

Abstract
Extracellular signal-regulated kinase 1 (ERK1) and ERK2 signaling in the spinal cord dorsal horn (SCDH) has been implicated in injury-induced pain hypersensitivity. Available ERK pathway inhibitors cannot distinguish between ERK1 and ERK2, nor can they differentially target the expression of neuronal or glial ERK1/2. We selectively inhibited the expression of ERK2 in neurons of the adult mouse SCDH by use of an ERK2 small interfering RNA (siRNA) delivered by a neurotropic adenoassociated viral vector. In situ hybridization revealed a siRNA vector-induced decrease in ERK2 mRNA in the ipsilateral SCDH. Immunohistochemistry showed a decreased neuronal phospho-ERK1/2 (pERK1/2), and Western blot analysis revealed that both ERK2 expression and phosphorylation were reduced by the siRNA vector. In contrast, basal ERK1 expression was not affected, although pERK1 was slightly increased. The siRNA vector-induced knockdown of ERK2 expression in the SCDH did not alter the baseline mechanical or thermal paw withdrawal thresholds. Hindpaw intraplantar injection of complete Freund's adjuvant (CFA) produced peripheral inflammation, mechanical allodynia, and thermal hyperalgesia in vector control animals that persisted for at least 96 h. It also caused an increase in SCDH ERK1 and ERK2 levels at 96 h and pERK1 and pERK2 levels at 1 and 96 h. The ERK2 siRNA vector prevented changes in ERK1, ERK2, and pERK2. In addition, the siRNA vector protected the animals from developing mechanical allodynia and thermal hyperalgesia throughout the 96 h after CFA. These findings indicate that ERK2 in the SCDH neurons is critical for the development of inflammatory pain hypersensitivity.
Keywords: ERK2, spatial knockdown, RNAi, inflammatory pain, spinal cord dorsal horn, rAAV vector
Introduction
Extracellular signal-regulated kinase 1 (ERK1) and ERK2 (Boulton et al., 1991), also referred to as p44 and p42 MAPK (mitogen-activated protein kinase), belong to a group of evolutionarily conserved serine/threonine protein kinases that play critical roles in cell proliferation, differentiation, and survival. They are activated by dual phosphorylation on their regulatory tyrosine and threonine residues by an upstream kinase, mitogen-activated protein kinase kinase (MEK). In the CNS, ERK1 and ERK2 have been linked to signal transduction cascades that regulate neuronal activity and plasticity. For example, ERK1/2 has been implicated in the enhanced excitability of spinal cord dorsal horn (SCDH) neurons in models of inflammatory pain (Ji et al., 1999, 2002; Galan et al., 2002; Dai et al., 2004; Cruz et al., 2005). These reports find that the injection of various proinflammatory substances into the hindpaw of rodents induces increases in ERK1/2 phosphorylation in the lumbar SCDH, which parallels the development of pain hypersensitivity. Prolonged ERK1/2 phosphorylation is also seen in neuropathic pain models (Zhuang et al., 2005; Tseng et al., 2007; Zhao et al., 2007). Intrathecal administration of U0126 [1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylmercapto)butadiene] or PD98059 [2-(2-amino-3-methoxyphenyl-4H-1-benzopyran-4-one], both inhibitors of MEK, attenuates pain hypersensitivity after peripheral tissue inflammation or nerve damage (Ji et al., 1999, 2002; Cruz et al., 2005; Karim et al., 2006; Zhao et al., 2007).
The high structural resemblance between ERK1 and ERK2 has limited studies of their individual contribution to inflammatory pain. ERK1 and ERK2 have similar sensitivities to activation by MEK (Zheng and Guan, 1993) and are often functionally redundant in vitro (Robbins et al., 1993). However, studies in knock-out mice indicate that ERK1 and ERK2 can play different roles in vivo. ERK1 knock-out mice are viable, fertile, and of normal size (Pagès et al., 1999), whereas ERK2 knock-out mice die before embryonic day 8.5 because of defects in trophoblast and placental development and in mesoderm differentiation (Hatano et al., 2003; Saba-El-Leil et al., 2003; Yao et al., 2003). These studies suggest that ERK1 is dispensable as long as ERK2 can compensate for its loss but not vice versa. Other studies indicate that ERK1 and ERK2 respond differently to growth factors and may regulate cell proliferation differently (Zeng et al., 2005; Li and Johnson, 2006; Frémin et al., 2007). Furthermore, ERK2 but not ERK1 is critical in the modulation of hippocampal long-term potentiation (English and Sweatt, 1996).
Mice expressing a neuron-specific dominant-negative MEK have impaired inflammatory pain and thermal hyperalgesia (Karim et al., 2006), suggesting that neuronal ERK1/2 plays a critical role in injury-induced pain. However, the contributions of ERK1 and ERK2 could not be separated. In this study, we used small interfering RNAs (siRNAs) to selectively knock down the expression of ERK2 in SCDH neurons. The siRNA is delivered by a neurotropic recombinant adenoassociated virus (rAAV), limiting the knockdown of ERK2 to neurons, which allowed us to examine the specific role of neuronal SCDH ERK2 in the development of injury-induced pain hypersensitivity in vivo.
Materials and Methods
Experimental animals and drugs.
Adult male C57BL/6 mice (The Jackson Laboratory) weighing 20–30 g were used for this study. Experiments were performed in accordance with National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The experimental protocol (no. 0508-392A) was approved by the Institutional Animal Care and Use Committee at Weill Cornell Medical College. Animals were housed under 12 h light/dark cycles in a pathogen-free room with ad libitum access to water and food.
Design and screening of siRNAs and viral vector production.
The approaches used to design and screen for siRNAs targeting the expression of a single gene were described previously (Garraway et al., 2007). Candidate siRNAs were selected by the siRNA selection program (http://jura.wi.mit.edu/siRNAext/) from the Whitehead Institute for Biomedical Research (Massachusetts Institute of Technology) (Yuan et al., 2004). The sense and antisense sequence of the siRNA were joined by a spacer (TTCAAGAGA) (Brummelkamp et al., 2002) to create a “stem-loop” sequence. Synthetic DNA oligomers with the corresponding sequences were ordered (Sigma-Genosys), cloned into a serotype-2 rAAV plasmid, and subjected to the psiCHECK Dual Luciferase Reporter Assay (Promega). Three plasmids expressing the most active siRNAs identified in the psiCHECK assay and one plasmid expressing a mismatch control siRNA were packaged into serotype-2 rAAV vectors (Musatov et al., 2002) and used in the in vivo studies.
The three active sequences (sense strand) identified by the psiCHECK assay were ERK2-5, 5′-GGAACAGGTTGTTCCCAAA; ERK2-7, 5′-GGAGCAGTATTATGACCCA; and ERK2-8, 5′-GACTGCTAGATTCCAGCCA, targeting regions spanning 1063–1081, 1178–1196, and 1289–1307 on the mouse ERK2 cDNA (GenBank accession number NM_011949), respectively. The ERK2-7 sequence was reversed to produce the mismatch (MM) sequence 5′-ACCCAGTATTATGACGAGG, which served as a control.
In vivo delivery of the rAAV vectors.
The viral vectors, ERK2-5, 2-7, 2-8, and MM were microinjected into the SCDH as described by South et al. (2003). The mice were anesthetized with ketamine/xylazine. A laminectomy was performed to remove part of the dorsal L2 and L3 spinous process, and the lumbar area of spinal cord was exposed for intraparenchymal injection (IPI). Three unilateral injections of 1 μl (1–3 × 109 viral particles/μl) were administered 0.5–0.7 mm apart, at a depth of 0.3 mm from the dorsal border and 0.5 mm from the midline, on the right side using a glass pipette with a 40-μm-diameter tip attached to a 5 μl Hamilton syringe. The syringe was mounted on a microinjector (David Kopf Instruments) attached to a stereotaxic unit (David Kopf Instruments). After the injection, the overlying muscles were closed with 5-0 chromic gut and the skins were closed with wound clips. Animals were allowed to recover for 3 weeks before undergoing behavioral tests or killed for histological and Western blot analysis.
