

Summary
Disruption of the TRPML1 channel results in the neurodegenerative disorder mucolipidosis type IV (MLIV), a lysosomal storage disease with severe motor impairments. The mechanisms underlying MLIV are poorly understood and there is no treatment. Here, we report a Drosophila MLIV model, which recapitulates the key disease features, including abnormal intracellular accumulation of macromolecules, motor defects and neurodegeneration. The basis for the buildup of macromolecules was defective autophagy, which resulted in oxidative stress and impaired synaptic transmission. Late-apoptotic cells accumulated in trpml mutant brains suggesting diminished cell clearance. The accumulation of late apoptotic cells and motor deficits were suppressed by expression of trpml+ in neurons, glia or hematopoietic cells. We conclude that the neurodegeneration and motor defects result primarily from decreased clearance of apoptotic cells. Since hematopoietic cells in humans are involved in clearance of apoptotic cells, our results raise the possibility that bone marrow transplantation may limit the progression of MLIV.
Introduction
The Transient Receptor Potential (TRP) channel superfamily participates in a remarkable diversity of processes in the nervous system (Venkatachalam and Montell, 2007). Nevertheless, the only neurodegenerative disease linked to a TRP channel is the early childhood disorder, mucolipidosis IV (MLIV). This highly debilitating autosomal recessive disease is characterized by severe motor deficits, mental retardation and neurodegeneration, including retinal degeneration (Bach, 2005). MLIV is a lysosomal storage disorder (LSD); one of ~40 LSDs, which together represent the most common cause of neurodegeneration during childhood (Cooper, 2003). As is typical of LSDs, cells from MLIV patients contain large vesicles and accumulate lysosomal storage components (Bach, 2005). Nevertheless, the underlying bases of the MLIV symptoms are not known and there is no effective treatment.
A key advance was the discovery that MLIV results from loss-of-function mutations in TRPML1 (Bargal et al., 2000; Bassi et al., 2000; Sun et al., 2000). TRPML1 appears to be widely expressed and consistent with the nature of MLIV, TRPML1 localizes to late endosomes and lysosomes (Manzoni et al., 2004). A C. elegans TRPML1 homolog, CUP-5, is also present in these organelles (Fares and Greenwald, 2001). Mutations in cup-5 result in maternal-effect lethality, excessive cell death and accumulation of large vacuoles (Hersh et al., 2002). However, a role for cup-5 in the nervous system has not been described. Recently, a mouse MLIV model has been developed, which recapitulates many features of the disorder (Venugopal et al., 2007). Nevertheless, many critical questions remain regarding the cause of the progressive motor defects, neurodegeneration and the mechanistic link to lysosomal dysfunction. Most importantly, no concept has emerged that offers potential for developing therapies for treating MLIV.
Here, we report the development of Drosophila as an animal model for MLIV. We found that trpml mutant flies exhibited a phenotype remarkably reminiscent of MLIV. Most importantly, we report insights into the cellular mechanism underlying the neurodegeneration and motor impairments. Our findings provide a conceptual framework for developing strategies for treating this neurodegenerative disease.
Results
Generation of mutations in Drosophila trpml
The Drosophila genome encodes one TRPML homolog (CG8743), which shares ~40% amino acid identities with human TRPML1-3 (Figure 1A). Human TRPML1 RNA is broadly expressed (Bargal et al., 2000; Bassi et al., 2000; Sun et al., 2000) and based on microarray studies, the fly TRPML RNA is also widely expressed, but at low levels (http://flyatlas.org/atlas.cgi?name=FBgn0036904). When expressed in HEK293 cells, YFP-tagged TRPML localized to lysosomes that were labeled by the low pH specific dye, LysoTracker (Figure S1). Like human TRPML1 (Venkatachalam et al., 2006), TRPML-YFP decorates the periphery of the lysosomes (Figure S1), indicating that it is a lysosomal membrane protein.
FIGURE 1. TRPML is required for normal viability and motor activity.
