

Abstract
Many cellular responses to corticosteroids involve the transcriptional modulation of target genes by a prototypical nuclear receptor, the glucocorticoid receptor (GR). In the classic model of steroid hormone action GR acts as ligand-dependent transcription factor by either activating or repressing gene expression through direct interactions with DNA or other transcription factors. Recent evidence suggests an important role for nontranscriptional effects of GR in the vascular system. The nontranscriptional actions of GR involve the rapid activation of protein kinases, such as phosphatidylinositol-3 kinase and Akt, leading to the activation of endothelial nitric oxide synthase. This novel pathway of steroid hormone action protects against ischemic injury by augmenting blood flow and decreasing vascular inflammation.
Keywords: Glucocorticoid receptor, Nontranscriptional, Nitric oxide, Ischemia, Akt
Introduction
The study of steroid hormone action has provided many important insights into the regulation of cellular functions by nuclear receptors and at the same time has revealed surprising levels of biological complexity. Corticosteroids are essential for normal development and the stress responses through the regulation of intermediary metabolism and the immune system [1, 2]. Produced by the adrenal glands under the regulation of the hypothalamus-pituitary gland axis, corticosteroids were among the first hormones to be identified and later found to exert their effects through a specific DNA-binding protein, the glucocorticoid receptor (GR) [3]. It was subsequently shown that GR can act as a ligand-dependent transcription factor that positively regulates genes through interaction with DNA enhancer sequences, called glucocorticoid response elements (GRE) [4, 5, 6]. Molecular analysis of the anti-inflammatory actions of GR later revealed a second important mechanism of GR function in the nucleus. Activated GR can negatively regulate expression of inflammatory genes through direct protein-protein interaction with proinflammatory transcription factors without DNA binding (termed transrepression) [7, 8]. Recent findings suggest that there is yet another level of GR action with particular importance for the vascular system. A rapid, nontranscriptional effect of GR was found to mediate tissue protection in myocardial infarction and stroke through activation of endothelial nitric oxide synthase (eNOS), which is mediated by the phosphatidylinositol 3-kinase (PI3K)/Akt pathway [9, 10].
In this contribution we review and discuss existing data on the use of corticosteroids in vascular inflammation and acute ischemia, provide an overview of the molecular mechanisms of GR action with emphasis on physiological and pharmacological significance, and discuss the implications for potential future therapeutic approaches.
Beneficial and harmful effects of corticosteroids in acute ischemia
Corticosteroids have been used in the treatment of cardiovascular disease, such as acute myocardial infarction and stroke, with conflicting results [11]. Several features make steroids attractive candidates for the treatment of acute ischemia. The degree of ischemic tissue damage is related to the extent of the inflammatory response, which is coordinated through the interaction of leukocytes with activated vascular endothelium. Corticosteroids inhibit endothelial cell activation and leukocyte-endothelial interaction, thereby exerting prominent, and very rapid, anti-inflammatory effects on the vasculature [12]. Steroid treatment improves survival of patients with acute myocardial infarction and protects the myocardium from experimental ischemic injury [13, 14, 15]. In ischemic stroke steroids can improve clinical outcome in a subset of patients with severe stroke and administration of high doses of corticosteroids substantially reduces tissue damage in experimental focal cerebral ischemia [16, 17, 18, 19]. In addition, our group has recently shown that high-dose steroid treatment decreases vascular inflammation and ischemic tissue damage after myocardial infarction and stroke through direct vascular effects involving the nontranscriptional activation of eNOS [9, 10].
The prolonged use of steroids, however, is often limited by their adverse effects attributed to a delayed genomic response initiated by GR. This is exemplified by the clinical syndrome of hyperglycemia, dyslipidemia, hypertension, osteoporosis, and impaired wound healing in patients with chronic corticosteroid excess (i.e., Cushing syndrome) [20]. Furthermore, down-regulation of eNOS is thought to contribute to steroid-induced hypertension [21]. Indeed, continued low-dose steroid administration increases ischemic injury after global ischemia of the brain [22]. In myocardial infarction impaired wound healing and cardiac remodeling can lead to cardiac rupture within 2 weeks of myocardial infarction [23, 24], and this has led to discontinuation of steroid treatment for acute ischemia. Therefore defining the exact molecular mechanisms involved in the beneficial and detrimental effects of GR could have important therapeutic implications.
