

Summary
Thimet oligopeptidase (EC 3.4.24.15; TOP) is a Zn(II) endopeptidase implicated in the processing of numerous physiological peptides. Though its role in selecting and processing peptides is not fully understood, it is believed that flexible loop regions lining the substrate-binding site allow the enzyme to conform to substrates of varying structure. The present study describes mutant forms of TOP in which Gly or Tyr residues in loop region 599-611 were replaced, individually and in combination, to elucidate the mechanism of substrate selection by TOP. Decreases in kcat observed upon mutation of Tyr605 and Tyr612 demonstrate that these residues contribute to the efficient cleavage of most substrates. Modeling studies showing that a hinge-bend movement would bring both Tyr612 and Tyr605 within H-bond distance of the cleaved peptide bond supports this role. Thus, molecular modeling studies support a key role in transition-state stabilization of this enzyme by Tyr605. Interestingly, kinetic parameters showed that a bradykinin derivative is processed distinctly from the other substrates tested suggesting an alternative catalytic mechanism may be employed for this particular substrate. The data demonstrate that neither Tyr605 nor Tyr612 are necessary for the hydrolysis of this substrate. Relative to other substrates, the bradykinin derivative is also unaffected by Gly mutations in the loop. This distinction suggests that the role of glycine residues in the loop is to properly orient these Tyr residues in order to accommodate varying substrate structures. This also opens the possibility that certain substrates may be cleaved by an open form of the enzyme.
Keywords: metallopeptidase, thimet oligopeptidase, substrate selectivity, enzyme flexibility, hydrogen bonding
Introduction
Thimet oligopeptidase (TOP, EC 3.4.24.15), a 78-kDa zinc dependent endopeptidase contains the HEXXH sequence in its active site common to other endopeptidases of the M3 family of metallopeptidases [1-3]. This zinc-binding motif causes attack of an activated water molecule at the carbonyl carbon of the scissile peptide bond and the formation of a tetrahedral oxyanion intermediate [4]. TOP is most closely related to neurolysin (EC 3.4.24.16), with which it shares 60% sequence identity, overall three dimensional structure, and the ability to target and hydrolyze numerous short peptides (<17 residues) involved in various physiological processes [3, 5-7]. Consistent with TOP's broad anatomic and subcellular distribution, it is implicated in hydrolyzing peptide substrates involved in vital functions such as blood pressure control, reproduction, nociception and antigen presentation, [8-13].
A distinguishing feature of the X-ray crystallographic-derived structures of the apo- (substrate free) forms of TOP [14] and neurolysin [4] is their catalytic site, located in a deep channel that limits the size and shape of accessible substrates [14]. At the base of the channel are conserved flexible loop regions that contribute to the specificity of these two enzymes. One particular loop in neurolysin, composed of residues 600-612 and located across from the enzyme's active site, appears to be highly mobile because it includes five glycine residues [4, 14, 15]. TOP's corresponding loop, residues 599-611, contains one fewer glycine residue. This loop region is of significance because of its proximity to the active site and because it contains two tyrosine residues, Tyr605 and Tyr612, shown to be important in substrate binding and catalysis [15-17].
Previous studies have demonstrated that the Tyr612 hydroxyl is required for efficient turnover of quenched fluorescent substrates [16, 17]. For instance, the kcat/Km for hydrolysis of mca-GlyProGlyPhe-dnp, a synthetic substrate, is reduced up to ~400-fold when Tyr612 is replaced with Phe [17]. The proposed role of Tyr612 of TOP is to stabilize the catalytic intermediate via hydrogen bond donation. This role is similar to that of other amino acid residues in peptidases, such as His231 in thermolysin [17, 18]. However, modeling suggests that Tyr612 of TOP is several angstroms too far from the substrate in the crystallized conformation of the enzyme to effectively form hydrogen bonds [14, 17].
It has been proposed that significant changes must occur, possibly upon substrate binding, for the Tyr605 and Tyr612 to be in position to play their proposed roles in substrate catalysis [14-17]. Recently, the structure of the substrate/inhibitor bound form of DcP, a bacterial dipeptidyl carboxypeptidase from E. coli bearing significant sequence similarity to TOP, has been elucidated [19]. Like TOP, DcP is bilobal, but unlike TOP the DcP structure is in a distinctly closed conformation. Using the structure of Dcpcarboxypeptidase, we have produced a model for the closed-form of TOP with bound substrate. The model has allowed for a more careful analyses of the residues that are in close proximity to bound substrate in TOP, including Tyr612 and residues contained in the loop region 599-611 that join domain I and II. Supported by computational studies, activity assays with several structurally distinct substrates reveal a more significant catalytic role for Tyr605 than previously supposed [15]. Furthermore, activity assays demonstrate the quenched-fluorescent analog of bradykinin requires neither Tyr residue for efficient turnover by TOP. This distinction among substrates has allowed for a careful analysis of the role of the conserved glycine residues in the 599-611 loop. The flexibility of the loop provides a means to bring Tyr612 and Tyr605 into close proximity to the bound substrate and allows optimal substrate positioning by the enzyme. The evidence suggests that certain substrates require formation of a closed form of the enzyme in order to be efficiently cleaved, while other substrates can be effectively utilized even by the open form of the enzyme. The possibility of alternate mechanisms of cleavage for different substrates has important implications for the physiological role of TOP and its wide distribution.
Results
Kinetic Studies: Tyr Mutants
The changes in enzyme kinetic parameters of TOP towards four structurally distinct substrates upon removal of the hydroxyl groups of Tyr605 and Tyr612 are shown in Table 1. The Y612F mutation resulted in a marked decrease in activity, as measured by changes in kcat/Km, with respect to wild type activity towards MCA, mcaNt, and mcaGnRH1-9. The decrease was 1000-2000-fold with respect to mcaNt and MCA and 200-fold with respect to mcaGnRH1-9 and these changes were mostly due to changes in kcat. The Y605F mutation (Table 1) resulted in a lesser, but still considerable, 100-fold decrease in activity towards MCA and mcaNt, and a 12-fold decrease towards GnRH1-9, again due to changes in kcat. Interestingly, the Y605F mutant did not show significant changes in kcat/Km with the mcaBk substrate; the parameters were very similar to that of the wild type. There were significant changes, however, in the kcat/Km with the double Y605/612F mutation and less change with the single Y612F, most notably due to changes in Km.
