

Abstract
Although apoptosis triggered by ultraviolet B (UVB)-mediated activation of the c-Jun N-terminal kinase (JNK) pathway is mediated by both intrinsic and extrinsic pathways, the mechanism of initiation of JNK activation remains obscure. Here, we report the characterization of the JNK-interacting protein 3 (JIP-3) scaffolding protein as an interacting partner of Rho-associated kinase 1 (ROCK1), as determined by tandem affinity protein purification. Upon UVB-induced stress in keratinocytes, ROCK1 was activated, bound to JIP-3, and activated the JNK pathway. Moreover, phosphorylation of JIP-3 by ROCK1 was crucial for the recruitment of JNK. Inhibition of the activity of ROCK1 in keratinocytes resulted in decreased activation of the JNK pathway and thus a reduction in apoptosis. ROCK1+/-mice exhibited decreased UVB-mediated activation of JNK and apoptosis relative to wild-type mice. Our findings present a new molecular mechanism by which ROCK1 functions as a UVB sensor that regulates apoptosis, an important event in the prevention of skin cancer.
INTRODUCTION
Mounting evidence implicates ultraviolet (UV) radiation as an important environmental carcinogen involved in the development of the most common skin cancers (1). UVB light induces the formation of cyclobutane pyrimidine dimers that cause DNA damage and result in chromatid breaks (2). Failure to initiate apoptosis of the affected cells results in uncontrolled cell proliferation through the accumulation of permanent mutations in genomic DNA (3). In recent years, there has been substantial progress in our understanding of UV radiation-mediated signal transduction pathways in mammalian cells. It is well established that UVB triggers the activation of members of the mitogen-activated protein kinase (MAPK) family, including extracellular signal-regulated kinase (ERK), JNK, and p38 MAPK (4). JNK is activated primarily by cytokines or exposure to environmental stresses such as UV irradiation. Components of the JNK pathway can be organized into signaling complexes by scaffold proteins such as the JNK-interacting proteins (JIPs) (5).
Rho-associated kinase (ROCK) was first identified as the serine/threonine kinase that binds to guanosine triphosphate (GTP)-bound RhoA, a small guanosine triphosphatase (6, 7). ROCK functions as a versatile kinase, phosphorylating various substrates such as myosin light chain (MLC) phosphatase (8), LIM kinase 3 (9), phosphatase and tensin homolog deleted from chromosome 10 (PTEN) (10), insulin receptor substrate (IRS) (11, 12), and ezrin/radixin/moesin (ERM) proteins (13). To date, two isoforms of ROCK have been cloned, ROCK1 and ROCK2 (14). Each isoform consists of a kinase domain near the N terminus of the protein, followed by a coiled-coil domain, a Rho-binding domain, a cysteine-rich domain located in the pleckstrin homology domain, and a zinc finger-binding domain. Both isoforms are about 160 kD with 92% homology at the kinase domain. The interaction of GTP-bound RhoA with the C terminus of ROCK creates an open conformation and, thus, an activated state of the kinase. Some studies have presented alternative means to activate ROCK1 that include binding of ROCK1 to arachidonic acid, oligomerization of ROCK1, or cleavage of ROCK1 by caspase-3 during apoptosis or differentiation (15). It is well known that ROCKs affect such cellular processes as apoptosis, adhesion, migration, proliferation, and metabolism (6-9, 11, 14, 16).
Because ROCK is involved in different biological processes, it is an important therapeutic target for the treatment of various human diseases including cancers, cardiovascular diseases, and neurological disorders. The function of ROCK in apoptosis has been extensively examined. Inhibition of ROCK1 reduces apoptosis of cardiomyocytes during ischemia-reperfusion injury (17), promotes embryonic stem cell survival (18), and inhibits both androgen-induced apoptosis of prostate cancer cells (19) and genotoxic stress-induced cell death (20). In various animal disease models, inhibition of ROCK promotes survival effects, including reduced apoptosis, which is accompanied in most cases by reduced inflammatory responses (21). ROCK also mediates the production of reactive oxygen species, which can in turn induce apoptosis (22, 23). Although ROCK plays an important role in apoptosis, the mechanism of its action is obscure.
Through tandem affinity protein (TAP) purification, we found that ROCK1 bound to JIP-3 and that this interaction was enhanced by UVB irradiation. Inhibition of the activity of ROCK1 during exposure to UVB light decreased the phosphorylation of JNK, which suggests that ROCK1 plays an important role in JNK-mediated cellular responses. ROCK1 phosphorylated JIP-3 in vitro and in vivo. Epidermis from ROCK1+/- mice was more resistant to UVB-induced cell death than was epidermis from wild-type (WT) mice. Therefore, we present evidence that ROCK1 acts as a key upstream regulator of the JIP-3 to JNK signaling pathway and as a sensor of UVB-induced cellular stress that is critical in mediating the intrinsic cell death pathway. These results suggest that ROCK1 signaling plays an important role in the prevention of skin cancer.
RESULTS
Identification of JIP-3 as a substrate of ROCK1
We proposed to elucidate the function of ROCK1 by studying its interacting partners. To understand how ROCK1 is involved in various cellular processes, we used the TAP purification approach (24) to identify cellular factors that interacted with ROCK1 upon UVB-induced stress. We expressed FLAG-hemagglutinin (HA)-double-tagged human full-length ROCK1 in 293ET cells. Cell extracts were subjected to sequential purification with anti-FLAG and anti-HA antibody resins. Several polypeptides, including filaggrin 2, insulysin, JIP-3, the arginine N-methyltransferase SKB1, and α-actinin, were identified by mass spectrometry as binding partners of ROCK1 (Fig. 1A, left panel). Among these potential ROCK1-interacting proteins, JIP-3 is of great interest. As a scaffold protein that promotes the activation of JNK, JIP-3 may serve as a mediator connecting ROCK1 and JIP-3 to the JNK signaling pathway in response to cellular stress (5).
Fig. 1.
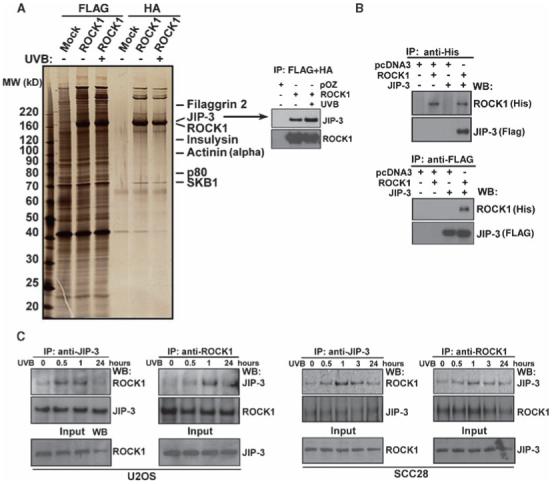
ROCK1 interacts with JIP-3, and UVB irradiation enhances this interaction. (A) Tandem affinity purification (TAP) of ROCK1. Cellular extracts from 293ET cells transiently transfected with FLAG-HA-tagged ROCK1, with or without UVB (30 mJ/cm2) irradiation, were sequentially immunoprecipitated with anti-FLAG and anti-HA antibody-conjugated affinity resins (lanes 2, 3, 5, and 6). The ROCK1-associated polypeptides were visualized by silver staining. Mock-transfected 293ET cells were used as a control (lanes 1 and 4). Selective data from mass spectrometry analysis are shown on the right. The right panel shows detection of JIP-3 in the TAP-purified samples by Western blotting. (B) Coimmunoprecipitation of JIP-3 by ROCK1. His-tagged ROCK1 and FLAG-tagged JIP-3 were transfected in 293ET cells. Immunoprecipitation (IP) and Western blotting (WB) were performed 36 hours after transfection as indicated. (C) Endogenous association between ROCK1 and JIP-3 in U2OS cells and SCC28 cells was analyzed by coimmunoprecipitation experiments and Western blots were performed for the indicated proteins. Cells were irradiated with UVB (30 mJ/cm2) for the indicated time intervals.
