on their basolateral margins and thus enter the CNS. As discussed in Section 1, low plasma DHA concentrations and competition with glucose for the GLUTs do not support a major role for DHA in supplying ascorbate to underlying tissues. However, humans and primates lacking the ability to synthesize ascorbate haves been recently shown to have GLUT1 in erythrocytes that take up DHA with high apparent affinity and little competition from glucose [66]. In human erythrocytes this mechanism appeared to require interaction of GLUT1 with the integral membrane protein stomatin [67]. If present in endothelial cells, such a mechanism might allow selective DHA uptake across the blood-brain barrier in humans. Further, even in mice that have not lost the ability to synthesize ascorbate, DHA uptake across the blood-brain barrier is only modestly suppressed by physiologic glucose concentrations [41]. It is thus possible that low plasma concentrations of DHA could enter the endothelial cells forming the blood-brain barrier. Since these cells would be expected to reduce the incoming DHA to ascorbate, there remains the problem of how that ascorbate would exit the cells into the brain interstitium. Nevertheless, under conditions of oxidant stress, enough DHA might be generated from ascorbate in the blood or even in the endothelial cells to allow its passage into the brain.
2.2 Entry of ascorbate into brain cells
Once in the CSF, ascorbate appears to enter the brain interstitium by diffusion, since interstitial brain ascorbate concentrations of 200-400 μM are similar to or only slightly higher than those in the CSF [53,54]. There are again two mechanisms by which ascorbate can enter neurons and glia from the interstitial space (Fig. 2): 1) transport of ascorbate on the SVCT2 and 2) oxidation of ascorbate to DHA and uptake of DHA on the GLUTs and subsequent intracellular reduction. Neurons likely use both mechanisms to maintain intracellular ascorbate, although transport on the SVCT2 contributes the most to amplifying the ascorbate concentration gradient from CSF to neurons. Neurons have long been known to accumulate ascorbate against a concentration gradient via a saturable, sodium-dependent mechanism [52,68,69]. That this gradient is due to the SVCT2 is supported by its high expression in brain relative to other organs [70] and by the finding that its absence in the SVCT2 knock-out mouse results in essentially undetectable low brain ascorbate content [71]. The SVCT2 is found in greater abundance in neuron-dense areas of the brain such as cortex, hippocampus and cerebellum [72] which correspond roughly with regional distributions of ascorbate in brain [73,74]. Further, cultured neurons derived from wild-type and heterozygous SVCT2-deficient mouse embryos have rates of ascorbate transport that are proportional to SVCT2 expression, implying a gene-dosage effect [75]. In most non-neuronal cell types, the SVCT2 has a high affinity for ascorbate (Km = 20-40 μM). This affinity corresponds well to plasma ascorbate concentrations of 30-60 μM. However, the apparent Km for ascorbate transport in cultured neuroblastoma cells is somewhat higher at 100-110 μM [76,77], which corresponds to higher CSF and interstitial ascorbate concentrations that supply the neurons with ascorbate (200-400 μM). Even if this relationship holds for the neuronal SVCT2 in vivo, the neuronal ascorbate transporter will still be nearly saturated by extracellular ascorbate. This suggests that ascorbate uptake and content in neurons will be limited by the number of SVCT2 proteins expressed, and affinity for ascorbate becomes relevant only when ascorbate deficiency is present.
In contrast to neurons, ascorbate uptake in supporting glial cells in the brain does not appear to involve the SVCT2. In situ hybridization studies in rodent brain show that SVCT2 mRNA is found only in neurons and not in astrocytes [78,79]. Also, most ascorbate is present in neurons and not supporting cells. This was first shown over 4 decades ago in histochemical studies [80]. Further, although total brain ascorbate concentrations in guinea pigs [81,82] and rats [83,84] are about 1-3 mM, intracellular concentrations of up to 10 mM have been calculated for cortical neurons in both post-natal and adult rats [85,86]. These concentrations are several-fold higher than those of GSH (2-3 mM) [87], which is usually the major low molecular weight intracellular antioxidant. Such high neonatal brain ascorbate concentrations also correlate well with the finding that SVCT2 mRNA is increased in fetal mouse brain compared to brain tissue from adult animals [88].
The difference in ascorbate content between neurons and glia is substantial. In post-natal rat brain, for example, areas rich in glia have ascorbate levels only about 10-20% those of areas with high neuronal density [89]. In astrocytes and glial supporting cells lacking the SVCT2, DHA uptake and reduction may be the only mechanism of ascorbate uptake. Even so, by analogy to ascorbate content in erythrocytes noted above, they would be expected to have ascorbate concentrations of 200-400 μM, which would facilitate intracellular functions of ascorbate discussed in the next section. Further, their ability to take up DHA generated in the brain interstitium during oxidant stress would create a “bystander effect” [90] that would help to prevent loss of ascorbate to the brain through ring-opening of DHA. Moreover, although glia may not normally express the SVCT2, oxidant stress due to ischemia-reperfusion injury has been shown to increase SVCT2 mRNA expression in both neurons and glia [91]. Indeed, primary culture astrocytes develop high affinity [92] sodium-dependent [93] ascorbate transport due to expression of the SVCT2 [94] that has been attributed to exposure to high oxygen concentrations in culture [95].
In addition to movement of ascorbate into neurons and glial cells, ascorbate is also released from both cell types. This release of ascorbate contributes to a homeostatic mechanism for maintaining extracellular rat brain ascorbate at 200-400 μM [53,96]. As reviewed most recently by Rice [97], ascorbate release from astrocytes and neurons has been linked in numerous studies to uptake of glutamate on one or more of its specific transporters [98-102]. This process is initiated either by activation of glutamatergic pathways or by direct infusion of glutamate [100]. A heteroexchange model has long been considered to explain the ability of glutamate uptake to cause ascorbate release [15,103]. However, as noted previously [104], heteroexchange on a single transporter is not supported, since ascorbate does not cause release of radiolabeled glutamate from synaptosomes. The mechanism of glutamate-induced ascorbate release was subsequently elucidated by Wilson and colleagues. It was known from earlier studies that glutamate transport, which also brings sodium into the cell, causes swelling of primary culture astrocytes [105,106]. Wilson's group showed that such astrocyte swelling in turn causes release of ascorbate from the cells, an effect due to opening of volume-sensitive organic anion channels [107]. A subsequent paper by the same group [108] showed clearly that glutamate uptake could be dissociated from ascorbate release by astrocytes in primary culture. First, both glutamate uptake and ascorbate efflux were attenuated when cell swelling was prevented by culture in hypertonic media. Second, ascorbate readily moved into glutamate-stimulated astrocytes down a concentration gradient, which would occur across a channel, but not on an exchange transporter. Third, inhibitors of volume-sensitive organic anion channels blocked ascorbate efflux, but had no effect on glutamate uptake. Finally, high intracellular ascorbate concentrations did not enhance glutamate uptake, as would be expected in a heteroexchange mechanism. Glutamate-induced ascorbate release has also been demonstrated in neuronal cells [109]. Ascorbate released into the brain interstitium and CSF in response to glutamate appears to have both antioxidant and neuromodulatory effects, as discussed in the next section.
3. Functions of vitamin C in the brain
The functions of ascorbate in the CNS and brain are numerous. Although all actions of ascorbate involve donation of a single electron, they can be divided into those considered antioxidant and non-antioxidant in nature. Many of the latter functions involve monovalent reduction of Fe3+ or Cu2+ at the active sites of dioxygenase enzymes in hydroxylation reactions. Regarding the antioxidant functions, ascorbate directly acts to scavenge oxygen- or nitrogen-based radical species generated during normal cellular metabolism. At the millimolar concentrations present in neurons in vivo, ascorbate will effectively scavenge superoxide [110], a major diffusible byproduct of rapid neuronal mitochondrial metabolism. Ascorbate in aqueous compartments can also recycle α-tocopherol in membranes by reducing the α-tocopheroxyl radical back to α-tocopherol. Ascorbate has been shown to spare/recycle α-tocopherol in lipid bilayers [111] and in erythrocytes [112]. Given the lipid-rich environment of the brain, the sparing or recycling α-tocopherol may be a very important role for ascorbate, as will be discussed below in regard to animal studies of combined vitamin C and E deficiencies. Ascorbate addition to cultured cells, brain slices, and brain microsomes prevents lipid peroxidation induced by various oxidizing agents [113,114], especially in combination with α-tocopherol [115,116]. However, results of ex vivo tissue and cell culture experiments are difficult to interpret, since the oxidant stress induced in these models may have little physiologic relevance, and since the presence of trace metals in buffers or tissue slices might result in apparent pro-oxidant effects of ascorbate. In vivo studies outline a more convincing role for an antioxidant effect of ascorbate in brain, especially those dealing with ischemia-reperfusion.
3.1 Ascorbate and neural maturation
The ascorbate content of developing rat brain doubles in late pregnancy [117,118], which fits well with results of studies showing that ascorbate enhances the differentiation of embryonic stem cells into neurons in culture [119]. This is associated with increases in expression of genes involved in neurogenesis, maturation, and neurotransmission [120]. Direct support for this concept was obtained by Lee, et al. [121]. They showed in that a single addition of ascorbate to 200 μM (in the low physiologic range for CSF) enhanced the differentiation of precursor cells into both neurons and astrocytes over several days in culture, an effect not due to changes in cell survival. Ascorbate at the same concentration also induced synaptic maturation of the neurons, based on finding increased numbers of miniature excitatory post-synaptic currents in the cultured neurons. The ascorbate effects were not mimicked by other antioxidants, such as GSH and vitamin E, leading to the conclusion that they occurred by some other mechanism. The cortical neuron precursors expressed mRNA for SVCT2, but not SVCT1, and this expression was not affected by differentiation. Although the effects of ascorbate were associated with an increase in intracellular ascorbate, this study did not distinguish between effects on intra- and extracellular ascorbate, nor did it consider whether neuronal differentiation and maturation could be induced by lower amounts of ascorbate. A subsequent study using cells prepared from mice lacking the SVCT2 showed that very low amounts of intracellular ascorbate (100 μM or less) were both sufficient and necessary to neuronal maturation, as measured by dendrite formation and increases in the number of miniature excitatory post-synaptic potentials [122].
3.2 Ascorbate and neurotransmission
The non-antioxidant functions of ascorbate in brain and neural-derived tissues center on neurotransmitters. For example, it is well established that ascorbate is essential for catecholamine biosynthesis in neural tissues, serving as a co-factor for dopamine β-hydroxylase in the conversion of dopamine to norepinephrine [123,124]. Moreover, an elegant series of studies by several groups documented a novel mechanism involving ascorbate and the AFR to enable transfer electrons across the chromaffin granule membrane [125-127]. Ascorbate inside the granule is oxidized to the AFR, either in hydroxylation reactions or in simply scavenging radicals that might otherwise react with redox-sensitive catechols in the granule. This AFR is then reduced back to ascorbate by interaction with the reduced form of a cytochrome b561 in the granule membrane, which is maintained in the reduced form by accepting an electron from cytoplasmic ascorbate. Ascorbate in chromaffin granules is then secreted concomitantly with catecholamines from cultured chromaffin cells [128] and in vivo by human adrenal glands, the latter in response to adrenocorticotrophin stimulation [129].