Table 1. Enzyme Kinetics.
Kinetic parameters of enzymes with four substrates.
| MCA | |||
|---|---|---|---|
| Enzyme | kcat (s-1) | Km(μM) | kcat/Km(μM-1s-1) |
| WT | 0.44±0.05 | 7.88±1.03 | 0.05±0.01 |
| G599A | 0.03±0.01 | 4.08±1.81 | 0.007±0.003 |
| G603A | 2.32±0.06 | 4.01±0.21 | 0.58±0.03 |
| G604A | 0.11±0.01 | 8.25±1.04 | 0.013±0.002 |
| G603A/G604A | 2.44±0.12 | 5.43±0.39 | 0.45±0.04 |
| G611A | 0.193±0.002 | 6.7±0.2 | 0.029±0.001 |
| G603P | 0.0026 ± 0.0003 | 6.43 ± 1.22 | 0.00041 ± 0.00009 |
| Y605F | 0.0086 ± 0.001 | 8.2 ± 1.6 | 0.0011 ± 0.0002 |
| Y612F | 0.00037 ±0.00004 | 9.0 ± 1.3 | 0.000041 ± 0.000007 |
| Y605/612F | 0.0023 ± 0.000002 | 8.1 ± 0.7 | 0.00028 ± 0.00002 |
| mcaBk | |||
|---|---|---|---|
| Enzyme | kcat (s-1) | Km(μM) | kcat/Km(μM-1s-1) |
| WT | 0.30±0.05 | 0.057±0.007 | 5.9±1.1 |
| G599A | 0.86±0.02 | 0.129±0.009 | 6.7±0.5 |
| G603A | 0.280±0.004 | 0.054±0.004 | 5.2±0.5 |
| G604A | 0.53±0.02 | 0.09±0.05 | 5.8±0.4 |
| G603A/G604A | 0.270±0.001 | 0.041±0.001 | 6.59±0.14 |
| G611A | 1.34±0.05 | 0.136±0.010 | 9.8±0.8 |
| G603P | 0.18 ± 0.05 | 2.1 ± 0.3 | 0.09 ± 0.02 |
| Y605F | 0.34 ± 0.01 | 0.08 ± 0.01 | 4.4 ± 0.4 |
| Y612F | 0.75 ± 0.02 | 0.57 ± 0.04 | 1.3 ± 0.11 |
| Y605/612F | 0.10 ± 0.003 | 0.51 ± 0.05 | 0.20 ± 0.02 |
| mcaNt | |||
|---|---|---|---|
| Enzyme | kcat (s-1) | Km(μM) | kcat/Km(μM-1s-1) |
| WT | 0.33±0.03 | 1.2±0.3 | 0.28±0.07 |
| G599A | 0.18±0.01 | 2.5±0.2 | 0.07±0.01 |
| G603A | 1.26±0.05 | 2.9±0.4 | 0.43±0.06 |
| G604A | 0.11±0.01 | 2.6±0.3 | 0.04±0.01 |
| G603A/G604A | 1.22±0.27 | 5.7±1.9 | 0.21±0.08 |
| G611A | 0.30±0.05 | 5.0±1.0 | 0.06±0.02 |
| G603P | 0.00449 ± 0.0005 | 2.9 ± 0.6 | 0.0016 ± 0.00037 |
| Y605F | 0.012 ± 0.002 | 4.2 ± 1.2 | 0.0029 ± 0.001 |
| Y612F | 0.00052 ± 0.00001 | 2.6 ± 0.1 | 0.0002 ± 0.00001 |
| Y605/612F | 0.0015 ± 0.0001 | 3.1 ± 0.38 | 0.00047 ± 0.00007 |
| mcaGnRH1-9 | |||
|---|---|---|---|
| Enzyme | kcat (s-1) | Km(μM) | kcat/Km(μM-1s-1) |
| WT | 11.2 ± 0.9 | 24 ± 4 | 0.47 ± 0.11 |
| Y612F | 0.061 ± 0.003 | 34 ± 3 | 0.0018 ± 0.0002 |
| Y605F | 1.4 ± 0.2 | 37 ± 10 | 0.038 ± 0.02 |
| Y605/612F | 0.025 ± 0.001 | 14 ± 1 | 0.0017 ± 0.002 |
Gly mutants
Wild type TOP has a clear preference for the mcaBk substrate over MCA and mcaNt, based on kcat/Km values (Table 1 and Figure 1). The majority of single substitutions of Ala for Gly in the loop region further increased this selectivity by considerably decreasing the activity towards the MCA and mcaNt substrates while generally having no effect or a slight improvement of activity towards mcaBk. This effect was observed for the MCA and mcaNt substrates with the G599A, G604A, and G611A mutant forms. For instance, each enzyme had decreased overall activity towards mcaNt due to decreased kcat values when compared to wild type, except for G611A which had a kcat value similar to that of wild type. In fact, both G599A and G604A had changes in Km that were consistent with the changes in kcat; about 3-fold for kcat and about 2-fold for Km. That is, changes in activity toward the mcaNt substrate were due to changes seen in both constants, although somewhat more for kcat, whereas those towards MCA were purely due to changes in kcat.
Fig. 1.
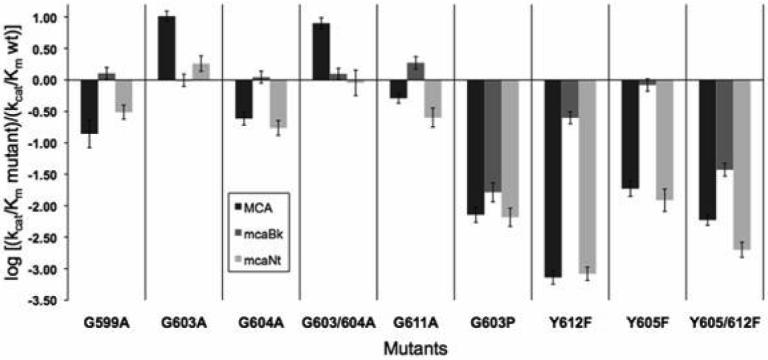
Comparison of kcat/Km of mutants to kcat/Km of wild type for three substrates. The kcat/Km for each mutant with MCA (dark gray), mcaBk (medium gray), and mcaNt (light gray) where wild type=0 on the log scale.