We first confirmed the presence of JIP-3 in ROCK1 purifications by Western blotting and found a slight increase in the abundance of JIP-3 in UVB-treated cells (Fig. 1A, right panel). The interaction between JIP-3 and ROCK1 was additionally confirmed by reciprocal immunoprecipitations from 293ET cells that were transiently transfected with His-ROCK1 and FLAG-tagged JIP-3 (FLAG-JIP-3) (Fig. 1B). To map the JIP-3-binding region(s) of ROCK1, we carried out pull-down experiments with constructs expressing different truncated forms of ROCK1, along with FLAG-JIP-3 or empty vector (pcDNA3), transfected into 293ET cells. JIP-3 bound to the N-terminus of ROCK1 containing the kinase domain (fig. S1), supporting our TAP and immunoprecipitation studies. To determine whether these two proteins interacted in vivo and whether UVB irradiation had any effect on the interaction, we performed coimmunoprecipitation assays with anti-ROCK1 and anti-JIP-3 antibodies on UVB-treated U2OS and SCC28 cells. We found that endogenous ROCK1 and JIP-3 interacted with each other and that this interaction was enhanced in both cell lines after UVB irradiation (Fig. 1C).
Because JIP-3 interacted with ROCK1, we determined whether JIP-3 was a target of its kinase activity. ROCK1 phosphorylates its substrates at the consensus sequences R/KXS/T or R/KXXS/T, in which the position of Arg (R) or Lys (K) residues is critical (15, 16, 25). We assessed ROCK1 activity by measuring the phosphorylation of the MYPT1 substrate protein in response to UVB treatment. UVB irradiation increased the phosphorylation of MYPT1 by more than a factor of 2 in human normal primary keratinocytes, 293ET cells, and SCC28 cells relative to that in untreated cells (Fig. 2A). We then measured the phosphorylation of FLAG-tagged JIP-3 in vitro by incubating recombinant ROCK1 with FLAG-JIP-3 purified from cell extracts. There was a striking increase in ROCK1-mediated phosphorylation of JIP-3 (Fig. 2B, right panel, lane 9) after 30 min incubation, and this effect was blocked by a ROCK inhibitor, Y-27632 (Fig. 2B, right panel, lane 10). Conversely, the phosphorylated form of JIP-3 was not detected in control FLAG-bound immunoprecipitated samples. Tandem mass spectrometry was performed to map the Ser/Thr phosphorylation sites in JIP-3. We found three Ser phosphorylation sites in JIP-3:Ser318, Ser368, and Ser369 from the phosphorylated peptides RDSRNMEVQVTQE (the tryptic peptide encompassing residues 316 to 328), and RTGSSPTQGIVNK (the tryptic peptide encompassing residues 365 to 377), respectively (Fig. 2B, lower panel).
Fig. 2.
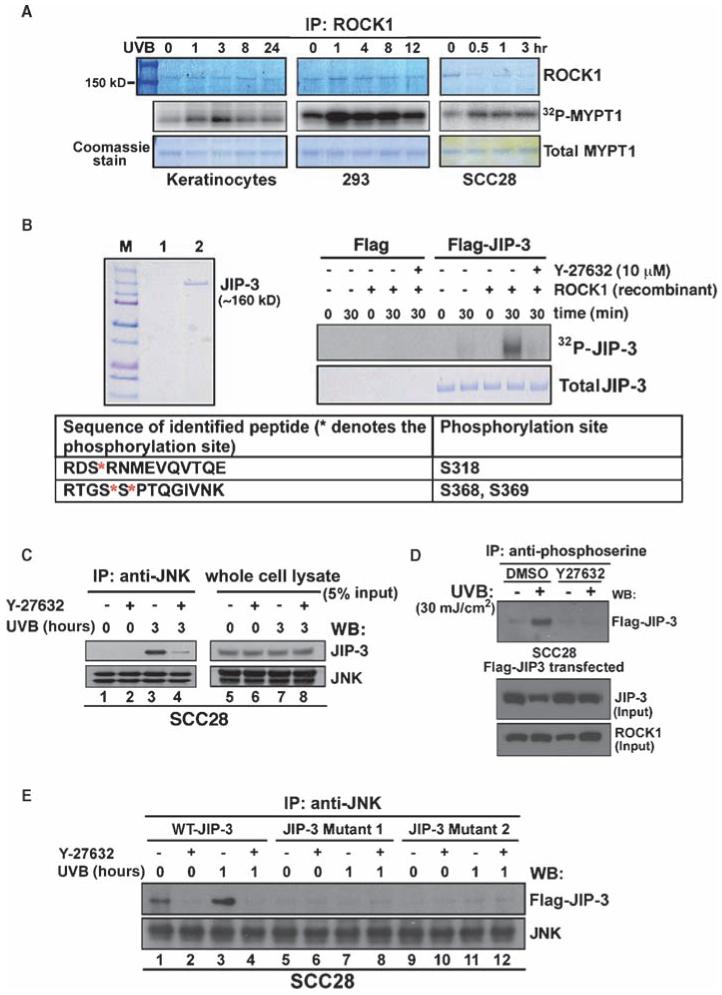
ROCK1-mediated phosphorylation of JIP-3 regulates the recruitment of JNK to JIP-3 upon UVB-induced stress. (A) An increase in ROCK1-mediated phosphorylation of MYPT1 in response to UVB irradiation. Normal human primary keratinocytes, 293ET cells, and SCC28 cells were irradiated with UVB for the indicated time points. Cell lysates were subjected to immunoprecipitation with an anti-ROCK1 antibody. The immunoprecipitated pellets were assayed for ROCK1-mediated phosphorylation of the substrate recombinant MYPT1. [32P]MYPT1 was autoradiographed, and total MYPT1 and precipitated ROCK1 proteins were stained with Coomassie blue. (B) Phosphorylation of JIP-3 by ROCK1. Purified FLAG-tagged JIP-3 (lane 2) and the control FLAG bead eluate (lane 1) were analyzed by SDS-PAGE and visualized by Coomassie blue staining (left panel). Ten micrograms of the JIP-3 protein was incubated with recombinant ROCK1 in the presence of 10 μCi [γ-32P]ATP, with or without Y-27632 (10 μM). Phosphorylated Ser residues in JIP-3 were identified by MALDI-TOF and tandem mass spectrometry; an asterisk denotes the phosphorylated residues. (C) Effect of ROCK inhibitor on the association between JNK and JIP-3. SCC28 cells were pretreated with a ROCK inhibitor, Y-27632 (10 μM), and then exposed to UVB irradiation (30 mJ/cm2). JNK was immunoprecipitated and complexes were analyzed by Western blotting with antibodies against JIP-3 and total JNK. (D) Inhibition of the kinase activity of ROCK1 prevents UVB-induced phosphorylation of JIP-3. FLAG-JIP-3-transfected SCC28 cells were treated with either dimethyl sulfoxide (DMSO) or Y-27632 (10 μM) and were exposed to UVB irradiation for 3 hours. Total Ser-phosphorylated proteins were immunoprecipitated with an antiphosphoserine antibody. Precipitated immunocomplexes (and input proteins) were analyzed by Western blotting with an anti-FLAG antibody. (E) Mutations of Ser residues in the phosphorylation sites to Ala residues inhibited the interaction between JIP-3 and JNK that occurred upon UVB irradiation. The JIP-3 mutant constructs used for this study included WT-JIP-3, JIP-3 Mutant 1(Ser318→Ala), and JIP-3 Mutant 2 (Ser368→Ala and Ser369→Ala).
