

Abstract
The capacity of certain pathogens to exploit innate immune receptors enables them to undermine immune clearance and persist in their host, often causing disease. Here we review subversive interactions of Porphyromonas gingivalis, a major periodontal pathogen, with the complement receptor-3 (CR3; CD11b/CD18) in monocytes/macrophages. Through its cell surface fimbriae, P. gingivalis stimulates Toll-like receptor-2 (TLR2) inside-out signaling which induces the high-affinity conformation of CR3. Although this activates CR3-dependent monocyte adhesion and transendothelial migration, P. gingivalis has co-opted this TLR2 proadhesive pathway for CR3 binding and intracellular entry. In CR3-deficient macrophages, the internalization of P. gingivalis is reduced 2-fold but its ability to survive intracellularly is reduced 1000-fold, indicating that CR3 is exploited by the pathogen as a relatively safe portal of entry. The interaction of P. gingivalis fimbriae with CR3 additionally inhibits production of bioactive (p70) interleukin-12, which mediates immune clearance. In vivo blockade of CR3 leads to reduced persistence of P. gingivalis in the mouse host and diminished ability to cause periodontal bone loss, the hallmark of periodontal disease. Strikingly, the ability of P. gingivalis to interact with and exploit CR3 depends upon quantitatively minor components (FimCDE) of its fimbrial structure, which predominantly consists of polymerized fimbrillin (FimA). Indeed, isogenic mutants lacking FimCDE but expressing FimA are dramatically less persistent and virulent than the wild-type organism both in vitro and in vivo. This model of immune evasion through CR3 exploitation by P. gingivalis supports the concept that pathogens evolved to manipulate innate immune function for promoting their adaptive fitness.
1 Introduction
Porphyromonas gingivalis is a predominant pathogen associated with periodontitis, an infection-driven chronic inflammatory disease that leads to destruction of the tooth-supporting tissues (Pihlstrom et al., 2005). This gram-negative anaerobic oral organism is moreover implicated as a contributory factor in several systemic conditions, including atherosclerosis (Gibson et al., 2006). The capacity of P. gingivalis to cause disease has been attributed to several virulence factors, such as fimbriae, LPS, hemagglutinins, and cysteine proteinases (gingipains), which in concert enable the pathogen to colonize and secure critical nutrients (Lamont and Jenkinson, 1998). However, a pathogen's ability to find a niche and establish chronic infection requires more than simple expression of appropriate adhesins or other factors for nutrient procurement. To persist in a hostile host environment, pathogens should be able to evade or subvert the host immune defense system aiming at controlling or eliminating them. Most of those successful pathogens which disable host defenses target preferentially innate immunity (Rosenberger and Finlay, 2003). This is not surprising given that innate defenses are the ones first encountered by the pathogens and, furthermore, subversion of innate immunity may additionally undermine the overall host defense. The latter is due to the significant instructive role of the innate response in the development of adaptive immunity (Pasare and Medzhitov, 2005). Although P. gingivalis is a successful pathogen that can also be found in systemic tissues (Kozarov et al., 2005), the mechanism(s) whereby P. gingivalis resists immune elimination are poorly understood. In this regard, the pathogen's in vitro ability to inhibit production of the IL-8 chemokine by gingival epithelial cells (Darveau et al., 1998) or to degrade secreted cytokines (Calkins et al., 1998) is thought to suppress host defenses. Here we summarize recent in vitro and in vivo work from our group indicating that at least one of the mechanisms whereby P. gingivalis escapes immunosurveillance involves exploitation of complement receptor 3 (CR3). The utilization of CR3 by P. gingivalis depends greatly upon expression of surface fimbriae, comprising polymerized fimbrillin (FimA) associated with quantitatively minor proteins (FimCDE) (Nishiyama et al., 2007, Wang et al., 2007).
2 CR3 in Innate Immunity
CR3 is a β2 integrin (CD11b/CD18) that is abundantly expressed by phagocytes, such as monocytes and neutrophils (Bhat et al., 1999; Yakubenko et al., 2002). This receptor recognizes a wide variety of structurally unrelated molecules from either the host (e.g., complement C3 fragment [iC3b], intercellular adhesion molecule-1 [ICAM-1], or fibrinogen) or pathogens (e.g., Bordetella pertussis filamentous hemagglutinin and Leishmania gp63) (Diamond et al., 1993; McGuirk and Mills, 2000; Russell and Wright, 1988). Similarly to other integrins, CR3 integrates the intracellular and extracellular environments through inside-out and outside-in bidirectional signaling. Inside-out signaling refers to the activation of the CR3 adhesive capacity from within the cell by means of signals generated by other receptors. Upon activation, CR3 binds ligands (or counter-receptors) resulting in stimulation of downstream signaling pathways, referred to as outside-in signaling (Shimaoka et al., 2002; Ginsberg et al., 2005). The ligand binding promiscuity of CR3 suggests that it may possess pattern recognition capabilities. In this context, CR3 has been shown to cluster with other pattern-recognition receptors (PRRs), such as CD14 and Toll-like receptors (TLRs), in membrane lipid rafts of activated cells (Triantafilou et al., 2004; Hajishengallis et al., 2006a).
CR3 plays diverse roles in immunity and inflammation, including iC3b-mediated phagocytosis, promotion of leukocyte transmigration to sites of extravascular inflammation, and induction of cytokine responses (Ehlers, 2000). Given these potentially protective roles in innate immune defense, it is puzzling, if not paradoxical, that CR3 appears to be a “preferred” receptor used by certain phylogenetically unrelated pathogens for promoting their survival in the mammalian host (Romani et al., 2002; Mosser and Edelson, 1987; Payne and Horwitz, 1987; Hellwig et al., 2001). In this chapter, we will present and discuss evidence that the interaction of CR3 with P. gingivalis leads to suppression of interleukin (IL)-12 in vitro and in vivo, increased intracellular survival of the pathogen in macrophages, as well as enhanced in vivo persistence and induction of periodontal bone loss.