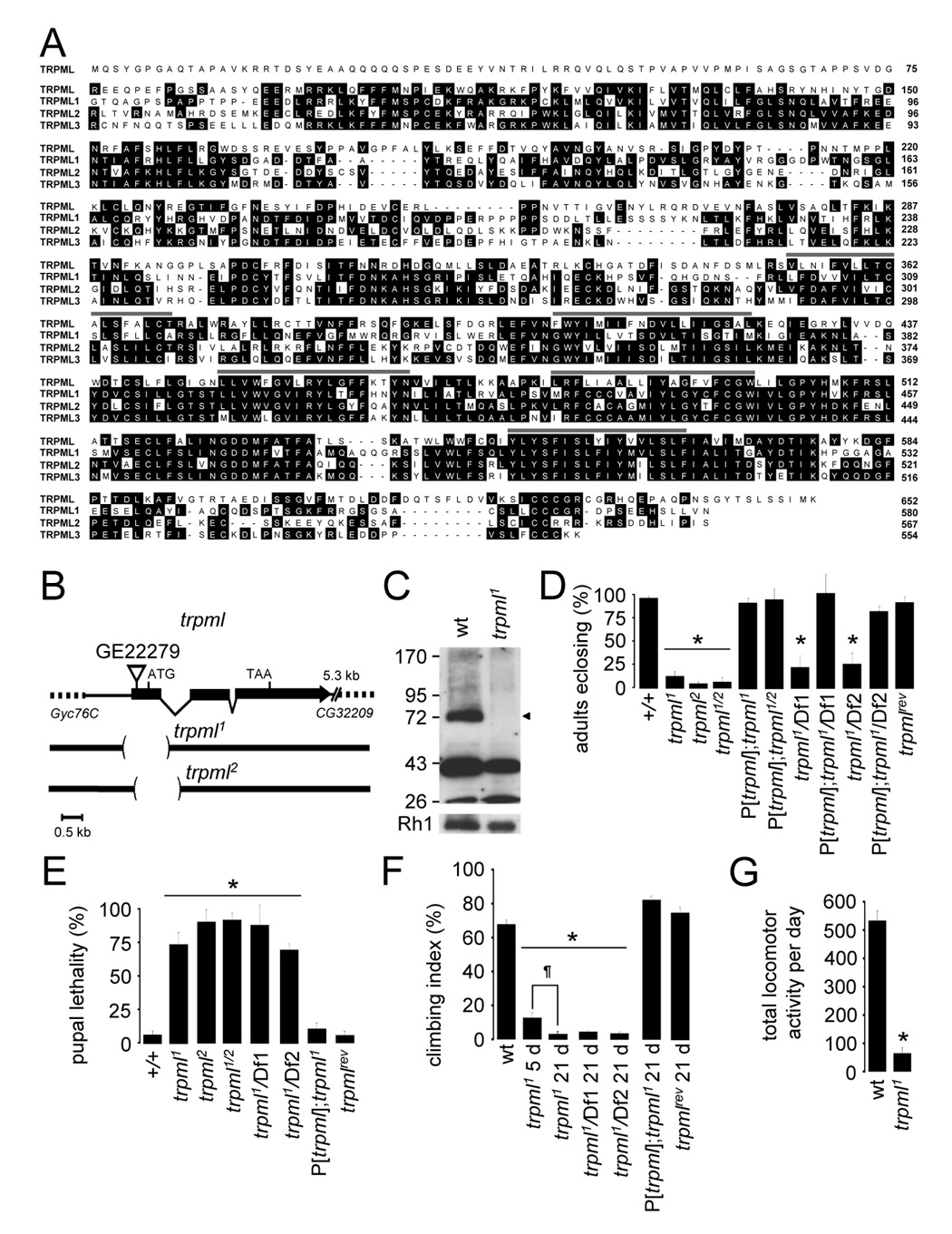
(A) Alignment of Drosophila and human TRPML proteins. Lines indicate TMDs.