However, the substitution of Ala for Gly at position 603 in either single or double mutations notably altered the preference of the enzyme (Figure 1 and Table 1). G603A had the effect of creating a greater preference for the 5-residue MCA substrate and to a lesser extent for the 10-residue mcaNt substrate compared to wild type and all other single mutants. The double mutant that combines the G603A substitution with a second Ala substitution (G604A) retained increased activity towards MCA. Activity for the double mutant towards the Nt derivative did not increase compared to wild type, although its activity was notably higher than the single G604A mutant.
Although the substitution of alanine for glycine at position 603 led to enhanced activity towards MCA and mcaNt, substitution of proline for the glycine caused a significant decrease in the kcat/Km with MCA and mcaNt. The decrease in activity was ~1000 fold with MCA and ~200 fold with mcaNt, both primarily due to decreases in kcat.
Data for the loop mutants further demonstrated that the mcaBk substrate is distinct (Table 1). This substrate showed only little to no change in activity with the loop glycine mutants. Only G611A, the mutation closest to Tyr612, resulted in any substantial effect on activity towards mcaBk. The G603A and G604A mutations, both of which lie close to Y605, caused no significant change in activity towards mcaBk. It is notable that Y612F and Y605F caused modest and no change, respectively, towards this same substrate. Substitution of proline for glycine at position 603 led to significant decreases in activity for the mcaBk substrate. In contrast to the other mutants, the change for the proline substitution was entirely due to changes in Km, not in kcat.
Denaturing activity trends
We previously reported the changes in activity of two substrates (MCA and mcaBk) at low urea concentrations [20]. Here we have expanded on that data with two additional structurally distinct and physiologically relevant, neuropeptide-based substrates (Figure 2). Similar to the Tyr mutations, urea had distinct effects on mcaBk not apparent for the other substrates tested. At low urea concentrations TOP lost activity towards MCA, mcaNt, and mcaGnRH1-9. However, the enzyme was fully active towards mcaBk even between 1 and 2 M urea. Interestingly, the trends in activity in urea paralleled the trends observed with the Y612F mutant. For mcaBk, which suffered an increase in Km with the Y612F mutant, low urea caused an increase in Km and kcat. Between 1-2 M urea, the Y612F enzyme also retained marked activity towards mcaGnRH1-9. Both MCA and mcaNt, the most sensitive to the Y612F mutation, showed the largest decrease in activity between 1-2 M urea. Above 3 M urea the enzyme lost activity to all substrates, due to enzyme denaturation and Zn(II) loss from the active site [20].
Fig. 2.

Percent activity of wt TOP with substrates MCA, mcaBk, mcaGnRH1-9, and mcaNt in the presence of increasing urea (M).
HPLC analysis
To determine if the change in activity towards the MCA and mcaNt substrates were due to a change in substrate recognition by the modified enzymes, resulting in an altered cleavage site, wild type TOP and MCA were incubated for 30 minutes and the products evaluated by HPLC. Two products at absorbance 330nm were detected, suggesting a single cleavage site in the MCA substrate. Extended incubation and examination of the products of mcaNt after 90 minutes revealed four products, leading to suggestions of additional cleavage sites for the mcaNt substrate. Identical results were obtained concerning the position of cleavage sites for the Gly mutants (data not shown).
Modeling and molecular simulations of wild type and mutant TOP
By analogy to the Dcp enzyme [19], the transition between the open (substrate free) and closed (substrate bound) forms of TOP likely occurs through a reorientation of domains I and II. Thus, a model of the closed form of TOP was created by separately fitting domains I and II of the open TOP crystal structure onto the structure of Dcp in its closed form [19]. The TOP domains superimposed very well on the Dcp structure, with RMS deviations of 1.50 Å and 1.21 Å for domains I and II respectively. After fitting and minimization, the closed model of TOP was quite similar to that of Dcp, indicating that the two domains of TOP form relatively rigid structures that change their relative orientation by pivoting on residues 156, 351, 544, and 616 connecting the two lobes. Modeling TOP onto Dcp moved several domain II Tyr residues of TOP known to be involved in catalysis or substrate-binding [17, 19] into positions analogous to those of the closed DcP and thus to the appropriate distances from the active site to perform such roles (Figure 3). Tyr605 and Tyr609 fall within the loop structure while Tyr612 is just at the end of the loop. The original substrate-free structure of TOP showed that Tyr612, an important catalytic residue based on mutagenesis studies [17], is more than 8 Å from the active site. The closed form orients the phenol oxygen of this residue within hydrogen bonding distance from the carboxyl group of the scissile peptide bond in a modeled substrate. Furthermore, Tyr605 and Tyr609, both implicated in substrate-binding are shown in Figure 3 to be within hydrogen bonding distance from the substrate.
Fig. 3.

Molecular model of TOP used as the initial structure for MD simulations of wt, G603A, and G604A TOP with the MCA substrate shown in space filling.
Since energy minimization only allows for limited conformational sampling, we also subjected our TOP model to an MD simulation in explicit solvent in order to sample additional conformations of the substrate and the enzyme. Although all residues were allowed to move freely in these simulations, the overall enzyme structure and the loop region maintained relatively low C α RMS deviation values from our intial model throughout this trajectory (<3.0 Å and <1.5 Å, respectively). The substrate also maintained its relative position in the active site during the simulation. These data do not preclude the existence of other possible conformations further away from the starting model that were not sampled during the MD simulation. However, significant structural homology between TOP and DcP around the active site residues of domain I and the loop and Tyr residues in domain II supports out initial conformation for the model. Furthermore, the experimental effect observed for Tyr 605 and 612 mutants on enzyme activity validate the close proximity of these residues to the substrate in the model.
The MD simulations on wild type TOP and all four glycine mutants (G599A, G603A, G604A, and G611A) also provide insight into how alanine mutations affect the structure and dynamics of the loop region. All of these simulations included a MCA-like substrate in the active site (Figure 3). As expected, based on the flexibility of glycine, all four alanine mutations led to decreased structural flexibility in the loop region. For example, the loop region in the wild type enzyme had an increased Cα RMS fluctuation over the final ns of the trajectories in the glycine-rich region of the loop between residues 599-604 (data not shown). As well, the wild type loop showed an ability to more readily access a wider variety of conformations. This was particularly true for the section of the loop between residues 605 and 612 that contains the tyrosine residues demonstrated to be important for catalysis in this study. This region had a greater average Cα RMS deviation (2.7 Å) from the initial model over the last ns of the simulation than observed in mutant simulations (1.2-1.95 Å). This increased conformational sampling also led the wild type simulation to show reduced hydrogen bonding between loop residues and the substrate (Table 2) at the end of the simulation despite having a close proximity between tyrosine hydroxyl groups (e.g. 2-3 Å) and the substrate in the initial model.