Immunohistochemistry.
Mice were anesthetized with pentobarbital and then perfused transcardially with 4% paraformaldehyde (PFA) in PBS with 1 mm NaF. Fifty milliliters of fixative was perfused over 5 min by using a peristaltic pump. The spinal cord was dissected and placed in 4% PFA for 1 h before being transferred to 30% sucrose for cryoprotection for 72 h. Lumbar spinal cord cryosections of 20 μm thick were obtained from a cryostat (Leica) for immunohistochemistry (IHC) and in situ hybridization.
For IHC, the spinal cord sections were incubated in blocking solution [3% normal goat serum, 0.1% Triton X-100 in Tris-buffered saline (TBS)] for 30 min. After washing in TBS, sections were incubated with one or two of the following primary antibodies: rabbit anti-green fluorescent protein (GFP) (1:1000; Invitrogen), rabbit anti-phospho-ERK1/2 (1:1000; Cell Signaling Technology), mouse anti-NeuN (1:400; Millipore), rabbit anti-c-fos (1:2000; Santa Cruz Biotechnology), rabbit anti-dynorphin A (1:2000; Bachem), mouse anti-GFAP (1:2000; Millipore), and rat anti-OX42 (1:1000; BD Biosciences) overnight in blocking solution at 4°C. The sections were washed in TBS and then incubated in appropriate fluorescent secondary antibodies or biotinylated goat anti-rabbit or anti-mouse IgG (1:250; Vector Laboratories) in blocking solution. Biotin slides were further incubated with 3,3-diaminobenzidinetetra-hydrochloride (DAB). Fluorescent slides were mounted in the anti-fading mounting medium GelMount (Invitrogen). DAB slides were dehydrated through a series of ethanol and xylenes, and then coverslipped in Permount (Thermo Fisher Scientific). To minimize variability in staining, tissues from all treatment groups were run in the same session. A negative control was performed using diluted normal goat serum instead of the primary antibody.
Nonradioactive in situ hybridization.
ERK2 mRNA expression in the SCDH was detected by nonradioactive in situ hybridization as described by Garraway et al. (2007). Digoxigenin (DIG)-labeled antisense or sense riboprobes were synthesized using an in vitro transcription kit (Roche Applied Science) from the mouse ERK2 cDNA (kindly provided by Dr. Michael J. Weber, University of Virginia, Charlottesville, VA). The sense riboprobe served as a background control. On day 1, slide mounted cryosections of spinal cord (20 μm) were incubated in the following: (1) 4% PFA in PBS for 20 min; (2) PBS, three times for 5 min each; (3) proteinase K solution (Sigma-Aldrich) for 2 min; (4) PBS, twice for 5 min each; (5) 4% PFA in PBS for 5 min; (6) PBS for 5 min; (7) 0.25% acetic anhydride in 0.1 m triethanolamine for 10 min; (8) 2× SSC, twice for 5 min each. Next, sections were incubated in prehybridization solution (50% formamide, 0.3 m NaCl, 10 mm Tris, pH 8.0, 1 mm EDTA, pH 8.0, 500 μg/ml salmon sperm DNA, and 500 μg/ml yeast tRNA) at 55°C in a chamber containing towels moistened with 4× SSC and 50% formamide. After incubation for 2 h, the prehybridization solution was drained and sections were hybridized with DIG-labeled antisense or sense probes for ERK2 (1:1000), coverslipped, and placed in a 55°C oven overnight. On day 2, the coverslips were removed and the hybridized spinal cord sections were sequentially incubated in the following: (1) 5× SSC at 55°C for 10 min; (2) 50% formamide in 2× SSC at 55°C for 20 min; (3) RNase buffer at 37°C, twice for 5 min each; (4) RNase A (50 μg/ml; Sigma-Aldrich) at 37°C for 30 min; (5) RNase buffer at 37°C for 15 min; (6) 50% formamide, 2× SSC at 55°C for 20 min; (7) 2× SSC, twice for 15 min each; (8) washing buffer for 10 min; (9) blocking solution for 30 min; (10) anti-DIG antisera conjugated to alkaline phosphatase (1:500) for 2 h; (11) washing buffer, twice for 15 min each; (12) detection buffer for 5 min; (13) NBT/BCIP (nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate) overnight. On day 3, slides were rinsed in distilled water and dehydrated through a graded ethanol series, xylenes, and coverslipped in Permount (Thermo Fisher Scientific).
Microscopic analysis.
Fluorescent IHC images were captured by a Zeiss LSM 510 laser-scanning confocal microscope. Bright-field IHC and in situ images were captured by a Nikon Eclipse 80i microscope (Nikon) equipped with a digital CoolSnap camera (Photometrics) through an interface between the camera and a Macintosh computer using the IPlab software (BD Biosciences Bioimaging). Quantitative analysis was performed by a blinded observer using MetaMorph software (Universal Imaging) as described previously (Garraway et al., 2007). A total of four to five sections spacing 400–500 μm apart were used for each animal. At least three animals were included in each treatment group.
Immunoblot.
Animals were deeply anesthetized by isoflurane and decapitated, and the lumbar SCDHs at the level of L4–L6 were rapidly dissected. The right and left dorsal horn were separated and immediately homogenized in modified RIPA buffer (50 mm Tris-HCl, pH 7.4, 1% NP-40, 1 mm EDTA, 150 mm NaCl) supplemented with protease inhibitor mixture (Sigma-Aldrich) diluted 1:10, 2 mm PMSF, 2 mm NaF, and phosphatase inhibitor mixture I and II (Sigma-Aldrich), and then frozen in liquid nitrogen. After all the samples were collected, tissues were thawed on ice, sonicated, and centrifuged at 4°C at 14,000 × g for 10 min to obtain the supernatant. The protein level of each sample was measured by the Bio-Rad DC assay. Samples were then diluted in Laemmli sample buffer on the same day to a final concentration of 2 μg/μl, boiled for 5 min, and stored at −80°C.
The SCDH samples were separated by SDS-PAGE gels (10% Tris-HCl gel; Bio-Rad) and transferred to polyvinylidene difluoride filters (Millipore), which were subsequently blocked in blocking solution (5% dry milk in TBS with 0.1% Tween 20) for at least an hour. Membranes were incubated with rabbit anti-phospho-ERK1/2 (pERK1/2) antibody (1:1000; 07-362; Cell Signaling Technology) in blocking solution overnight at 4°C, washed in TBS with 0.1% Tween 20 (TBST), and then incubated with HRP-conjugated anti-rabbit IgG (1:1000; Pierce Biotechnology) in blocking solution for 1 h. Membranes were washed with TBST followed by TBS and developed using SuperSignal West Pico ECL kit (Pierce Biotechnology), and then exposed to film (Kodak) for various times. Next, membranes were stripped (Pierce) and reprobed for ERK1/2 using rabbit anti-ERK1/2 antibody (1:5000; Cell Signaling Technology) followed by HRP-conjugated anti-rabbit secondary antibody (1:10,000; Pierce Biotechnology). For loading control, blots were further stripped and reprobed for β-actin using a mouse monoclonal antibody (1:50,000; Sigma-Aldrich) followed by HRP-conjugated anti-mouse secondary antibody (1:200,000; Pierce Biotechnology). Exposures yielding signal intensity in the linear range without saturation were used for densitometry analysis with Fluorchem 9900 (Alpha Innotech). Ratios of intensity of pERK1/2 or ERK1/2 to β-actin were calculated, normalized to the control samples, and subjected to statistical analysis. At least four animals were included in each treatment group.