3 P. gingivalis interacts with CR3 through its cell surface fimbriae
The Hanazawa group was the first to show that P. gingivalis interacts with CR3 though its cell surface fimbriae. Specifically, these investigators showed that the fimbriae of P. gingivalis bind mouse macrophage CR3 resulting in induction of tumor necrosis factor-α (TNF-α) and IL-1β production (Takeshita et al., 1998). Subsequently, our group established that mouse or human CR3 does not work in isolation for innate recognition of P. gingivalis. Rather, CR3 engages in cooperative interactions with the CD14/TLR2 signaling complex in response to P. gingivalis fimbriae. (Hajishengallis et al., 2005; Hajishengallis et al., 2006a; Hajishengallis et al., 2006b; Harokopakis and Hajishengallis, 2005). According to this model, P. gingivalis fimbriae bind CD14 and activate TLR2- and phosphatidylinositol 3-kinase (PI3K)-mediated inside-out signaling leading to activation of the ligand-binding capacity of CR3 (Harokopakis and Hajishengallis, 2005) (Fig. 1). These interactions take place in membrane lipid rafts where CD14 is constitutively found, whereas TLR2 and CR3 are recruited there following cell stimulation with P. gingivalis fimbriae (Hajishengallis et al., 2006a; Hajishengallis et al., 2006b). Unlike wild-type P. gingivalis, the ability of nonfimbriated mutants to activate and interact with CR3 is relatively poor (Hajishengallis et al., 2006b; Harokopakis et al., 2006).
Fig. 1. P. gingivalis exploits TLR2 and CR3 and undermines innate immunity.
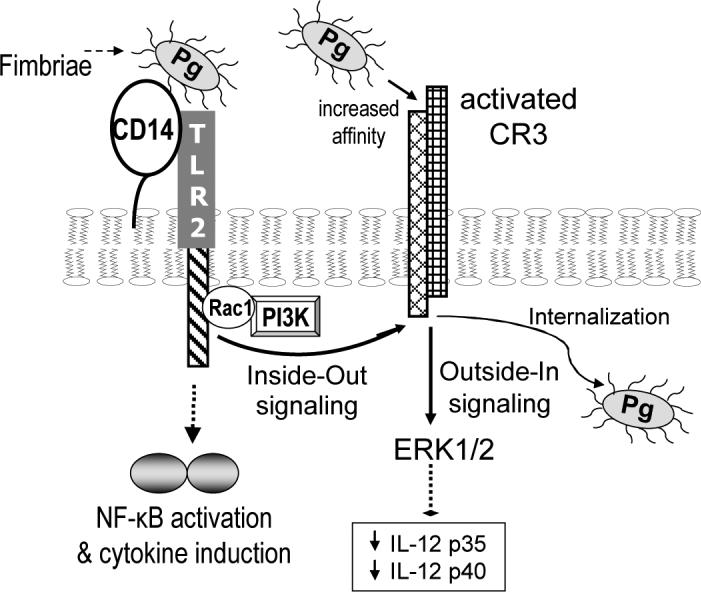
P. gingivalis exploits its fimbrial-mediated interaction with macrophage TLR2 to activate the high-affinity conformation of CR3. This pathway proceeds through Rac1/PI3K-mediated inside-out signaling and requires CD14 for facilitating the fimbria-TLR2 interaction (Harokopakis and Hajishengallis, 2005; Harokopakis et al., 2006). TLR2 stimulation by P. gingivalis also induces NF-κB activation and cytokine production (Hajishengallis et al., 2005; Hajishengallis et al., 2006a). Upon CR3 activation, fimbriated P. gingivalis can readily interact with CR3 leading to outside-in signaling, which via ERK1/2 downregulates IL-12 p35 and p40 and consequently inhibits production of bioactive (p70) IL-12 (Hajishengallis et al., 2007). This in turn undermines IL-12-mediated immune clearance, leading to persistence of the pathogen and enhanced virulence in the mouse periodontitis model (Hajishengallis et al., 2007). Moreover, CR3 uptake of P. gingivalis promotes its intracellular persistence in macrophages (Wang et al., 2007).
4 Biological Significance of CR3-P. gingivalis interactions: In Vitro mechanistic studies
We investigated the biological significance of P. gingivalis interactions with CR3 as they relate to three distinct functions of this integrin, i.e., as an adhesion molecule involved in transmigration, as a phagocytic receptor, and as an outside-in signaling receptor for modulation of cytokine responses. These findings are summarized and discussed below.
4.1 P. gingivalis stimulates CR3-dependent transendothelial migration of monocytes
Monocyte transmigration is mediated by interacting sets of cell adhesion molecules, including the CR3 - ICAM-1 pair (Libby, 2002). The ability of P. gingivalis fimbriae to activate TLR2 inside-out signaling for CR3 activation suggested that this pathogen may stimulate monocyte transmigration. Indeed, we found that fimbriated P. gingivalis (or purified fimbriae) stimulate CR3-dependent monocyte adhesion to endothelial ICAM-1 and transmigration across endothelial cell monolayers (Harokopakis et al., 2006). This may represent a potentially protective mechanism which can contribute to monocyte recruitment to sites of P. gingivalis infection. However, since the adhesion of monocytes to the arterial endothelium and their subsequent migration into the subendothelial area is a hallmark of early atherogenesis (Libby, 2002), P. gingivalis-induced CR3 activation may constitute a mechanistic basis linking this pathogen to inflammatory atherosclerotic processes. In this regard, viable P. gingivalis has been found in atherosclerotic plaques (Kozarov et al., 2005), although it is uncertain how the pathogen resists immune elimination and relocates there from the oral environment. An interesting hypothesis is that P. gingivalis not only stimulates the transmigratory activity of monocytes/macrophages but also exploits them as “Trojan horses” for disseminating to systemic tissues. Despite the lack of supporting evidence for this intriguing mechanism, additional work by our group has shown that P. gingivalis can indeed persist within macrophages if it is taken up through CR3 (below).