(B) trpml genomic locus. The deletions in trpml1 and trpml2 are shown.
(C) Western blot of extracts from wild-type (wt) and trpml1 flies probed with anti-TRPML antibodies and reprobed with anti-Rh1 antibodies (see Supplemental Experimental Procedures). The ~75 kDa band (arrowhead) corresponds to TRPML. The lower bands were due to non-specific interactions with anti-TRPML.
(D) Percentage of pharate adults without the TM3 balancer after inter se crosses. n≥4 vials of each genotype; *, p≤5×10−6, ANOVA. Df1, Df(3L)ED228; Df2, Df(3L)Exel6135; trpml1/2, trpml1/trpml2; P[trpml], genomic rescue; trpmlrev, precise excision.
(E) Percentage of dead pupae. n=4 vials; *, p≤0.005, ANOVA.
(F) Climbing indices. Percentages of flies in a 50 ml glass cylinder that climb to the 25 ml mark in 15 s after being tapped down. n≥13, 10–20 flies each; *, p≤×10−12, ANOVA; ¶, p≤5×10−4, t-test.
(G) Number of crossings of an infra-red beam (24 h period) in an actometer. n=3, 13–14 individual 5 day-old flies each; *, p≤5×10−11, t-test.
To generate a mutation in trpml, we obtained flies with a P-element insertion (GE22279), 242 bases 5’ of the translation initiation site (Figure 1B). GE22279 flies had no obvious phenotype. We mobilized the transposon and identified two imprecise excision lines, with distinct 1.1 kb deletions, extending past the region encoding the first TMD (Figure 1B): trpml1 and trpml2 (−456 - +641 and −234 - +860 base-pairs relative to the translation start site, respectively). We raised antibodies to TRPML, which were ineffective for immunostaining, but on Western blots recognized the predicted 75 kD protein in wild-type fly extracts (~75 kD), which was undetectable in trpml1 (Figure 1C).
Reduced viability and locomotor activity in the trpml mutants
To assess whether trpml was required for viability, we placed the mutations in trans with a balancer chromosome and performed inter se crosses. The percentages of viable adults without the balancer were significantly lower than in wild-type (Figure 1D; expected maximum of 33.3% scaled to 100%). When we placed trpml1 in trans with trpml2 (trpml1/2) or either of two deficiencies spanning the trpml locus (76C3), the percentages of adults were similar to those obtained with the homozygous mutants (Figure 1D). The reduced viability was rescued by a wild-type trpml transgene (P[trpml]) and a line with a precise-excision of the P-element (trpmlrev) showed wild-type viability (Figure 1D). The reduced viability was due to pupal semi-lethality, since the percentage of dead pupae rose from <7% in wild-type, to >70% in the different trpml mutant combinations (Figure 1E).
A primary clinical manifestation of MLIV is a progressive deficit in motor function. To assess whether there was impaired motor function in the trpml mutants, we assayed negative geotaxis. After a tap to the bottom of a cylinder, 69.6±3.0% of 5 day-old wild-type flies climbed to the top half within 15 seconds (Figure 1F). By 21 days, there was only a modest decrement in this activity (59.4±3.5%). In contrast, only 12±3.2% of 5 day-old trpml1 flies ascended to the top half and by 21 days, the negative geotaxis was nearly eliminated (Figure 1F; 2.4±1.2%), demonstrating a progressive loss of motor function. 21 day-old trpml1 in trans with either deficiency displayed similarly reduced climbing, and the defect was rescued by the wild-type transgene (Figure 1F). We also assayed motor activity by placing 5 day-old flies in tubes for 24 hours, and tabulated the number of times they crossed an infrared beam. In this assay, trpml1 flies also showed a significant reduction in activity (Figure 1G).