Table 2. Percent hydrogen bonding distances of mutants.
The percentage of hydrogen bonding that occurred in the last nanosecond (ns) was calculated by looking at every ten picosecond frame. Average minimum distance between the side chain hydroxyl oxygen of Tyr residues and MCA in parenthesis. These data were calculated from the MD simulations described in Figure 3. Distances and percent hydrogen bonding for all mutants were calculated based on last (ns) of trajectories. All simulations were run for 10ns except for G604A which ran for 15ns.
| % Hydrogen Bonding in Last ns (Average Distance (nm) Between Tyrosines and MCA-like substrate) | |||
|---|---|---|---|
| Y605-MCA | Y609-MCA | Y612-MCA | |
| WT | 11% (0.30) | 0% (0.70) | 0% (0.34) |
| G599A | 5% (0.34) | 0% (0.44) | 66% (0.23) |
| G603A | 3% (0.33) | 73% (0.31) | 69% (0.21) |
| G604A | 83% (0.26) | 73% (0.21) | 0% (0.32) |
| G611A | 0% (0.41) | 0% (0.37) | 0% (0.31) |
Beyond reducing the flexibility of the loop, different alanine substitutions led to different hydrogen bonding patterns between tyrosine residues in the loop and the substrate (Table 2). Thus, in addition to generally decreasing flexibility the alanine mutants may restrict the loop to different conformations relative to the substrate. It would be tenuous to interpret these hydrogen bonding results too strongly in terms of catalysis since the simulations have a relatively short timescale (10-15 ns) and include a substrate-like molecule that would not necessarily mimic enzyme interactions in the transition state. For example, tyrosine residues in the loop of wild type TOP clearly have the ability to interact with the substrate during catalysis, although the loop sampled conformations farther from the substrate in the wild type simulation. Nonetheless, these results imply that conformational differences caused by different alanine substitutions could lead to differences in experimentally observed kinetic data, such as the increased activity of G603A towards MCA compared to the adjacent G604A mutation. As well, Tyr609 formed hydrogen bonds with substrate in several trajectories. It would be interesting for future studies to consider the possible role of this residue in catalysis in more detail.
Discussion
A major finding of the present study is that the bradykinin analogue mcaBk can still be efficiently cleaved after removal of the tyrosine hydroxyls of Y605 and Y612 from the wild type form of the enzyme, thus making this substrate distinct among the four substrates tested. This discovery helped reveal the primary role of glycine residues in the 599-611 loop in positioning the Tyr605 and Tyr612 residues needed for substrate hydrolysis. Additionally, our data indicate that Tyr605 is responsible for transition state stabilization by hydrogen bonding interactions with the substrate.
Role of Tyr605 and Tyr612 in catalysis
Activity assays and molecular modeling support a direct role for both Tyr605 and Tyr612 in peptide hydrolysis by TOP. Previous data demonstrating the crucial role of Tyr612 in cleaving the quenched fluorescent substrate MCA [16, 17] was corroborated and expanded upon in the present study with two additional physiologically related substrates, mcaNt and mcaGnRH1-9. Removal of the Tyr hydroxyl in the Tyr612Phe mutant resulted in 500-2000 fold decreases in kcat for these three substrates (see Table 1). Molecular modeling of the closed-form of TOP showed that the hydroxyl of Tyr612 is within H-bonding distance of the carbonyl carbon of the cleaved peptide bond. Tyr605 also seems to play a significant, though lesser role than Tyr612. The Tyr605Phe mutant suffered a 10-200 fold decrease in kcat for hydrolysis of MCA, mcaNt, and mcaGnRH1-9. In a previous study [16], Oliveira et al determined that Tyr605 drives substrate specificity via an interaction at the P1 residue of the bradykinin-based substrate Abz-GFSXFRQEDDnp. However no clear effect on kcat was observed and thus no direct catalytic role was assigned to Tyr605. From the present study, it appears that Tyr605 does play a significant role, as evidenced in the large decrease in kcat with MCA and mcaNt. It is possible that Tyr605 may position some substrates; without Tyr605 the peptide is no longer in the proper position with respect to Tyr612. Alternatively, Tyr605 may be more directly involved in catalysis, as evidenced by the changes seen in the kcat/Km with the single Tyr mutant. Molecular modeling and molecular dynamics indeed suggest that the Tyr605 hydroxyl is in close range to the carbonyl of the scissile peptide bond. Therefore, Tyr605 is probably also responsible for transition stabilization suggested previously for the Tyr612 residue [16]. This coordinated effort is similar to that of His231 and Tyr157 in thermolysin [21, 22]. His231 (analogous to Tyr612 in TOP) and Tyr157 (analogous to Tyr605 in TOP) work together in the transition state stabilization of this enzyme, both forming H-bonds to the transition state intermediate [21, 22]. In thermolysin, His231 plays the dominant role to Tyr157, the removal of which results in a ~200-fold decrease in activity. This is comparable to the relative roles Tyr612 and Tyr605 mutants of TOP play where Tyr612 suffered a 500-2000 fold decrease in activity relative to wild type, and Tyr605Phe a 10-200 fold decrease. This rotation and approach of hydrogen bonds by Tyr residues has been seen in other peptidases such as T. acidophilum aminopeptidase factor F3, Saccaromyces cervisiae, human dipeptidyl peptidase III (DPP III) among others, indicating similar transition state stabilizations during the catalytic event [21, 23-26].