Behavioral testing.
All behavioral testing was conducted in groups of 10 mice per treatment by a blinded observer. Motor function tests were performed before mechanical stimulus threshold or thermal paw withdrawal threshold were measured (Garraway et al., 2007). No deficits in reflexes for surface righting, placing/stepping, and grasping/climbing were found in animals treated with either the control rAAV vector or rAAV viral vectors.
Mechanical stimulus threshold to a nonnoxious mechanical stimulus was determined by paw withdrawal to the application of a series of calibrated von Frey filaments to the surface of the hindpaws. The animals were placed in a Plexiglas cage with mesh flooring suspended above the researcher and left to acclimate for 30 min. von Frey filaments were applied perpendicularly against the midplantar surface of the foot. The “up-down” method of Dixon (Chaplan et al., 1994) was used to determine the value at which paw withdrawal occurred 50% of the time, interpreted as the mechanical threshold.
Thermal paw withdrawal threshold was assessed using a thermal nociceptive stimulus apparatus (Hargreaves et al., 1988). Before the test, the animals were allowed to acclimate for 30 min to the test chamber on a preheated glass plate maintained at 30°C. A radiant thermal stimulus (5.10 amp) was applied to the midplantar surface of the hindpaw through the glass plate. The latency, in seconds, for the withdrawal of the paw from the heat source was determined automatically. If no response was elicited, the heat source was automatically shut off at 20 s to prevent tissue injury. A minimum break of 5 min was allowed between each trial. Three to six trials were performed for each paw.
Fifteen microliters of complete Freund's adjuvant (CFA) (Sigma-Aldrich) was injected into the right hindpaws of lightly restrained mice. The mice had received either the control vector MM or siRNA vector 2-7 in the right SCDH at least 3 weeks before the intraplantar injection of CFA. Mechanical stimulus threshold, thermal paw withdrawal thres-hold, and paw size were measured before (baseline) and at 24, 48, and 96 h after CFA.
Statistical analysis.
The immunohistochemical, in situ hybridization, Western blot, and behavioral data were analyzed by one-way ANOVA followed by the Student–Newman–Keuls test (multiple groups) or the t test (two groups) using the InStat software (GraphPad; version 3.00). The data are represented as mean ± SEM.
Results
Knockdown of ERK2 expression in the lumbar SCDH neurons by rAAV vectors expressing active siRNAs
Three serotype-2 rAAV vectors (vectors 2-5, 2-7, and 2-8) expressing ERK2 siRNAs and one control vector expressing a mismatch siRNA (vector MM) were used in the current study. The psiCHECK Dual Luciferase Assay was performed before the virus preparation and injection to examine the ability of vectors to inhibit ERK2 expression in cultured cells. Compared with a control rAAV plasmid not expressing a siRNA, all three active siRNA vector plasmids significantly inhibited the activity of Renilla luciferase, which was translated from a fusion mRNA containing the ERK2 and luciferase cDNA sequences. The control MM vector was ineffective in this assay (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
The rAAV vectors were injected intraparenchymally into the SCDH of adult mice. Three weeks after IPI of the rAAV vectors, a robust and spatially localized expression of GFP was observed on the ipsilateral side of lumbar SCDH (Fig. 1A–D). The expression of GFP resulting from each of the four rAAV vectors extended for >3 mm rostrocaudally, encompassing the full L4, L5, and L6 spinal segments in mouse. In situ hybridization was performed on slides adjacent to the GFP slides to examine the mRNA expression of ERK2. On the side contralateral to the vector injection, ERK2 mRNA was observed in the gray matter. Vector MM did not alter the ERK2 mRNA level (Fig. 1E) in the ipsilateral dorsal horn. In contrast, there was a nearly complete depletion of ERK2 mRNA in the region corresponding to the GFP immunolabeling in animals treated with vector 2-5, 2-7, or 2-8 (Fig. 1F–H). A densitometry analysis of the in situ images showed that the extent of ERK2 mRNA knockdown was similar among the three active siRNA vectors, ranging from 75 to 80% reduction on the ipsilateral side compared with the contralateral side (Fig. 1I) (p < 0.05 vs vector MM).
Figure 1.

Expression of GFP and the knockdown of ERK2 mRNA in the mouse SCDH at 3 weeks after the IPI of a rAAV vector expressing an ERK2 siRNA. The expression of GFP in the ipsilateral SCDH after administration of a rAAV vector expressing a control siRNA (MM) (A) or an active ERK2 siRNA, 2-5 (B), 2-7 (C), or 2-8 (D). E–H, In situ hybridization revealed no change in ERK2 mRNA level in the ipsilateral SCDH compared with the contralateral side in animals treated with vector MM (E). In contrast, each of the three active siRNA vectors induces a significant decrease in ERK2 mRNA in the ipsilateral SCDH (F–H). Scale bar, 500 μm. I, ERK2 mRNA expression was estimated from in situ images. The ratio of the integrated optical density of in situ labeling in the ipsilateral SCDH to that in the contralateral SCDH was calculated. Data are mean ± SEM (n = 3). Compared with the control vector MM, vector 2-5, 2-7, and 2-8 induced a significant decrease (*p < 0.05) in the ERK2 mRNA expression in the ipsilateral SCDH. There was no difference in the ratio among the three active siRNA vectors.
To determine whether vector administration resulted in glial activation, we compared sections of SCDH from mice that were untreated or had received either the vector MM or the active siRNA, vector 2-7 at 3 weeks before the analysis. One example of the immunolabeling is shown in supplemental Figure 2 (available at www.jneurosci.org as supplemental material). Compared with sections from untreated mice, there was no evidence of hypertrophy, thickened processes, or enlarged cell bodies in GFAP-labeled astrocytes (Guo et al., 2007) or the shortened, thickened processes seen with activated microglia labeled with OX42 (Raghavendra et al., 2004). Examination at a higher magnification than presented in supplemental Figure 2 (available at www.jneurosci.org as supplemental material) supported these observations (data not shown). There was no evidence of neuronal cell loss, a finding consistent with a previous observation that vector-derived siRNAs did not result in loss of nuclei in SCDH (Garraway et al., 2007).
GFP immunolabeling is colocalized with NeuN, a neuronal marker, but not GFAP, an astroglial marker, or OX42, a microglial marker, as revealed by confocal fluorescent IHC in animals that received vector MM (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material) or vector 2-7 (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material).
Because of the high sequence similarity between the ERK1 and ERK2 proteins and the lack of a specific ERK2 antiserum, IHC is unable to distinguish ERK1 and ERK2 expression. Therefore, Western blot was used to quantify the knockdown of ERK2 expression. Compared with the control vector MM, vector 2-7 induced a >50% reduction in the expression of ERK2 in the ipsilateral SCDH (Fig. 2A,B) (p < 0.05), whereas the expression of ERK1 was not affected (Fig. 2A,C). This knockdown was not present on the contralateral side (Fig. 2A,B). As expected, the level of pERK2, the active form of ERK2, was also decreased on the ipsilateral side in animals treated with vector 2-7 compared with vector MM (Fig. 2D,E) (p < 0.05). Interestingly, there was an accompanying increase in pERK1 in blots from these animals (Fig. 2D,F) (p < 0.05), suggesting a loss of competition between ERK1 and ERK2 for their binding and activation by MEK. Vector MM did not have any effect on the expression or phosphorylation of ERK1 and ERK2 compared with animals that did not receive IPI of a vector (supplemental Fig. 4A–E, available at www.jneurosci.org as supplemental material).