Progressive neurodegeneration in the trpml mutants
MLIV causes progressive neurodegeneration, including retinal degeneration. The fly compound eye consists of ~800 ommatidia, each of which includes seven photoreceptor cells in each plane of section. Photoreceptor cells include a microvillar compartment, the rhabdomere, which is the site of phototransduction (Wang and Montell, 2007). In contrast to wild-type ommatidia, all of which had a full set of photoreceptor cells regardless of age, a significantly lower fraction of the ommatidia in 21 day-old trpml1 flies retained all seven rhabdomeres (Figures 2A–2C; wild-type, 100%; trpml1, 53.8±9.9%; p≤0.005). This cell death was activity-dependent as 21 day-old dark-reared trpml1 had a significantly higher fraction of ommatidia with all seven rhabdomeres (Figure 2C; 84.5±4.8%; p≤0.01). This phenotype was progressive as all ommatidia in newly eclosed trpml1 contained the full set of rhabdomeres (Figure 2C).
FIGURE 2. Neurodegeneration in trpml retina and brain.
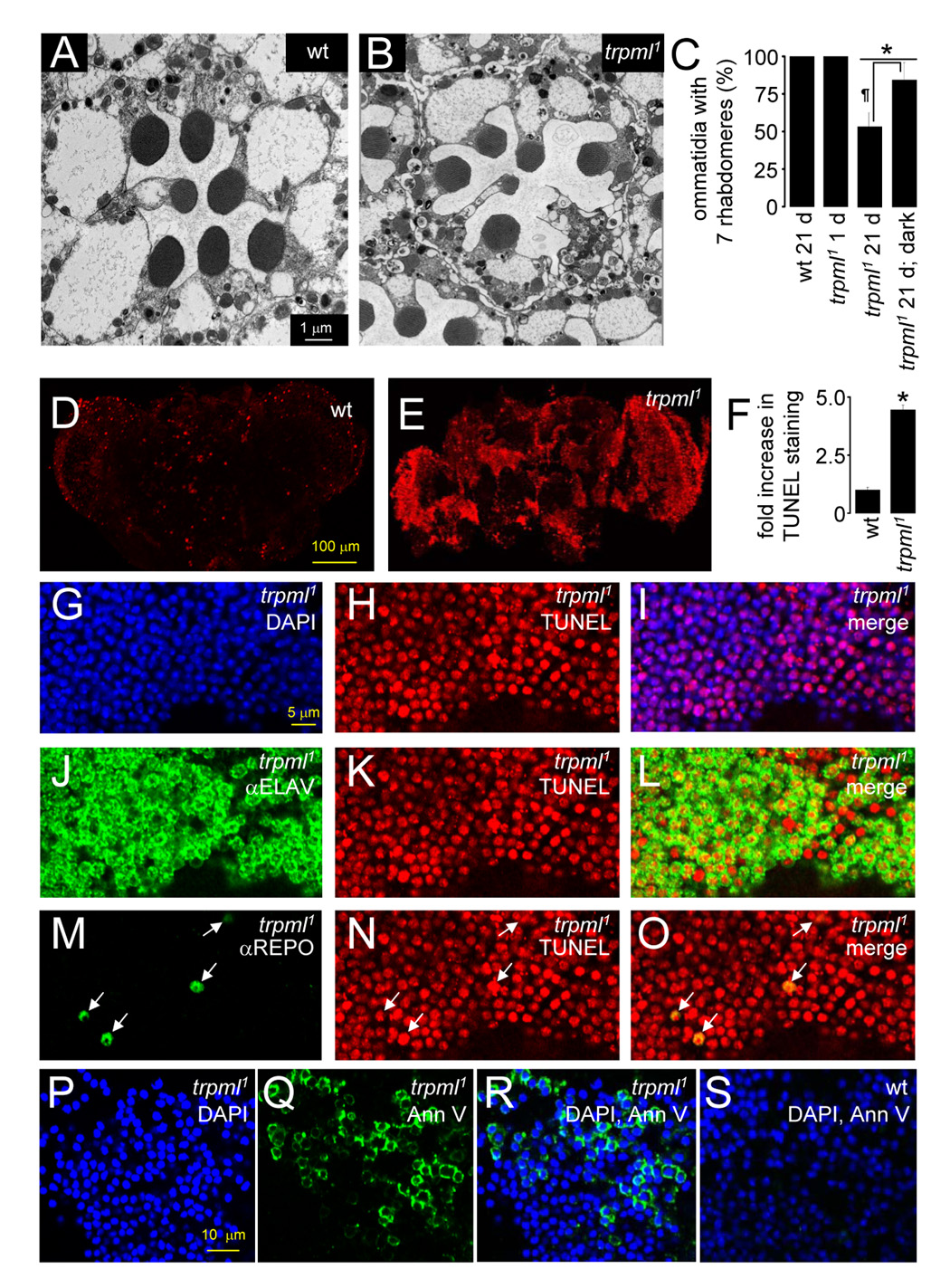
(A and B) Transmission EMs of photoreceptor cells (21 day-old flies): (A) wt, and (B) trpml1.