Possibility that mcaBk could be cleaved by an open form of the enzyme
The most surprising finding from this study was that TOP does not require either Tyr605 or, more significantly, Tyr612 for significant activity towards the mcaBk substrate (see Table 1). Tyr612Phe caused only a slight increase in Km for this substrate. Virtually no change in the kinetic parameters for TOP with mcaBk was detected upon removal of the Tyr605 hydroxyl, especially when compared to the 10-100 fold change with the other substrates tested (see Table 1). While the enzyme did show a significant decrease in activity towards the mcaBk substrate when both Tyr605 and 612 hydroxyls were removed, it still retained considerable activity. kcat/Km for mcaBk with the double Tyr mutant was 0.20 uM-1s-1 (Table 1), comparable to the rate constants for mcaNt and mcaGnRH1-9 with wild type (0.28 and 0.37 uM-1s-1). Clearly, hydrolysis of mcaBk does not absolutely require these Tyr residues. This observation suggests: 1) the enzyme may not need to be in the closed conformation to process mcaBk and 2) the mechanism of cleavage of mcaBk is altered with respect to other substrates. The first suggestion is supported by the significant activity retained towards mcaBk in the presence of low concentrations (1-2 M) of urea (Figure 2). Previous fluorescence data implied that this concentration of urea favors either denaturation of domain II or at least an open conformation of the enzyme [20]. This finding is significant, as the open-closed hinge mechanism is likely to be the key factor in limiting substrate length.
Movement of flexible hinge regions to modulate the open-closed scenario has been demonstrated in a variety of metallopeptidases and their intermediate forms [27-29]. The majority of TOP substrates tested can only be hydrolyzed when the loop region is in the closed conformation that brings Tyr605 and 612 into the proper position. No other Tyr residues or possible H-bond donors are apparent in the structure of the TOP enzyme. Based on the structure of Carboxypeptidase Dcp [19], this complete closure of the crevice is needed for efficient catalysis because it causes the internal crevice to be inaccessible from outside. However, if TOP can remain in the open position for certain substrates, as suggested in this study with mcaBk, then it may be possible that under certain conditions this enzyme can cleave larger (>17 aa) substrates such as peptides that function in cell signaling [30].
The open/closed conformational change also opens the possibility for an additional mechanism to regulate TOP's activity. Certain Cys residues of TOP are known to be involved in thiol activation/S-glutathionylation, promoting an oligomerized enzyme with reduced enzyme activity [31-33]. It is possible that oxidation and the open-closed transition are connected, and that thiol oxidation forces the enzyme into a closed state.
Role of Gly residues of the 599-611 loop in positioning Tyr605 and Tyr612
Previous work has suggested that the flexible loop region of TOP is responsible for this enzyme's positioning of substrates for catalysis [15]. The present results clarify the primary role of the Gly residues of TOP to be positioning of Tyr605 and Tyr612. This is supported by the fact that hydrolysis of mcaBk, which changed very little when the Tyr residues were mutated, was also relatively unaffected by the mutation of Gly residues in the 599-611 loop (Table 1).
This is in contrast to the other substrates used in this study, all of which showed significant decreases in kcat upon the removal of either Tyr605 or Tyr612. Further, activities against MCA and mcaNt were affected to a significant degree by either the single or double Gly mutations in the loop. The Gly604Ala and Gly603Ala mutations as well as Tyr605Phe single mutation had no effect on activity towards mcaBk, while the change in activity of Gly611 towards mcaBk was mirrored by the small change in activity of the Tyr612Phe mutant. These results may point to a specific role for the Gly residues in positioning Tyr605 and Tyr612, and also suggest a coordinated role these loop glycine residues play in the selection of substrates. The MD simulations also support a role of these glycine residues in positioning the catalytic Tyr residues.
Previous work [16], in which Ala607 of the 599-611 substrate binding loop in TOP was changed to Gly (the corresponding residue in neurolysin), demonstrated that this residue may be important in governing the differences in substrate selection by these two enzymes. However, it seems unlikely that the residue in this position of the loop is responsible for allowing TOP to adopt an active conformation, since this position is not conserved between the two enzymes. Both enzymes bind and hydrolyze a diverse array of peptides, often at the same cleavage site. Rather, the evidence in this paper supports a role for the conserved Gly residues (599, 603, 604, 611) in the loop, particularly Gly603, in maintaining the plasticity of the active site and the full range of function of TOP. Most recently [30] potential new substrates adhering to the size specificity ascribed to TOP were described. These are consistent with the findings of the role of the Gly substrate binding loop.
To conclude, our study presents evidence that particular amino acids in the catalytic loop region of TOP are crucial for positioning important Tyr residues involved in the catalysis of physiological relevant peptides. Additionally, the mechanism for catalysis employed by TOP determines this enzyme's success with a wide variety of substrates.
Materials and methods
Reagents
The quenched fluorescent substrates MCA (7-methoxycoumarin-4-acetyl-Pro-Leu-Gly-Pro-Lys-dinitrophenol) and modified bradykinin, mcaBk (7-methoxycoumarin-4-acetyl)-[Ala7, Lys(dinitrophenol)9)]-bradykinin) were purchased from Bachem (King of Prussia, PA). Modified neurotensin (Mca-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Lys(Dnp)-OH) and mcaGnRH1-9 (Mca-Glu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-OH) were synthesized by AnaSpec (San Jose, CA). Tris (2-carboxyethyl)phosphine hydrocholoride (TCEP) was obtained from Pierce Chemical Co. (Rockford, IL). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Mutagenesis and protein expression
Site-directed mutagenesis of rat EP24.15 was performed on the expression vector pGEX-24.15 as a template [34]. Oligonucleotide primers were synthesized with mismatches, coding for the appropriate amino acid change following prokaryotic codon usage rules to obviate the use of rare codons. Mutations were performed using separate forward (Fw) and reverse (Rv) primers.
FwRepG611A=TACGACGCTCAGTACTATGCTTACTTGTGGAGTGAGGTG, RvRepG611A=CACCTCACTCCACAAGTAAGCATAGTACTGAGCGTCGTA; FwRepG603A=CTTTTGGCCACCTCGCTGCTGGCTACGACGCTCAGTAC RvRepG603A=GTACTGAGCGTCGTAGCCAGCAGCGAGGTGGCCAAAAG; FwRepG604A=GGCCACCTCGCTGGTGCCTACGACGCTCAGTAC, RvRepG604A=GTACTGAGCGTCGTAGGCACCAGCGAGGTGGCC; FwRepG599A=CAACATGCCAGCCACTTTTGCCCACCTCGCTGGTGGCTACG, RvRepG599A=CAACATGCCAGCCACTTTTGCCCACCTCGCTGGTGGCTACG. FwRepY605F=CCACCTCGCTGGTGGC TTCGACGCTCAGTACTATG, RvRepY605F=CATAGTACTGAGCGTCGAAGCCACCAGCGAGGTGG; FwRepY609F=GGCTACGACGCTCAGTTCTATGGCTACTTGTGG RvRepY609F=CCACAAGTAGCCATAGAACTGAGCGTCGTAGCC FwRepY612F=GCTCAGTACTATGGCTTCTTGTGGAGTGAGGTG, RvRepY612F=CACCTCACTCCACAAGAAGCCATAGTACTGAGC; FwRepG603P=CTTTTGGCCACCTCGCTCCCGGCTACGACGCTCAGTA, RvRepG603P=TACTGAGCGTCGTAGCCGGGAGCGAGGTGGCCAAAAG. All constructs were sequenced to ensure the correct mutation was created.