Figure 2.

Inhibition of ERK2 expression and phosphorylation in the ipsilateral SCDH of mice treated with vector 2-7, an active ERK2 siRNA. An example of the blot is shown in A, the relative protein levels of ERK2 are shown in B, and ERK1 is shown in C. A, B, Western blot analysis shows a reduction (*p < 0.05 vs the control vector MM group) of ERK2 expression only on the ipsilateral side of the SCDH after vector 2-7 treatment. A, C, ERK1 expression was unaffected by vector 2-7. Western blot analysis (D, E) showed a decrease (*p < 0.05 vs the control vector MM group) in pERK2 and an increase (*p < 0.05 vs the control vector MM group) in pERK1 (D, F) in the ipsilateral SCDH after vector 2-7 treatment. A blot is shown in D, and the relative levels of pERK2 and pERK1 are shown in E and F, respectively. The data are the averages from four separate gel determinations. Error bars indicate SEM. C, Contralateral; I, ipsilateral.
We examined the cellular localization of pERK1/2 on spinal sections using an antibody that recognizes both pERK1 and pERK2. In animals that received the control vector MM, pERK1/2 was observed in the lumbar SCDH, mainly in laminas I and II (Fig. 3A). Confocal double fluorescent IHC revealed that pERK1/2 immunolabeling was colocalized with NeuN (Fig. 3A–C) but not with GFAP or OX42 (data not shown). In animals treated with vector 2-7, the pERK1/2 immunolabeling was dramatically decreased in the ipsilateral dorsal horn (Fig. 3D–F), suggesting that pERK1/2 immunolabeling is mainly composed of pERK2. As stated previously, we noticed an increase in pERK1 accompanying ERK2 knockdown. However, the increase in phosphorylation of ERK1 was apparently not sufficient to overcome the knockdown of ERK2 expression and phosphorylation in these SCDH neurons.
Figure 3.
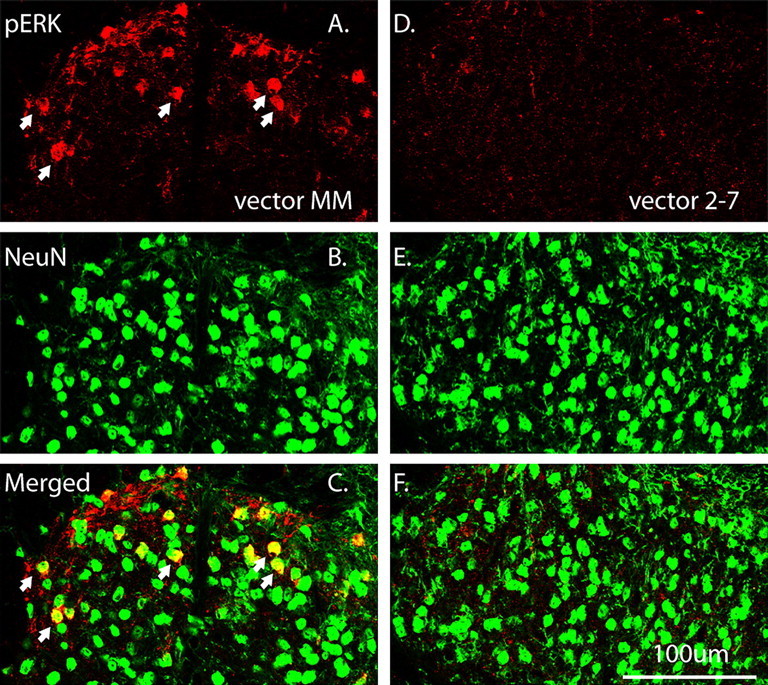
The cellular localization of pERK1/2 immunolabeling in the ipsilateral SCDH at 3 weeks after rAAV vector administration. A, pERK1/2 immunolabeling was detected in the ipsilateral SCDH after the control vector, MM (red). B, NeuN labeling revealed typically distributed neuronal morphologies (green). C, Merged image shows that pERK1/2 was strongly colocalized with NeuN (yellow, arrows). D, pERK1/2 immunolabeling was significantly reduced by vector 2-7 compared with vector MM (A). E, The NeuN labeling pattern is similar to that seen with vector MM (B). F, Merged image shows almost no colocalization of pERK1/2 and NeuN labeling after vector 2-7 compared with vector MM (C). Scale bar, 100 μm.
Knockdown of ERK2 expression prevented ERK activation in the SCDH after intraplantar CFA administration
It has been established that the injection of CFA into the hindpaw of mouse induces a rapid increase in the phosphorylation of ERK1 and ERK2 in the ipsilateral lumbar SCDH, which is then maintained for at least 7 d (Ji et al., 2002; Adwanikar et al., 2004). This induction is associated with the development of hyperalgesia and allodynia in the injected paw. ERK1/2 may regulate their targets by either posttranslational or transcriptional mechanisms, presumably at different stages of the injury-induced pain. Therefore, we examined ERK1/2 phosphorylation in the lumbar SCDH at both 1 and 96 h after CFA.
As shown in Figure 4A, Western blot analysis showed a significant reduction in ERK2 in the ipsilateral SCDH after vector 2-7 compared with the control MM vector. The reduction was observed at 1 h and maintained at 96 h after CFA (p < 0.05 vs vector MM). In the control animals, ERK2 was increased at 96 h after CFA in the ipsilateral SCDH compared with no treatment (NT) or 1 h (Fig. 4A,B) (p < 0.05). There was also an increase in ERK1 expression at 96 h in the control animals compared with NT and 1 h (Fig. 4A,C) (p < 0.05). Vector 2-7 did not alter ERK1 expression and prevented the ERK1 increase induced by CFA (Fig. 4A,C). In control mice (no vector treatment), CFA induced an increase in pERK2 at 1 h that persisted at 24 and 96 h after CFA (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Subsequent figures present data on the 1 and 96 h post-CFA time points. There was a decrease in basal pERK2 after vector 2-7 and this reduction persisted at 1 and 96 h after CFA (Fig. 4D,E) (p < 0.05 vs vector MM). In contrast, pERK2 was increased at 1 and 96 h after CFA in the control animals treated with vector MM (Fig. 4D,E) (p < 0.05 vs vector MM/NT). Basal pERK1 was increased as a result of IPI of vector 2-7. This increase was not altered by CFA at either 1 or 96 h. In contrast, pERK1 was increased by CFA in the vector MM group at 1 and 96 h (Fig. 4D,F) (p < 0.05 vs vector MM/NT). These results clearly demonstrated that the increases in ERK2, ERK1, and pERK2 after CFA were prevented by vector 2-7, although vector 2-7 induced an increase in basal pERK1 level.
Figure 4.

Vector 2-7 but not vector MM reduces ERK2 expression in the ipsilateral SCDH and prevents the phosphorylation of ERK2 induced by intraplantar CFA. A, B, Western blot analysis shows a significant reduction in ERK2 in the ipsilateral SCDH after vector 2-7 compared with the control, vector MM in the absence of intraplantar treatment (NT) (*p < 0.05 vs vector MM/NT). This reduction in ERK2 persists at 1 and 96 h after CFA (*p < 0.05 vs vector MM). At 96 h after CFA, ERK2 is increased in the ipsilateral SCDH of the vector MM group NT or 1 h after CFA (#p < 0.05). A, C, ERK1 expression is also increased at 96 h after CFA (#p < 0.05), and this increase is prevented by vector 2-7 treatment. D, E, pERK2 is decreased after vector 2-7 and the reduction persists at 1 and 96 h after CFA (*p < 0.05 vs vector MM). For the vector MM, pERK2 was increased at 1 and 96 h after CFA compared with the NT control (#p < 0.05). D, F, pERK1 was increased after vector 2-7 (*p < 0.05 vs vector MM/NT). This increase in pERK1 was not altered after CFA in the vector 2-7 group. In contrast, pERK1 was increased at 1 and 96 h in the vector MM group (#p < 0.05 vs vector MM/NT). The data are the averages from four separate gel determinations. Error bars indicate SEM.