4.2 P. gingivalis enters macrophages via CR3 and resists intracellular killing
Our initial report that P. gingivalis fimbriae stimulate TLR2 inside-out signaling for CR3 activation (Harokopakis and Hajishengallis, 2005) was published concomitantly with a study by an independent group which showed that mycobacterial lipoarabinomannan also activates this proadhesive pathway (Sendide et al., 2005). Strikingly, mycobacteria exploit the TLR2/CR3 pathway for promoting their entry into monocytes/macrophages (Sendide et al., 2005) where they can parasitize (Ernst, 1998). The potential for CR3 exploitation by certain pathogens may, at least partly, be related to the notion that CR3 is not linked to vigorous microbicidal mechanisms, in contrast to most phagocytic receptors (Lowell, 2006; Wright and Silverstein, 1983; Yamamoto and Johnston, 1984; Caron and Hall, 1998; Rosenberger and Finlay, 2003). Consistent with this concept, the in vivo phagocytic uptake of Bordetella pertussis through the Fcγ receptor III (CD16) facilitates its clearance in contrast to CR3-mediated uptake (Hellwig et al., 2001). It is thus intriguing to speculate that pathogen-induced TLR2 inside-out signaling for CR3 activation may be a general pathway exploited by certain pathogens. We therefore investigated whether P. gingivalis can similarly induce its uptake through CR3 resulting in intracellular persistence rather than post-phagocytosis killing.
We first determined whether CR3 mediates P. gingivalis internalization by macrophages and found that CR3-deficient (CD11b−/−) mouse macrophages display significantly reduced capacity in the uptake of fimbriated P. gingivalis, compared to normal macrophages (Hajishengallis et al., 2006b) (Fig. 2). In contrast, no significant differences were observed regarding the uptake of nonfimbriated (FimA-deficient) mutants, suggesting that CR3 preferentially takes up fimbriated P. gingivalis (Hajishengallis et al., 2006b). Although TLR2 is not a phagocytic receptor, TLR2 deficiency similarly inhibits the uptake of P. gingivalis, consistent with its role in inside-out signaling for CR3 activation (Hajishengallis et al., 2006b) (Fig. 2).
Fig. 2. P. gingivalis uptake by macrophages depends on TLR2 and CR3.

Peritoneal macrophages from wild-type mice or mice deficient in TLR2, TLR4 (control), or CR3 (CD11b), were incubated with FITC-labeled P. gingivalis at a MOI of 10:1 for the indicated times at 37°C. Internalization was assessed by flow cytometry after washing the macrophages and quenching extracellular fluorescence, and was expressed as % FITC-positive macrophages (A). The mean fluorescence intensity (MFI) at the 60-min time-point is also shown (B) as a relative measure of the number of internalized bacteria. Results are means ± SD (n = 3; for clarity only the upper or lower SD is shown in A). Asterisks indicate statistically significant (p < 0.05) differences between PRR deficiencies and wild-type controls. Reproduced from Hajishengallis et al. 2006b. Copyright 2006 American Society for Microbiology.
We next followed the fate of internalized P. gingivalis in mouse macrophages or human monocytes by monitoring the recovery of viable internalized cells over time. P. gingivalis persisted intracellularly in a viable state for at least 72 hrs, in contrast to Aggregatibacter (Actinobacillus) actinomycetemcomitans, another periodontal pathogen (Socransky et al., 1998), which was readily killed (Wang et al., 2007). Unlike wild-type P. gingivalis, a nonfimbriated mutant was not recovered at 72 hrs but viable counts were obtained after 24 and 48 hrs, albeit at significantly lower levels. Strikingly, two other isogenic mutants which express a defective form of fimbriae, comprising FimA but lacking the FimCDE components (Fig. 3), were cleared even more rapidly than the nonfimbriated mutant (Wang et al., 2007). Because none of the mutants interact efficiently with CR3, these data imply that CR3 may, at least partly, be responsible for the enhanced persistence of wild-type P. gingivalis. Indeed, CR3 deficiency results in dramatic reduction of the intracellular survival of wild-type P. gingivalis by a factor of 103. In contrast, a FimCDE mutant displays limited intracellular persistence which is not affected CR3 deficiency (Wang et al., 2007) (Fig. 4A).
Fig. 3. The P. gingivalis fimbrial gene cluster and related mutants.