Assessment of purification to homogeneity, yield, and proper folding of expressed proteins was by native polyacrylamide gel electrophoresis (PAGE) on an 8 % gel under reducing conditions as previously described [35]. Yields of expressed protein were similar for all of the mutations.
To determine if gross structural alterations occurred during mutagenesis and subsequent protein expression, mutants were compared to wild type by CD spectroscopy. CD spectra were collected in the wavelength range of 300 nm to 185 nm at 1 nm intervals with a Jasco 715 spectropolarimeter (Jasco, Easton, MD). The instrument wavelength was checked with benzene vapor. Optical rotation was calibrated by measuring the ellipticity of d-10 camphorsulfonic acid at 192.5 and 290 nm. Measurements of optical ellipticity were made at 25° C using a 0.1 cm path length quartz cell. At least eight reproducible scans were collected for each sample. Buffer alone was used for a control blank in these experiments, and the averaged buffer spectrum was subtracted from each averaged protein spectrum. The contribution due to the polypeptide component alone, were similar for all of the mutations compared to the wild type protein.
Kinetic assays
Kinetic assays were performed as previously described [20]. Cleavage of the fluorogenic MCA [36], mcaBk, and mcaNt substrates were monitored by the increase in emission at 400 nm over time using a λexcitation of 325 nm. The mcaGnRH1-9 substrate was monitored by HPLC (Agilent 1100) using increase in peak area for the emission of Mca at 400 nm using a λexcitation of 325 nm. Assays were performed at least in duplicate at 23° C in 25 mM Tris/HCl at pH 7.8, containing 1 mM TCEP, 1 μM ZnCl2, and 10 % glycerol, adjusted to a conductivity of 12 mS/cm2 with NaCl.
Kinetic parameters were determined using a hyperbolic fit to the plot of substrate concentration vs. rate of product formation. All curve fitting procedures were performed using the program T-curve 2D (SPSS Inc, Chicago, IL.)
HPLC analysis
Products of the enzymatic reaction of wild type and mutant TOP with substrates MCA and mcaNt were analyzed using HPLC (Hewlett Packard 1090). The reaction mixture, 50 μl total volume in Tris buffer, contained either MCA (350 μM) and 0.4 μM enzyme, or mcaNt (100 μM) and 0.9 μM of Enzyme. A sample was taken at 0 min (before initiation of the reaction) and after reacting for 90 minutes at room temperature. Each reaction was terminated with the addition of equal volumes of 0.1 % trifluoroacetic acid in methanol.
A 20 μl aliquot of the reaction mixture was subjected to reverse-phase HPLC using a C18 3 μ column (150 mm × 4.6 mm; Alltech, Bannockburn, IL) at a flow rate of 1 ml/min with a linear gradient of 10-66 % acetonitrile in 0.1 % trifluoroacetic acid. Elution of substrates and products was monitored by absorbance at 330 nm [20].
Modeling and molecular dynamics (MD) simulations
An initial model of TOP with bound MCA (Pro-Leu-Gly-Pro) substrate was based on the TOP crystal structure [PDB ID # 1s4b] [14] with the loop conformation modified analogous to a structure of dipeptidyl carboxypeptidase [Dcp; PDB ID # 1y79] that has product bound in the active site [19]. Specifically, a model of the closed-form of TOP was generated by clipping TOP at the division between domains I and II (residues Leu156, Val351, Gln544 and Glu616), and separately fitting domains I and II to the structure of Dcp. Identification of the appropriate clipping points was aided by using the Alternate Domain Fit tool from the suite of tools within the SWISS-PdbViewer (http://www.expasy.org/spdbv/) software version 3.7 and 3.9b2 [37]. The fitting procedure could be accomplished by two methods with similar overall results. Fitting the entirety of the domains using the Bestfit with structure alignment resulted in a total RMS backbone deviation of 1.52 Å. After fitting the domains, the TOP backbone was religated. Alternatively, the Zn and active site residues could be overlayed to fit domains I and the conserved His600, Tyr605, and Tyr612 of TOP used to fit domains II. The second procedure resulted in a similar RMS backbone deviation of 1.51 Å with a slightly better fit of the active site residues. G603A and G604A mutations were made to this minimized model.
MD simulations of wt, G599A, G603A, G604A, and G611A TOP were performed and analyzed with the GROMACS 3.3.1 suite [38]. TOP models were solvated in a cubic box of 41111 SPC water molecules with Na+ and Cl- ions to neutralize the system and provide a salt concentration of 100 mM. These solvated models were subjected to 50 steps of steepest descents minimization and were heated to 298 K over 20 ps. Initial position restraints on all Cα atoms were released in gradual steps over the first 275 ps of the 10 ns trajectories. Temperature (298 K) and pressure (1 bar) were controlled using Berendsen coupling protocols with time constants of 0.1 ps and 1.0 ps, respectively [39]. Electrostatic and Lennard-Jones interactions were cutoff at 10 Å with long range electrostatics computed using PME [40]. Bonds were constrained with the LINCS algorithm [41]. Distance restraints analogous to those used for other metalloenzyme simulations [42] were used to maintain interactions between Zn2+ and His473, His477, and Glu502. Properties were averaged over the last ns of trajectories, and hydrogen bonds were defined geometrically with a donor-acceptor distance cutoff of 3.5 Å and an angle cutoff of 30°.