Next, DAB IHC was performed to quantify and localize pERK1/2 changes in the SCDH after CFA. In the control vector MM group, immunolabeling of pERK1/2 was observed mainly in laminas I and II in the ipsilateral SCDH. Intraplantar CFA injection induced a significant increase in the pERK1/2 labeling at 1 and 96 h, measured as the number of pERK of labeled neurons in laminas I and II or percentage of field (Fig. 5A–C,G,H) (p < 0.05 vs vector MM/NT). The level of pERK1/2 immunolabeling in the vector 2-7 group was significantly lower than the control group at each corresponding time point (Fig. 5D–G,H) (p < 0.05 vs vector MM).
Figure 5.
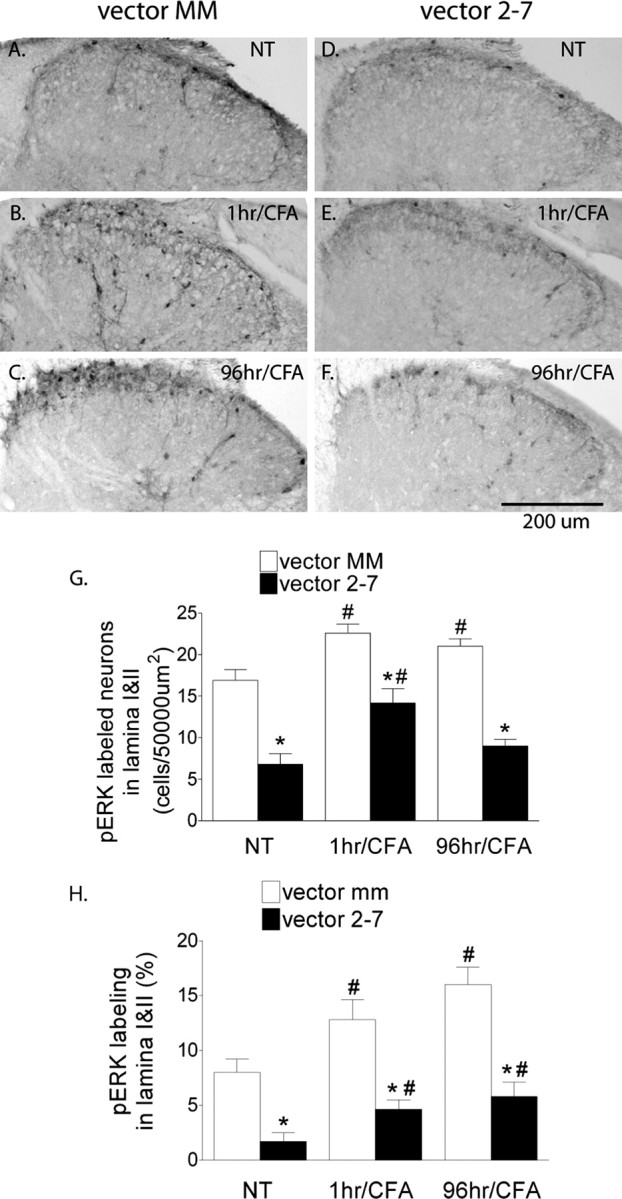
Vector 2-7 prevents the increased phosphorylation of ERK1/2 that is induced in the ipsilateral SCDH by intraplantar CFA. A, The basal level expression of pERK1/2 can be seen mainly in lamina I–II neurons in the control mice after vector MM and in the absence of intraplantar CFA treatment (NT). B, C, pERK1/2 immunolabeling was increased at 1 h (B) and 96 h (C) after CFA. D–F, pERK1/2 immunolabeling in the SCDH of mice treated with vector 2-7 is reduced compared with MM control, before (NT) and after CFA. G, H, Quantification of pERK1/2 as labeled neuron density (G) or percentage of field (H) in laminas I–II revealed a significant increase at 1 and 96 h after CFA in the MM control mice (#p < 0.05 vs vector MM/NT). vector 2-7 reduced pERK1/2 immunolabeling at each corresponding time point compared with vector MM (G, H) (*p < 0.05 vs vector MM). CFA induced an increase in pERK1/2 labeling as measured by neuron density or percentage of field at 1 and 96 h in the vector MM group (G, H) (#p < 0.05). For the vector 2-7 group, CFA produced increases in labeling although less than the corresponding increases in the vector MM group. Scale bar, 500 μm. Error bars indicate SEM.
Effects of the knockdown of neuronal ERK2 in the SCDH on motor reflexes, acute thermal and mechanical thresholds
To investigate the functional consequences of the ERK2 knockdown, we first examined the motor reflexes, hindpaw thermal withdrawal latency, and mechanical withdrawal threshold in animals before and after IPI of each viral vector. We found that none of the animals that received either vector MM or vector 2-7 exhibited any signs of motor deficits (data not shown). No change was observed in heat withdrawal latency or mechanical withdrawal threshold, when tested at least 3 weeks after the vector administration (Fig. 6A,B), indicating that neither the IPI procedure nor knockdown of neuronal ERK2 in the SCDH affected acute pain thresholds.
Figure 6.
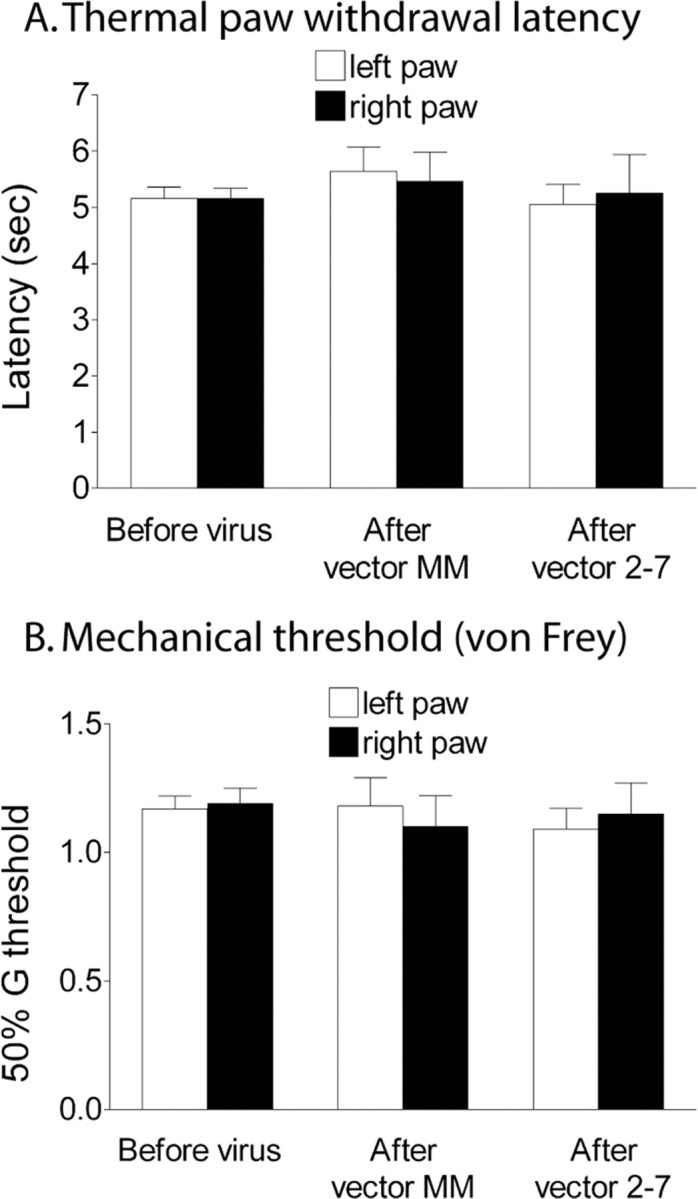
A spatial knockdown of ERK2 in the SCDH by vector 2-7 does not affect thermal (A) or mechanical (B) paw withdrawal thresholds. A brief thermal stimulus was applied to the paw (A), or mechanical (tactile) stimuli were applied using von Frey hairs (B). These thresholds were measured before and 3 weeks after the ipsilateral IPI vector administration into the right SCDH of either the control vector (MM) or the siRNA vector 2-7 (n = 10 per treatment group). Error bars indicate SEM.