Formation of the ROCK1-JIP-3 complex was confirmed by glycerol-gradient sedimentation experiments with partially purified His-tagged ROCK1 from 293ET cells. JIP-3 and ROCK1 peaked in fraction 12 (fig. S2), and cosedimentation of JNK, RhoA, and ROCK1 was observed in fractions 15 to 17, suggesting that ROCK1 and JIP-3 formed a complex with a link to JNK. We next tested the hypothesis that ROCK1-mediated phosphorylation of JIP-3 is required for the interaction between JIP-3 and JNK. We found that UVB irradiation stimulated the JNK-JIP-3 interaction in SCC28 cells (Fig. 2C, left panel, lane 3); however, inhibition of ROCK1 with Y-27632 dramatically reduced this interaction (Fig. 2C, left panel, lane 4). Furthermore, JIP-3 was phosphorylated in response to UVB irradiation in cells transfected with FLAG-JIP-3 (Fig. 2D), and JIP-3 phosphorylation at Ser residues was inhibited by Y-27632 (Fig. 2D), thus confirming JIP-3 as a ROCK1 substrate in vivo. The functional importance of the newly identified phosphorylation sites of JIP-3 was tested by generating point mutations of the target Ser residues in JIP-3. These mutations prevented the formation of interactions between the mutated JIP-3 proteins and JNK regardless of exposure to UVB radiation (Fig. 2E, lanes 5 to 12). Together, these data indicate that ROCK1 binds to JIP-3 and phosphorylates JIP-3 in response to UVB-induced stress, thus providing a biochemical route by which JNK can be activated.
ROCK1 mediates the activation of JNK and its downstream signaling after UVB-induced stress
JIP-3 is implicated in apoptosis by regulating JNK activation and its downstream signaling (5, 26-28). Activation of JNK is a critical regulator of various aspects of mammalian physiology, including cell proliferation, cell survival, DNA repair, and autophagy (4, 27, 29-32). JNK undergoes dynamic phosphorylation when mammalian cells are exposed to DNA-damaging stress, including UVB irradiation (4, 33). Previous studies showed that ROCK1-mediated apoptosis is one of the apoptotic pathways triggered during genotoxic stress (20). Because the UVB-enhanced interaction between ROCK1 and JIP-3 was crucial for the recruitment of JNK, we investigated whether the ROCK1-JIP-3 complex accounts for sustained JNK activation and apoptosis. First, we evaluated the role of ROCK1 in JNK downstream signaling and the JNK-mediated cell death response. Knockdown of ROCK1 by short hairpin RNA (shRNA) substantially decreased the abundance of phosphorylated JNK (the activated form of JNK) in UVB-exposed cell lines, including U2OS, 293ET, and HaCaT cells, relative to that in control shRNA-transfected cells (Fig. 3A). Inhibition of ROCK1 did not, however, affect other stress-responsive kinases such as ERK and p38 (Fig. 3A, left panel). As predicted, knockdown of JIP-3 reduced UVB-induced JNK phosphorylation in HaCaT cells (Fig. 3A) relative to that in control small interfering RNA (siRNA)-transfected cells. Furthermore, treatment of cells with Y-27632 before UVB exposure resulted in a decrease in the abundance of phosphorylated JNK (most evidently JNK2) and JNK targets such as Jun and γ-H2AX (Fig. 3B). Inhibition of ROCK1 thus resulted in attenuation of the signaling pathway downstream of JNK in response to UVB-induced stress.
Fig. 3.
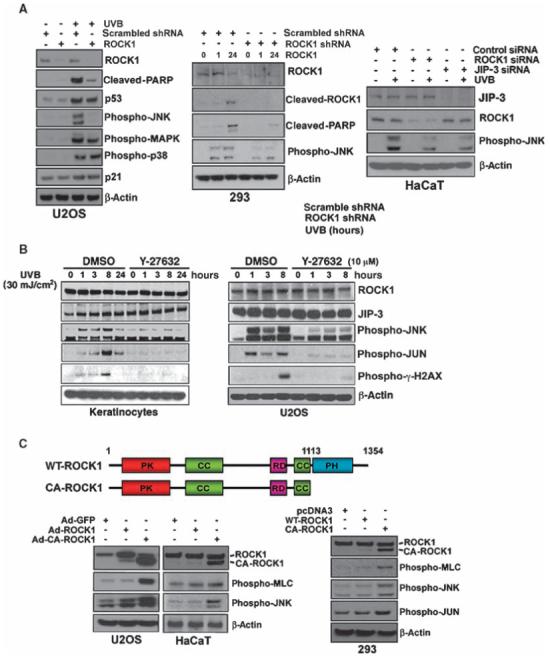
ROCK1 acts as an activator of the pathway downstream of JNK in response to UVB-induced stress. (A) Depletion of ROCK1 abrogates the activation of the UVB-induced JNK pathway. U2OS and 293ET cells were transfected with either scrambled shRNA or ROCK1-specific shRNA followed by UVB irradiation (30 mJ/cm2). HaCaT cells were transfected with scrambled or ROCK1-specific siRNA and were then irradiated by UVB (30 mJ/cm2). Transfected cell extracts were analyzed by Western blotting with the indicated antibodies. (B) Effect of the ROCK1 inhibitor, Y-27632, on UVB-induced JNK pathway activation in normal human primary keratinocytes and U2OS cells. (C) Constitutively active ROCK1 increased the abundance of phosphorylated JNK and its target, phosphorylated c-Jun. Adenoviruses (Ad) or expression vectors expressing WT-ROCK1 or CA-ROCK1 (constitutively active) were infected or transfected into U2OS, HACAT, and 293ET cells. Cell extracts were analyzed by Western blotting with antibodies against ROCK1, phospho-γH2AX, phospho-MLC, phosphoJNK, phospho-JUN, and β-actin.