Ascorbate has been proposed to function as a neuromodulator of both dopamine- and glutamate-mediated neurotransmission (reviewed in [15,104]). More recent studies relating ascorbate modulation of glutamate dynamics with changes in rat behavior show that such modulation is complex, since it depends on the site in the brain studied, level of behavioral activity, and level of extracellular ascorbate [102,130,131]. The mechanism(s) by which ascorbate affects neuronal transmission have not been established, but could relate in part to redox changes in the N-methyl-D-aspartate (NMDA) receptor. For example, ascorbate has been shown to protect neurons from excitotoxicity induced by activation of the NMDA receptor and it prevented glutamate-induced cell damage and death in cultured cerebellar granule cells [132,133]. This could be due to redox modulation of the receptor itself by ascorbate [134,135], or to direct scavenging of ROS generated by receptor activation. A more severe stress of ischemia releases large amounts of ascorbate from brain cells [136], which is associated with glutamate uptake by neurons and glia [99,137]. Removal of extracellular glutamate by such a process, would also decrease excitotoxicity caused by activation of cell surface and synaptic glutamate receptors [15,104,138].
It was hoped that knowledge of the regional distribution of ascorbate throughout the brain would provide vital clues as to its neuromodulatory function. Areas such as the forebrain that typically show the highest levels of ascorbate are rich in catecholamine innervation [139]. However, there does not appear to be any clear relationship between extracellular ascorbate levels and any neurotransmitters, and the question as to how such modulation is achieved has yet to be clarified. Ascorbate deficiency in guinea pigs is associated with significant increases (∼25%) in dopamine levels and similar relative decreases in norepinephrine [140], presumably because normal amounts of dopamine could not be metabolized into norepinephrine by dopamine β-hydroxylase in the absence of ascorbate as an essential co-factor. In the animals with the most severe ascorbate deficiency, neurotransmitter levels never normalized. However, with only slightly higher brain ascorbate levels, dopamine and norepinephrine contents slowly returned to control levels over the course of eight weeks. This recovery indicates an excellent plasticity of the brain in dealing with a low ascorbate environment, but shows that there are situations in which minimal ascorbate is sufficient to maintain apparently healthy growth, but in which brain function can be compromised. Following an even longer period of ascorbate deficiency, depressed levels of both dopamine and norepinephrine were reported along with increased catecholamine oxidation [141].
Pharmacological manipulations highlight the strength of the relationship between ascorbate and dopaminergic function. Treatment with amphetamine, which affects dopaminergic, cholinergic and glutamatergic activity, increased levels of both dopamine and ascorbate in the caudate nucleus [142]. Apomorphine (a dopamine receptor agonist) increased striatal dopamine and ascorbate, whereas the inverse is true of haloperidol (a dopamine receptor antagonist) which decreased both. These results reflect a well-regulated interaction between dopamine and ascorbate function, especially in the striatum [143]. The increases in extracellular ascorbate levels that are seen following activity or amphetamine administration are not uniform across brain areas and may implicate the heteroexchange system between ascorbate and glutamate in the control of these changes. There are rich glutamatergic projections from the cortex to the neostriatum. When the cortex is damaged, severing these connections, basal ascorbate levels decrease in the neostriatum and activity-induced increases in extracellular release are also diminished [144]. A large body of evidence (reviewed in Rebec and Pierce, [15]) suggests that the substantia nigra may be critically involved in dopaminergic and ascorbate interactions in the neostriatum which would implicate a highly complex pathway of innervation and synaptic signaling governing the relationship.
There is also evidence that ascorbate is involved in the regulation of both acetylcholine and catecholamine release from synaptic vesicles [145]. Scopolamine is a muscarinic receptor antagonist that also inhibits the action of acetylcholinesterase to break down acetylcholine in the synaptic cleft. In rats fed an ascorbate-enriched diet, the acetylcholinesterase inhibition caused by scopolamine was reversed [146], whereas in a guinea pig vitamin C deficiency model, brain acetylcholinesterase was found to be decreased relative to control animals [141]. Conversely, scopolamine treatment decreased ascorbate levels in rat striatum [147]. In vivo voltammetry studies of amphetamine-induced activity enhancement showed increased levels of ascorbate in the caudate nucleus. Parenteral administration of scopolamine and MK801 (an NMDA receptor blocker) both diminished the increase in ascorbate, further supporting relationship between ascorbate release and cholinergic receptor stimulation [148]. In addition to the noted effects of ascorbate on neurotransmitters, ascorbate also has been shown to be involved in neuropeptide amidation [149] and release in brain [150,151].
3.3. Ascorbate effects on behavior: learning, memory, and locomotion
Animal behavior studies involving ascorbate treatments have focused on two major areas, learning/memory and locomotor activity. Most of the positive effects of ascorbate follow parenteral administration of the vitamin (typically intraperitoneal injection), since much higher effective doses can be given by this route compared to oral administration. Intraperitoneal ascorbate (125 mg/kg) reversed memory deficits in mice induced both by age and scopolamine treatment in a transfer latency task in the elevated plus maze and a passive avoidance task [152] and in a habituation-based task in a light-dark paradigm [155]. When ascorbate was given orally for 30 days (300mg/kg) in conjunction with vitamin E in aged mice (15 months), it also improved performance on a passive avoidance task [156]. Improvement was not noted in 3 month old mice or when ascorbate was administered alone in that study. It should be noted, however, that these are atypical tests of learning and memory. The first two are variants of tasks more often used to measure anxiety than learning, and the third relies on learned associations between a response and an electric shock.
Less agreement has been found in rats using a shock avoidance shuttle-box paradigm. In one study, repeated ascorbate treatments (60-120mg/kg) either intraperitoneally for 14 days or orally for 30 days improved both acquisition and retention in this passive avoidance task [157]. This contrasts with results of an earlier study in which five days of acute pre-test ascorbate dosing (1g/kg intraperioneally) actually led to poorer performance [158]. The shock-avoidance test is highly stressful, and in the latter study involved up to 100 electric shocks per day. Even in the former study, where as few as one or two shocks may have been administered to the rat, the stress response may have been a confounding factor in the results. Ascorbate provided in drinking water has been shown to reduce the fear response in Japanese quail chicks tested in a less stressful light-dark emergence paradigm [159]. If ascorbate altered anxiety levels or the stress response in any of the above tasks, then task motivation, and therefore performance, may have been affected rather than cognitive ability. Long-term low levels of dietary ascorbate did not lead to impairments in learning and memory or anxiety in gulonolactone oxidase knockout mice that are unable to synthesize their own vitamin C [160] despite an approximately 75% reduction in brain ascorbate. Further, different doses of ascorbate and different dosing regimens used within these experiments rarely reflected a clear pattern of results or obvious dose-response relationships. Notwithstanding the negative results, it seems possible that ascorbate could be a mediator of learning and memory, especially stress-related learning, although the exact circumstances under which it has nootropic (cognitive enhancing) abilities are as yet unclear.
As with the effects on neurotransmitter levels noted above, a modulatory role of ascorbate on behavior can be more clearly seen in animals undergoing pharmacological challenge. Treatment with amphetamine increased the number of avoidance responses made in the shuttle box task describe above [161,162], and this effect was blocked by ascorbate treatment. The testing procedure itself led to decreased dopamine concentrations in the striatum, hippocampus and hypothalamus. These decreases were not seen in amphetamine-treated animals. Following ascorbate treatment, striatal dopamine was further decreased, although it remained unchanged relative to untested controls in hypothalamus and hippocampus. Amphetamine also increases locomotor activity and stereotypic behaviors (e.g. rearing, grooming, and sniffing). Very large (2g/kg) doses of vitamin C alone had no effect on these behaviors in C57Bl/6 mice, but blocked an amphetamine-induced increase in stereotypy whilst locomotor activity remained unchanged [163]. The behavioral effects of amphetamine were also attenuated either by intraventricular or striatal infusions of ascorbate. The results following ascorbate administration were similar to those of haloperidol (a dopamine receptor antagonist) and larger effects were found when both agents were combined [164,165]. Scopolamine had almost identical results to apomorphine (a dopamine receptor agonist) in that both lead to increases in motor activity and stereotypy [166]. They also inhibit ascorbate turnover and increase ascorbate catabolism in the striatum [167]. In rats with nigrostriatal lesions, treatment with amphetamine led to dopamine release contralateral to the lesioned area and results in circling behaviors. High (1g/kg) doses of ascorbate blocked dopamine-mediated circling behavior, something that may also be achieved through dopamine receptor blockers [168]. Although there is clearly a complex relationship between ascorbate and the cholinergic and dopaminergic systems, available data suggests that ascorbate can behave as a dopamine receptor antagonist. In fact ascorbate has been shown to inhibit binding of specific D1 ([3H] SKF 38393) and D2 ([3H] N-0437) receptor agonists [169].
3.4 Ascorbate and collagen synthesis
Less is known about the role of ascorbate in collagen synthesis in brain than in other organs, but minimal amounts seem essential for blood vessel and neural sheath formation. Mice deficient in the ascorbate transporter SVCT2 have undetectable levels of ascorbate in brain; they die with capillary hemorrhage in penetrating vessels of the brain [170]. This pattern of hemorrhage is reminiscent of that seen in mice with porencephaly, a disease caused by a gene defect in the synthesis of type IV collagen α1 [171]. Milder variants of this defect in humans have been associated with small vessel disease and hemorrhagic stroke [172]. Ascorbate-dependent collagen synthesis has also been linked to formation of the myelin sheath that surrounds many neuronal processes: ascorbate added to a mixed culture of rat Schwann cells and dorsal root ganglion neurons promoted myelin formation and differentiation by Schwann cells during formation of the basal lamina of the myelin sheath [173]. The latter process was considered linked to the ability of the Schwann cells to generate collagen.
4. Importance of Vitamin C in the brain: neuroprotective functions of ascorbate
4.1 Brain ascorbate deficiency
Scurvy causes severe lassitude and asthenia in humans. Although the disease has been associated with paraparesis in humans, death appears to be due more to complications of systemic collagen dysfunction rather than to a distinct neurologic syndrome [174]. This likely relates to the fact that ascorbate is avidly retained by the CNS during ascorbate deficiency [175]. Indeed, as described by James Lind in his Treatise on Scurvy in 1772 [176], even in sailors whose organs were ravaged by hemorrhage and edema in scurvy, Lind noted: “…the brains of these poor people were always sound and entire…” Perhaps reflecting this observation, guinea pigs near death from scurvy had undetectable ascorbate in liver, but the brain content of ascorbate was still about 25% of normal [177]. Whereas this suggests that decreases in CNS ascorbate do not play a major role in the signs and symptoms of generalized scurvy, it also suggests that the strong retention of ascorbate in the CNS reflects its importance to neuronal function. This hypothesis is supported by studies in guinea pigs with moderate vitamin E deficiency in which an acute deficiency of ascorbate was superimposed [178]. After 2 weeks of vitamin E deprivation, plasma and brain α-tocopherol concentrations were decreased by 65% and 32%, respectively. These animals gained weight and appeared completely normal. However, within 5-6 days of removing vitamin C from their diet, most of the animals developed a progressive ascending paralysis and died within 24 hours. No neurologic signs were apparent in animals with single deficiencies of vitamins C or E. Death occurred in the doubly deficient animals despite only 37% and 67% decreases in brain and spinal cord ascorbate concentrations, respectively. These animals had no signs of scurvy (no skin or hair changes, no hemarthroses), and only small increases in liver and brain F2-isoprostanes (a marker of oxidant stress). Standard hematoxylin and eosin staining of the brains and spinal cords showed no abnormalities. However, Nissl and silver degeneration stains revealed widespread neuronal loss and degeneration in the pons and long motor tracts of the spinal cord [179]. Thus, even a modest decrease in CNS ascorbate accelerated signs of vitamin E deficiency in this model and led to significant neuronal loss.