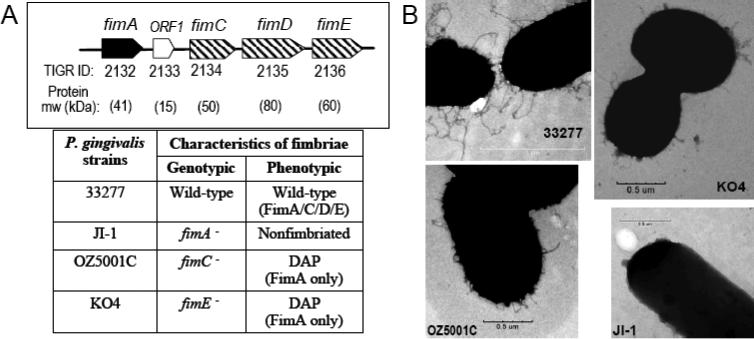
(A) The P. gingivalis 33277 fimbrial gene cluster contains fimA, encoding the main fimbrillin subunit, and fimCDE encoding accessory proteins associated with fimbriae (the role of ORF1 is unknown, although its product is not associated with fimbriae) (Watanabe et al., 1996; Nishiyama et al., 2007). The direction of transcription for each ORF is shown with the TIGR designation below the ORF. Strains OZ5001C and KO4 lacking fimC and fimE, respectively, express fimbriae devoid of all accessory proteins (DAP), whereas lack of fimA in strain JI-1 abrogates expression of both FimA and FimCDE resulting in a non-fimbriated state (Nishiyama et al., 2007; Wang et al., 2007). (B) Wild-type and mutant P. gingivalis surface structures visualized by transmission electron microscopy (Wang et al., 2007). Reproduced from Wang et al. 2007. Copyright 2007 The American Association of Immunologists, Inc.
Fig. 4. P. gingivalis exploits CR3-mediated internalization to persist in macrophages.
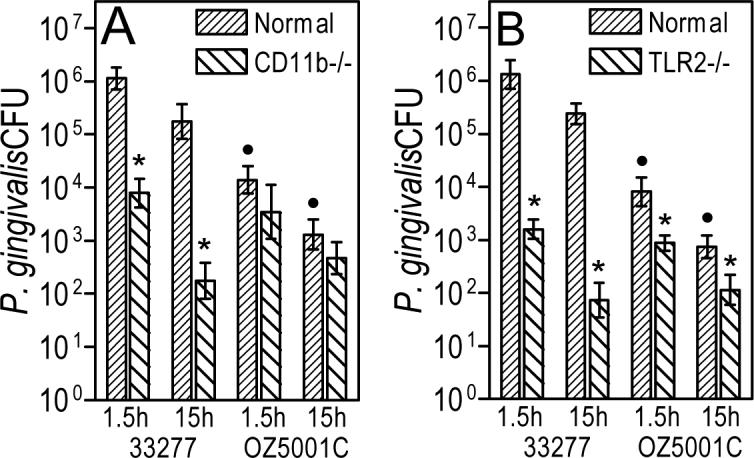
The persistence of viable internalized P. gingivalis 33277 or OZ5001C (FimCDE mutant) in normal, CR3-deficient (CD11b−/−) [A], or TLR2-deficient [B] macrophages was determined by an antibiotic protection-based intracellular survival assay. Data are shown as means ± SD (n = 3). Asterisks indicate significant (p < 0.05) differences between receptor-deficient and wild-type macrophages, whereas black circles denote significant (p < 0.05) differences between 33277 and OZ5001C. Reproduced from Wang et al. 2007. Copyright 2007 The American Association of Immunologists, Inc.
The implications of the ability of P. gingivalis to resist intracellular killing are currently uncertain. However, the differential susceptibility of wild-type P. gingivalis and the FimA or FimCDE mutants in intracellular killing correlates with their in vivo virulence in a mouse periodontitis model (Wang et al., 2007) (Fig. 5A). In addition, it is conceivable that the persistence of P. gingivalis in macrophages may be sufficient for co-opting the migration potential of these cells, facilitating relocation to systemic tissues, as alluded to above.
Fig. 5. Involvement of CR3 in induction of periodontal bone loss by P. gingivalis.

Wild-type P. gingivalis (strain 33277) induces significantly higher levels of periodontal bone resorption compared to mutants that do not efficiently interact with CR3 (A), while a CR3 antagonist inhibits the ability of P. gingivalis 33277 to cause periodontal bone loss (B). In panel A, BALB/c mice were orally infected or not with P. gingivalis 33277 (wild-type fimbriae), OZ5001C (fimbriae lacking FimCDE), KO4 (fimbriae lacking FimCDE) or JI-1 (nonfimbriated). In panel B, the mice were pretreated with a CR3 antagonist (XVA143) or PBS control prior to infection with P. gingivalis 33277 (P.g.) or vehicle only (Sham). The mm distance from the cementoenamel junction to the alveolar bone crest was measured at 14 predetermined sites in defleshed maxillae and the data were transformed to indicate bone loss (Hajishengallis et al., 2007; Wang et al., 2007). Results are shown as means ± SD (n = 5) and negative values indicate bone loss. In panel A, asterisks show significant (p < 0.05) differences between infected and sham-infected mice. The sign “x” indicates significant difference between 33277 and JI-1. In panel B, asterisks denote significant (p < 0.05) differences between PBS-treated/P. gingivalis-infected mice and the rest of the groups, among which no significant differences were found. Reproduced from Wang et al. 2007 (A) and Hajishengallis et al. 2007 (B). Copyright 2007 The American Association of Immunologists, Inc.