Molecular structure of GR
GR is expressed ubiquitously and belongs to the nuclear receptor superfamily, which also includes receptors for the mineralocorticoids, estrogens (ER), progestins, and androgens, as well as receptors for peroxisome proliferators, vitamin D, and thyroid hormones. Phylogenetic and sequence analysis has shown that GR with the receptors for progestins, mineralocorticoids, and androgens form a subfamily of oxosteroid receptors that is distinct from the ER subfamily [25]. GR has the prototypical modular structure of nuclear receptors: an N-terminal transcriptional activation function 1 domain, a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) harboring a ligand-dependent transcriptional activation function-2 domain [6, 26]. Alternative mRNA splicing results in a second GR isoform, GRβ, that is defective in steroid binding and acts as a dominant negative inhibitor of GRα in vitro [3, 27, 28]. However, a clear functional role for GRβ has yet to be established.
The activation of GR is regulated by the LBD. In the absence of ligand, GR is assembled into a multiprotein complex that includes heat shock proteins and immunophilins. This retains GR in the cytoplasm but enables high-affinity ligand binding [29, 30]. Hormone binding to the LBD leads to a conformational change in the receptor followed by dissociation of the multiprotein complex and rapid nuclear entry. Nuclear translocation is mediated by two nuclear localization sequences located adjacent to the DBD, and in the LBD, respectively [31, 32]. Interestingly, hormone-bound GR is not permanently localized to the nucleus. Rather, GR rapidly and continuously shuttles between the nucleus and the cytoplasm [33, 34].
Transcriptional modulation by GR: DNA binding vs. protein-protein interaction
In the nucleus GR regulates transcription by two distinct mechanisms: DNA binding-dependent transcriptional modulation or DNA binding-independent modulation mediated by protein-protein interaction. In the first instance, activated GR binds to GRE present in the regulatory regions of target genes and activates transcription through recruitment of transcriptional coactivators, such as steroid receptor coactivator-1, and the basic transcription machinery [6, 35]. Transcriptional activation by GR requires homodimerization through two different dimerization interfaces. A five amino acid sequence (D loop) in the second zinc-finger of the DBD mediates cooperative DNA-binding to the palindromic motive of the GRE, and mutations in the D loop abrogate transactivation by GR [36, 37]. In addition, dimerization requires reciprocal interactions of two hydrophobic amino acids in the LBD. Consequently mutations in this region also interfere with transactivation [38]. By the same mechanism of direct interaction with DNA, GR has also been reported to repress transcription through negative GREs [39].
The second instance of GR nuclear action is independent of DNA binding and involves modulation of transcriptional activity through direct protein-protein interaction with inducible transcription factors [40]. An important example of DNA-independent actions of steroids is transrepression of the proinflammatory transcription factors activator protein (AP) 1 and nuclear transcription factor κB (NF-κB). GR weakly interacts with and inhibits AP-1-dependent transcription without altering its DNA binding [7, 41, 42]. Importantly, DNA binding inactive mutants of GR are fully capable of AP-1 transrepression [37]. Interaction between activated GR and NF-κB is mediated by the second zinc-finger in the DBD of GR, and transrepression does not require DNA binding of GR. Instead, GR interferes with the transactivation potential of the p65/RelA subunit of NF-κB (see [43] and references herein). In addition to transrepression, protein-protein interaction can also lead to synergistic induction of promoter activity, as shown for interaction of GR with Stat-5 on the β-casein promoter [44].
To link the pleiotropic molecular actions of GR to biologically significant functions, Reichardt and Schutz [45] have generated different genetically modified mice. Mice lacking GR (GR−/−) die shortly after birth due to impaired maturation of several organs including the lungs [46]. In contrast, overexpression of GR renders mice resistant to stress and endotoxic shock, which seem to involve a decrease in the inflammatory response [47]. While the absence of GR is incompatible with life, DNA binding and transactivation of target genes by GR is not essential for development or survival. Mice with a targeted mutation in the D loop dimerization domain of GR (GRdim), which impairs cooperative DNA binding and transactivation by GR, lack GRE-dependent gene expression but survive to adulthood [48]. GRdim mice, however, do not respond to corticosteroids with induction of gluconeogenic enzymes, such as tyrosine aminotransferase, demonstrating that they are defective in mounting a positive GRE-mediated transcriptional response. Interestingly, the anti-inflammatory actions of steroids are preserved in these mice. Steroids potently suppress the local and systemic inflammatory response in GRdim mice, and repression of AP-1 and NF-κB-dependent genes, such as matrix metalloproteinase-3 and collagenase-3, is comparable to wild-type mice [48, 49, 50]. Indeed, analysis of GRdim mice has recently provided insight into the mechanism of impaired wound repair [51]. After skin injury there is enhanced granulation tissue and increased numbers of fibroblasts in GRdim mice as compared to control, suggesting that the inhibitory effect of steroids on wound healing is mediated by the DNA binding activity of GR (see Table 1).
Table 1.