To further study the consequence of ROCK1 activation on JNK activation, we generated a constitutively active, 130-kD, cleaved mutant ROCK1 (CA-ROCK1) (20, 34). U2OS, HaCaT, and 293ET cells were either transfected or infected with adenoviruses expressing CA-ROCK1 or WT-ROCK1. Overexpression of CA-ROCK1 resulted in a marked increase in the phosphorylation of JNK and the ROCK1 target MLC relative to that in cells expressing WT-ROCK1 (Fig. 3C). We next verified whether depletion or inhibition of ROCK1 modulated UVB-induced apoptosis. Depletion of ROCK1 caused a considerable decrease in UVB-mediated apoptosis as compared with that observed in control shRNA-transfected cells (fig. S3A). We also studied whether Y-27632 could affect the UVB-induced apoptotic response. Y-27632 prevented both the activation of JNK and UVB-induced apoptosis (fig. S3B). Overexpression of ROCK1 in ROCK1-depleted cells overcame the effect of inhibition of ROCK1 activity and resulted in an increase in UVB-induced apoptosis and the abundance of phosphorylated JNK relative to that in cells not reconstituted with ROCK1 (fig. S4).
Activation of the ROCK1→JIP-3→JNK axis by UVB irradiation induces the intrinsic apoptosis pathway
We examined the effect of depletion of ROCK1 on the activation of three major caspases (caspase-3, caspase-8, and caspase-9) to determine whether UVB-induced apoptosis mediated by the ROCK1→JIP-3→JNK pathway functioned through the intrinsic or extrinsic apoptosis pathway. Knockdown of ROCK1 in HaCaT cells notably inhibited the activities of caspase-3 and caspase-9, but not caspase-8, in response to UVB irradiation (Fig. 4A). We also observed that knockdown of ROCK1 by shRNA resulted in the inhibition of the release of cytochrome c from mitochondria upon UVB irradiation (Fig. 4B). These results strongly suggest that UVB-induced activation of ROCK1 and the resulting JIP-3-mediated activation of JNK promoted apoptosis through the intrinsic mitochondrial pathway.
Fig. 4.
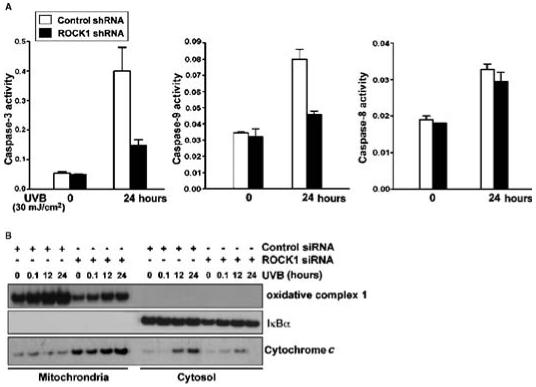
Effect of ROCK1 on the intrinsic cell death pathway. (A)Caspase-3, caspase-8, and caspase-9 activity assays were performed in cell lysates from control or ROCK1 knockdown HaCaT cells after 24 hours of UVB treatment. The results for the caspase assays are presented as the mean ± SEM (n = 3). (B) Control and ROCK1 knockdown HaCaT cells were irradiated with UVB at 30 mJ/cm2, harvested at 0 and 24 hours, and whole lysates were fractionated. Fractions were assessed for the presence of cytochrome c, oxidative complex 1 (mitochondrial fraction), and IκBα (cytosolic fraction) by Western blotting analysis.
ROCK1 is essential for UVB-induced apoptosis in mouse epidermis
To understand the biological significance of ROCK1 in the cellular response to UVB-induced stress, we investigated the effect of the activation of ROCK1 on the JNK pathway in vivo. ROCK1-/- mice are markedly underrepresented in litters on a C57Bl/6 background, suggesting that lethality occurs in utero or postnatally (35). Thus, we examined the effect of the partial deletion of ROCK1 expression on the UVB damage-induced JNK pathway in the dorsal skin of ROCK1+/- mice (36). First, we tested whether the activity of ROCK changed in the skin epidermis upon exposure to UVB radiation. An increase in ROCK activity was detected in WT mouse skin upon challenge with UVB (Fig. 5A). Topical application of a chemical inhibitor of ROCK, fasudil (HA1077), to the skin abolished UVB-induced ROCK activity in vivo, as indicated by the abundance of phosphorylated myosin-binding subunit (MBS), a substrate of ROCK (Fig. 5A, right panel). Furthermore, fasudil also decreased the abundance of phosphorylated γ-H2AX and JNK. In addition, a reduction in the number of terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL)-positive cells was observed in the epidermis of inhibitor-treated mice, which indicated the presence of fewer apoptotic cells (Fig. 5B, right panel).
Fig. 5.
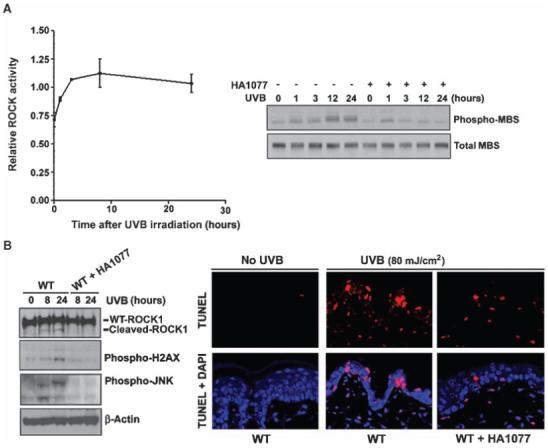
Inhibition of ROCK activity in the epidermal skin of WT mice compromises UVB-induced phosphorylation of JNK and γ-H2AX phosphorylation and apoptosis. (A) An ELISA-based assay of ROCK activity was performed on skin extracts from UVB-irradiated mice. Results are presented as the mean ± SEM (n = 3) (left panel). ROCK activity was also determined by assessing the abundance of phospho-MBS, a ROCK1 target, with or without treatment by the ROCK inhibitor HA1077 in extracts from UVB-irradiated epidermal skin (right panel). (B) HA1077 repressed UVB-induced activation of JNK and γ-H2AX in epidermal skin. WT and HA1077-treated mouse skin extracts were analyzed by Western blotting with antibodies against phospho-γ-H2AX, β-actin, phospho-JNK, and ROCK1. TUNEL and kinase assays were performed on the skin after UVB exposure (80 mJ/cm2) for 24 hours.
We also examined the effect of the partial deletion of ROCK1 expression on the UVB damage-induced JNK pathway in the dorsal skin from ROCK1+/- mice (36). As expected, the abundance of ROCK1 protein in the skin of ROCK1+/- mice was about half of that in the skin of WT mice (Fig. 6A). There was a substantial reduction in the abundance of the phosphorylated forms of JNK (phospho-JNK) and γ-H2AX (phospho-γ-H2AX) in UVB-irradiated skin from ROCK1+/- mice relative to WT mice (Fig. 6A). Moreover, ROCK1+/- mice exposed to UVB radiation had a lower number of TUNEL-positive cells in the epidermis than did their UVB-irradiated WT littermates (Fig. 6B). Decreases in the abundance of phospho-JNK and phospho-γ-H2AX were observed in the epidermis of ROCK1+/- mice after UVB irradiation (Fig. 6B). No apoptotic cells were detected in mice that were not exposed to UVB irradiation. We propose, therefore, that activation of ROCK1 upon UVB-induced stress is essential for activation of the JNK signaling pathway and UVB-induced apoptosis in vivo.