If the SVCT2 transporter is responsible for maintaining high CSF and neuronal ascorbate concentrations, what happens when the protein is knocked out in the mouse? Targeted deletion of the SVCT2 protein results in homozygotes that die shortly after birth: they never take a breath and have diffuse cerebral hemorrhage [180]. Whereas the latter could reflect increased capillary fragility, the late-stage embryos do not show hemorrhage elsewhere and lack signs of generalized scurvy. The proximal cause of death is unclear. Catecholamine synthesis is an ascorbate-dependent function and ascorbate levels are known to be in the millimolar range in adrenal gland (Fig. 3). Although catecholamine synthesis in the SVCT2 knockout mouse is decreased, but this does not appear to be the cause of death [181]. Although the lungs failed to expand, their architecture was normal, as were surfactant levels [182]. As noted earlier, ascorbate levels in brain and pituitary of the homozygous knockout embryos were undetectable. This observation provides strong evidence that uptake on the SVCT2 is the only route for ascorbate entry into neural tissues. The case for substantial DHA uptake across the blood-brain barrier is also not supported by the finding that blood ascorbate concentrations, although significantly decreased from about 70 μM to 30 μM [183], should still have been sufficient to generate some DHA for uptake into the brain in the stressed embryos.
4.2 Ascorbate in cerebral ischemia
4.2.1 Ascorbate versus DHA in cerebral ischemia
Perhaps the most dramatic acute oxidant stress in the CNS is the ischemia-reperfusion injury that occurs with ischemic stroke. Ischemia initially depletes intracellular GSH [184] and ascorbate [185] in brain. If reperfusion with oxygen rich blood occurs, the ROS generated due to abnormal mitochondrial metabolism will extend tissue damage to areas with decreased oxidant defenses [186]. Parenteral ascorbate injections decreased infarct size in both primate and rodent models of middle cerebral artery ischemia-reperfusion. When monkeys were given 1 g/day of ascorbate parenterally for 6 days before middle cerebral artery occlusion, brain infarct size was decreased by 50% in the ascorbate-treated group compared to the control group not treated with ascorbate [187]. Ascorbate was measured in the basal ganglia of these animals and was found to increase 50% above levels in the treated compared to the non-treated monkeys [187]. Thus, relatively modest increases in brain ascorbate content due to several days of parenteral administration were associated with neuroprotection against ischemia-reperfusion injury. In another primate study, high dose ascorbate (500 mg/kg up to 2 g) was administered into the middle cerebral artery just before occlusion, which was maintained for 4 h. At 24 h after reperfusion, the mean infarct size in ascorbate-treated monkeys was only 1/3 of that in non-treated controls. This dramatic decrease in infarct size with acute dosing was surprising, given that ascorbate normally enters the CNS slowly through the choroid plexus, as discussed above. Indeed, in a subsequent study in mice with middle cerebral artery occlusion, high dose DHA given by intra-luminal infusion injection just before, 15 min after, or 3 h after occlusion markedly decreased infarct volume, mortality, and neurological deficits in mice [188]. It is important to note that improvement occurred despite the fact that such high DHA doses may be toxic to some cell types, such as insulin-secreting cells of islets [189]. In contrast to results with DHA, no effects were observed for comparable injections of ascorbate. Protection by DHA infusion was observed in both ischemia alone and ischemia-reperfusion models. A subsequent study by the same group [190] replicated the initial results in mice and further showed that post-ischemic DHA infusion prevented the 50% decrease in brain ascorbate and also largely prevented the increase in lipid peroxidation due to ischemia-reperfusion injury. Why ascorbate infusion decreased lesion size in the primate study but not in the mouse studies is unclear, but it could relate to breakdown of the blood-brain barrier in ischemic regions, thus allowing entry of both ascorbate and DHA, or to differences in the ability of ascorbate to gain access to the CNS in the two species. That is, loss of the ability to synthesize ascorbate may have favored development of alternate routes or of more rapid transit of ascorbate into the CNS in primates.
4.2.2 Possible risks of ascorbate therapy in cerebral ischemia
The studies in which brain ascorbate was measured clearly support the notion that increases in brain ascorbate concentrations can be beneficial in ischemia or ischemia-reperfusion models of stroke. They also allay to some extent the concern raised by a human study of acute parenteral ascorbate prophylaxis before ischemia-reperfusion related to abdominal vascular surgery [191]. In that study intravenous ascorbate infusion (2 g given 2 h before ischemia was induced) increased tissue release of ROS, possibly due to a Fenton-type reaction of ascorbate with free iron in ischemic areas. This was thought in turn to account for the increased generation of lipid peroxides and inflammatory cytokines observed. That opposite results were seen following middle cerebral artery occlusion suggests that ascorbate did not encounter significant amounts of free iron in the brain. Such iron would likely be outside living cells, where it would encounter only non-reactive DHA in the DHA infusion studies. This DHA would be taken up and to ascorbate intracellularly, where it would not have exposure to free iron.
4.3 Primacy of intracellular ascorbate and the SVCT2 in neuroprotection
The results discussed thus far suggest that ascorbate plays a role in both sustaining normal function of the CNS and in ameliorating damage induced by pathological conditions that increase generation of ROS (e.g., severe antioxidant vitamin depletion, lack of the SVCT, and ischemia-reperfusion injury). As alluded to above, a consistent factor in these pathologies is the need to maintain intracellular, as opposed to extracellular, ascorbate. The concept that intracellular ascorbate is crucial for protection against oxidant stress is also supported by results of several in vivo studies. For example, an increase in intracellular (but not extracellular) ascorbate content of rat brain slices decreased the swelling induced by oxidant stress [192]. Increases in intracellular ascorbate were also found to prevent loss of α-tocopherol and decrease lipid peroxidation caused by culture of neurons in oxygenated medium [193].
Finally, the neuroprotective role of intracellular ascorbate highlights a crucial role for the SVCT2 in preserving intracellular ascorbate. This was evident in recent studies using in primary cultures prepared from hippocampal neurons derived from embryos that either lacked the SVCT2 or expressed normal levels of the protein [194]. After 7-14 days of growth in defined media lacking ascorbate, intracellular ascorbate was very low in SVCT2-expressing neurons (100 μM or less), and undetectable in SVCT2-deficient neurons. Excitotoxic challenge with NMDA or direct addition of the oxidant hydrogen peroxide markedly enhanced death of the cells lacking the SVCT2 compared to cells from wild-type mice. Moreover, following middle cerebral artery occlusion in rats, SVCT2 mRNA increased over several hours in the peri-infarct penumbra, both in neurons and in glia [195]. Together, the results of these studies strongly support a protective role for ascorbate and for the SVCT2 in brain during acute ROS generation.
5. Potential therapeutic functions of vitamin C in neurodegenerative disorders
Oxidative stress in the brain with focus on neurodegenerative diseases has been extensively reviewed [196], so only aspects relevant to ascorbate will be considered here. Neurons appear to be especially sensitive to ascorbate deficiency, perhaps because they have 10-fold higher rates of oxidative metabolism than supporting glia [197,198]. This neuronal sensitivity is most apparent when ascorbate supply is low in conditions in which there is excess oxidant stress. The involvement of reactive oxygen species in neurodegenerative disorders explains the enthusiasm for ascorbate as an antioxidant therapeutic approach, although its complicated interactions with neurotransmitter systems as described above make it difficult to discern the specific mechanisms involved.
5.1 Alzheimer's disease
Alzheimer's disease is caused by a combination of genetic and lifestyle factors and it is established that oxidative stress plays a key role in the pathogenesis of the disease [199,200] contributing to the degeneration of the basal forebrain cholinergic system and general cell death. Alzheimer's disease patients have been found to have lower plasma [201] and CSF [202] ascorbate levels despite adequate nutritional intake. Positive relationships have been shown between ascorbate supplement use and reduced disease incidence [203,204] and also with disease-related markers of oxidative stress [205], although these beneficial results are not universal [206,207]. Interpretation of the data is hindered by the nature of the studies as population or epidemiological studies have high levels of variability inherent in their design. Despite the large amount of information that can be collected from participants, and complicated statistical techniques that can be used to control for variability in the data, individual differences can still have a significant influence on results. The most important contributors to variability are education and career, diet, exercise, alcohol consumption, cigarette smoking and general health and illness across the lifetime. Any combination of the above factors may contribute to the lack of consistency in findings among these studies. Nevertheless, there is further evidence to support the potential for vitamin C as a therapeutic avenue for Alzheimer's disease. Orally administered ascorbate protected the CA1 area of the hippocampus in rats against oxidative stress and cytokine release induced by injection of fibrillar β-amyloid [208]. It also protected SH-SY5Y neuroblastoma cells from β-amyloid induced apoptosis [209]. Scopolamine (which impairs memory in rodents) is often used as a pharmacological model for Alzheimer's disease. As noted earlier, ascorbate blocked or attenuated the effects of scopolamine in several different types of studies. Furthermore, ascorbate has been shown to be an effective acetylcholinesterase inhibitor [210], the most common form of treatment used for Alzheimer's disease. These results may seem contradictory, since scopolamine can also inhibit acetylcholinesterase, however, the net effect of vitamin C appears to be a boost to cholinergic system functioning, although this relationship needs further investigation.
5.2 Parkinson's disease
Parkinson's disease involves severe decreases in dopaminergic signaling in the central nervous system, specifically the motor cortex. Oxidative injury is also thought to play a key role in the pathogenesis of the disease. Given the range of data presented above concerning the relationship between ascorbate and dopaminergic function it is understandable that ascorbate is being investigated for its therapeutic potential in this disease. Although population studies concerning ascorbate intake show no protection against development of Parkinson's disease [211], there are a number of lines of evidence that suggest that ascorbate as a pharmacological agent may be of more benefit. Ascorbate has been shown to improve bioavailability of levodopa (which can then be converted into dopamine) in members of an elderly Parkinson's disease population with low baseline bioavailability for levodopa [212]. MPTP (1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine) causes massive decreases in dopamine in striatum and leads to Parkinsonian like symptoms in humans if ingested [213]. MPTP, while not toxic itself, crosses the blood brain barrier where it is metabolized to MPP+ (1-methyl-4-phenylpyridinium) which is highly toxic especially to dopaminergic neurons of the substantia nigra. As such it is often used to model Parkinsonian symptoms in animals. MPTP administration also increases ascorbate oxidation [214] and inversely, co-administration of ascorbate reduces its effect by about 20% [215]. This is due, at least in part, to the antioxidant properties of ascorbate providing protection against the oxidative damage induced by MPP+.