Different GR actions mediate the response to steroids. The mode of GR action mediating the response to steroids is indicated (D DNA binding-dependent action, I DNA binding-independent action, NT nontranscriptional action) (adapted from [1, 87])
| Organ/system | Effect | GR action |
|---|---|---|
| Brain | Anxiety reaction | ? |
| Hippocampal neuron response | D | |
| Lung | Postnatal maturation | I |
| Cardiovascular system | Ischemia protection | NT |
| Suppression of vascular inflammation | NT | |
| Hematopoietic system | Erythroblast proliferation | D |
| Liver | Induction of gluconeogenic enzymes | D |
| Bone | Osteoporosis | ? |
| Immune system | Thymocyte apoptosis | D |
| Inflammation | Suppression of local and systemic inflammatory response | I |
Rapid nontranscriptional effects of GR
Some effects of corticosteroids are very rapid, making primarily transcriptional mechanisms of action unlikely and hence have been referred to as “nontranscriptional” effects [52]. However, it should be noted that this concept does not exclude the possibility of indirect downstream transcriptional modulation secondary to the initiated signaling pathways. Corticosteroids alter amphibian behavior and increase inositol trisphosphate in vascular smooth muscle cells within minutes of administration [53, 54]. In mice the antianaphylactic effect of high-dose corticosteroids occurs acutely and is unaltered by the transcriptional inhibitor actinomycin D [55]. Furthermore, in patients with active rheumatoid arthritis steroids rapidly inhibit leukocyte recruitment into inflamed joints [56]. In myocardial infarction and ischemic stroke, high-dose steroids cause a transient decrease in blood pressure and systemic vascular resistance accompanied by an increase in coronary and cerebral blood flow within minutes of administration [10, 14, 57].
Although some effects of steroids do not seem to be mediated by GR, and are therefore designated nonspecific, there is growing evidence for rapid nontranscriptional actions of GR, especially with regard to inflammation [58, 59]. The synthetic corticosteroid dexamethasone (Dex) rapidly inhibits cytosolic phospholipase A2 activity and release of arachidonic acid, which are important mediators of inflammation. Inhibition was reversed by the GR antagonist RU486 (mifepristone), and by pharmacological inhibition of Src, suggesting that rapid inhibition of cytosolic phospholipase A2 by GR is mediated by Src [60]. Interestingly, a nonclassical effect of GR on rapid kinase signaling is also involved in the inhibition of AP-1 by steroids. The transcriptional activity of the AP-1 subunit c-Jun is enhanced through phosphorylation on Ser63/73 by members of the Jun N-terminal kinase (JNK) subfamily in response to inflammatory cytokines [61]. Activated GR prevented phosphorylation of c-Jun, and transcriptional enhancement of AP-1, by blocking the JNK signaling cascade [62]. Acute Dex treatment interfered with JNK activation in the cytoplasm and nucleus without altering JNK subcellular distribution. These effects did not involve direct interaction between JNK and GR and were independent of DNA binding, since a dimerization defective GR mutant, which lacks transcriptional capacity, is still able to suppress JNK activation in response to Dex [63].
There is growing evidence that the nontranscriptional actions of several other steroid hormone receptors regulate physiologically important processes [64]. For example, osteocyte apoptosis and bone loss is prevented by the androgen and estrogen steroid receptors through nontranscriptional activation of protein kinase Src and mitogen-activated protein kinase [65, 66]. Vascular nitric oxide production and vasodilation by estrogen depends on nontranscriptional activation of eNOS and are mediated by ERα-dependent activation of the PI3 K/Akt pathway [67, 68, 69]. Indeed, the vascular protective effects of estrogen are dependent on the nontranscriptional activation of eNOS via PI3 K/Akt [68].
Nontranscriptional activation of eNOS by corticosteroids
Many beneficial actions of corticosteroids in vivo involve the vascular endothelium. An important endogenous mediator of vascular integrity is endothelium-derived NO [70]. NO produced by eNOS possesses anti-inflammatory, antiatherogenic, and anti-ischemic properties [71, 72]. Enhanced NO production by administration of the eNOS substrate l-arginine or upregulation of eNOS by statins confers stroke protection [73, 74, 75], and transgenic mice overexpressing eNOS show decreased leukocyte accumulation and reduced vascular lesion formation following vascular injury [76]. Conversely, mice with targeted disruption of eNOS (eNOS−/−) exhibit increased vascular inflammation and larger cerebral infarctions following experimental ischemia [77, 78], while inhibition of NOS activity decreases cerebral blood flow and increases infarct size after ischemia [79].
In two different mouse models of ischemic injury, transient myocardial ischemia and transient focal cerebral ischemia, the protective effects of steroids were mediated by the nontranscriptional activation of eNOS by GR [9, 10]. High-dose corticosteroids significantly reduced ischemic tissue damage to the heart and the brain, while low doses were ineffective. Cotreatment with the GR antagonist RU486 completely reversed the effects of Dex, suggesting that GR mediated tissue protection. Furthermore, ischemia protection was mediated by NO, since the beneficial actions of steroids were absent in eNOS−/− mice or blocked by cotreatment with the NOS inhibitor Nω-Nitro-l-arginine methylester. Steroid treatment acutely increased eNOS activity in vitro and in vivo, which led to decreased vascular inflammation and increased vasorelaxation and regional cerebral blood flow in a NO-dependent manner.