(C) Percentages of ommatidia with full set of 7 rhabdomeres. n≥3 animals, ≥30 ommatidia per animal; *, difference from wt, p≤0.01, ANOVA; ¶, p≤0.05, t-test.
(D and E) Confocal images of TUNEL-labeled brains (21 day-old flies): (D) wt, and (E) trpml1.
(F) Fold increase in TUNEL labeling in trpml1 normalized to wt mean. n=3; *, p≤5×10−4, t-test.
(G–I) Confocal images of brains from 21 day-old trpml1 viewed at 310 nm to detect DAPI (G) and 568 nm to detect TUNEL (H). (I) Merge.
(J–L) Same as (G–I) but brains viewed at 633 nm to detect ELAV (J).
(M–O) Same as (G–I) but brains viewed at 488 nm to detect REPO (M). Arrows indicate glia.
(P–R) Confocal images of 21 day-old trpml1 brains viewed at 310 nm to detect DAPI (P) and 488 nm to detect Annexin V-FITC (Q). (R) Merge.
(S) Same as (R) but with 21 day-old wt brains.
To determine whether there was progressive degeneration in trpml brains, we performed TUNEL staining. Old wild-type or young trpml1 brains showed only minimal TUNEL staining (Figure 2D and Figure S2A). In contrast, 21 day-old trpml1 brains displayed increased TUNEL staining (Figure 2D–2F and Figure S2E; arbitrary units: wild-type, 5.7±0.7; trpml1, 25.3±1.2), which was rescued by the wild-type transgene (Figures S2C–S2D and S2F). To identify the cell types that were dying, we co-stained 21 day-old trpml1 and trpml1/Df brains with TUNEL and antibodies that label the nuclei of neurons (αELAV) and glia (αREPO). Both neurons (Figure 2J–2L and FigureS3D–S3F) and glia (Figure 2M–2O and FigureS3G–S3I) were TUNEL positive.
Drosophila mutants displaying degeneration in the brain typically accumulate large vacuoles (Kretzschmar et al., 1997). 21 day-old trpml1 brains also showed a 4-fold increase in vacuoles (Figures S4A–S4C; wild-type, 1.8±0.6; trpml1, 7.4±1.3). However, the vacuoles did not progress to the massive holes typical of other neurodegeneration mutants, potentially due to a defect in clearance of dead cells (see below).
Apoptotic cells have elevated phosphatidylserine (PS) in the outer plasma membrane leaflet due to increased translocation of PS from the inner leaflet during early apoptosis. Annexin A5 binds PS, providing a specific marker for apoptotic cells (van den Eijnde et al., 1998). Wild-type brains showed no Annexin V-FITC staining. In contrast, 21 day-old trpml1 brains displayed 10-fold elevated Annexin V-FITC labeling (Figure 2P–2S and FigureS5; wild-type, 3.4±1.8%; trpml1, 35.1±10.9%; p≤0.05).