Fig. 6.
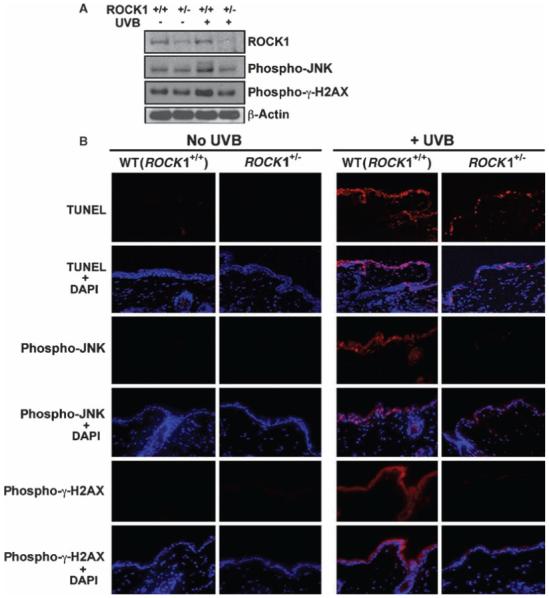
ROCK1 activation is essential for UVB-mediated apoptosis and JNK activation in skin in vivo. (A) UVB-mediated phosphorylation of JNK is ROCK1 dependent in vivo. Western blot analysis of the abundance of ROCK1 and phosphorylated JNK and phospho-γ-H2AX with or without UVB irradiation in the skin of WT and ROCK1+/- mice. ROCK1+/- mice and their WT littermates were irradiated with UVB at 160 mJ/cm2. After 12 hours, mouse skins were harvested and cell lysates were analyzed by Western blotting with antibodies against ROCK1, phospho-γ-H2AX, phospho-JNK, and β-actin. (B) ROCK1+/- mice have compromised phosphorylation of JNK in response to UVB-induced stress in the skin. Representative TUNEL staining of skin from ROCK1+/- and WT littermates, analyzed 12 hours after the mice received 160 mJ/cm2 of UVB irradiation. Immunofluorescence staining of phosphorylated JNK and phosphorylated γ-H2AX was performed on the epidermal skin from ROCK1+/- and WT mice, with or without UVB irradiation.
DISCUSSION
We have shown that ROCK1-mediated activation of JNK is required for UVB-induced apoptosis. It is well established that exposure of mammalian cells to DNA damage, including that caused by UVB irradiation, activates JNK. However, a key question that remains unresolved concerns the upstream regulator(s) of JNK activation and the signaling that leads to the DNA-damage response and apoptosis. Here, we identified ROCK as a key regulator of the JNK signaling pathway. We showed that upon exposure to UVB-induced stress, ROCK1 was activated, bound to and phosphorylated JIP-3, and consequently activated JNK. Our findings suggest, therefore, that ROCK1 may act as a cellular switch that triggers cells to undergo apoptosis after UVB-induced stress.
A hallmark of exposure to UVB radiation is the activation of Ser/Thr kinases that eventually lead to cell cycle arrest or apoptosis (4). Growing evidence has linked ROCK1 to various forms of cell death (14, 34). We and others have shown that inhibition of ROCK1 activity can protect cells or tissue from apoptosis (16). Activation of ROCK1 by caspase-3-mediated cleavage during apoptosis is important for regulating the succeeding morphological events, including loss of cell-to-cell adhesion, membrane blebbing, membrane disintegration, and formation of apoptotic bodies destined for phagocytosis (34, 37). Thus far, the role of ROCK1 activation in earlier signaling events governing apoptosis has remained elusive. In our studies, we investigated UVB-induced stress because it elicits rapid phosphorylation of JNK before apoptosis. In previous studies, we have shown that increased expression of RhoE by p53 during genotoxic stress inhibits the activation of ROCK1 (20). In addition, a study has shown the increase in c-Jun expression through RhoA and ROCK, which is independent of ROCK-initiated actin polymerization (38).
Using TAP purification, we have identified the JIP-3 scaffold protein as a target of the kinase activity of ROCK1 and established that the ROCK1-JIP-3 interaction is required for the activation of the JNK pathway in response to UVB-induced damage. JIP-3 is a multifunctional protein; it is a cargo adaptor protein mediating axonal transport of kinesin (5, 39, 40) and a scaffold protein for the JNK pathway (26, 41). Several JNK scaffold proteins such as POSH, JIP1, and β-arrestin-2 have been identified (5). Scaffold proteins permit the precise assembly of proteins to form JNK signaling modules and consequently facilitate protein activation. JIP-3 is known to activate JNK signaling through the coordination of MAPK kinase (MEK), MAPK kinase 7 (MKK7), JNK, and c-Jun. JIP-3 is found in neuronal cells and we detected it at high abundance in human keratinocytes and other cell types used in our studies (42). JIP-3-/- mice exhibit difficulty in breathing after birth and have defects in development of the telencephalon. The genes encoding RhoA, the Rho guanine nucleotide exchange factor (GEF) Net1, and ROCK are down-regulated in the brains of JIP-3-/- mice (42). In contrast, we did not observe any changes in the expression of JIP-3 upon inhibiting ROCK1 activity. Further understanding of the control of ROCK, RhoA, and Rho-GEF gene expression by JIP-3 would be invaluable. To date, several new binding partners of JIP-3 have been found, including Pin1 (43) and Toll-like receptor 4 (44).
Phosphorylation of JNK has been extensively studied as an apoptotic stimulus (45). Mitochondrial-mediated cell death signaling is defective in Jnk1-/-,Jnk2-/- mouse embryonic fibroblasts (33). γ-H2AX is a known JNK target and is important in the generation of DNA ladders during apoptosis (46). We observed that inhibition of ROCK1 activity inhibited the intrinsic cell death pathway elicited by UVB-induced stress. Together, these data support a model in which activation of ROCK1 induces apoptosis through the phosphorylation of JIP-3, which enhances the activation of JNK and its downstream targets (Fig. 7). The next series of challenges presented by the elucidation of this previously unidentified pathway includes understanding the precise regulation of proteins that form complexes with JNK and of downstream targets of JNK in response to UVB-induced stress and apoptosis. Future studies are also needed to elucidate how the ROCK1-JNK pathway mediates apoptosis in various cell types and how it is regulated by other stresses.
Fig. 7.