Knockdown of ERK2 in the SCDH prevented CFA-induced pain
After intraplantar CFA administration, there was an equal increase in paw size in animals treated with either the control vector MM or the siRNA vector 2-7 (Fig. 7A) (p < 0.05 vs baseline; n = 10 per treatment group), indicating a similar degree of peripheral inflammation. However, only the vector control animals exhibited significant decreases in the thermal withdrawal latency (thermal hyperalgesia) (Fig. 7B) (p < 0.05) and mechanical withdrawal threshold (mechanical allodynia) (Fig. 7C) (p < 0.05) at 24, 48, and 96 h after CFA. The ERK2 knockdown in the SCDH protected the animals from developing thermal hyperalgesia and mechanical allodynia for up to 96 h after CFA (Fig. 7B,C).
Figure 7.
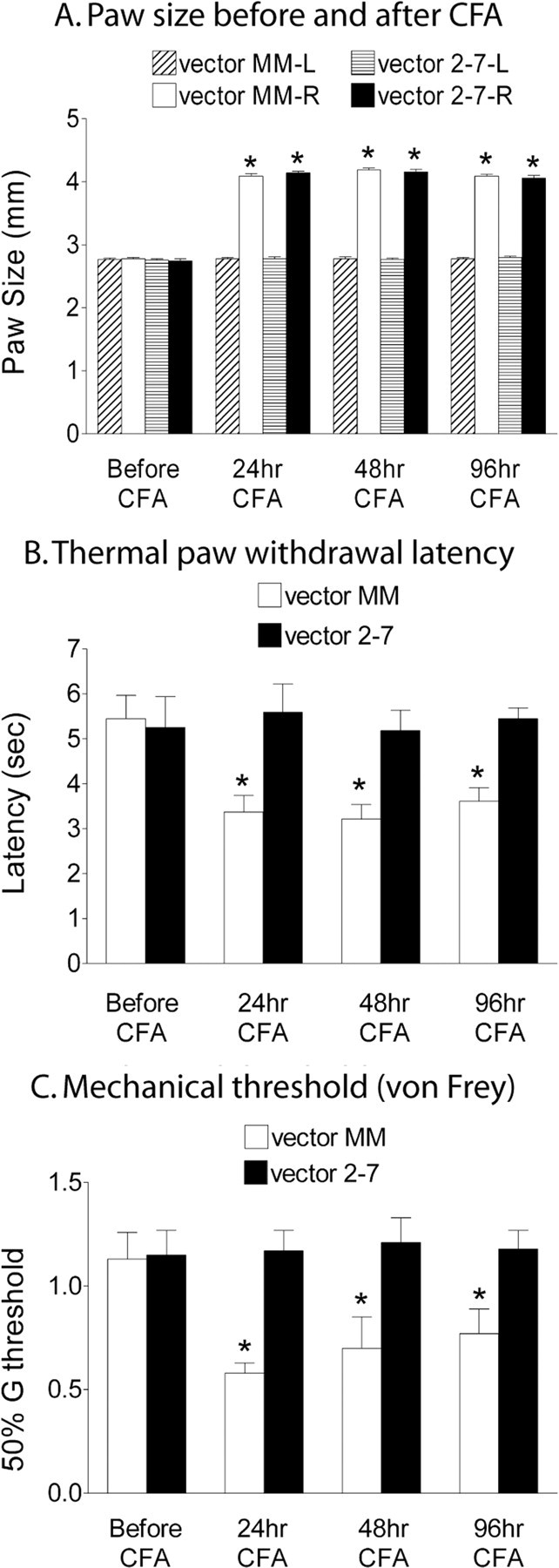
A spatial knockdown of ERK2 in the SCDH by vector 2-7 significantly reduces thermal hyperalgesia (B) and mechanical allodynia (C) resulting from the intraplantar injection of the inflammatory agent, CFA. A, The CFA resulted in equivalent inflammation as measured by paw size at 24, 48, and 96 h (*p < 0.05 vs baseline; n = 10 per treatment group). The control vector MM group showed thermal hyperalgesia as measured as a reduction in the paw withdrawal threshold using a thermal stimulus (B) and mechanical allodynia as measured as a reduction in the mechanical threshold (50% G threshold) using von Frey hairs (C) applied to the paw at 24, 48, and 96 h after intraplantar CFA compared with the baseline (*p < 0.05 vs baseline). The mice that received vector 2-7 were protected from CFA-induced thermal hyperalgesia (B) and mechanical allodynia (C) (n = 10 per treatment group). Error bars indicate SEM.
The expression of c-fos and dynorphin A after CFA
To investigate how ERK2 knockdown may prevent CFA-induced inflammatory pain, we examined the expression of c-fos (supplemental Fig. 6, available at www.jneurosci.org as supplemental material) and dynorphin A (supplemental Fig. 7, available at www.jneurosci.org as supplemental material), two genes that could be regulated by ERK via a cAMP response element (CRE)-mediated mechanism and lead to long-lasting synaptic modifications in pain (Ji et al., 2002; Obata et al., 2003). We found there was a very low level of c-fos immunolabeling before CFA. At 1 h, the expression of c-fos was strongly induced in the ipsilateral SCDH. This induction was equal in both groups of animals and diminished at 96 h. Dynorphin A immunolabeling was not different in animals that received vector 2-7 compared with the control animals. In addition, we did not observe any changes in the level of dynorphin A at either 1 or 96 h after CFA. These results suggest that the expression of c-fos or dynorphin A is unlikely to be regulated by neuronal ERK2 at either 1 or 96 h after CFA.
Discussion
We report the first spatial-temporal knockdown of ERK2 gene expression mediated by a siRNA in the SCDH of adult mice. The ERK2 siRNAs delivered by a neurotropic rAAV vector produced a localized reduction in the basal level of both ERK2 and its phosphorylated form (pERK2) in SCDH neurons. The increase in both ERK2 and pERK2 induced by intraplantar CFA in the SCDH of control mice was prevented by the ERK2 siRNA. In addition, the ERK2 siRNA vector protected the mice from CFA-induced thermal hyperalgesia and mechanical allodynia for at least 96 h.
The rAAV vector-based siRNA approach presents a potent and facile tool to produce a spatial and temporal knockdown of the expression of a gene of interest (Garraway et al., 2007). Several factors dictated the choice of a rAAV vector for the delivery of the ERK2 siRNA. First, the serotype-2 rAAV vector used in the current study selectively transduces neurons in vivo (Kaspar et al., 2002). Second, rAAV is able to mediate long-term siRNA expression and gene knockdown in the transduced cells. Although we only examined the GFP and ERK expression for 6 weeks, our previous study demonstrated that a single administration of a rAAV vector resulted in the knockdown of NR1 gene expression that persisted for at least 6 months (Garraway et al., 2007). Third, rAAV is safe and therefore convenient to use in behavioral experiments requiring repeated measurements. Fourth, rAAV-mediated gene knockdown could be controlled both temporally and spatially. This conditional approach avoids embryonic lethality associated with a constitutive knock-out of ERK2 (Hatano et al., 2003; Saba-El-Leil et al., 2003; Yao et al., 2003).