5.3 Huntington's disease
Huntington's disease involves a loss of neurons in the striatum leading to severe motor and cognitive deficits. Mice that express the gene for Huntington's disease show a deficit in striatal ascorbate release during periods of behavioral activity [216]. This deficit was reversed with ascorbate injections which also improved the behavioral phenotype of repetitive movements [217].
6. Conclusions
That ascorbate is important for neuronal maturation and function, as well as for protection of the brain against oxidant stress is well supported by the evidence presented in this review. The vitamin is maintained at high concentrations in brain and in neurons in particular relative to other organs. In addition, strong homeostatic mechanisms maintain brain and neuronal ascorbate concentrations within very tight limits. Thus, not only is it difficult to deplete brain ascorbate, but it is also difficult if not impossible to increase levels for more than a short period above those set by uptake and recycling mechanisms. Whereas oral supplements generally increase brain ascorbate by only 20% at most, larger relative increases may occur if significant oxidant stress has caused localized ascorbate deficiency in brain areas affected by neurodegeneration or inflammation. Study of the role of ascorbate in human brain function has been limited, but with the availability of suitable mouse models, ascorbate deficiency or excess can be studied in more detail, particularly with regard to effects of the vitamin on brain development, neurotransmitter function and responses to inflammatory or oxidant stresses, such as might exist in cerebral atherosclerosis or in several neurodegenerative diseases.
Acknowledgments
This work was supported by NIH grant DK 50435.
Abbreviations
- AFR
ascorbate free radical
- CNS
central nervous system
- CSF
cerebrospinal fluid
- DHA
dehydroascorbic acid
- GLUT
glucose transporter
- GSH
gluthathione
- HIF-1α
hypoxia-inducible factor 1α
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine
- NMDA
N-methyl-D-aspartate
- ROS
reactive oxygen species
- SVCT1&2
sodium-dependent vitamin C transporter types 1 & 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chatterjee IB, Majumder AK, Nandi BK, Subramanian N. Synthesis and some major functions of vitamin C in animals. Ann N Y Acad Sci. 1975;258:24–47. doi: 10.1111/j.1749-6632.1975.tb29266.x. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 3.Lumper L, Schneider W, Staudinger H. Untersuchungen zur Kinetik der mikrosomalen NADH:Semidehydroascorbat-Oxydoreduktase. Hoppe Seylers Z Physiol Chem. 1967;348:323–328. [PubMed] [Google Scholar]
- 4.Schulze HU, Gallenkamp H, Staudinger H. Untersuchungen zum mikrosomalen NADH-abhängigen Elektronentransport. Hoppe Seylers Z Physiol Chem. 1970;351:809–817. [PubMed] [Google Scholar]
- 5.Wakefield LM, Cass AE, Radda GK. Electron transfer across the chromaffin granule membrane. Use of EPR to demonstrate reduction of intravesicular ascorbate radical by the extravesicular mitochondrial NADH:ascorbate radical oxidoreductase. J Biol Chem. 1986;261:9746–9752. [PubMed] [Google Scholar]
- 6.Bielski BH, Allen AO, Schwarz HA. Mechanism of disproportionation of ascorbate radicals. J Am Chem Soc. 1981;103:3516–3518. [Google Scholar]
- 7.Tolbert BM, Ward JB. Dehydroascorbic acid. In: Seib PA, Tolbert BM, editors. Ascorbic Acid: Chemistry, Metabolism, and Uses. Washington, D.C.: American Chemical Society; 1982. pp. 101–123. [Google Scholar]
- 8.Koshiishi I, Mamura Y, Liu J, Imanari T. Degradation of dehydroascorbate to 2,3-diketogulonate in blood circulation. Biochim Biophys Acta. 1998;1425:209–214. doi: 10.1016/s0304-4165(98)00073-7. [DOI] [PubMed] [Google Scholar]
- 9.Wells WW, Xu DP. Dehydroascorbate reduction. J Bioenerg Biomembr. 1994;26:369–377. doi: 10.1007/BF00762777. [DOI] [PubMed] [Google Scholar]
- 10.May JM, Asard H. Ascorbate Recycling. In: Asard H, May JM, Smirnoff N, editors. Vitamin C Functions and biochemistry in animals and plants. London: Bios Scientific Publishers; 2004. pp. 139–158. [Google Scholar]
- 11.May JM, Qu ZC, Cobb CE. Extracellular reduction of the ascorbate free radical by human erythrocytes. Biochem Biophys Res Commun. 2000;267:118–123. doi: 10.1006/bbrc.1999.1906. [DOI] [PubMed] [Google Scholar]
- 12.VanDuijn MM, Tijssen K, VanSteveninck J, van den Broek PJA, Van der Zee J. Erythrocytes reduce extracellular ascorbate free radicals using intracellular ascorbate as an electron donor. J Biol Chem. 2000;275:27720–27725. doi: 10.1074/jbc.M910281199. [DOI] [PubMed] [Google Scholar]
- 13.Upston JM, Karjalainen A, Bygrave FL, Stocker R. Efflux of hepatic ascorbate: a potential contributor to the maintenance of plasma vitamin C. Biochem J. 1999;342:49–56. [PMC free article] [PubMed] [Google Scholar]
- 14.Davis KA, Samson SE, Best K, Mallhi KK, Szewczyk M, Wilson JX, Kwan CY, Grover AK. Ca(2+)-mediated ascorbate release from coronary artery endothelial cells. Br J Pharmacol. 2006;147:131–139. doi: 10.1038/sj.bjp.0706492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rebec GV, Pierce RC. A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog Neurobiol. 1994;43:537–565. doi: 10.1016/0301-0082(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 16.Mendiratta S, Qu ZC, May JM. Erythrocyte ascorbate recycling: Antioxidant effects in blood. Free Radic Biol Med. 1998;24:789–797. doi: 10.1016/s0891-5849(97)00351-1. [DOI] [PubMed] [Google Scholar]
- 17.Jackson TS, Xu AM, Vita JA, Keaney JF., Jr Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ Res. 1998;83:916–922. doi: 10.1161/01.res.83.9.916. [DOI] [PubMed] [Google Scholar]
- 18.Arrigoni O, De Tullio MC. Ascorbic acid: much more than just an antioxidant. Biochim Biophys Acta Gen Subj. 2002;1569:1–9. doi: 10.1016/s0304-4165(01)00235-5. [DOI] [PubMed] [Google Scholar]
- 19.May JM, Qu ZC. Transport and intracellular accumulation of vitamin C in endothelial cells: relevance to collagen synthesis. Arch Biochem Biophys. 2005;434:178–186. doi: 10.1016/j.abb.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 20.Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang YX, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 21.Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang YX, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 22.Corpe C, Tu H, Wang J, Eck P, Wang Y, Schnermann J, Faulhaber-Walter R, Nussbaum R, Levine M. SVCT1 (Slc23a1) knockout mice: Slc23a1 as the vitamin C kidney reabsorptive transporter. The FASEB Journal. 2008;21:LB111. Meeting abstract. [Google Scholar]
- 23.Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang YX, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 24.Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang YX, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 25.Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang YX, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 26.DiLabio GA, Wright JS. Hemiketal formation of dehydroascorbic acid drives ascorbyl radical anion disproportionation. Free Radic Biol Med. 2000;29:480–485. doi: 10.1016/s0891-5849(00)00357-9. [DOI] [PubMed] [Google Scholar]
- 27.Pastore P, Rizzetto T, Curcuruto O, Cin MD, Zaramella A, Marton D. Characterization of dehydroascorbic acid solutions by liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2001;15:2051–2057. doi: 10.1002/rcm.476. [DOI] [PubMed] [Google Scholar]
- 28.Hughes RE, Maton SC. The passage of vitamin C across the erythrocyte membrane. Br J Haematol. 1968;14:247–253. doi: 10.1111/j.1365-2141.1968.tb01494.x. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch JC. Dehydroascorbic acid. J Chromatogr A. 2000;881:299–307. doi: 10.1016/s0021-9673(00)00166-7. [DOI] [PubMed] [Google Scholar]
- 30.Vera JC, Rivas CI, Fischbarg J, Golde DW. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature. 1993;364:79–82. doi: 10.1038/364079a0. [DOI] [PubMed] [Google Scholar]
- 31.Dhariwal KR, Hartzell WO, Levine M. Ascorbic acid and dehydroascorbic acid measurements in human plasma and serum. Am J Clin Nutr. 1991;54:712–716. doi: 10.1093/ajcn/54.4.712. [DOI] [PubMed] [Google Scholar]
- 32.Okamura M. Uptake of L-ascorbic acid and L-dehydroascorbic acid by human erythrocytes and HeLa cells. J Nutr Sci Vitaminol (Tokyo) 1979;25:269–279. doi: 10.3177/jnsv.25.269. [DOI] [PubMed] [Google Scholar]
- 33.Bigley R, Wirth M, Layman D, Riddle M, Stankova L. Interaction between glucose and dehydroascorbate transport in human neutrophils and fibroblasts. Diabetes. 1983;32:545–548. doi: 10.2337/diab.32.6.545. [DOI] [PubMed] [Google Scholar]
- 34.Washko P, Rotrosen D, Levine M. Ascorbic acid in human neutrophils. Am J Clin Nutr. 1991;54:1221S–1227S. doi: 10.1093/ajcn/54.6.1221s. [DOI] [PubMed] [Google Scholar]
- 35.Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br J Nutr. 1982;47:473–482. doi: 10.1079/bjn19820059. [DOI] [PubMed] [Google Scholar]
- 36.Helgerson AL, Carruthers A. Equilibrium Ligand Binding to the Human Erythrocyte Sugar Transporter. J Biol Chem. 1987;262:5464–5475. [PubMed] [Google Scholar]
- 37.Montel-Hagen A, Kinet S, Manel N, Mongellaz C, Prohaska R, Battini JL, Delaunay J, Sitbon M, Taylor N. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 38.May JM, Qu ZC, Qiao H, Koury MJ. Maturational loss of the vitamin C transporter in erythrocytes. Biochem Biophys Res Commun. 2007;360:295–298. doi: 10.1016/j.bbrc.2007.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 40.Nualart FJ, Rivas CI, Montecinos VP, Godoy AS, Guaiquil VH, Golde DW, Vera JC. Recycling of vitamin C by a bystander effect. J Biol Chem. 2003;278:10128–10133. doi: 10.1074/jbc.M210686200. [DOI] [PubMed] [Google Scholar]