Activation of PI3 K/Akt by corticosteroids
Corticosteroids have recently been reported to activate protein kinase Akt [9, 10, 68, 80], which is an important regulator of cell cycle progression and mediator of cellular survival downstream of PI3 K [81]. Phosphorylation of eNOS by Akt leads to increase endothelial NO release [82, 83]. In a ligand-dependent manner corticosteroids activate PI3 K and Akt, which is blocked by cotreatment with RU486 or the PI3 K inhibitor LY294002 but not by transcriptional inhibitors [9, 10] (Fig. 1). Although induction of nontranscriptional actions requires high doses of steroids, these effects are nevertheless specifically mediated by GR. This was demonstrated in transfection studies in COS7 cells, which lack endogenous GR. Whereas Dex did not activate PI3 K or Akt in the absence of GR, it readily induced Akt kinase activation after transfection with GR, or the dimerization defective GR mutant (A458T), which is unable to transactivate target genes [10]. This suggests a DNA binding independent mechanism of activation. The fact that PI3 K activation can be suppressed by the GR antagonist RU486 further argues against the involvement of transrepression in this process, since the RU486 compound still induces transrepression of AP-1 dependent gene expression in reporter assays [37]. Akt
Fig. 1.
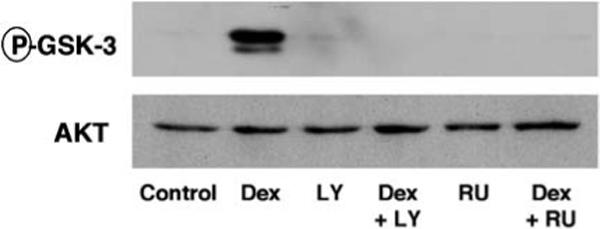
GR activates Akt through PI3 K. Above Effect of Dex with and without RU486 (RU) and LY294002 (LY) on Akt kinase activity, measured by phosphorylation of the Akt downstream target GSK-3 by immunoblotting (P-GSK-3). Below Immunoblotting for total Akt levels in the kinase reaction. (Reprinted from [9])
The mechanism of PI3 K activation by GR is currently unknown but seems to involve association of GR with the regulatory p85α subunit of PI3 K. Also, activation does not require dimerization of the receptor since the dimerization defective mutant is equally active [10]. A model of the nontranscriptional actions of GR is depicted in Fig. 2. Analogies might be drawn to PI3 K activation by another steroid receptor, ERα, although there is only modest sequence homology between the two receptors. Activation of PI3 K by ERα involves activation of Src and formation of a ternary complex of p85 and Src with the activated receptor, which was mediated by the C-terminus of ERα including the LBD [84]. Since positive cross talk between Src and PI3 K has already been described it might be speculated that this mechanism is also involved in activation of PI3 K by GR [85].
Fig. 2.
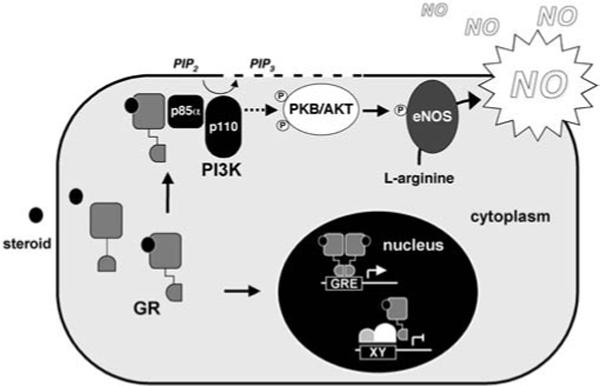
Nuclear and nonnuclear actions of the glucocorticoid receptor. Association of GR with the regulatory p85 subunit of PI3 K stimulates formation of 3′-phosphorylated phosphatidylinositols (e.g., phosphatidylinositol 3,4,5-trisphosphate, PIP3) from phosphatidylinositol phosphate precursors (e.g., phosphatidylinositol 4,5-trisphosphate, PIP2). This leads to the subsequent recruitment and activation of protein kinase Akt, which enhances NO release through phosphorylation of eNOS. In the nucleus GR binds either directly to glucocorticoid response elements (GRE) or modulates the function of other transcription factors
Corticosteroids as anti-ischemic therapy
The study of corticosteroid action in acute ischemia has yielded conflicting results, which may reflect differences in dosage regimens, time to onset of treatment, or the specific clinical or experimental condition. Several new aspects concerning the mechanism of ischemia protection have emerged from recent studies, which might allow for optimized treatment strategies.