Increased lysosomal storage and lipofuscin in trpml mutant flies
Many cell types in MLIV patients accumulate lysosome-like vesicles and autofluorescence (Goldin et al., 1995). Therefore, we evaluated whether trpml mutants displayed these features. All trpml mutant tissues examined had significantly elevated autofluorescence at 488 nm, including hemocytes (Figures S6A and S6D; arbitrary units: wild-type, 5.5±0.5; trpml1, 13.8±1.7; p≤0.01), photoreceptor cells (Figures S6G and S6J; 14 day-old: wild-type, 2.4±0.3; trpml1, 9.3±1.1; p≤0.001) and brains (Figures S6M–S6N; wild-type, 19.6±0.6; trpml1, 47.8±4.8; p≤0.005). The autofluorescence was progressive, with a pronounced increase in trpml ommatidia and brains between days 1 and 14 (Figures S7A and S7B; arbitrary units, trpml1 brains: 1 day, 20.2±0.8; 14 days, 47.8±4.8; p≤0.005).
To determine whether the autofluorescence in trpml1 cells was within lysosomes, we stained hemocytes and photoreceptor cells with LysoTracker and examined the staining at a wavelength (568 nm) at which autofluorescence was not detected. The autofluorescence colocalized with LysoTracker in hemocytes (Figures S6A–S6F) and photoreceptor cells (Figures S6G–S6L). The numbers of lysosomes also increased in the trpml mutants (Figures S6A–S6L; lysosomes/ommatidia: wild-type, 5.0±0.8; trpml1 14.5±2.1; p≤0.0005).
Lysosomal autofluorescence is indicative of lipofuscin (polymerized non-degradable protein and lipid-containing material in the lysosomal lumen; Terman and Brunk, 2004). To address whether the autofluorescence in trpml cells was from lipofuscin, we examined the spectral properties of extracted lipids. The peaks characteristic of lipofuscin are distinct from other types of autofluorescence, such as porphyrins. In trpml1, the excitation and emission maxima matched lipofuscin (Figure S6O; ~380 nm and 460 nm) (Fletcher et al., 1973).
Defective autophagy and mitochondrial dysfunction in trpml neurons
Lipofuscin accumulation indicates disrupted autophagy, a lysosomal degradation process impaired in many neurodegenerative diseases (Klionsky, 2007). Defective autophagy can lead to either an accumulation of autophagosomes, which contain components that are normally degraded after these vesicles fuse with lysosomes, or an accumulation of autolysosomes if fusion with lysosomes proceeds normally but lysosomal proteolysis is disrupted (Klionsky, 2007). To determine whether trpml cells amassed autophagosome-like vesicles, we performed transmission EM on retinal sections. In photoreceptor cell bodies from 21 day-old trpml1, there were 6.5-fold more cytoplasmic membrane inclusions than in wild-type (Figures 3A–3E; vesicles/cell body: wild-type, 0.85±0.2; trpml1, 5.6±0.9). These membranous inclusions varied in diameter (0.2–1.0 µm) and consisted of multilamellar and multivesicular bodies (Figure 3D). The multilamellar vesicles displayed the morphological hallmarks of autophagosomes — membranous compartments containing internal membranous structures and cytoplasmic material (Mizushima, 2004). The multivesicular bodies may be late endosomes (Dermaut et al., 2005). The accumulation of the cytoplasmic membrane inclusions trpml1 was age-dependent (vesicles/cell body: 1 day-old, 0.5±0.2; 21 day-old, 5.6±0.9; p≤0.0001).
FIGURE 3. Disruption of autophagy in neural tissues in trpml.