- 41.Agus DB, Gambhir SS, Pardridge WM, Speilholz C, Baselga J, Vera JC. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J Clin Invest. 1997;100:2842–2848. doi: 10.1172/JCI119832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam DK, Daniel PM. The influx of ascorbic acid into the rat's brain. Q J Exp Physiol. 1986;71:483–489. doi: 10.1113/expphysiol.1986.sp003007. [DOI] [PubMed] [Google Scholar]
- 43.Angelow S, Haselbach M, Galla HJ. Functional characterisation of the active ascorbic acid transport into cerebrospinal fluid using primary cultured choroid plexus cells. Brain Res. 2003;988:105–113. doi: 10.1016/s0006-8993(03)03350-x. [DOI] [PubMed] [Google Scholar]
- 44.Hakvoort A, Haselbach M, Galla HJ. Active transport properties of porcine choroid plexus cells in culture. Brain Res. 1998;795:247–256. doi: 10.1016/s0006-8993(98)00284-4. [DOI] [PubMed] [Google Scholar]
- 45.Spector R. Vitamin homeostasis in the central nervous system. N Engl J Med. 1977;296:1393–1398. doi: 10.1056/NEJM197706162962409. [DOI] [PubMed] [Google Scholar]
- 46.García ML, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, Silva C, Cortes C, Reinicke K, Vera JC, Aguayo LG, Olate J, Molina B, Nualart F. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- 47.Angelow S, Haselbach M, Galla HJ. Functional characterisation of the active ascorbic acid transport into cerebrospinal fluid using primary cultured choroid plexus cells. Brain Res. 2003;988:105–113. doi: 10.1016/s0006-8993(03)03350-x. [DOI] [PubMed] [Google Scholar]
- 48.Angelow S, Haselbach M, Galla HJ. Functional characterisation of the active ascorbic acid transport into cerebrospinal fluid using primary cultured choroid plexus cells. Brain Res. 2003;988:105–113. doi: 10.1016/s0006-8993(03)03350-x. [DOI] [PubMed] [Google Scholar]
- 49.Hakvoort A, Haselbach M, Galla HJ. Active transport properties of porcine choroid plexus cells in culture. Brain Res. 1998;795:247–256. doi: 10.1016/s0006-8993(98)00284-4. [DOI] [PubMed] [Google Scholar]
- 50.Angelow S, Haselbach M, Galla HJ. Functional characterisation of the active ascorbic acid transport into cerebrospinal fluid using primary cultured choroid plexus cells. Brain Res. 2003;988:105–113. doi: 10.1016/s0006-8993(03)03350-x. [DOI] [PubMed] [Google Scholar]
- 51.Hakvoort A, Haselbach M, Galla HJ. Active transport properties of porcine choroid plexus cells in culture. Brain Res. 1998;795:247–256. doi: 10.1016/s0006-8993(98)00284-4. [DOI] [PubMed] [Google Scholar]
- 52.Spector R, Lorenzo AV. Ascorbic acid homeostasis in the central nervous system. Am J Physiol. 1973;225:757–763. doi: 10.1152/ajplegacy.1973.225.4.757. [DOI] [PubMed] [Google Scholar]
- 53.Miele M, Fillenz M. In vivo determination of extracellular brain ascorbate. J Neurosci Methods. 1996;70:15–19. doi: 10.1016/S0165-0270(96)00094-5. [DOI] [PubMed] [Google Scholar]
- 54.Schenk JO, Miller E, Gaddis R, Adams RN. Homeostatic control of ascorbate concentration in CNS extracellular fluid. Brain Res. 1982;253:353–356. doi: 10.1016/0006-8993(82)90709-0. [DOI] [PubMed] [Google Scholar]
- 55.Reiber H, Ruff M, Uhr M. Ascorbate concentration in human cerebrospinal fluid (CSF) and serum. Intrathecal accumulation and CSF flow rate. Clin Chim Acta. 1993;217:163–173. doi: 10.1016/0009-8981(93)90162-w. [DOI] [PubMed] [Google Scholar]
- 56.Lönnrot K, Metsä-Ketelä T, Molnár G, Ahonen JP, Latvala M, Peltola J, Pietilä T, Alho H. The effect of ascorbate and ubiquinone supplementation on plasma and CSF total antioxidant capacity. Free Radic Biol Med. 1996;21:211–217. doi: 10.1016/0891-5849(95)02207-4. [DOI] [PubMed] [Google Scholar]
- 57.García ML, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, Silva C, Cortes C, Reinicke K, Vera JC, Aguayo LG, Olate J, Molina B, Nualart F. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- 58.García ML, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, Silva C, Cortes C, Reinicke K, Vera JC, Aguayo LG, Olate J, Molina B, Nualart F. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- 59.Pardridge WM. Brain metabolism: a perspective from the blood-brain barrier. Physiol Rev. 1983;63:1481–1535. doi: 10.1152/physrev.1983.63.4.1481. [DOI] [PubMed] [Google Scholar]
- 60.García ML, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, Silva C, Cortes C, Reinicke K, Vera JC, Aguayo LG, Olate J, Molina B, Nualart F. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- 61.Qiao H, May JM. Development of ascorbate transport in brain capillary endothelial cells in culture. Brain Res. 2008;1208:79–86. doi: 10.1016/j.brainres.2008.02.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.García ML, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, Silva C, Cortes C, Reinicke K, Vera JC, Aguayo LG, Olate J, Molina B, Nualart F. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- 63.García ML, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, Silva C, Cortes C, Reinicke K, Vera JC, Aguayo LG, Olate J, Molina B, Nualart F. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- 64.Farrell CL, Yang J, Pardridge WM. GLUT-1 glucose transporter is present within apical and basolateral membranes of brain epithelial interfaces and in microvascular endothelia with and without tight junctions. J Histochem Cytochem. 1992;40:193–199. doi: 10.1177/40.2.1552163. [DOI] [PubMed] [Google Scholar]
- 65.Rumsey SC, Kwon O, Xu GW, Burant CF, Simpson I, Levine M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem. 1997;272:18982–18989. doi: 10.1074/jbc.272.30.18982. [DOI] [PubMed] [Google Scholar]
- 66.Montel-Hagen A, Kinet S, Manel N, Mongellaz C, Prohaska R, Battini JL, Delaunay J, Sitbon M, Taylor N. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 67.Montel-Hagen A, Kinet S, Manel N, Mongellaz C, Prohaska R, Battini JL, Delaunay J, Sitbon M, Taylor N. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 68.Wilson JX. Vitamin C transport in animals and plants. In: Asard H, May JM, Smirnoff N, editors. Vitamin C Functions and biochemistry in animals and plants. London: Bios Scientific Publishers; 2004. pp. 97–113. [Google Scholar]
- 69.Patel M, McIntosh L, Bliss T, Ho D, Sapolsky R. Interactions among ascorbate, dehydroascorbate and glucose transport in cultured hippocampal neurons and glia. Brain Res. 2001;916:127–135. doi: 10.1016/s0006-8993(01)02877-3. [DOI] [PubMed] [Google Scholar]
- 70.Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang YX, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 71.Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 72.Mun GH, Kim MJ, Lee JH, Kim HJ, Chung YH, Chung YB, Kang JS, Hwang YI, Oh SH, Kim JG, Hwang DH, Shin DH, Lee WJ. Immunohistochemical study of the distribution of sodium-dependent vitamin C transporters in adult rat brain. J Neurosci Res. 2006;83:919–928. doi: 10.1002/jnr.20751. [DOI] [PubMed] [Google Scholar]
- 73.Mefford IN, Oke AF, Adams RN. Regional distribution of ascorbate in human brain. Brain Res. 1981;212:223–226. doi: 10.1016/0006-8993(81)90056-1. [DOI] [PubMed] [Google Scholar]
- 74.Harrison FE, Yu SS, Van Den Bossche KL, Li L, May JM, McDonald MP. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J Neurochem. 2008;106:1198–1208. doi: 10.1111/j.1471-4159.2008.05469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qiu S, Li L, Weeber EJ, May JM. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J Neurosci Res. 2007;85:1046–1056. doi: 10.1002/jnr.21204. [DOI] [PubMed] [Google Scholar]
- 76.Castro M, Caprile T, Astuya A, Millán C, Reinicke K, Vera JC, Vásquez O, Aguayo LG, Nualart F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J Neurochem. 2001;78:815–823. doi: 10.1046/j.1471-4159.2001.00461.x. [DOI] [PubMed] [Google Scholar]
- 77.May JM, Li L, Hayslett K, Qu ZC. Ascorbate Transport and Recycling by SH-SY5Y Neuroblastoma Cells: Response to Glutamate Toxicity. Neurochem Res. 2006;31:785–794. doi: 10.1007/s11064-006-9077-z. [DOI] [PubMed] [Google Scholar]
- 78.Castro M, Caprile T, Astuya A, Millán C, Reinicke K, Vera JC, Vásquez O, Aguayo LG, Nualart F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J Neurochem. 2001;78:815–823. doi: 10.1046/j.1471-4159.2001.00461.x. [DOI] [PubMed] [Google Scholar]
- 79.Berger UV, Lu XC, Liu W, Tang Z, Slusher BS, Hediger MA. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J Neurochem. 2003;86:896–906. doi: 10.1046/j.1471-4159.2003.01891.x. [DOI] [PubMed] [Google Scholar]
- 80.Shimizu N, Matsunami T, Onishi S. Histochemical demonstration of ascorbic acid in the locus coeruleus of the mammalian brain. Nature. 1960;186:479–480. doi: 10.1038/186479a0. [DOI] [PubMed] [Google Scholar]
- 81.Hill KE, Montine TJ, Motley AK, Li X, May JM, Burk RF. Combined deficiency of vitamins E and C causes paralysis and death in guinea pigs. Am J Clin Nutr. 2003;77:1484–1488. doi: 10.1093/ajcn/77.6.1484. [DOI] [PubMed] [Google Scholar]
- 82.Hughes RE, Hurley RJ, Jones PR. The retention of ascorbic acid by guinea-pig tissues. Br J Nutr. 1971;26:433–438. doi: 10.1079/bjn19710048. [DOI] [PubMed] [Google Scholar]
- 83.Kratzing CC, Kelly JD, Kratzing JE. Ascorbic acid in fetal rat brain. J Neurochem. 1985;44:1623–1624. doi: 10.1111/j.1471-4159.1985.tb08804.x. [DOI] [PubMed] [Google Scholar]
- 84.Oke AF, May L, Adams RN. Ascorbic acid distribution patterns in human brain. A comparison with nonhuman mammalian species. Ann N Y Acad Sci. 1987;498:1–12. doi: 10.1111/j.1749-6632.1987.tb23747.x. [DOI] [PubMed] [Google Scholar]