A consistent feature of the beneficial actions of corticosteroids is the requirement for high-dose steroids [9, 10, 14, 19, 57]. Ischemia protection usually requires acute treatment (e.g., less than 6 h after the onset of ischemia). These features are in agreement with a nontranscriptional mechanism of ischemia protection by GR mediated by NO. By enhancing endothelial NO production corticosteroids decrease postischemic vascular inflammation and increase ischemic blood flow [9, 10]. The therapeutic use of corticosteroids is limited by side effects, which are typically associated with chronic treatment. Thus it appears that these detrimental effects are related to the genomic actions of corticosteroids. By exploring the rapid, nontranscriptional actions of GR the biological and pharmacological actions of corticosteroids could be considerably broadened. This could lead to the development of a novel class of drugs that selectively activates the nontranscriptional actions of GR. Indeed, synthetic compounds that separate specific GR functions, such as transactivation from transrepression by GR, have recently been identified [86]. It remains to be determined whether these compounds are capable of separating the beneficial from the detrimental effects of corticosteroids.
Abbreviations
- AP
Activator protein
- DBD
DNA-binding domain
- Dex
Dexamethasone
- eNOS
Endothelial nitric oxide synthase
- ER
Estrogen receptor
- GR
Glucocorticoid receptor
- GRE
Glucocorticoid response elements
- JNK
Jun N-terminal kinase
- LBD
Ligand-binding domain
- NF-κB
Nuclear transcription factor κB
- PI3K
Phosphatidylinositol 3-kinase
Biography
 Florian P. Limbourg received his M.D. degree from Freiburg University Medical School in Germany. He is currently a postdoctoral research fellow at the Brigham and Women's Hospital and Harvard Medical School, Boston, Mass., USA. His fellowship is supported by the Deutsche Forschungsgemeinschaft.
Florian P. Limbourg received his M.D. degree from Freiburg University Medical School in Germany. He is currently a postdoctoral research fellow at the Brigham and Women's Hospital and Harvard Medical School, Boston, Mass., USA. His fellowship is supported by the Deutsche Forschungsgemeinschaft.
 James K. Liao received his M.D. degree from the University of California at San Francisco School of Medicine. He is presently Associate Professor of Medicine at Harvard Medical School and the Director of Vascular Medicine Research at Brigham and Women's Hospital in Boston, Mass., USA.
James K. Liao received his M.D. degree from the University of California at San Francisco School of Medicine. He is presently Associate Professor of Medicine at Harvard Medical School and the Director of Vascular Medicine Research at Brigham and Women's Hospital in Boston, Mass., USA.
Contributor Information
Florian P. Limbourg, Vascular Medicine Research, Cardiovascular Division, Brigham and Women's Hospital, Harvard Medical School, 65 Landsdowne Street, Cambridge, MA 02139 USA Tel.: +1−617−768−8424, Fax: +1−617−768−8425
James K. Liao, Vascular Medicine Research, Cardiovascular Division, Brigham and Women's Hospital, Harvard Medical School, 65 Landsdowne Street, Cambridge, MA 02139 USA e-mail: jliao@rics.bwh.harvard.edu Tel.: +1−617−768−8424, Fax: +1−617−768−8425
References
- 1.Tronche F, Kellendonk C, Reichardt HM, Schutz G. Genetic dissection of glucocorticoid receptor function in mice. Curr Opin Genet Dev. 1998;8:532–538. doi: 10.1016/s0959-437x(98)80007-5. [DOI] [PubMed] [Google Scholar]
- 2.McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338:171–179. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- 3.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. Functional domains of the human glucocorticoid receptor. Cell. 1986;46:645–652. doi: 10.1016/0092-8674(86)90339-9. [DOI] [PubMed] [Google Scholar]
- 5.Hollenberg SM, Evans RM. Multiple and cooperative trans-activation domains of the human glucocorticoid receptor. Cell. 1988;55:899–906. doi: 10.1016/0092-8674(88)90145-6. [DOI] [PubMed] [Google Scholar]
- 6.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 7.Jonat C, Rahmsdorf HJ, Park KK, Cato AC, Gebel S, Ponta H, Herrlich P. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62:1189–1204. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- 8.Karin M. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell. 1998;93:487–490. doi: 10.1016/s0092-8674(00)81177-0. [DOI] [PubMed] [Google Scholar]