4.3 P. gingivalis interaction with CR3 downregulates IL-12 induction
Consistent with earlier results (Takeshita et al., 1998), we found that mouse or human CR3 contributes to induction of several proinflammatory cytokines by P. gingivalis fimbriae, including TNF-α, IL-1β, and IL-6 (Hajishengallis et al., 2005; Hajishengallis et al., 2006a; Hajishengallis et al., 2007). Strikingly, however, the binding of P. gingivalis fimbriae to activated CR3 results in reduced production of bioactive (p70) IL-12 (Hajishengallis et al., 2005; Hajishengallis et al., 2007), a key cytokine involved in intracellular bacterial clearance (Trinchieri, 2003). At the mechanistic level, suppression of IL-12p70 production is mediated by CR3-dependent phosphorylation of ERK1/2 which leads to downregulation of IL-12 p35 and p40 subunits (Hajishengallis et al., 2007) (Fig. 1). Because the ability of mouse macrophages to elicit IL-12p70 in response to P. gingivalis fimbriae is upregulated by CR3 deficiency but is abrogated by TLR2 deficiency (Hajishengallis et al., 2005), it can be concluded that CR3 binding of fimbriae inhibits TLR2-dependent induction of IL-12p70. Moreover, P. gingivalis fimbriae can block IL-12p70 induction by other bacterial stimuli which activate TLR4. Indeed, the capacity of LPS from E. coli or A. actinomycetemcomitans to induce IL-12p70 in IFN-γ-primed monocytes is suppressed by P. gingivalis fimbriae, although other proinflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-8) are upregulated (Hajishengallis et al., 2007). Similar downregulation of LPS-induced IL-12p70 is observed when whole cells of P. gingivalis are used, provided that the bacteria express fully mature fimbriae containing the FimCDE accessory proteins (Wang et al., 2007). This inhibitory activity is CR3-dependent but is irrelevant to P. gingivalis internalization since pretreating cells with cytochalasin D does not reverse the effect (Wang et al., 2007). IL-12p70 production by macrophages is significant for host defense in that it activates cytotoxic T lymphocytes cells and natural killer cells to produce IFN-γ, which in turn activates the bactericidal function of macrophages (Trinchieri, 2003). The ability of P. gingivalis to inhibit IL-12 induction may be particularly relevant to oral disease. In this context, P. gingivalis readily takes intracellular refuge in permissive cells, such as epithelial cells (Lamont et al., 1995) and endothelial cells (Progulske-Fox et al., 1999), and a reduction in IL-12-dependent stimulation of cell-mediated immunity may compromise the killing of these P. gingivalis-infected cells. This may consequently allow the pathogen a window of opportunity to establish infection and create a niche that is appropriate for its survival and growth. Moreover, since P. gingivalis inhibits IL-12 induction by other organisms, this mechanism may promote the survival of both P. gingivalis and co-habiting organisms in the subgingival pocket.
5 In vivo evidence for CR3 exploitation by P. gingivalis and implications in periodontitis
Based on the concept that inhibition of IL-12p70 may constitute a microbial tactic to evade immunity, we reasoned that CR3 blockade with a small-molecule antagonist (XVA143; m.w. 585) would upregulate induction of IL-12p70 and IFN-γ in response to P. gingivalis and facilitate its clearance by the host. This notion was experimentally confirmed in a peritonitis model of P. gingivalis infection (Hajishengallis et al., 2007), suggesting that CR3 antagonists may be used therapeutically for controlling P. gingivalis infection. CR3 was conclusively implicated as an exploited receptor in additional experiments demonstrating that CR3-deficient mice elicit higher IL-12p70 and IFN-γ levels and display enhanced clearance of P. gingivalis compared to wild-type mice (Hajishengallis et al., 2007).
It seems curious why the host would “allow” a key receptor, such as CR3, become an Achilles’ heel to infection by at least some pathogens. From the host point of view, however, CR3-dependent inhibition of IL-12 appears to serve a physiological role. In this regard, the phagocytosis of apoptotic cells by macrophages is heavily dependent upon CR3 and is associated with inhibition of IL-12p70, since apoptotic cells are not normally recognized as danger that would justify induction of cell-mediated immunity (Kim et al., 2004; Mevorach et al., 1998). Moreover, in the case of extracellular pathogens that can readily be controlled with complement activation and humoral immunity, the phagocytosis of iC3b-coated bacteria by CR3 would help control potentially destructive inflammation through IL-12 downregulation. In fact, inhibition of IL-12 would not only suppress T helper type 1 cell-mediated immunity but would also upregulate T helper type 2 responses required for effective humoral (antibody) responses (Trinchieri, 1998). It is thus possible that P. gingivalis has co-opted a physiological anti-inflammatory CR3-dependent mechanism to evade innate immune clearance. This mechanism may be exploited also by other pathogens. For instance, the interaction of Bordetella pertussis filamentous hemagglutinin with CR3 similarly leads to inhibition of IL-12p70 (McGuirk and Mills, 2000) and the in vivo phagocytic uptake of B. pertussis via CR3 fails to promote its clearance (Hellwig et al., 2001).
In the context of periodontal disease, the virulence of P. gingivalis can be measured by its capacity to induce periodontal bone resorption in animal models. Using a validated model of mouse periodontitis (Baker et al., 2000), we demonstrated that CR3 blockade inhibits the ability of P. gingivalis to persist in the mouse host and to induce periodontal bone loss (Hajishengallis et al., 2007) (Fig. 5B). Since FimA- or FimCDE-deficient mutants of P. gingivalis cannot effectively interact with CR3, they would be expected to display relatively reduced persistence and virulence in the bone loss model. Although our findings confirmed this hypothesis as mentioned above (Fig. 5A) (Wang et al., 2007), additional defects could have contributed to the results. This is based on the notion that the FimCDE accessory proteins mediate binding to certain extracellular matrix proteins which may facilitate optimal P. gingivalis colonization in the oral cavity (Nishiyama et al., 2007). Interestingly, however, although the FimCDE-deficient mutants display enhanced adhesive properties compared to the FimA-deficient mutant, the former are less virulent in inducing periodontal bone loss (Fig. 5A) or in resisting intracellular killing by mouse macrophages or human monocytes (Wang et al., 2007). It is possible that expression of FimA devoid of the accessory proteins may elicit robust host responses that could eliminate P. gingivalis. In this regard, the FimCDE mutants are stronger inducers of NF-κB activation in vitro than both wild-type and FimA-deficient P. gingivalis (Wang et al., 2007). In general, increased microbial immunostimulatory potential correlates with reduced microbial survival in the host, as exemplified by genetically modified Yersenia pestis expressing an immunostimulatory version of LPS (Montminy et al., 2006). It is uncertain at the moment why FimCDE-deficient mutants are more proinflammatory than wild-type P. gingivalis. However, at least in part, this could be explained by the lack of efficient interactions with CR3. Thus, diminished CR3 outside-in signaling by the FimCDE mutants would not only result in higher IL-12 induction, but also reduced ERK1/2 activation downstream of CR3. Reduced ERK1/2 activation may in turn result in decreased production of the anti-inflammatory IL-10 (Martin et al., 2003).