- 85.Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1998;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- 86.Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- 87.Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1998;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- 88.Castro M, Caprile T, Astuya A, Millán C, Reinicke K, Vera JC, Vásquez O, Aguayo LG, Nualart F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J Neurochem. 2001;78:815–823. doi: 10.1046/j.1471-4159.2001.00461.x. [DOI] [PubMed] [Google Scholar]
- 89.Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1998;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- 90.Nualart FJ, Rivas CI, Montecinos VP, Godoy AS, Guaiquil VH, Golde DW, Vera JC. Recycling of vitamin C by a bystander effect. J Biol Chem. 2003;278:10128–10133. doi: 10.1074/jbc.M210686200. [DOI] [PubMed] [Google Scholar]
- 91.Berger UV, Lu XC, Liu W, Tang Z, Slusher BS, Hediger MA. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J Neurochem. 2003;86:896–906. doi: 10.1046/j.1471-4159.2003.01891.x. [DOI] [PubMed] [Google Scholar]
- 92.Wilson JX, Jaworski EM, Kulaga A, Dixon SJ. Substrate regulation of ascorbate transport activity in astrocytes. Neurochem Res. 1990;15:1037–1043. doi: 10.1007/BF00965751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Astuya A, Caprile T, Castro M, Salazar K, García MD, Reinicke K, Rodríguez F, Vera JC, Millán C, Ulloa V, Low M, Martínez F, Nualart F. Vitamin C uptake and recycling among normal and tumor cells from the central nervous system. J Neurosci Res. 2005;79:146–156. doi: 10.1002/jnr.20326. [DOI] [PubMed] [Google Scholar]
- 94.Astuya A, Caprile T, Castro M, Salazar K, García MD, Reinicke K, Rodríguez F, Vera JC, Millán C, Ulloa V, Low M, Martínez F, Nualart F. Vitamin C uptake and recycling among normal and tumor cells from the central nervous system. J Neurosci Res. 2005;79:146–156. doi: 10.1002/jnr.20326. [DOI] [PubMed] [Google Scholar]
- 95.Berger UV, Lu XC, Liu W, Tang Z, Slusher BS, Hediger MA. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J Neurochem. 2003;86:896–906. doi: 10.1046/j.1471-4159.2003.01891.x. [DOI] [PubMed] [Google Scholar]
- 96.Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- 97.Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- 98.Cammack J, Ghasemzadeh B, Adams RN. The pharmacological profile of glutamate-evoked ascorbic acid efflux measured by in vivo electrochemistry. Brain Res. 1991;565:17–22. doi: 10.1016/0006-8993(91)91731-f. [DOI] [PubMed] [Google Scholar]
- 99.Miele M, Boutelle MG, Fillenz M. The physiologically induced release of ascorbate in rat brain is dependent on impulse traffic, calcium influx and glutamate uptake. Neuroscience. 1994;62:87–91. doi: 10.1016/0306-4522(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 100.O'Neill RD, Fillenz M, Sundstrom L, Rawlins JN. Voltammetrically monitored brain ascorbate as an index of excitatory amino acid release in the unrestrained rat. Neurosci Lett. 1984;52:227–233. doi: 10.1016/0304-3940(84)90166-6. [DOI] [PubMed] [Google Scholar]
- 101.Rebec GV, Witowski SR, Sandstrom MI, Rostand RD, Kennedy RT. Extracellular ascorbate modulates cortically evoked glutamate dynamics in rat striatum. Neurosci Lett. 2005;378:166–170. doi: 10.1016/j.neulet.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 102.Sandstrom MI, Rebec GV. Extracellular ascorbate modulates glutamate dynamics: role of behavioral activation. BMC Neurosci. 2007;8:32. doi: 10.1186/1471-2202-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cammack J, Ghasemzadeh B, Adams RN. The pharmacological profile of glutamate-evoked ascorbic acid efflux measured by in vivo electrochemistry. Brain Res. 1991;565:17–22. doi: 10.1016/0006-8993(91)91731-f. [DOI] [PubMed] [Google Scholar]
- 104.Grünewald RA. Ascorbic acid in the brain. Brain Res Rev. 1993;18:123–133. doi: 10.1016/0165-0173(93)90010-w. [DOI] [PubMed] [Google Scholar]
- 105.Staub F, Peters J, Kempski O, Schneider GH, Schurer L, Baethmann A. Swelling of glial cells in lactacidosis and by glutamate: significance of Cl(-)-transport. Brain Res. 1993;610:69–74. doi: 10.1016/0006-8993(93)91218-h. [DOI] [PubMed] [Google Scholar]
- 106.Koyama Y, Sugimoto T, Shigenaga Y, Baba A, Iwata H. A morphological study on glutamate-induced swelling of cultured astrocytes: involvement of calcium and chloride ion mechanisms. Neurosci Lett. 1991;124:235–238. doi: 10.1016/0304-3940(91)90102-y. [DOI] [PubMed] [Google Scholar]
- 107.Siushansian R, Dixon SJ, Wilson JX. Osmotic swelling stimulates ascorbate efflux from cerebral astrocytes. J Neurochem. 1996;66:1227–1233. doi: 10.1046/j.1471-4159.1996.66031227.x. [DOI] [PubMed] [Google Scholar]
- 108.Wilson JX, Peters CE, Sitar SM, Daoust P, Gelb AW. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000;858:61–66. doi: 10.1016/s0006-8993(99)02433-6. [DOI] [PubMed] [Google Scholar]
- 109.May JM, Li L, Hayslett K, Qu ZC. Ascorbate transport and recycling by SH-SY5Y neuroblastoma cells: Response to glutamate toxicity. Neurochem Res. 2006;31:785–794. doi: 10.1007/s11064-006-9077-z. [DOI] [PubMed] [Google Scholar]
- 110.Jackson TS, Xu AM, Vita JA, Keaney JF., Jr Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ Res. 1998;83:916–922. doi: 10.1161/01.res.83.9.916. [DOI] [PubMed] [Google Scholar]
- 111.Niki E, Noguchi N, Tsuchihashi H, Gotoh N. Interaction among vitamin C, vitamin E, and β-carotene. Am J Clin Nutr. 1995;62(Suppl):1322S–1326S. doi: 10.1093/ajcn/62.6.1322S. [DOI] [PubMed] [Google Scholar]
- 112.May JM, Qu ZC, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 113.Seregi A, Schaefer A, Komlós M. Protective role of brain ascorbic acid content against lipid peroxidation. Experientia. 1978;34:1056–1057. doi: 10.1007/BF01915344. [DOI] [PubMed] [Google Scholar]
- 114.Kovachich GB, Mishra OP. The effect of ascorbic acid on malonaldehyde formation, K+, Na+ and water content of brain slices. Exp Brain Res. 1983;50:62–68. doi: 10.1007/BF00238232. [DOI] [PubMed] [Google Scholar]
- 115.Sato K, Saito H, Katsuki H. Synergism of tocopherol and ascorbate on the survival of cultured brain neurones. Neuroreport. 1993;4:1179–1182. [PubMed] [Google Scholar]
- 116.Bano S, Parihar MS. Reduction of lipid peroxidation in different brain regions by a combination of α-tocopherol and ascorbic acid. J Neural Transm. 1997;104:1277–1286. doi: 10.1007/BF01294728. [DOI] [PubMed] [Google Scholar]
- 117.Kratzing CC, Kelly JD, Kratzing JE. Ascorbic acid in fetal rat brain. J Neurochem. 1985;44:1623–1624. doi: 10.1111/j.1471-4159.1985.tb08804.x. [DOI] [PubMed] [Google Scholar]
- 118.Kratzing CC, Kelly JD. Tissue levels of ascorbic acid during rat gestation. Int J Vitam Nutr Res. 1982;52:326–332. [PubMed] [Google Scholar]
- 119.Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol. 2000;18:675–679. doi: 10.1038/76536. [DOI] [PubMed] [Google Scholar]
- 120.Shin DM, Ahn JI, Lee KH, Lee YS, Lee YS. Ascorbic acid responsive genes during neuronal differentiation of embryonic stem cells. Neuroreport. 2004;15:1959–1963. doi: 10.1097/00001756-200408260-00025. [DOI] [PubMed] [Google Scholar]
- 121.Lee JY, Chang MY, Park CH, Kim HY, Kim JH, Son H, Lee YS, Lee SH. Ascorbate-induced differentiation of embryonic cortical precursors into neurons and astrocytes. J Neurosci Res. 2003;73:156–165. doi: 10.1002/jnr.10647. [DOI] [PubMed] [Google Scholar]
- 122.Qiu S, Li L, Weeber EJ, May JM. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J Neurosci Res. 2007;85:1046–1056. doi: 10.1002/jnr.21204. [DOI] [PubMed] [Google Scholar]
- 123.Diliberto EJ, Jr, Allen PL. Semidehydroascorbate as a product of the enzymic conversion of dopamine to norepinephrine. Coupling of semidehydroascorbate reductase to dopamine-beta-hydroxylase. Mol Pharmacol. 1980;17:421–426. [PubMed] [Google Scholar]
- 124.Diliberto EJ, Jr, Allen PL. Mechanism of dopamine-beta-hydroxylation. Semidehydroascorbate as the enzyme oxidation product of ascorbate. J Biol Chem. 1981;256:3385–3393. [PubMed] [Google Scholar]
- 125.Flatmark T, Terland O. Cytochrome b561 of the bovine adrenal chromaffin granules. A high potential b-type cytochrome. Biochim Biophys Acta. 1971;253:487–491. doi: 10.1016/0005-2728(71)90052-1. [DOI] [PubMed] [Google Scholar]
- 126.Njus D, Knoth J, Cook C, Kelley PM. Electron transfer across the chromaffin granule membrane. J Biol Chem. 1983;258:27–30. [PubMed] [Google Scholar]
- 127.Wakefield LM, Cass AE, Radda GK. Electron transfer across the chromaffin granule membrane. Use of EPR to demonstrate reduction of intravesicular ascorbate radical by the extravesicular mitochondrial NADH:ascorbate radical oxidoreductase. J Biol Chem. 1986;261:9746–9752. [PubMed] [Google Scholar]
- 128.Levine M, Asher A, Pollard H, Zinder O. Ascorbic acid and catecholamine secretion from cultured chromaffin cells. J Biol Chem. 1983;258:13111–13115. [PubMed] [Google Scholar]
- 129.Padayatty SJ, Doppman JL, Chang R, Wang Y, Gill J, Papanicolaou DA, Levine M. Human adrenal glands secrete vitamin C in response to adrenocorticotrophic hormone. Am J Clin Nutr. 2007;86:145–149. doi: 10.1093/ajcn/86.1.145. [DOI] [PubMed] [Google Scholar]
- 130.Kiyatkin EA, Rebec GV. Ascorbate modulates glutamate-induced excitations of striatal neurons. Brain Res. 1998;812:14–22. doi: 10.1016/s0006-8993(98)00814-2. [DOI] [PubMed] [Google Scholar]
- 131.Rebec GV, Witowski SR, Sandstrom MI, Rostand RD, Kennedy RT. Extracellular ascorbate modulates cortically evoked glutamate dynamics in rat striatum. Neurosci Lett. 2005;378:166–170. doi: 10.1016/j.neulet.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 132.Ciani E, Groneng L, Voltattorni M, Rolseth V, Contestabile A, Paulsen RE. Inhibition of free radical production or free radical scavenging protects from the excitotoxic cell death mediated by glutamate in cultures of cerebellar granule neurons. Brain Res. 1996;728:1–6. [PubMed] [Google Scholar]