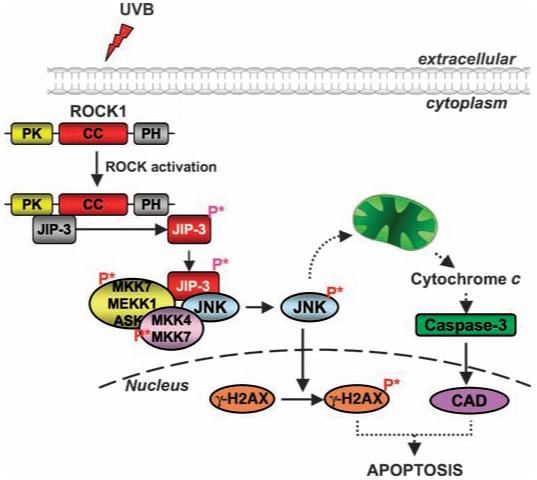
Model showing the activation of the JNK pathway upon challenge by UVB. Upon UVB irradiation, ROCK1 is activated and recruits and phosphorylates JIP-3. Phosphorylated JIP-3 (P* denotes phosphorylation) triggers the phosphorylation and activation of JNK. Activated JNK causes the release of cytochrome c from the mitochondria and stimulates the intrinsic cell death pathway. Activated JNK also phosphorylates γ-H2AX, which is essential for apoptosis to occur.
Our study sheds new light on the mechanism of upstream regulation of JNK-mediated apoptosis, an area of emerging interest with clinical relevance to common human diseases such as diabetes (47) and cancer (48). It is well established that deregulation of Rho-ROCK signaling in tumors has been linked to increased invasion and metastasis (49-51). A recent study identified phosphoinositide-dependent protein kinase 1 (PDK1) as a stimulator of ROCK1 at the plasma membrane, which regulates cancer cell motility by antagonizing inhibition of ROCK1 by RhoE (52). Our findings suggest, therefore, that activation of the ROCK1-JIP-3-JNK signaling cascade in response to UVB-induced damage is an alternate mode of maintaining normal tissue homeostasis.
MATERIALS AND METHODS
Cell culture and cell lines
U2OS cells and 293ET cells were grown in the presence of 5% CO2 and 20% O2 at 37°C in humidified chambers. U2OS cells, 293ET cells, and MCF7 cells were grown in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. Adenoviruses expressing His-tagged ROCK1 were generated, amplified, purified, and titrated as previously reported (20). The human ROCK1 sequences GTA CTT GTA TGA AGA TGA C (shRNA 1), GTA AAC GAG CTT TGG TTA C (shRNA2), GCC AAG CCA TAT TGA GTT ATT (shRNA 3), and GAG GTT TGT TGG ACT TTC ATA (shRNA 4) were targeted for RNA interference. U2OS cells stably expressing the ROCK1 siRNA were generated by annealing the shRNA, targeted to a respective sequence, to the BamH1 and Xho1 sites of the pBabe-U6 vector that harbors a puromycin-resistance gene. The pBabe-ROCK1 transfectants were selected with 2 μg/ml of puromycin. JIP-1 and JIP-3 siRNA oligonucleotides were purchased from Santa Cruz Biotechnology.
ROCK1 complex purification
A detailed TAP purification procedure has been described previously (24). pOZ-FLAG-HA-tagged human WT-ROCK1, pOZ-FLAG-HA vector, and pOZ-FLAG-HA-short-ROCK1 were transfected into 293ET cells and the resulting proteins were purified with anti-FLAG M2 monoclonal antibody (mAb)-conjugated agarose beads (Sigma) followed by anti-HA 12CA5 mAb-conjugated agarose beads in Buffer A [25 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2 mM MgCl2, 2 mM NaF, 2 mM Na3VO4, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1% Triton X-100].
In vitro ROCK assay
Cell lysates (500 μg), either treated or untreated with UVB or CPT, were mixed with 2 μg of anti-ROCK1 antibody (Santa Cruz) for 4 hours and then with 20 μl of protein G-beads for 1 hour. Kinase assays were performed in 50 μl of kinase mixture [20 mM Tris, pH 7.5, 5 mM MgCl2, 100 mM KCl, 0.1 mM dithiothreitol (DTT), 100 μM adenosine triphosphate (ATP), 1 mM EDTA, 1 μM microcystin-LR, 1 μg MYPT1 substrate protein (Upstate Biotechnology), and 1 μCi [γ-32P]ATP] and incubated at 30°C for 30 min. Immunoprecipitates were separated by SDS-polyacrylamide gel electrophoresis (PAGE). The gel was dried and exposed to film. The gel slice was excised for radioactive counting.
ROCK-mediated phosphorylation of JIP-3
FLAG-JIP-3 and FLAG control vectors were transfected into 293ET cells and the resulting proteins were purified with anti-FLAG M2 mAb-conjugated agarose beads (Sigma). Ten micrograms of the FLAG-purified material was incubated with recombinant ROCK1 (Upstate Biotechnology) in 50 μl of kinase mixture (20 mM Tris, pH 7.5, 5 mM MgCl2, 100 mM KCl, 0.1 mM DTT, 100 μM ATP, 1 mM EDTA, 1 μM microcystin-LR, and 1 μCi [γ-32P]ATP) and incubated at 30°C for 30 min. Ten microliters of kinase reaction was subjected to SDS-PAGE. For JIP-3 phosphorylation site analysis, the kinase reaction was performed without [γ-32P]ATP and the bands were excised and analyzed by the Harvard Medical School Taplin Biological Mass Spectrometry Facility.
Mass spectrometry and glycerol gradient centrifugation
FLAG-HA double-affinity-purified materials were separated by 4 to 20% gradient SDS-PAGE and were silver stained (Invitrogen). The proteins were analyzed by the Mass Spectrometry Facility at Emory University. For glycerol gradient sedimentation, 200 μl of Ni2+ column eluate from His-ROCK1-transfected 293ET cells was loaded on the top of a 4.5-ml 10 to 40% glycerol gradient cushion in Buffer A containing 100 mM KCl and centrifuged for 10 hours at 50,000 rpm (Beckman, SW55Ti). After centrifugation, individual fractions (200 μl) were collected from top to bottom. Five microliters of each fraction was analyzed by Western blotting.
Western blotting and immunoprecipitation
Cells were lysed in 20 mM Hepes, pH 7.5, 1 mM EDTA, 1 mM EGTA, 120 mM NaCl, 100 mM NaF, 2 mM Na3VO4, 1% Triton X-100, 1 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. Proteins were quantified with the Bio-Rad Protein Assay Kit. Equal amounts of protein per sample were subjected to SDS-PAGE and transferred to nitrocellulose membranes (Invitrogen). Immunoprecipitations were performed with 500 μg of cell extract incubated with anti-His-tag polyclonal antibody (Santa Cruz), anti-ROCK1 goat polyclonal antibody (Santa Cruz), anti-FLAG antibody (Sigma), anti-JIP-3 antibody (Santa Cruz), anti-RhoA antibody (Santa Cruz), or anti-phosphoserine antibodies (ZYMED Laboratories). The immunocomplexes were precipitated with protein G-conjugated beads (Sigma). Antibodies used for immunoblotting include anti-ROCK1 (Santa Cruz), anti-phospho-H2AX (Ser139, clone JBW301, Millipore, cat. no. P16104), anti-phospho-MBS (Upstate), anti-phospho-MLC, anti-phospho-JNK, anti-phospho-Akt, anti-phospho-p38, anti-phospho-MAPK (CST), and anti-caspase-3, all from CST.
Cell fractionation
Cells were lysed in buffer (250 mM sucrose, 20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin at pH 7.5) and incubated for 30 min on ice. Cells were homogenized by 30 strokes through a 22-gauge needle. Homogenates were centrifuged at 750g for 10 min at 4°C to collect the mitochondrial pellets.