Although the concept that CR3 plays a role in periodontitis is recent, it is intriguing to speculate that CR3 may, at least partly, be related to the age-related alterations associated with this disease. Periodontitis and other infection-driven chronic inflammatory diseases generally appear rather late in life, but it is not clear whether, or what kind of, age-related alterations in innate immune function are responsible. Interestingly, although phagocytosis generally declines with aging (Sebastian et al., 2005; Butcher et al., 2000), CR3 (CD11b/CD18)-dependent phagocytosis is intact (Butcher et al., 2001). Specifically, unlike FcγRIIIa (CD16)-mediated phagocytosis which declines because of age-related downregulation of CD16 expression, CD11b expression is preserved at old age (Butcher et al., 2001). Interestingly, CD16-mediated phagocytosis readily induces the oxidative burst response, in contrast to phagocytosis through CR3 (Payne and Horwitz, 1987; Wright and Silverstein, 1983; Lowell, 2006). In this respect, studies in macrophages have shown that CD16-derived phagosomes fuse more readily with lysosomes than CR3-derived phagosomes, suggesting association of CD16 with enhanced microbial killing (Vieira et al., 2002). It seems possible, therefore, that CR3-mediated internalization of P. gingivalis may stay intact with aging, whereas alternative uptake of the pathogen by strongly microbicidal pathways may decline. In relative terms, this means that CR3-mediated internalization of P. gingivalis may increase with aging. It is not known at the moment whether CR3 in advanced age is more readily exploitable by P. gingivalis leading to increased disease activity, but it is certainly a testable hypothesis.
6 CR3 exploitation by P. gingivalis depends on TLR2
CR3 exploitation by P. gingivalis is initiated at the level of TLR2, since TLR2 inside-out signaling is required for effective interaction of P. gingivalis fimbriae with CR3 (Harokopakis et al., 2006; Harokopakis and Hajishengallis, 2005). Consistent with this, we have now found that the intracellular survival of fimbriated P. gingivalis is dramatically reduced in TLR2-deficient macrophages relative to normal controls (Wang et al., 2007) (Fig. 4B). These results are in line with a study by an independent group that TLR2-deficient mice are more resistant to P. gingivalis-induced periodontal bone loss than wild-type controls (Burns et al., 2006). Although TLR2 deficiency limits efficient activation of CR3, which in turn cannot be readily exploited by P. gingivalis, this does not necessarlily rule out the possibility that TLR2 may be exploited by P. gingivalis in CR3-independent ways. In this regard, TLR2 signaling has been implicated in immune evasion by Yersinia enterocolitica through induction of IL-10-mediated immunosuppression (Sing et al., 2005).
A recent report has presented evidence suggesting that CR3 and TLR4 cooperate for the uptake and intracellular killing of Salmonella enterica serovar Typhimurium (van Bruggen et al., 2007). Although this was shown in neutrophils, rather than in macrophages, it could be speculated that CR3 may be exploited in a contextual way, i.e., dependent upon which TLR is predominantly activated by the pathogen. Interestingly, P. gingivalis appears to be biased toward preferentially activating TLR2 both in vitro and in vivo (Burns et al., 2006; Hajishengallis et al., 2006a). Although bacterial LPS in general is a strong TLR4 agonist, P. gingivalis seems to deviate from the norm in that it expresses a heterogeneous mixture of lipid A species, which can induce cell activation through TLR2 or TLR4 (weakly) or even antagonize TLR4-induced cell activation (Darveau et al., 2004; Dixon and Darveau, 2005). Therefore, by altering the proportions of its different lipid A moieties, P. gingivalis may increase its virulence through manipulation of the innate response in ways that predominant activation of TLR2 over TLR4, may allow effective exploitation of CR3.
7 Conclusion
The interactions of P. gingivalis with CR3 is but one of the ways this pathogen interacts with the complement system in general. Interestingly, P. gingivalis is very resistant to killing by complement; this is attributable to the ability of its gingipain proteases to degrade C3 and C5 and thereby prevent deposition of C3b on the bacterial cell surface, which moreover contains a complement-resistant anionic polysaccharide (Popadiak et al., 2007; Slaney et al., 2006). Intriguingly, degradation of C5 by P. gingivalis leads to generation of a biologically active C5a-like fragment (Wingrove et al., 1992). This bioactive fragment activates a chemotactic response in neutrophils, presumably through the C5a receptor (C5aR), but the implications for periodontal disease are uncertain. In the light of recent developments that C5aR cross-talks with TLR signaling pathways and inhibits IL-12 (Hawlisch et al., 2005; Zhang et al., 2007), it could be speculated that P. gingivalis may use more than one complement-related mechanisms for escaping IL-12-mediated clearance. These considerations along with our findings regarding CR3 exploitation by P. gingivalis suggest that this pathogen “prefers” to manipulate the complement response rather than to merely inactivate it. The elucidation of complement-dependent immune evasion strategies of P. gingivalis may help control periodontitis, or other systemic conditions associated with P. gingivalis infections, through the application of appropriate complement inhibitors.