- 133.Atlante A, Gagliardi S, Minervini GM, Ciotti MT, Marra E, Calissano P. Glutamate neurotoxicity in rat cerebellar granule cells: a major role for xanthine oxidase in oxygen radical formation. J Neurochem. 1997;68:2038–2045. doi: 10.1046/j.1471-4159.1997.68052038.x. [DOI] [PubMed] [Google Scholar]
- 134.Majewska MD, Bell JA. Ascorbic acid protects neurons from injury induced by glutamate and NMDA. Neuroreport. 1990;1:194–196. doi: 10.1097/00001756-199011000-00004. [DOI] [PubMed] [Google Scholar]
- 135.Majewska MD, Bell JA, London ED. Regulation of the NMDA receptor by redox phenomena: inhibitory role of ascorbate. Brain Res. 1990;537:328–332. doi: 10.1016/0006-8993(90)90379-p. [DOI] [PubMed] [Google Scholar]
- 136.Hillered L, Persson L, Bolander HG, Hallstrom A, Ungerstedt U. Increased extracellular levels of ascorbate in the striatum after middle cerebral artery occlusion in the rat monitored by intracerebral microdialysis. Neurosci Lett. 1988;95:286–290. doi: 10.1016/0304-3940(88)90672-6. [DOI] [PubMed] [Google Scholar]
- 137.Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- 138.Yusa T. Increased extracellular ascorbate release reflects glutamate re-uptake during the early stage of reperfusion after forebrain ischemia in rats. Brain Res. 2001;897:104–113. doi: 10.1016/s0006-8993(01)02099-6. [DOI] [PubMed] [Google Scholar]
- 139.Mefford IN, Oke AF, Adams RN. Regional distribution of ascorbate in human brain. Brain Res. 1981;212:223–226. doi: 10.1016/0006-8993(81)90056-1. [DOI] [PubMed] [Google Scholar]
- 140.Hoehn SK, Kanfer JN. Effects of chronic ascorbic acid deficiency on guinea pig lysosomal hydrolase activities. J Nutr. 1980;110:2085–2094. doi: 10.1093/jn/110.10.2085. [DOI] [PubMed] [Google Scholar]
- 141.Deana R, Bharaj BS, Verjee ZH, Galzigna L. Changes relevant to catecholamine metabolism in liver and brain of ascorbic acid deficient guinea-pigs. Int J Vitam Nutr Res. 1975;45:175–182. [PubMed] [Google Scholar]
- 142.Saponjic RM, Mueller K, Krug D, Kunko PM. The effects of haloperidol, scopolamine, and MK-801 on amphetamine-induced increases in ascorbic and uric acid as determined by voltammetry in vivo. Pharmacol Biochem Behav. 1994;48:161–168. doi: 10.1016/0091-3057(94)90512-6. [DOI] [PubMed] [Google Scholar]
- 143.Desole MS, Miele M, Enrico P, Esposito G, Fresu L, De Natale G, Miele E. Investigations into the relationship between the dopaminergic system and ascorbic acid in rat striatum. Neurosci Lett. 1991;127:34–38. doi: 10.1016/0304-3940(91)90888-z. [DOI] [PubMed] [Google Scholar]
- 144.Basse-Tomusk A, Rebec GV. Corticostriatal and thalamic regulation of amphetamine-induced ascorbate release in the neostriatum. Pharmacol Biochem Behav. 1990;35:55–60. doi: 10.1016/0091-3057(90)90204-u. [DOI] [PubMed] [Google Scholar]
- 145.Kuo CH, Hata F, Yoshida H, Yamatodani A, Wada H. Effect of ascorbic acid on release of acetylcholine from synaptic vesicles prepared from different species of animals and release of noradrenaline from synaptic vesicles of rat brain. Life Sci. 1979;24:911–915. doi: 10.1016/0024-3205(79)90341-2. [DOI] [PubMed] [Google Scholar]
- 146.Lee L, Kang SA, Lee HO, Lee BH, Jung IK, Lee JE, Hoe YS. Effect of supplementation of vitamin E and vitamin C on brain acetylcholinesterase activity and neurotransmitter levels in rats treated with scopolamine, an inducer of dementia. J Nutr Sci Vitaminol (Tokyo) 2001;47:323–328. doi: 10.3177/jnsv.47.323. [DOI] [PubMed] [Google Scholar]
- 147.Desole MS, Miele M, Enrico P, Esposito G, Fresu L, De Natale G, Miele E. Investigations into the relationship between the dopaminergic system and ascorbic acid in rat striatum. Neurosci Lett. 1991;127:34–38. doi: 10.1016/0304-3940(91)90888-z. [DOI] [PubMed] [Google Scholar]
- 148.Saponjic RM, Mueller K, Krug D, Kunko PM. The effects of haloperidol, scopolamine, and MK-801 on amphetamine-induced increases in ascorbic and uric acid as determined by voltammetry in vivo. Pharmacol Biochem Behav. 1994;48:161–168. doi: 10.1016/0091-3057(94)90512-6. [DOI] [PubMed] [Google Scholar]
- 149.Eipper BA, Mains RE, Glembotski CC. Identification in pituitary tissue of a peptide alpha-amidation activity that acts on glycine-extended peptides and requires molecular oxygen, copper, and ascorbic acid. Proc Natl Acad Sci U S A. 1983;80:5144–5148. doi: 10.1073/pnas.80.16.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Glembotski CC. The role of ascorbic acid in the biosynthesis of the neuroendocrine peptides alpha-MSH and TRH. Ann N Y Acad Sci. 1987;498:54–62. doi: 10.1111/j.1749-6632.1987.tb23750.x. [DOI] [PubMed] [Google Scholar]
- 151.Miller BT, Cicero TJ. Ascorbic acid enhances the release of luteinizing hormone-releasing hormone from the mediobasal hypothalamus in vitro. Life Sci. 1986;39:2447–2454. doi: 10.1016/0024-3205(86)90487-x. [DOI] [PubMed] [Google Scholar]
- 152.Parle M, Dhingra D. Ascorbic Acid: a promising memory-enhancer in mice. J Pharmacol Sci. 2003;93:129–135. doi: 10.1254/jphs.93.129. [DOI] [PubMed] [Google Scholar]
- 153.De Angelis L, Furlan C. The effects of ascorbic acid and oxiracetam on scopolamine-induced amnesia in a habituation test in aged mice. Neurobiol Learn Mem. 1995;64:119–124. doi: 10.1006/nlme.1995.1050. [DOI] [PubMed] [Google Scholar]
- 154.Parle M, Dhingra D. Ascorbic Acid: a promising memory-enhancer in mice. J Pharmacol Sci. 2003;93:129–135. doi: 10.1254/jphs.93.129. [DOI] [PubMed] [Google Scholar]
- 155.De Angelis L, Furlan C. The effects of ascorbic acid and oxiracetam on scopolamine-induced amnesia in a habituation test in aged mice. Neurobiol Learn Mem. 1995;64:119–124. doi: 10.1006/nlme.1995.1050. [DOI] [PubMed] [Google Scholar]
- 156.Arzi A, Hemmati AA, Razian A. Effect of vitamins C and E on cognitive function in mouse. Pharmacol Res. 2004;49:249–252. doi: 10.1016/j.phrs.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 157.Shahidi S, Komaki A, Mahmoodi M, Atrvash N, Ghodrati M. Ascorbic acid supplementation could affect passive avoidance learning and memory in rat. Brain Res Bull. 2008;76:109–113. doi: 10.1016/j.brainresbull.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 158.Desole MS, Anania V, Esposito G, Carboni F, Senini A, Miele E. Neurochemical and behavioural changes induced by ascorbic acid and d-amphetamine in the rat. Pharmacol Res Commun. 1987;19:441–450. doi: 10.1016/0031-6989(87)90083-x. [DOI] [PubMed] [Google Scholar]
- 159.Jones RB, Satterlee DG, Cadd GG. Timidity in Japanese quail: effects of vitamin C and divergent selection for adrenocortical response. Physiol Behav. 1999;67:117–120. doi: 10.1016/s0031-9384(99)00039-6. [DOI] [PubMed] [Google Scholar]
- 160.Harrison FE, Yu SS, Van Den Bossche KL, Li L, May JM, McDonald MP. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J Neurochem. 2008;106:1198–1208. doi: 10.1111/j.1471-4159.2008.05469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Desole MS, Anania V, Esposito G, Carboni F, Senini A, Miele E. Neurochemical and behavioural changes induced by ascorbic acid and d-amphetamine in the rat. Pharmacol Res Commun. 1987;19:441–450. doi: 10.1016/0031-6989(87)90083-x. [DOI] [PubMed] [Google Scholar]
- 162.Desole MS, Miele M, Demontis P, Carboni F, Senini A, Esposito G, Anania V. d-amphetamine and active behavior--induced changes of regional brain ascorbic acid levels in the rat. Pharmacol Res Commun. 1988;20:499–509. doi: 10.1016/s0031-6989(88)80077-8. [DOI] [PubMed] [Google Scholar]
- 163.Tolbert LC, Thomas TN, Middaugh LD, Zemp JW. Effect of ascorbic acid on neurochemical, behavioral, and physiological systems mediated by catecholamines. Life Sci. 1979;25:2189–2195. doi: 10.1016/0024-3205(79)90091-2. [DOI] [PubMed] [Google Scholar]
- 164.White LK, Carpenter M, Block M, Basse-Tomusk A, Gardiner TW, Rebec GV. Ascorbate antagonizes the behavioral effects of amphetamine by a central mechanism. Psychopharmacology (Berl) 1988;94:284–287. doi: 10.1007/BF00176860. [DOI] [PubMed] [Google Scholar]
- 165.White LK, Maurer M, Kraft ME, Oh C, Rebec GV. Intrastriatal infusions of ascorbate antagonize the behavioral response to amphetamine. Pharmacol Biochem Behav. 1990;36:485–489. doi: 10.1016/0091-3057(90)90245-d. [DOI] [PubMed] [Google Scholar]
- 166.Desole MS, Miele M, Esposito G, Enrico P, Fresu L, De Natale G, Miele E. Further investigations into the relationship between the dopaminergic system, ascorbic acid and uric acid in the rat striatum. Eur J Pharmacol. 1991;205:97–100. doi: 10.1016/0014-2999(91)90777-n. [DOI] [PubMed] [Google Scholar]
- 167.Desole MS, Miele M, Esposito G, Enrico P, Fresu L, De Natale G, Miele E. Further investigations into the relationship between the dopaminergic system, ascorbic acid and uric acid in the rat striatum. Eur J Pharmacol. 1991;205:97–100. doi: 10.1016/0014-2999(91)90777-n. [DOI] [PubMed] [Google Scholar]
- 168.Tolbert LC, Thomas TN, Middaugh LD, Zemp JW. Ascorbate blocks amphetamine-induced turning behavior in rats with unilateral nigro-striatal lesions. Brain Res Bull. 1979;4:43–48. doi: 10.1016/0361-9230(79)90056-x. [DOI] [PubMed] [Google Scholar]
- 169.Tolbert LC, Morris PE, Jr, Spollen JJ, Ashe SC. Stereospecific effects of ascorbic acid and analogues on D1 and D2 agonist binding. Life Sci. 1992;51:921–930. doi: 10.1016/0024-3205(92)90400-j. [DOI] [PubMed] [Google Scholar]