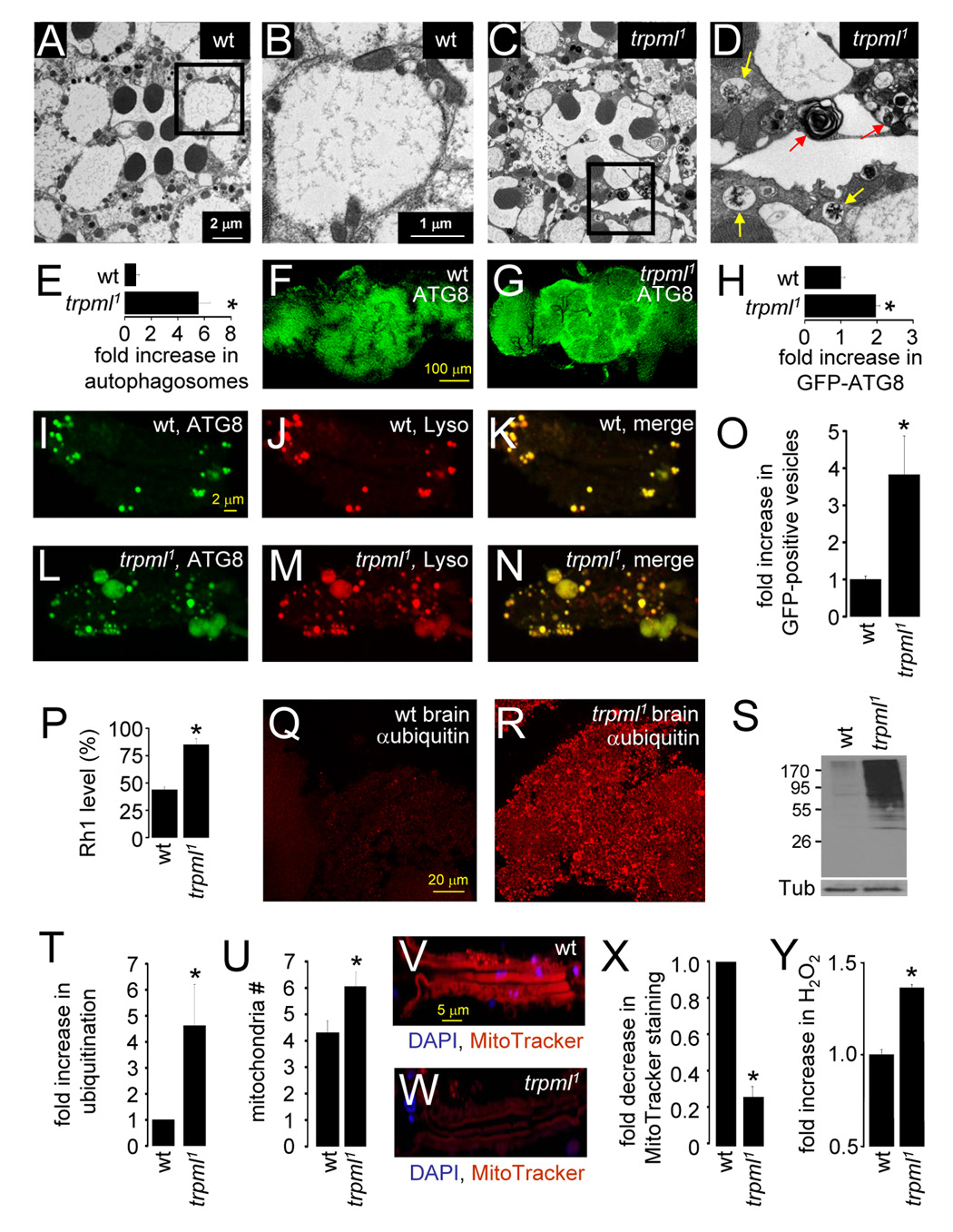
(A–D) Transmission EMs of 21 day-old photoreceptor cells: (A and B) wt, (C and D) trpml1. (B and D) Magnifications of the boxed regions in (A and C). The red and yellow arrows in (D) indicate multilamellar and multivesicular bodies respectively.
(E) Fold-increase in autophagosome-like vesicles in trpml1 photoreceptors. n=5 animals, ≥30 ommatidia per animal; *, p≤10−4.