- 9.Hafezi-Moghadam A, Simoncini T, Yang E, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limbourg F, Huang Z, Plumier J, Simoncini T, Fujioka M, Tuckermann J, Schütz G, Moskowitz M, Liao J. Rapid non-transcriptional activation of endothelial nitric oxide synthase mediates increase in cerebral blood flow and stroke protection by corticosteroids. J Clin Invest. 2002;110:1729–1738. doi: 10.1172/JCI15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiemermann C. Corticosteroids and cardioprotection. Nat Med. 2002;8:453–455. doi: 10.1038/nm0502-453. [DOI] [PubMed] [Google Scholar]
- 12.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelialleukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci U S A. 1992;89:9991–9995. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barzilai D, Plavnick J, Hazani A, Einath R, Kleinhaus N, Kanter Y. Use of hydrocortisone in the treatment of acute myocardial infarction. Summary of a clinical trial in 446 patients. Chest. 1972;61:488–491. doi: 10.1378/chest.61.5.488. [DOI] [PubMed] [Google Scholar]
- 14.Libby P, Maroko PR, Bloor CM, Sobel BE, Braunwald E. Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest. 1973;52:599–607. doi: 10.1172/JCI107221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spath JA, Jr, Lane DL, Lefer AM. Protective action of methylprednisolone on the myocardium during experimental myocardial ischemia in the cat. Circ Res. 1974;35:44–51. doi: 10.1161/01.res.35.1.44. [DOI] [PubMed] [Google Scholar]
- 16.Patten BM, Mendell J, Bruun B, Curtin W, Carter S. Double-blind study of the effects of dexamethasone on acute stroke. Neurology. 1972;22:377–383. doi: 10.1212/wnl.22.4.377. [DOI] [PubMed] [Google Scholar]
- 17.Bertorelli R, Adami M, Di Santo E, Ghezzi P. MK 801 and dexamethasone reduce both tumor necrosis factor levels and infarct volume after focal cerebral ischemia in the rat brain. Neurosci Lett. 1998;246:41–44. doi: 10.1016/s0304-3940(98)00221-3. [DOI] [PubMed] [Google Scholar]
- 18.Courten-Myers GM de, Kleinholz M, Wagner KR, Xi G, Myers RE. Efficacious experimental stroke treatment with high-dose methylprednisolone. Stroke. 1994;25:487–492. doi: 10.1161/01.str.25.2.487. [DOI] [PubMed] [Google Scholar]
- 19.Slivka AP, Murphy EJ. High-dose methylprednisolone treatment in experimental focal cerebral ischemia. Exp Neurol. 2001;167:166–172. doi: 10.1006/exnr.2000.7532. [DOI] [PubMed] [Google Scholar]
- 20.Braunwald E, Zipes DP, Libby P. Heart disease. Saunders; Philadelphia: 2001. [Google Scholar]
- 21.Wallerath T, Witte K, Schafer SC, Schwarz PM, Prellwitz W, Wohlfart P, Kleinert H, Lehr HA, Lemmer B, Forstermann U. Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc Natl Acad Sci U S A. 1999;96:13357–13362. doi: 10.1073/pnas.96.23.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sapolsky RM, Pulsinelli WA. Glucocorticoids potentiate ischemic injury to neurons: therapeutic implications. Science. 1985;229:1397–1400. doi: 10.1126/science.4035356. [DOI] [PubMed] [Google Scholar]
- 23.Roberts R, DeMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 1976;53:I204–I206. [PubMed] [Google Scholar]
- 24.Bulkley BH, Roberts WC. Steroid therapy during acute myocardial infarction. A cause of delayed healing and of ventricular aneurysm. Am J Med. 1974;56:244–250. doi: 10.1016/0002-9343(74)90603-2. [DOI] [PubMed] [Google Scholar]
- 25.Nuclear Receptors Nomenclature Committee A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97:161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 26.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95:2435–2441. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. J Biol Chem. 1999;274:27857–27866. doi: 10.1074/jbc.274.39.27857. [DOI] [PubMed] [Google Scholar]
- 29.Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- 30.Pratt WB, Silverstein AM, Galigniana MD. A model for the cytoplasmic trafficking of signalling proteins involving the hsp90-binding immunophilins and p50cdc37. Cell Signal. 1999;11:839–851. doi: 10.1016/s0898-6568(99)00064-9. [DOI] [PubMed] [Google Scholar]
- 31.Picard D, Yamamoto KR. Two signals mediate hormone-dependent nuclear localization of the glucocorticoid receptor. EMBO J. 1987;6:3333–3340. doi: 10.1002/j.1460-2075.1987.tb02654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Hache RJ, Lefebvre YA. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol. 1999;19:1025–1037. doi: 10.1128/mcb.19.2.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sackey FN, Hache RJ, Reich T, Kwast-Welfeld J, Lefebvre YA. Determinants of subcellular distribution of the glucocorticoid receptor. Mol Endocrinol. 1996;10:1191–1205. doi: 10.1210/mend.10.10.9121487. [DOI] [PubMed] [Google Scholar]