8 Acknowledgments
The authors acknowledge support by U.S. Public Health Service Grants DE015254 and DE018292 (to G.H.), and DE14605 (to D.R.D.) from the National Institutes of Health; Grants-in-Aid for Scientific Research (15591957 to F.Y. and 17791318 to S.N.) from the Japan Society for the Promotion of Science; and the AGU High-Tech Research Center Project from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to F.Y.).
References
- Baker PJ, Dixon M, Roopenian DC. Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun. 2000;68:5864–5868. doi: 10.1128/iai.68.10.5864-5868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat N, Perera P-Y, Carboni JM, Blanco J, Golenbock DT, Mayadas TN, Vogel SN. Use of a photoactivatable taxol analogue to identify unique cellular targets in murine macrophages: Identification of murine CD18 as a major taxol-binding protein and a role for Mac-1 in taxol-induced gene expression. J Immunol. 1999;162:7335–7342. [PubMed] [Google Scholar]
- Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- Butcher S, Chahel H, Lord JM. Ageing and the neutrophil: no appetite for killing? Immunology. 2000;100:411–416. doi: 10.1046/j.1365-2567.2000.00079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, O'Mahony D, Lord JM. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol. 2001;70:881–886. [PubMed] [Google Scholar]
- Calkins CC, Platt K, Potempa J, Travis J. Inactivation of tumor necrosis factor-α by proteinases (gingipains) from the periodontal pathogen, Porphyromonas gingivalis. Implications of immune evasion. J Biol Chem. 1998;273:6611–6614. doi: 10.1074/jbc.273.12.6611. [DOI] [PubMed] [Google Scholar]
- Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660–1665. doi: 10.1128/iai.66.4.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J Cell Biol. 1993;120:1031–1043. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon DR, Darveau RP. Lipopolysaccharide heterogeneity: innate host responses to bacterial modification of lipid a structure. J Dent Res. 2005;84:584–595. doi: 10.1177/154405910508400702. [DOI] [PubMed] [Google Scholar]
- Ehlers MRW. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2000;2:289–294. doi: 10.1016/s1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
- Ernst JD. Macrophage receptors for Mycobacterium tuberculosis. Infect Immun. 1998;66:1277–1281. doi: 10.1128/iai.66.4.1277-1281.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J Dent Res. 2006;85:106–121. doi: 10.1177/154405910608500202. [DOI] [PubMed] [Google Scholar]
- Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Ratti P, Harokopakis E. Peptide mapping of bacterial fimbrial epitopes interacting with pattern recognition receptors. J Biol Chem. 2005;280:38902–38913. doi: 10.1074/jbc.M507326200. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Shakhatreh M-AK, Wang M, Liang S. Complement receptor 3 blockade Promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007;179:2359–2367. doi: 10.4049/jimmunol.179.4.2359. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama S-I, Ratti P, Schifferle RE, Lyle EA, Triantafilou M, Triantafilou K, Yoshimura F. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006a;8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Wang M, Harokopakis E, Triantafilou M, Triantafilou K. Porphyromonas gingivalis fimbriae proactively modulate β2 integrin adhesive activity and promote binding to and internalization by macrophages. Infect Immun. 2006b;74:5658–5666. doi: 10.1128/IAI.00784-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. TLR2 trans-modulates monocyte adhesion and transmigration via Rac1- and PI3K-mediated inside-out signaling in response to Porphyromonas gingivalis fimbriae. J Immunol. 2006;176:7645–7656. doi: 10.4049/jimmunol.176.12.7645. [DOI] [PubMed] [Google Scholar]
- Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur J Immunol. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Kohl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–426. doi: 10.1016/j.immuni.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Hellwig SM, van Oirschot HF, Hazenbos WL, van Spriel AB, Mooi FR, van De Winkel JG. Targeting to Fcγ receptors, but not CR3 (CD11b/CD18), increases clearance of Bordetella pertussis. J Infect Dis. 2001;183:871–879. doi: 10.1086/319266. [DOI] [PubMed] [Google Scholar]
- Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Jr., Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. 2005;25:e17–e18. doi: 10.1161/01.ATV.0000155018.67835.1a. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885. doi: 10.1128/iai.63.10.3878-3885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RJ, Jenkinson HF. Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Lowell CA. Rewiring phagocytic signal transduction. Immunity. 2006;24:243–245. doi: 10.1016/j.immuni.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- McGuirk P, Mills KH. Direct anti-inflammatory effect of a bacterial virulence factor: IL-10-dependent suppression of IL-12 production by filamentous hemagglutinin from Bordetella pertussis. Eur J Immunol. 2000;30:415–422. doi: 10.1002/1521-4141(200002)30:2<415::AID-IMMU415>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. 1998;188:2313–2320. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, Fukase K, Kusumoto S, Sweet C, Miyake K, Akira S, Cotter RJ, Goguen JD, Lien E. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccha-ride response. Nat Immunol. 2006;7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edelson PJ. The third component of complement (C3) is responsible for the intracellular survival of Leishmania major. Nature. 1987;327:329–331. doi: 10.1038/327329b0. [DOI] [PubMed] [Google Scholar]