Flow cytometry and apoptosis assays
Cell death as determined by the fragmentation of DNA was measured by photometric enzyme immunoassay with the Cell Death Detection ELISA kit (Roche Applied Science) following the manufacturer’s suggested protocol. Briefly, cells were plated in six-well plates and, after the indicated treatments, all cells, including those in suspension, were collected by centrifugation at 1500g, lysed, and centrifuged at 20,000g to obtain the cytoplasmic fractions. The cytoplasmic fractions containing the fragmented DNA were transferred to microtiter plates that had been coated with a monoclonal anti-histone antibody. The amount of fragmented DNA consisting of nucleosomes bound to the anti-histone antibody was evaluated by peroxidase-conjugated monoclonal anti-DNA antibody with ABTS [2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt] as a substrate at 405 nm. For fluorescence-activated cell sorting (FACS) analysis, cells were pelleted and washed once with PBS. The cells were fixed for 3 hours with 2% paraformaldehyde in PBS at room temperature. TUNEL assays were performed with the TUNEL assay kit (In Situ Cell Death Detection Kit, TMR Red, Roche) to quantify apoptotic cells visualized by fluorescence microscopy and FACS analysis.
Caspase assay
Caspase activities were determined with caspase assay kits for caspase-3 and caspase-8 (Sigma) and with the caspase-9 assay kit (Calbiochem) for caspase-9. The assays were carried out according to manufacturers’ protocols. Briefly, 5 × 106 cells were seeded in p150 dishes. After 24 hours, cells were transfected with control siRNA or ROCK1-specific siRNA. Two dishes were used for each data point and cells were harvested at different time intervals after UVB irradiation. Cells were trypsinized and washed once with PBS followed by resuspension of 1 × 107 cells per 100 μl of lysis buffer. The cells were incubated on ice for 20 min. Supernatant was obtained from the cell lysates by centrifuging at 20,000g for 15 min. The cell lysates were incubated with either the caspase-3 substrate (Ac-DEVD-pNA), the caspase-8 substrate (Ac-IETD-pNA), or the caspase-9 substrate (LEHD-pNA). As appropriate, the caspase-3 inhibitor AC-DEVD-CHO and the caspase-8 inhibitor Ac-IETD-CHO were used as controls. The plates were incubated at 37°C for 90 min and the signal was read at 405 nm.
Rho kinase assay
Twenty micrograms of protein was used for each assay. Rho kinase activities were measured by a Rho kinase assay kit (MBL International Corporation). Briefly, 10 μl containing 20 μg of protein extract was incubated with 90 μl of kinase buffer containing 0.1 mM ATP. The entire reaction was added to the well of a 96-well plate precoated with the detector antibody AF20, an antibody that specifically detects only the phosphorylated form of Thr696 on human MBS. The quantity of phosphorylated substrate was measured by binding it to a horseradish peroxidase conjugate of AF20, which catalyzed the conversion of the chromogenic substrate tetramethyl-benzidine from a colorless solution to a blue solution (OD = 450 nm), or a yellow solution after the addition of the stopping reagent.
ROCK1+/- mice
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital. Eight-week-old ROCK1+/- mice and their WT littermates were used for UVB-exposure experiments. Mice were shaved and the remaining hairs were removed by hair remover (Nair, NJ) 48 hours before UVB treatment. On the next day, the anesthetized animals were irradiated by UVB with the use of a custom-made UVB-irradiation apparatus with four photochemical lamps (RPR 3000, Southern N.E. Ultraviolet Co., Bradford, CT). The delivered UVB dose was measured each time with a photometer (model IL 1400A, International Light Inc., Newburyport, MA). The animals were killed at 8 and 24 hours after UVB exposure. The back skin was harvested and frozen in liquid nitrogen or fixed in 2% methanol-free formaldehyde (Polysciences Inc., PA). The skin was embedded in tissue-freezing medium (TBS, NC) and 7-μm frozen sections of tissue were obtained from the skins of animals subjected to different doses of UVB exposure. About six representative sections were analyzed.
Acknowledgments
We thank L. Brown and Cutaneous Biology Research Center investigators for helpful discussions. We thank A. J. Ridley for suggestions and encouragement and S. Narumiya for the ROCK1 cDNA. This work was supported by NIH grants (CA127247, CA 80058, CA085681, HL052233, and CA85214), the American Diabetes Association (7-05-PPG-02), and Shiseido Research Core funding.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencesignaling.org/cgi/content/full/1/47/ra14/DC1
Fig. S1. The kinase domain of ROCK1 is involved in binding to JIP-3.
Fig. S2. Glycerol gradient analysis of ROCK1 and JIP-3.
Fig. S3. Depletion of ROCK1 compromises UVB-induced apoptosis.
Fig. S4. Ectopic expression of ROCK1 in ROCK1-depleted HACAT cells rescued the ROCK1-suppression phenotype.
REFERENCES AND NOTES
- 1.Ananthaswamy HN, Loughlin SM, Cox P, Evans RL, Ullrich SE, Kripke ML. Sunlight and skin cancer: Inhibition of p53 mutations in UV-irradiated mouse skin by sunscreens. Nat. Med. 1997;3:510–514. doi: 10.1038/nm0597-510. [DOI] [PubMed] [Google Scholar]
- 2.Trempus CS, Mahler JF, Ananthaswamy HN, Loughlin SM, French JE, Tennant RW. Photocarcinogenesis and susceptibility to UV radiation in the v-Ha-ras transgenic Tg.AC mouse. J. Invest. Dermatol. 1998;111:445–451. doi: 10.1046/j.1523-1747.1998.00237.x. [DOI] [PubMed] [Google Scholar]
- 3.Nataraj AJ, Trent JC, II, Ananthaswamy HN. p53 gene mutations and photocarcinogenesis. Photochem. Photobiol. 1995;62:218–230. doi: 10.1111/j.1751-1097.1995.tb05262.x. [DOI] [PubMed] [Google Scholar]
- 4.Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci. STKE. 2003;2003:re2. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- 5.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 6.Leung T, Chen XQ, Manser E, Lim L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 1996;16:5313–5327. doi: 10.1128/mcb.16.10.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- 8.Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 2000;150:797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amano T, Tanabe K, Eto T, Narumiya S, Mizuno K. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem. J. 2001;354:149–159. doi: 10.1042/0264-6021:3540149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, Wu D. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- 11.Furukawa N, Ongusaha P, Jahng WJ, Araki K, Choi CS, Kim HJ, Lee YH, Kaibuchi K, Kahn BB, Masuzaki H, Kim JK, Lee SW, Kim YB. Role of Rho-kinase in regulation of insulin action and glucose homeostasis. Cell Metab. 2005;2:119–129. doi: 10.1016/j.cmet.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 12.Farah S, Agazie Y, Ohan N, Ngsee JK, Liu XJ. A Rho-associated protein kinase, ROK alpha, binds insulin receptor substrate-1 and modulates insulin signaling. J. Biol. Chem. 1998;273:4740–4746. doi: 10.1074/jbc.273.8.4740. [DOI] [PubMed] [Google Scholar]
- 13.Kosako H, Yoshida T, Matsumura F, Ishizaki T, Narumiya S, Inagaki M. Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene. 2000;19:6059–6064. doi: 10.1038/sj.onc.1203987. [DOI] [PubMed] [Google Scholar]