- 34.Madan AP, DeFranco DB. Bidirectional transport of glucocorticoid receptors across the nuclear envelope. Proc Natl Acad Sci U S A. 1993;90:3588–3592. doi: 10.1073/pnas.90.8.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 36.Dahlman-Wright K, Wright A, Gustafsson JA, Carlstedt-Duke J. Interaction of the glucocorticoid receptor DNA-binding domain with DNA as a dimer is mediated by a short segment of five amino acids. J Biol Chem. 1991;266:3107–3112. [PubMed] [Google Scholar]
- 37.Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, Cato AC. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994;13:4087–4095. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 39.Drouin J, Sun YL, Chamberland M, Gauthier Y, De Lean A, Nemer M, Schmidt TJ. Novel glucocorticoid receptor complex with DNA element of the hormone-repressed POMC gene. EMBO J. 1993;12:145–156. doi: 10.1002/j.1460-2075.1993.tb05640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gottlicher M, Heck S, Herrlich P. Transcriptional crosstalk, the second mode of steroid hormone receptor action. J Mol Med. 1998;76:480–489. doi: 10.1007/s001090050242. [DOI] [PubMed] [Google Scholar]
- 41.Schule R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, Verma IM, Evans RM. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 1990;62:1217–1226. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- 42.Konig H, Ponta H, Rahmsdorf HJ, Herrlich P. Interference between pathway-specific transcription factors: glucocorticoids antagonize phorbol ester-induced AP-1 activity without altering AP-1 site occupation in vivo. EMBO J. 1992;11:2241–2246. doi: 10.1002/j.1460-2075.1992.tb05283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 44.Stocklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 45.Reichardt HM, Schutz G. Glucocorticoid signalling-multiple variations of a common theme. Mol Cell Endocrinol. 1998;146:1–6. doi: 10.1016/s0303-7207(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 46.Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schutz G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- 47.Reichardt HM, Umland T, Bauer A, Kretz O, Schutz G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol Cell Biol. 2000;20:9009–9017. doi: 10.1128/mcb.20.23.9009-9017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 49.Tuckermann JP, Reichardt HM, Arribas R, Richter KH, Schutz G, Angel P. The DNA binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J Cell Biol. 1999;147:1365–1370. doi: 10.1083/jcb.147.7.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schutz G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–7173. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grose R, Werner S, Kessler D, Tuckermann J, Huggel K, Durka S, Reichardt HM. A role for endogenous glucocorticoids in wound repair. EMBO Rep. 2002;3:575–582. doi: 10.1093/embo-reports/kvf119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wehling M. Specific, nongenomic actions of steroid hormones. Annu Rev Physiol. 1997;59:365–393. doi: 10.1146/annurev.physiol.59.1.365. [DOI] [PubMed] [Google Scholar]
- 53.Orchinik M, Murray TF, Moore FL. A corticosteroid receptor in neuronal membranes. Science. 1991;252:1848–1851. doi: 10.1126/science.2063198. [DOI] [PubMed] [Google Scholar]
- 54.Steiner A, Vogt E, Locher R, Vetter W. Stimulation of the phosphoinositide signalling system as a possible mechanism for glucocorticoid action in blood pressure control. J Hypertens Suppl. 1988;6:S366–S368. doi: 10.1097/00004872-198812040-00114. [DOI] [PubMed] [Google Scholar]
- 55.Inagaki N, Miura T, Nakajima T, Yoshida K, Nagai H, Koda A. Studies on the anti-allergic mechanism of glucocorticoids in mice. J Pharmacobiodyn. 1992;15:581–587. doi: 10.1248/bpb1978.15.581. [DOI] [PubMed] [Google Scholar]
- 56.Smith MD, Ahern MJ, Brooks PM, Roberts-Thomson PJ. The clinical and immunological effects of pulse methylprednisolone therapy in rheumatoid arthritis. III. Effects on immune and inflammatory indices in synovial fluid. J Rheumatol. 1988;15:238–241. [PubMed] [Google Scholar]
- 57.Vyden JK, Nagasawa K, Rabinowitz B, Parmley WW, Tomoda H, Corday E, Swan HJ. Effects of methylprednisolone administration in acute myocardial infarction. Am J Cardiol. 1974;34:677–686. doi: 10.1016/0002-9149(74)90157-x. [DOI] [PubMed] [Google Scholar]
- 58.Buttgereit F, Scheffold A. Rapid glucocorticoid effects on immune cells. Steroids. 2002;67:529–534. doi: 10.1016/s0039-128x(01)00171-4. [DOI] [PubMed] [Google Scholar]
- 59.Pitzalis C, Pipitone N, Perretti M. Regulation of leukocyte-endothelial interactions by glucocorticoids. Ann N Y Acad Sci. 2002;966:108–118. doi: 10.1111/j.1749-6632.2002.tb04208.x. [DOI] [PubMed] [Google Scholar]