Consistent with the observations in this study, several reports (Kaspar et al., 2002; South et al., 2003; Garraway et al., 2007) have provided evidence at the ultrastructural and light microscope levels as well as direct behavioral threshold evidence that the injection of AAV into the brain or SCDH does not result in significant immune or glial activation or behavioral sensitization. It has been reported that a high dose of a siRNA might induce nonspecific and off-target effects (Bridge et al., 2003; Sledz et al., 2003). Our previous study indicated that neither an NR1 siRNA nor a control siRNA delivered by the rAAV vector induced detectable cellular toxicity (Garraway et al., 2007). Transduced neurons exhibited unaltered expression of NeuN compared with the contralateral side. We also did not see any signs of gliosis or neuronal damage (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The three vectors expressing different siRNAs induced a similar degree of GFP expression and knockdown of the ERK2 mRNA in the SCDH, suggesting the knockdown is unlikely to be induced by nonspecific or off-target effects. The knockdown clearly targets the ERK2 mRNA and protein as revealed by in situ hybridization and Western blot. The closely related ERK1 was unaffected at the protein level.
The ERK2 knockdown was accompanied by an increase in ERK1 phosphorylation. This has been observed in cultured hepatocytes and led to the suggestion that ERK1 and ERK2 may compete for activation by MEK (Frémin et al., 2007), so that a siRNA-mediated knockdown of ERK2 favors the increased phosphorylation of ERK1.
ERK1 and ERK2 are highly homologous in their exon–intron coding sequences (Hanks and Hunter, 1995). However, the 5′-flanking regions of the two genes have no apparent homology (Pagès et al., 1995; Sugiura et al., 1997). This divergence in the 5′-regulatory sequence is likely to enable distinct patterns of gene expression and the possibility of different roles for each ERK protein. Although the reason for the preferential phosphorylation of ERK2 in SCDH neurons is not known, our data clearly show that the ERK2 siRNA vector 2-7 greatly reduced basal and induced pERK immunolabeling in the SCDH (Fig. 3A,D) and that this pERK species was pERK2 as measured by Western blot (Fig. 2D).
In the absence of injury, baseline behavioral responses are comparable in the knockdown and the control animals. Thus, ERK2 does not appear to mediate these baseline responses; rather they may be mediated by AMPA receptors (South et al., 2003).
Transgenic mice expressing neuron-specific dominant-negative MEK exhibited a decrease in the induction of formalin-induced phase II spontaneous pain behavior and thermal hyperalgesia (Karim et al., 2006). In our study, the ERK2 knockdown completely blocked CFA-induced thermal hyperalgesia and mechanical allodynia. Additional experiments examining inflammatory pain behaviors earlier than 24 h after CFA are required to demonstrate the role of ERK2 in the early induction of inflammatory pain.
Injury-inducing stimuli such as intraplantar CFA injection result in a rapid activation of ERK1/2 in the SCDH. NMDA receptors play a major role in ERK1/2 activation (Ji et al., 1999; Cheng et al., 2008), although other players are also involved (Kawasaki et al., 2004). This activation of ERK1/2 after CFA is sustained at 24 h and persists for at least 96 h. Several sources may contribute to the prolonged ERK1/2 activation, such as sustained primary afferent input from the periphery. The injected hindpaw remains swollen at 96 h, suggesting the presence of an ongoing peripheral inflammation. Another source could be the descending excitatory pathway from supraspinal sites (Svensson et al., 2006).
ERK1/2 can phosphorylate several pain-related proteins including the NR1 subunit of the NMDA receptor (Krapivinsky et al., 2003) and the Kv4.2 potassium channel (Hu et al., 2006). Phosphorylation of these proteins can contribute to the central sensitization in spinal neurons after peripheral injury, which leads to increased membrane excitability in the affected neurons. In addition to its role in the posttranslational regulation, ERK1/2 may also maintain pain hypersensitivity by promoting transcription of genes that are important for neuronal plasticity. A major transcription factor activated by ERK1/2 is cAMP response element-binding protein (CREB), which in turn induces transcriptional activation of many genes such as c-fos, TrkB (Obata et al., 2003), NK-1, and prodynorphin (Ji et al., 2002) via CRE-mediated mechanism. We found the immediate-early gene c-fos was activated in the SCDH in control animals at 1 h but not 96 h after CFA. However, we observed a comparable change in the ERK2 knockdown animals, indicating c-fos was activated by ERK2-independent mechanisms. In addition, we were unable to detect any changes in the level of dynorphin A at 1 or 96 h after CFA. Ji et al. (2002) found that prodynorphin mRNA levels are increased at 24 and 48 h after CFA and this increase as well as the inflammatory pain is attenuated by pretreatment with a MEK inhibitor. Therefore, we may have missed the critical period for expression of dynorphin.
ERK1 and ERK2 mRNA levels are upregulated at 12 h after formalin (Li et al., 2004). We found the expression of ERK1 and ERK2 protein remains upregulated at 96 h after intraplantar CFA in control animals, but was prevented by the ERK2 siRNA vector. Our data suggest that ERK2 may be involved in regulating its own transcription. The promoter region of ERK2 contains a CRE site (Sugiura et al., 1997), allowing the involvement of the transcription factor CREB, although how ERK2 might regulate ERK1 expression is not clear.
One intriguing question left is the role of glial ERK1/2 in injury-induced pain hypersensitivity. ERK1/2 activation in inflammatory pain models has been reported to be primarily neuronal (Ji et al., 1999, 2002). In contrast, glial ERK1/2 activation has been widely reported in neuropathic pain models (Zhuang et al., 2005; Zhao et al., 2007), suggesting major mechanistic differences between these two types of pain. Glial ERK1/2 activation usually occurs several days after the initial injury, and regulates glia–neuron communication by promoting prostaglandin E2 (PGE2) release from microglia, which can subsequently act on neurons that express PGE2 receptor E-prostanoid 2 (Zhao et al., 2007). However, even in neuropathic pain models in which glia activation appears to play a more prominent role, neuronal ERK1/2 activation is maintained in the SCDH for several weeks, which can be reinduced at later stages of nerve injury, enhancing response to both innocuous and noxious peripheral stimuli (Wang et al., 2004). The continuous involvement of neuronal ERK is also supported by a study that showed that sustained neuronal ERK1/2 activation correlated with neuropathic pain behavior in the chronic constriction injury model (Tseng et al., 2007). Although it is clear that ERK2 is involved in mediating injury-induced pain, to further elucidate the exact mechanism, it will be necessary to identify the downstream target proteins that are regulated by neuronal ERK2. Some of these [e.g., the Kv4.2 potassium channel (Hu et al., 2006)] have been identified, whereas others can be identified using the selective attenuation of ERK2.
In conclusion, we have demonstrated that ERK2 siRNAs derived from a rAAV vector selectively inhibited the expression of neuronal ERK2 and blocked the CFA-induced phosphorylation of ERK2 in the mouse SCDH. Although knockdown of ERK2 expression in the SCDH neurons does not affect baseline thermal or mechanical threshold, it does protect animals from CFA-induced mechanical allodynia and thermal hyperalgesia. Our data suggest that neuronal ERK2 is critical in the development of CFA-induced pain hypersensitivity. Targeting this signaling protein at spinal sites may provide successful management for injury-induced pain.