Acknowledgements
We thank Meera Srikanthan, Lindsay Kua, Connie Wu, Susan Kim, and Sabina Khan for technical assistance. We also thank Didem Vardar-Ulu for technical advice. This study was supported by a Howard Hughes Medical Institute Undergraduate Education Program Grant, a National Science Foundation (NSF) Research Experiences for Undergraduate Award to Wellesley College (CHE-0353813), the National Institute for Neurological Disorders and Stroke (NS39892) of the National Institutes of Health (MJG), and the Camille and Henry Dreyfus Supplemental Research Grant under the Scholar/Fellow Program (JAS).
Abbreviations
- TOP
thimet oligopeptidase
- MCA
7-methoxycoumarin-4-acetyl-Pro-Leu-Gly-Pro-Lys-dinitrophenol
- TCEP
Tris(2-Carboxyethyl) Phosphine Hydrochloride
References
- 1.Rawlings ND, Barrett AJ. Evolutionary Families of Peptidases. Biochemical Journal. 1993;290:205–218. doi: 10.1042/bj2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierotti A, Dong KW, Glucksman MJ, Orlowski M, Roberts JL. Molecular-Cloning and Primary Structure of Rat Testes Metalloendopeptidase Ec 3.4.24.15. Biochemistry. 1990;29:10323–10329. doi: 10.1021/bi00497a006. [DOI] [PubMed] [Google Scholar]
- 3.Dauch P, Vincent JP, Checler F. Molecular-Cloning and Expression of Rat-Brain Endopeptidase-3.4.24.16. Journal of Biological Chemistry. 1995;270:27266–27271. doi: 10.1074/jbc.270.45.27266. [DOI] [PubMed] [Google Scholar]
- 4.Brown CK, Madauss K, Lian W, Beck MR, Tolbert WD, Rodgers DW. Structure of neurolysin reveals a deep channel that limits substrate access. Proc Natl Acad Sci U S A. 2001;98:3127–32. doi: 10.1073/pnas.051633198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ray K, Hines CS, Rodgers DW. Mapping sequence differences between thimet oligopeptidase and neurolysin implicates key residues in substrate recognition. Protein Sci. 2002;11:2237–46. doi: 10.1110/ps.0216302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dando PM, Brown MA, Barrett AJ. Human thimet oligopeptidase. Biochem J. 1993;294(Pt 2):451–7. doi: 10.1042/bj2940451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rioli V, Gozzo FC, Heimann AS, Linardi A, Krieger JE, Shida CS, Almeida PC, Hyslop S, Eberlin MN, Ferro ES. Novel natural peptide substrates for endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. J Biol Chem. 2003;278:8547–55. doi: 10.1074/jbc.M212030200. [DOI] [PubMed] [Google Scholar]
- 8.Smith AI, Lew RA, Shrimpton CN, Evans RG, Abbenante G. A novel stable inhibitor of endopeptidases EC 3.4.24.15 and 3.4.24.16 potentiates bradykinin-induced hypotension. Hypertension. 2000;35:626–630. doi: 10.1161/01.hyp.35.2.626. [DOI] [PubMed] [Google Scholar]
- 9.Smith AI, Shrimpton CN, Norman UM, Clarke IJ, Wolfson AJ, Lew RA. Neuropeptidases regulating gonadal function. Biochem Soc Trans. 2000;28:430–4. [PubMed] [Google Scholar]
- 10.Akil H, Watson SJ, Young E, Lewis ME, Khachaturian H, Walker JM. Endogenous Opioids - Biology and Function. Annual Review of Neuroscience. 1984;7:223–255. doi: 10.1146/annurev.ne.07.030184.001255. [DOI] [PubMed] [Google Scholar]
- 11.Portaro FC, Gomes MD, Cabrera A, Fernandes BL, Silva CL, Ferro ES, Juliano L, de Camargo AC. Thimet oligopeptidase and the stability of MHC class I epitopes in macrophage cytosol. Biochem Biophys Res Commun. 1999;255:596–601. doi: 10.1006/bbrc.1999.0251. [DOI] [PubMed] [Google Scholar]
- 12.Silva CL, Portaro FC, Bonato VL, de Camargo AC, Ferro ES. Thimet oligopeptidase (EC 3.4.24.15), a novel protein on the route of MHC class I antigen presentation. Biochem Biophys Res Commun. 1999;255:591–5. doi: 10.1006/bbrc.1999.0250. [DOI] [PubMed] [Google Scholar]
- 13.Kim SI, Pabon A, Swanson TA, Glucksman MJ. Regulation of cellsurface major histocompatibility complex class I expression by the endopeptidase EC3.4.24.15 (thimet oligopeptidase) Biochem J. 2003;375:111–20. doi: 10.1042/BJ20030490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray K, Hines CS, Coll-Rodriguez J, Rodgers DW. Crystal structure of human thimet oligopeptidase provides insight into substrate recognition, regulation, and localization. J Biol Chem. 2004;279:20480–9. doi: 10.1074/jbc.M400795200. [DOI] [PubMed] [Google Scholar]
- 15.Machado MF, Rioli V, Dalio FM, Castro LM, Juliano MA, Tersariol IL, Ferro ES, Juliano L, Oliveira V. The role of Tyr605 and Ala607 of thimet oligopeptidase and Tyr606 and Gly608 of neurolysin in substrate hydrolysis and inhibitor binding. Biochem J. 2007;404:279–88. doi: 10.1042/BJ20070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliveira V, Araujo MC, Rioli V, de Camargo AC, Tersariol IL, Juliano MA, Juliano L, Ferro ES. A structure-based site-directed mutagenesis study on the neurolysin (EC 3.4.24.16) and thimet oligopeptidase (EC 3.4.24.15) catalysis. FEBS Lett. 2003;541:89–92. doi: 10.1016/s0014-5793(03)00310-7. [DOI] [PubMed] [Google Scholar]
- 17.Sigman JA, Edwards SR, Pabon A, Glucksman MJ, Wolfson AJ. pH dependence studies provide insight into the structure and mechanism of thimet oligopeptidase (EC 3.4.24.15) FEBS Letters. 2003;545:224–228. doi: 10.1016/s0014-5793(03)00548-9. [DOI] [PubMed] [Google Scholar]