- 60.Croxtall JD, Choudhury Q, Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br J Pharmacol. 2000;130:289–298. doi: 10.1038/sj.bjp.0703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333:F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 62.Caelles C, Gonzalez-Sancho JM, Munoz A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 1997;11:3351–3364. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez MV, Jimenez B, Berciano MT, Gonzalez-Sancho JM, Caelles C, Lafarga M, Munoz A. Glucocorticoids antagonize AP-1 by inhibiting the Activation/phosphorylation of JNK without affecting its subcellular distribution. J Cell Biol. 2000;150:1199–1208. doi: 10.1083/jcb.150.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cato AC, Nestl A, Mink S. Rapid actions of steroid receptors in cellular signaling pathways. Sci STKE. 2002;2002:RE9. doi: 10.1126/stke.2002.138.re9. [DOI] [PubMed] [Google Scholar]
- 65.Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- 66.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O'Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by non-genotropic signaling of sex steroids. Science. 2002;298:843–846. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 67.Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. 2000;87:677–682. doi: 10.1161/01.res.87.8.677. [DOI] [PubMed] [Google Scholar]
- 70.Loscalzo J. Nitric oxide and vascular disease. N Engl J Med. 1995;333:251–253. doi: 10.1056/NEJM199507273330410. [DOI] [PubMed] [Google Scholar]
- 71.De Caterina R, Gimbrone MAJr. Leukocyte-endothelial interactions and the pathogenesis of atherosclerosis. In: Kristensen SD, Schmidt EB, De Caterina R, Endres S, editors. n-3 fatty acids – prevention and treatment in vascular disease. Springer; Berlin Heidelberg New York: 1995. pp. 9–24. [Google Scholar]
- 72.Ishida A, Sasaguri T, Kosaka C, Nojima H, Ogata J. Induction of the cyclin-dependent kinase inhibitor p21 (Sdi1/Cip1/Waf1) by nitric oxide-generating vasodilator in vascular smooth muscle cells. J Biol Chem. 1997;272:10050–10057. doi: 10.1074/jbc.272.15.10050. [DOI] [PubMed] [Google Scholar]
- 73.Dalkara T, Morikawa E, Panahian N, Moskowitz MA. Blood flow-dependent functional recovery in a rat model of focal cerebral ischemia. Am J Physiol. 1994;267:H678–H683. doi: 10.1152/ajpheart.1994.267.2.H678. [DOI] [PubMed] [Google Scholar]
- 74.Morikawa E, Moskowitz MA, Huang Z, Yoshida T, Irikura K, Dalkara T. L-Arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke. 1994;25:429–435. doi: 10.1161/01.str.25.2.429. [DOI] [PubMed] [Google Scholar]
- 75.Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:8880–8885. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kawashima S, Yamashita T, Ozaki M, Ohashi Y, Azumi H, Inoue N, Hirata K, Hayashi Y, Itoh H, Yokoyama M. Endothelial NO synthase overexpression inhibits lesion formation in mouse model of vascular remodeling. Arterioscler Thromb Vasc Biol. 2001;21:201–207. doi: 10.1161/01.atv.21.2.201. [DOI] [PubMed] [Google Scholar]
- 77.Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, Huang PL. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J Clin Invest. 1998;101:1225–1232. doi: 10.1172/JCI1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 79.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 80.Langdown ML, Holness MJ, Sugden MC. Early growth retardation induced by excessive exposure to glucocorticoids in utero selectively increases cardiac GLUT1 protein expression and Akt/protein kinase B activity in adulthood. J Endocrinol. 2001;169:11–22. doi: 10.1677/joe.0.1690011. [DOI] [PubMed] [Google Scholar]
- 81.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 82.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 83.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, Fiorentino R, Varricchio L, Barone MV, Auricchio F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001;20:6050–6059. doi: 10.1093/emboj/20.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, Choi Y. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4:1041–1049. doi: 10.1016/s1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- 86.Vayssiere BM, Dupont S, Choquart A, Petit F, Garcia T, Marchandeau C, Gronemeyer H, Resche-Rigon M. Synthetic glucocorticoids that dissociate transactivation and AP-1 transrepression exhibit antiinflammatory activity in vivo. Mol Endocrinol. 1997;11:1245–1255. doi: 10.1210/mend.11.9.9979. [DOI] [PubMed] [Google Scholar]
- 87.Newton R. Molecular mechanisms of glucocorticoid action: what is important? Thorax. 2000;55:603–613. doi: 10.1136/thorax.55.7.603. [DOI] [PMC free article] [PubMed] [Google Scholar]