- Nishiyama S-I, Murakami Y, Nagata H, Shizukuishi S, Kawagishi I, Yoshimura F. Involvement of minor components associated with the FimA fimbriae of Porphyromonas gingivalis in adhesive functions. Microbiology. 2007;153:1916–1925. doi: 10.1099/mic.0.2006/005561-0. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol. 2005;560:11–18. doi: 10.1007/0-387-24180-9_2. [DOI] [PubMed] [Google Scholar]
- Payne NR, Horwitz MA. Phagocytosis of Legionella pneumophila is mediated by human monocyte complement receptors. J Exp Med. 1987;166:1377–1389. doi: 10.1084/jem.166.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol. 2007;178:7242–7250. doi: 10.4049/jimmunol.178.11.7242. [DOI] [PubMed] [Google Scholar]
- Progulske-Fox A, Kozarov E, Dorn B, Dunn W, Jr., Burks J, Wu Y. Porphyromonas gingivalis virulence factors and invasion of cells of the cardiovascular system. J. Periodontal. Res. 1999;34:393–399. doi: 10.1111/j.1600-0765.1999.tb02272.x. [DOI] [PubMed] [Google Scholar]
- Romani L, Bistoni F, Puccetti P. Fungi, dendritic cells and receptors: a host perspective of fungal virulence. Trends Microbiol. 2002;10:508–514. doi: 10.1016/s0966-842x(02)02460-5. [DOI] [PubMed] [Google Scholar]
- Rosenberger CM, Finlay BB. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol. 2003;4:385–396. doi: 10.1038/nrm1104. [DOI] [PubMed] [Google Scholar]
- Russell DG, Wright SD. Complement receptor type 3 (CR3) binds to an Arg-Gly-Asp-containing region of the major surface glycoprotein, gp63, of Leishmania promastigotes. J Exp Med. 1988;168:279–292. doi: 10.1084/jem.168.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian C, Espia M, Serra M, Celada A, Lloberas J. MacrophAging: a cellular and molecular review. Immunobiology. 2005;210:121–126. doi: 10.1016/j.imbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Sendide K, Reiner NE, Lee JS, Bourgoin S, Talal A, Hmama Z. Crosstalk between CD14 and complement receptor 3 promotes phagocytosis of mycobacteria: regulation by phosphatidylinositol 3-kinase and cytohesin-1. J Immunol. 2005;174:4210–4219. doi: 10.4049/jimmunol.174.7.4210. [DOI] [PubMed] [Google Scholar]
- Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct. 2002;31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922. [DOI] [PubMed] [Google Scholar]
- Sing A, Reithmeier-Rost D, Granfors K, Hill J, Roggenkamp A, Heesemann J. A hypervariable N-terminal region of Yersinia LcrV determines Toll-like receptor 2-mediated IL-10 induction and mouse virulence. Proc Natl Acad Sci U S A. 2005;102:16049–16054. doi: 10.1073/pnas.0504728102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaney JM, Gallagher A, Aduse-Opoku J, Pell K, Curtis MA. Mechanisms of resistance of Porphyromonas gingivalis to killing by serum complement. Infect Immun. 2006;74:5352–5361. doi: 10.1128/IAI.00304-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- Takeshita A, Murakami Y, Yamashita Y, Ishida M, Fujisawa S, Kitano S, Hanazawa S. Porphyromonas gingivalis fimbriae use β2 integrin (CD11/CD18) on mouse peritoneal macrophages as a cellular receptor, and the CD18 β chain plays a functional role in fimbrial signaling. Infect Immun. 1998;66:4056–4060. doi: 10.1128/iai.66.9.4056-4060.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Brandenburg K, Kusumoto S, Fukase K, Mackie A, Seydel U, Triantafilou K. Combinational clustering of receptors following stimulation by bacterial products determines lipopolysaccharide responses. Biochem J. 2004;381:527–536. doi: 10.1042/BJ20040172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Immunobiology of interleukin-12. Immunol Res. 1998;17:269–278. doi: 10.1007/BF02786451. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- van Bruggen R, Zweers D, van Diepen A, van Dissel JT, Roos D, Verhoeven AJ, Kuijpers TW. Complement receptor 3 and Toll-like receptor 4 act sequentially in uptake and intracellular killing of unopsonized Salmonella enterica serovar Typhimurium by human neutrophils. Infect Immun. 2007;75:2655–2660. doi: 10.1128/IAI.01111-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J. 2002;366:689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Shakhatreh M-AK, James D, Liang S, Nishiyama S.-i., Yoshimura F, Demuth DR, Hajishengallis G. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Onoe T, Ozeki M, Shimizu Y, Sakayori T, Nakamura H, Yoshimura F. Sequence and product analyses of the four genes downstream from the fimbrilin gene (fimA) of the oral anaerobe Porphyromonas gingivalis. Microbiol Immunol. 1996;40:725–734. doi: 10.1111/j.1348-0421.1996.tb01133.x. [DOI] [PubMed] [Google Scholar]
- Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem. 1992;267:18902–18907. [PubMed] [Google Scholar]
- Wright SD, Silverstein SC. Receptors for C3b and C3bi promote phagocytosis but not the release of toxic oxygen from human phagocytes. J Exp Med. 1983;158:2016–2023. doi: 10.1084/jem.158.6.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubenko VP, Lishko VK, Lam SC, Ugarova TP. A molecular basis for integrin αMβ2 ligand binding promiscuity. J Biol Chem. 2002;277:48635–48642. doi: 10.1074/jbc.M208877200. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Johnston RB., Jr. Dissociation of phagocytosis from stimulation of the oxidative metabolic burst in macrophages. J Exp Med. 1984;159:405–416. doi: 10.1084/jem.159.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, Wetsel RA, Miwa T, Song WC. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–236. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]