- 170.Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 171.Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, Aguglia U, van der Knaap MS, Heutink P, John SW. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 172.Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, John SW. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 173.Eldridge CF, Bunge MB, Bunge RP, Wood PM. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J Cell Biol. 1987;105:1023–1034. doi: 10.1083/jcb.105.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895–906. doi: 10.1016/s0190-9622(99)70244-6. [DOI] [PubMed] [Google Scholar]
- 175.Hornig D. Distributin of ascorbic acid, metabolites and analogues in man and animals. Ann NY Acad Sci. 1975;258:103–118. doi: 10.1111/j.1749-6632.1975.tb29271.x. [DOI] [PubMed] [Google Scholar]
- 176.Lind J. A Treatise on the Scurvy. Birmingham, AL: The Classics of Medicine Library, Gryphon Editions, Ltd; 1772. [Google Scholar]
- 177.Hughes RE, Hurley RJ, Jones PR. The retention of ascorbic acid by guinea-pig tissues. Br J Nutr. 1971;26:433–438. doi: 10.1079/bjn19710048. [DOI] [PubMed] [Google Scholar]
- 178.Hill KE, Montine TJ, Motley AK, Li X, May JM, Burk RF. Combined deficiency of vitamins E and C causes paralysis and death in guinea pigs. Am J Clin Nutr. 2003;77:1484–1488. doi: 10.1093/ajcn/77.6.1484. [DOI] [PubMed] [Google Scholar]
- 179.Burk RF, Christensen JM, Maguire MJ, Austin LM, Whetsell WO, Jr, May JM, Hill KE, Ebner FF. A combined deficiency of vitamins E and C causes severe central nervous system damage in guinea pigs. J Nutr. 2006;136:1576–1581. doi: 10.1093/jn/136.6.1576. [DOI] [PubMed] [Google Scholar]
- 180.Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 181.Bornstein SR, Yoshida-Hiroi M, Sotiriou S, Levine M, Hartwig HG, Nussbaum RL, Eisenhofer G. Impaired adrenal catecholamine system function in mice with deficiency of the ascorbic acid transporter (SVCT2) FASEB J. 2003;17:1928–1930. doi: 10.1096/fj.02-1167fje. [DOI] [PubMed] [Google Scholar]
- 182.Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 183.Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 184.Rehncrona S, Folbergrova J, Smith DS, Siesjo BK. Influence of complete and pronounced incomplete cerebral ischemia and subsequent recirculation on cortical concentrations of oxidized and reduced glutathione in the rat. J Neurochem. 1980;34:477–486. doi: 10.1111/j.1471-4159.1980.tb11170.x. [DOI] [PubMed] [Google Scholar]
- 185.Flamm ES, Demopoulos HB, Seligman ML, Poser RG, Ransohoff J. Free radicals in cerebral ischemia. Stroke. 1978;9:445–447. doi: 10.1161/01.str.9.5.445. [DOI] [PubMed] [Google Scholar]
- 186.Rice ME, Lee EJ, Choy Y. High levels of ascorbic acid, not glutathione, in the CNS of anoxia-tolerant reptiles contrasted with levels in anoxia-intolerant species. J Neurochem. 1995;64:1790–1799. doi: 10.1046/j.1471-4159.1995.64041790.x. [DOI] [PubMed] [Google Scholar]
- 187.Ranjan A, Theodore D, Haran RP, Chandy MJ. Ascorbic acid and focal cerebral ischaemia in a primate model. Acta Neurochir (Wien) 1993;123:87–91. doi: 10.1007/BF01476291. [DOI] [PubMed] [Google Scholar]
- 188.Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, McTaggart RA, Choudhri TF, Kim LJ, Mocco J, Pinsky DJ, Fox WD, Israel RJ, Boyd TA, Golde DW, Connolly ES., Jr Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci USA. 2001;98:11720–11724. doi: 10.1073/pnas.171325998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Patterson JW. The diabetogenic effect of dehydroascorbic and dehydroisoascorbic acids. J Biol Chem. 1950;183:81–88. [Google Scholar]
- 190.Mack WJ, Mocco J, Ducruet AF, Laufer I, King RG, Zhang Y, Guo W, Pinsky DJ, Connolly ES., Jr A cerebroprotective dose of intravenous citrate/sorbitol-stabilized dehydroascorbic acid is correlated with increased cerebral ascorbic acid and inhibited lipid peroxidation after murine reperfused stroke. Neurosurgery. 2006;59:383–388. doi: 10.1227/01.NEU.0000223496.96945.A7. [DOI] [PubMed] [Google Scholar]
- 191.Bailey DM, Raman S, McEneny J, Young IS, Parham KL, Hullin DA, Davies B, McKeeman G, McCord JM, Lewis MH. Vitamin C prophylaxis promotes oxidative lipid damage during surgical ischemia-reperfusion. Free Radic Biol Med. 2006;40:591–600. doi: 10.1016/j.freeradbiomed.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 192.Brahma B, Forman RE, Stewart EE, Nicholson C, Rice ME. Ascorbate inhibits edema in brain slices. J Neurochem. 2000;74:1263–1270. doi: 10.1046/j.1471-4159.2000.741263.x. [DOI] [PubMed] [Google Scholar]
- 193.Li X, Huang J, May JM. Ascorbic acid spares alpha-tocopherol and decreases lipid peroxidation in neuronal cells. Biochem Biophys Res Commun. 2003;305:656–661. doi: 10.1016/s0006-291x(03)00836-2. [DOI] [PubMed] [Google Scholar]
- 194.Qiu S, Li L, Weeber EJ, May JM. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J Neurosci Res. 2007;85:1046–1056. doi: 10.1002/jnr.21204. [DOI] [PubMed] [Google Scholar]
- 195.Berger UV, Lu XC, Liu W, Tang Z, Slusher BS, Hediger MA. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J Neurochem. 2003;86:896–906. doi: 10.1046/j.1471-4159.2003.01891.x. [DOI] [PubMed] [Google Scholar]
- 196.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 197.Hediger MA. New view at C. Nat Med. 2002;8:445–446. doi: 10.1038/nm0502-445. [DOI] [PubMed] [Google Scholar]
- 198.Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- 199.Montine T, Neely M, Quinn J, Beal M, Markesbery W, Roberts L, Morrow J. Lipid peroxidation in aging brain and Alzheimer's disease. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 200.Pratico D. Alzheimer's disease and oxygen radicals: new insights. Biochem Pharmacol. 2002;63:563–567. doi: 10.1016/s0006-2952(01)00919-4. [DOI] [PubMed] [Google Scholar]
- 201.Riviere S, Birlouez-Aragon I, Nourhashemi F, Vellas B. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749–754. doi: 10.1002/(sici)1099-1166(1998110)13:11<749::aid-gps860>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 202.Schippling S, Kontush A, Arlt S, Buhmann C, Sturenburg HJ, Mann U, Muller-Thomsen T, Beisiegel U. Increased lipoprotein oxidation in Alzheimer's disease. Free Radic Biol Med. 2000;28:351–360. doi: 10.1016/s0891-5849(99)00247-6. [DOI] [PubMed] [Google Scholar]
- 203.Morris MC, Beckett LA, Scherr PA, Hebert LE, Bennett DA, Field TS, Evans DA. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:121–126. doi: 10.1097/00002093-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 204.Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223–3229. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 205.Quinn JF, Montine KS, Moore M, Morrow JD, Kaye JA, Montine TJ. Suppression of longitudinal increase in CSF F2-isoprostanes in Alzheimer's disease. J Alzheimers Dis. 2004;6:93–97. doi: 10.3233/jad-2004-6110. [DOI] [PubMed] [Google Scholar]
- 206.Luchsinger JA, Tang MX, Shea S, Mayeux R. Antioxidant vitamin intake and risk of Alzheimer disease. Arch Neurol. 2003;60:203–208. doi: 10.1001/archneur.60.2.203. [DOI] [PubMed] [Google Scholar]
- 207.Fillenbaum GG, Kuchibhatla MN, Hanlon JT, Artz MB, Pieper CF, Schmader KE, Dysken MW, Gray SL. Dementia and Alzheimer's disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann Pharmacother. 2005;39:2009–2014. doi: 10.1345/aph.1G280. [DOI] [PubMed] [Google Scholar]
- 208.Rosales-Corral S, Tan DX, Reiter RJ, Valdivia-Velazquez M, Martinez-Barboza G, Acosta-Martinez JP, Ortiz GG. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: a comparative, in vivo study versus vitamin C and E. J Pineal Res. 2003;35:80–84. doi: 10.1034/j.1600-079x.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 209.Huang J, May JM. Ascorbic acid protects SH-SY5Y neuroblastoma cells from apoptosis and death induced by beta-amyloid. Brain Res. 2006;1097:52–58. doi: 10.1016/j.brainres.2006.04.047. [DOI] [PubMed] [Google Scholar]
- 210.Dhingra D, Parle M, Kulkarni SK. Comparative brain cholinesterase-inhibiting activity of Glycyrrhiza glabra, Myristica fragrans, ascorbic acid, and metrifonate in mice. J Med Food. 2006;9:281–283. doi: 10.1089/jmf.2006.9.281. [DOI] [PubMed] [Google Scholar]
- 211.Zhang SM, Hernan MA, Chen H, Spiegelman D, Willett WC, Ascherio A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology. 2002;59:1161–1169. doi: 10.1212/01.wnl.0000028688.75881.12. [DOI] [PubMed] [Google Scholar]
- 212.Nagayama H, Hamamoto M, Ueda M, Nito C, Yamaguchi H, Katayama Y. The effect of ascorbic acid on the pharmacokinetics of levodopa in elderly patients with Parkinson disease. Clin Neuropharmacol. 2004;27:270–273. doi: 10.1097/01.wnf.0000150865.21759.bc. [DOI] [PubMed] [Google Scholar]
- 213.Langston JW, Langston EB, Irwin I. MPTP-induced parkinsonism in human and non-human primates--clinical and experimental aspects. Acta Neurol Scand Suppl. 1984;100:49–54. [PubMed] [Google Scholar]
- 214.Desole MS, Esposito G, Fresu L, Migheli R, Enrico P, Miele M, De Natale G, Miele E. Correlation between 1-methyl-4-phenylpyridinium ion (MPP+) levels, ascorbic acid oxidation and glutathione levels in the striatal synaptosomes of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated rat. Neurosci Lett. 1993;161:121–123. doi: 10.1016/0304-3940(93)90274-o. [DOI] [PubMed] [Google Scholar]
- 215.Wagner GC, Carelli RM, Jarvis MF. Ascorbic acid reduces the dopamine depletion induced by methamphetamine and the 1-methyl-4-phenyl pyridinium ion. Neuropharmacology. 1986;25:559–561. doi: 10.1016/0028-3908(86)90184-x. [DOI] [PubMed] [Google Scholar]
- 216.Rebec GV, Barton SJ, Ennis MD. Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington's disease gene. J Neurosci. 2002;22:RC202. doi: 10.1523/JNEUROSCI.22-02-j0006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rebec GV, Barton SJ, Marseilles AM, Collins K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport. 2003;14:1263–1265. doi: 10.1097/00001756-200307010-00015. [DOI] [PubMed] [Google Scholar]