- 18.Holland DR, Hausrath AC, Juers D, Matthews BW. Structural-Analysis of Zinc Substitutions in the Active-Site of Thermolysin. Protein Science. 1995;4:1955–1965. doi: 10.1002/pro.5560041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Comellas-Bigler M, Lang R, Bode W, Maskos K. Crystal structure of the E. coli dipeptidyl carboxypeptidase Dcp: further indication of a ligand-dependent hinge movement mechanism. J Mol Biol. 2005;349:99–112. doi: 10.1016/j.jmb.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 20.Sigman JA, Patwa TH, Tablante AV, Joseph CD, Glucksman MJ, Wolfson AJ. Flexibility in substrate recognition by thimet oligopeptidase as revealed by denaturation studies. Biochem J. 2005;388:255–61. doi: 10.1042/BJ20041481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marie-Claire C, Ruffet E, Tiraboschi G, Fournie-Zaluski MC. Differences in transition state stabilization between thermolysin (EC 3.4.24.27) and neprilysin (EC 3.4.24.11) Febs Letters. 1998;438:215–219. doi: 10.1016/s0014-5793(98)01267-8. [DOI] [PubMed] [Google Scholar]
- 22.Holden HM, Matthews BW. The Binding of L-Valyl-L-Tryptophan to Crystalline Thermolysin Illustrates the Mode of Interaction of a Product of Peptide Hydrolysis. Journal of Biological Chemistry. 1988;263:3256–3260. doi: 10.2210/pdb3tmn/pdb. [DOI] [PubMed] [Google Scholar]
- 23.Yiallouros I, Berkhoff EG, Stocker W. The roles of Glu93 and Tyr149 in astacin-like zinc peptidases. Febs Letters. 2000;484:224–228. doi: 10.1016/s0014-5793(00)02163-3. [DOI] [PubMed] [Google Scholar]
- 24.Kyrieleis OJ, Goettig P, Kiefersauer R, Huber R, Brandstetter H. Crystal structures of the tricorn interacting factor F3 from Thermoplasma acidophilum, a zinc aminopeptidase in three different conformations. J Mol Biol. 2005;349:787–800. doi: 10.1016/j.jmb.2005.03.070. [DOI] [PubMed] [Google Scholar]
- 25.Thompson MW, Archer ED, Romer CE, Seipelt RL. A conserved tyrosine residue of Saccharomyces cerevisiae leukotriene A(4) hydrolase stabilizes the transition state of the peptidase activity. Peptides. 2006;27:1701–1709. doi: 10.1016/j.peptides.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Salopek-Sondi B, Vukelic B, Spoljaric J, Simaga S, Vujaklija D, Makarevic J, Jajcanin N, Abramic M. Functional tyrosine residue in the active center of human dipeptidyl peptidase III. Biological Chemistry. 2008;389:163–167. doi: 10.1515/BC.2008.021. [DOI] [PubMed] [Google Scholar]
- 27.Okoniewska M, Tanaka T, Yada RY. The pepsin residue glycine-76 contributes to active-site loop flexibility and participates in catalysis. Biochem J. 2000;349:169–77. doi: 10.1042/0264-6021:3490169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kempner ES. Movable Lobes and Flexible Loops in Proteins - Structural Deformations That Control Biochemical-Activity. Febs Letters. 1993;326:4–10. doi: 10.1016/0014-5793(93)81749-p. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka T, Yamaguchi H, Kato H, Nishioka T, Katsube Y, Oda J. Flexibility impaired by mutations revealed the multifunctional roles of the loop in glutathione synthetase. Biochemistry. 1993;32:12398–404. doi: 10.1021/bi00097a018. [DOI] [PubMed] [Google Scholar]
- 30.Cunha FM, Berti DA, Ferreira ZS, Klitzke CF, Markus RP, Ferro ES. Intracellular peptides as natural regulators of cell signaling. J Biol Chem. 2008 doi: 10.1074/jbc.M801252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shrimpton CN, Glucksman MJ, Lew RA, Tullai JW, Margulies EH, Roberts JL, Smith AI. Thiol activation of endopeptidase EC 3.4.24.15. A novel mechanism for the regulation of catalytic activity. J Biol Chem. 1997;272:17395–9. doi: 10.1074/jbc.272.28.17395. [DOI] [PubMed] [Google Scholar]
- 32.Demasi M, Piassa Filho GM, Castro LM, Ferreira JC, Rioli V, Ferro ES. Oligomerization of the cysteinyl-rich oligopeptidase EP24.15 is triggered by S-glutathionylation. Free Radic Biol Med. 2008;44:1180–90. doi: 10.1016/j.freeradbiomed.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Sigman JA, Sharky ML, Walsh ST, Pabon A, Glucksman MJ, Wolfson AJ. Involvement of surface cysteines in activity and multimer formation of thimet oligopeptidase. Protein Eng. 2003;16:623–8. doi: 10.1093/protein/gzg073. [DOI] [PubMed] [Google Scholar]
- 34.Glucksman M, Roberts JL. Strategies for characterizing, cloning and expressing soluble endopeptidases. Methods in Neurosciences. 1995;23:296–316. [Google Scholar]
- 35.Cummins PM, Pabon A, Margulies EH, Glucksman MJ. Zinc coordination and substrate catalysis within the neuropeptide processing enzyme endopeptidase EC 3.4.24.15. Identification of active site histidine and glutamate residues. J Biol Chem. 1999;274:16003–9. doi: 10.1074/jbc.274.23.16003. [DOI] [PubMed] [Google Scholar]
- 36.Wolfson AJ, Shrimpton CN, Lew RA, Smith AI. Differential activation of endopeptidase EC 3.4.24.15 toward natural and synthetic substrates by metal ions. Biochem Biophys Res Commun. 1996;229:341–8. doi: 10.1006/bbrc.1996.1803. [DOI] [PubMed] [Google Scholar]
- 37.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 38.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Journal of Molecular Modeling. 2001;7:306–317. [Google Scholar]
- 39.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-Dynamics with Coupling to an External Bath. Journal of Chemical Physics. 1984;81:3684–3690. [Google Scholar]
- 40.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. Journal of Chemical Physics. 1993;98:10089–10092. [Google Scholar]
- 41.Hess B, Bekker H, Berendsen HJC, Fraaije J. LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry. 1997;18:1463–1472. [Google Scholar]
- 42.Manzetti S, McCulloch DR, Herington AC, van der Spoel D. Modeling of enzyme-substrate complexes for the metalloproteases MMP-3, ADAM-9 and ADAM-10. Journal of Computer-Aided Molecular Design. 2003;17:551–565. doi: 10.1023/b:jcam.0000005765.13637.38. [DOI] [PubMed] [Google Scholar]