- 14.Riento K, Ridley AJ. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell. Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 15.Noma K, Oyama N, Liao JK. Physiological role of ROCKs in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2006;290:C661–C668. doi: 10.1152/ajpcell.00459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J, Wei L. Rho kinase in the regulation of cell death and survival. Arch. Immunol. Ther. Exp. (Warsz.) 2007;55:61–75. doi: 10.1007/s00005-007-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, Ma XL, Willette RN, Yue TL. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc. Res. 2004;61:548–558. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Muguruma K, Sasai Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 19.Papadopoulou N, Charalampopoulos I, Alevizopoulos K, Gravanis A, Stournaras C. Rho/ROCK/actin signaling regulates membrane androgen receptor induced apoptosis in prostate cancer cells. Exp. Cell Res. 2008;314:3162–3174. doi: 10.1016/j.yexcr.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Ongusaha PP, Kim HG, Boswell SA, Ridley AJ, Der CJ, Dotto GP, Kim YB, Aaronson SA, Lee SW. RhoE is a pro-survival p53 target gene that inhibits ROCK I-mediated apoptosis in response to genotoxic stress. Curr. Biol. 2006;16:2466–2472. doi: 10.1016/j.cub.2006.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Tura A, Schuettauf F, Monnier PP, Bartz-Schmidt KU, Henke-Fahle S. The H-1152P-mediated inhibition of Rho-kinase (ROCK) reduces reactive gliosis and promotes cell survival in the rodent retina. Invest. Ophthalmol. Vis. Sci. 2008 doi: 10.1167/iovs.08-1973. 10.1167/iovs.08-1973. [DOI] [PubMed] [Google Scholar]
- 22.Sun Q, Yue P, Ying Z, Cardounel AJ, Brook RD, Devlin R, Hwang JS, Zweier JL, Chen LC, Rajagopalan S. Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of Rho/ROCK. Arterioscler. Thromb. Vasc. Biol. 2008;28:1760–1766. doi: 10.1161/ATVBAHA.108.166967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajimoto H, Hashimoto K, Bonnet SN, Haromy A, Harry G, Moudgil R, Nakanishi T, Rebeyka I, Thebaud B, Michelakis ED, Archer SL. Oxygen activates the Rho/Rho-kinase pathway and induces RhoB and ROCK-1 expression in human and rabbit ductus arteriosus by increasing mitochondria-derived reactive oxygen species: A newly recognized mechanism for sustaining ductal constriction. Circulation. 2007;115:1777–1788. doi: 10.1161/CIRCULATIONAHA.106.649566. [DOI] [PubMed] [Google Scholar]
- 24.Shi Y, Sawada J, Sui G, Affar EB, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 25.Liao JK, Seto M, Noma K. Rho kinase (ROCK) inhibitors. J. Cardiovasc. Pharmacol. 2007;50:17–24. doi: 10.1097/FJC.0b013e318070d1bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelkar N, Gupta S, Dickens M, Davis RJ. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 2000;20:1030–1043. doi: 10.1128/mcb.20.3.1030-1043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, Hibi M, Nakabeppu Y, Shiba T, Yamamoto KI. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol. Cell. Biol. 1999;19:7539–7548. doi: 10.1128/mcb.19.11.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 29.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: From junk to gold? Nat. Rev. Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 32.Bode AM, Dong Z. The functional contrariety of JNK. Mol. Carcinog. 2007;46:591–598. doi: 10.1002/mc.20348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cyto-chrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 34.Leverrier Y, Ridley AJ. Apoptosis: Caspases orchestrate the ROCK ‘n’ bleb. Nat. Cell Biol. 2001;3:E91–E93. doi: 10.1038/35070151. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, Noda Y, Matsumura F, Taketo MM, Narumiya S. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J. Cell Biol. 2005;168:941–953. doi: 10.1083/jcb.200411179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/- haploinsufficient mice. Circulation. 2005;112:2959–2965. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ndozangue-Touriguine O, Hamelin J, Breard J. Cytoskeleton and apoptosis. Biochem. Pharmacol. 2008;76:11–18. doi: 10.1016/j.bcp.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Marinissen MJ, Chiariello M, Tanos T, Bernard O, Narumiya S, Gutkind JS. The small GTP-binding protein RhoA regulates c-Jun by a ROCK-JNK signaling axis. Mol. Cell. 2004;14:29–41. doi: 10.1016/s1097-2765(04)00153-4. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein LS. Kinesin molecular motors: Transport pathways, receptors, and human disease. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6999–7003. doi: 10.1073/pnas.111145298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 2001;152:959–970. doi: 10.1083/jcb.152.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuura H, Nishitoh H, Takeda K, Matsuzawa A, Amagasa T, Ito M, Yoshioka K, Ichijo H. Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1-JNK signaling pathway. A new mode of regulation of the MAP kinase cascade. J. Biol. Chem. 2002;277:40703–40709. doi: 10.1074/jbc.M202004200. [DOI] [PubMed] [Google Scholar]
- 42.Kelkar N, Delmotte MH, Weston CR, Barrett T, Sheppard BJ, Flavell RA, Davis RJ. Morphogenesis of the telencephalic commissure requires scaffold protein JNK-interacting protein 3 (JIP3) Proc. Natl. Acad. Sci. U.S.A. 2003;100:9843–9848. doi: 10.1073/pnas.1733944100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becker EB, Bonni A. Pin1 mediates neural-specific activation of the mitochondrial apoptotic machinery. Neuron. 2006;49:655–662. doi: 10.1016/j.neuron.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 44.Matsuguchi T, Masuda A, Sugimoto K, Nagai Y, Yoshikai Y. JNK-interacting protein 3 associates with Toll-like receptor 4 and is involved in LPS-mediated JNK activation. EMBO J. 2003;22:4455–4464. doi: 10.1093/emboj/cdg438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Lin A. Role of JNK activation in apoptosis: A double-edged sword. Cell Res. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 46.Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY, Bode AM, Dong Z. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell. 2006;23:121–132. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vallerie SN, Furuhashi M, Fucho R, Hotamisligil GS. A predominant role for parenchymal c-Jun amino terminal kinase (JNK) in the regulation of systemic insulin sensitivity. PLoS ONE. 2008;3:e3151. doi: 10.1371/journal.pone.0003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell. 2007;26:675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 49.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat. Rev. Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 50.Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J. Cell Biol. 2007;178:23–30. doi: 10.1083/jcb.200701120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 52.Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat. Cell Biol. 2008;10:127–137. doi: 10.1038/ncb1675. [DOI] [PubMed] [Google Scholar]
- Citation.Ongusaha PP, Qi HH, Raj L, Kim Y-B, Aaronson SA, Davis RJ, Shi Y, Liao JK, Lee SW. Identification of ROCK1 as an upstream activator of the JIP-3 to JNK signaling axis in response to UVB damage. Sci. Signal. 2008;1:ra14. doi: 10.1126/scisignal.1161938. [DOI] [PMC free article] [PubMed] [Google Scholar]