Footnotes
This work was supported in part by National Institute on Drug Abuse (NIDA) Grants DA001457 and DA000198 (C.E.I.), NIDA Training Grant DA007274 (A.R.W.) and Minority Supplement Grant DA001457 (S.M.G.), and NIDA Center Grant DA005130. We thank Dr. Ann Gregus, Dr. Deborah M. Hegarty, Paigee Chou, Justin Granstein, Michelle Contreras, and Zach Epstein-Peterson for advice and/or assistance.
References
- Adwanikar H, Karim F, Gereau RW., 4th Inflammation persistently enhances nocifensive behaviors mediated by spinal group I mGluRs through sustained ERK activation. Pain. 2004;111:125–135. doi: 10.1016/j.pain.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheng HT, Suzuki M, Hegarty DM, Xu Q, Weyerbacher AR, South SM, Ohata M, Inturrisi CE. Inflammatory pain-induced signaling events following a conditional deletion of the N-methyl-d-aspartate receptor in spinal cord dorsal horn. Neuroscience. 2008;155:948–958. doi: 10.1016/j.neuroscience.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CD, Neto FL, Castro-Lopes J, McMahon SB, Cruz F. Inhibition of ERK phosphorylation decreases nociceptive behaviour in monoarthritic rats. Pain. 2005;116:411–419. doi: 10.1016/j.pain.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Dai Y, Fukuoka T, Wang H, Yamanaka H, Obata K, Tokunaga A, Noguchi K. Contribution of sensitized P2X receptors in inflamed tissue to the mechanical hypersensitivity revealed by phosphorylated ERK in DRG neurons. Pain. 2004;108:258–266. doi: 10.1016/j.pain.2003.12.034. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Frémin C, Ezan F, Boisselier P, Bessard A, Pagès G, Pouysségur J, Baffet G. ERK2 but not ERK1 plays a key role in hepatocyte replication: an RNAi-mediated ERK2 knockdown approach in wild-type and ERK1 null hepatocytes. Hepatology. 2007;45:1035–1045. doi: 10.1002/hep.21551. [DOI] [PubMed] [Google Scholar]
- Galan A, Lopez-Garcia JA, Cervero F, Laird JM. Activation of spinal extracellular signaling-regulated kinase-1 and -2 by intraplantar carrageenan in rodents. Neurosci Lett. 2002;322:37–40. doi: 10.1016/s0304-3940(02)00078-2. [DOI] [PubMed] [Google Scholar]
- Garraway SM, Xu Q, Inturrisi CE. Design and evaluation of small interfering RNAs that target expression of the N-methyl-d-aspartate receptor NR1 subunit gene in the spinal cord dorsal horn. J Pharmacol Exp Ther. 2007;322:982–988. doi: 10.1124/jpet.107.123125. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hatano N, Mori Y, Oh-hora M, Kosugi A, Fujikawa T, Nakai N, Niwa H, Miyazaki J, Hamaoka T, Ogata M. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells. 2003;8:847–856. doi: 10.1046/j.1365-2443.2003.00680.x. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW., 4th The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Hu HJ, Adwanikar H, Kaplan D, Gereau RW., 4th Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron-specific dominant negative mitogen activated protein kinase kinase (MEK) Mol Pain. 2006;2:2. doi: 10.1186/1744-8069-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Vissel B, Bengoechea T, Crone S, Randolph-Moore L, Muller R, Brandon EP, Schaffer D, Verma IM, Lee KF, Heinemann SF, Gage FH. Adeno-associated virus effectively mediates conditional gene modification in the brain. Proc Natl Acad Sci U S A. 2002;99:2320–2325. doi: 10.1073/pnas.042678699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Li J, Johnson SE. ERK2 is required for efficient terminal differentiation of skeletal myoblasts. Biochem Biophys Res Commun. 2006;345:1425–1433. doi: 10.1016/j.bbrc.2006.05.051. [DOI] [PubMed] [Google Scholar]
- Li X, Lighthall G, Liang DY, Clark JD. Alterations in spinal cord gene expression after hindpaw formalin injection. J Neurosci Res. 2004;78:533–541. doi: 10.1002/jnr.20274. [DOI] [PubMed] [Google Scholar]
- Musatov S, Roberts J, Pfaff D, Kaplitt M. A cis-acting element that directs circular adeno-associated virus replication and packaging. J Virol. 2002;76:12792–12802. doi: 10.1128/JVI.76.24.12792-12802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–4126. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès G, Stanley ER, Le Gall M, Brunet A, Pouysségur J. The mouse p44 mitogen-activated protein kinase (extracellular signal-regulated kinase 1) gene. Genomic organization and structure of the 5′-flanking regulatory region. J Biol Chem. 1995;270:26986–26992. doi: 10.1074/jbc.270.45.26986. [DOI] [PubMed] [Google Scholar]
- Pagès G, Guérin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouysségur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Zhen E, Cheng M, Xu S, Vanderbilt CA, Ebert D, Garcia C, Dang A, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1, 2, and 3. J Am Soc Nephrol. 1993;4:1104–1110. doi: 10.1681/ASN.V451104. [DOI] [PubMed] [Google Scholar]
- Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque N, Ang SL, Meloche S. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003;4:964–968. doi: 10.1038/sj.embor.embor939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- South SM, Kohno T, Kaspar BK, Hegarty D, Vissel B, Drake CT, Ohata M, Jenab S, Sailer AW, Malkmus S, Masuyama T, Horner P, Bogulavsky J, Gage FH, Yaksh TL, Woolf CJ, Heinemann SF, Inturrisi CE. A conditional deletion of the NR1 subunit of the NMDA receptor in adult spinal cord dorsal horn reduces NMDA currents and injury-induced pain. J Neurosci. 2003;23:5031–5040. doi: 10.1523/JNEUROSCI.23-12-05031.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura N, Suga T, Ozeki Y, Mamiya G, Takishima K. The mouse extracellular signal-regulated kinase 2 gene. Gene structure and characterization of the promoter. J Biol Chem. 1997;272:21575–21581. doi: 10.1074/jbc.272.34.21575. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Tran TK, Fitzsimmons B, Yaksh TL, Hua XY. Descending serotonergic facilitation of spinal ERK activation and pain behavior. FEBS Lett. 2006;580:6629–6634. doi: 10.1016/j.febslet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng TJ, Hsieh YL, Hsieh ST. Reversal of ERK activation in the dorsal horn after decompression in chronic constriction injury. Exp Neurol. 2007;206:17–23. doi: 10.1016/j.expneurol.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Wang H, Dai Y, Fukuoka T, Yamanaka H, Obata K, Tokunaga A, Noguchi K. Enhancement of stimulation-induced ERK activation in the spinal dorsal horn and gracile nucleus neurons in rats with peripheral nerve injury. Eur J Neurosci. 2004;19:884–890. doi: 10.1111/j.0953-816x.2004.03203.x. [DOI] [PubMed] [Google Scholar]
- Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, Boucher DM. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci U S A. 2003;100:12759–12764. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. siRNA selection server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32:W130–W134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng P, Wagoner HA, Pescovitz OH, Steinmetz R. RNA interference (RNAi) for extracellular signal-regulated kinase 1 (ERK1) alone is sufficient to suppress cell viability in ovarian cancer cells. Cancer Biol Ther. 2005;4:961–967. doi: 10.4161/cbt.4.9.1912. [DOI] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci. 2007;27:2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng CF, Guan KL. Properties of MEKs, the kinases that phosphorylate and activate the extracellular signal-regulated kinases. J Biol Chem. 1993;268:23933–23939. [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]