(F and G) Brains (21 day-old flies) expressing GFP-ATG8 and viewed at 488 nm: (F) wt, (G) trpml1.
(H) Fold-increase in GFP-ATG8 fluorescence in trpml1 brains. n=3; *, p≤5×10−4.
(I–N) Ommatidia from 21 day-old flies expressing GFP-ATG8: (I–K) control, (L–N) trpml1. Ommatidia were viewed at 488 nm to detect GFP-ATG8 (I–L) and 568 nm to detect LysoTracker (Lyso; J–M). (K and N) Merges.
(O) Fold-increase in GFP-positive vesicles in trpml1 ommatidia. n=3 independent experiments; *, p≤5×10−5.
(P) Rh1 remaining after 6 h exposure to blue light (480 nm) in 5 day-old flies. Means are relative to the starting value (defined as 100%). n=3; *, p≤0.05.
(Q and R) 4 week-old brains stained with anti-ubiquitin antibodies (Supplemental Experimental Procedures): (Q) wt, (R) trpml1.
(S) Western blot of head extracts (4 week-old flies) probed with anti-ubiquitin antibodies and reprobed with anti-Tubulin antibodies.
(T) Fold increase of ubiquitination in head extracts based on Western blotting. Data were normalized to wt (1.0). n=3; *, p ≤0.05.
(U) Mitochondria per photoreceptor cell in 21 day-old wt flies. n=5 animals; *, p≤0.05.
(V and W) Ommatidia from 21 day-old wt (V) and trpml1 (W) flies viewed at 310 nm to detect DAPI (blue) and 568 nm to detect MitoTracker-orange CM-H2TMRos (red).
(X) Fold decrease in MitoTracker-orange CM-H2TMRos staining in trpml1 head extracts, normalized to wt (set at 1.0). n=3; *, p≤5×10−6.
(Y) Relative H2O2 levels in whole fly extracts. Data were normalized to wt (set at 1.0). n≥6, 10 flies/experiment; *, p≤5×10−6. Dissected brains and ommatidia were viewed by confocal microscopy.
All statistical analyses, t-test.
To assay further whether there was an increase in autophagosomes in trpml1, we used an autophagosomal marker, GFP-ATG8 (Scott et al., 2004) and performed imaging under conditions in which autofluorescence had dissipated. 21 day-old trpml1 brains showed double the GFP-ATG8 fluorescence compared to wild-type (Figures 3F–3H; 1.96±0.1-fold elevation) while the number of GFP-ATG8 positive vesicles in 21 day-old mutant ommatidia increased nearly four-fold (Figures 3I–3O; 3.8±1.0).
In some LSDs, the buildup of autophagosomes results from a disruption in fusion of autophagosomes with lysosomes (Settembre et al., 2007). To address whether there was a defect in fusion in trpml1, we tested for colocalization of markers for autophagosomes (GFP-ATG8) and lysosomes (LysoTracker). In contrast to other LSDs, the autophagosome and lysosome markers overlapped (Figures 3I–N; wild-type, 91±4.6%; trpml1, 89±2.9%). Thus, it appeared that there was no reduction in autophagosome-lysosome fusion. Rather, trpml1 neurons accumulated autolysosomes, consistent with a defect in proteolytic degradation following fusion of lysosomes and autophagosomes.
To test for a defect in lysosomal degradation, we evaluated whether blue (480 nm) light-induced degradation of rhodopsin1 (Rh1) was reduced in trpml1 photoreceptor cells, since blue light exposure leads to constitutive activation of Rh1, endocytosis and degradation in lysosomes (Xu et al., 2004). A 6 hour exposure to blue light caused a ~55% reduction of Rh1 levels in wild-type, while trpml1 showed only a ~15% decrease (Figure 3P; Rh1 remaining: wild-type, 43.8±0.1%; trpml1, 85±0.1%; p≤0.05).
Mammalian TRPML1 has been reported to be a proton-permeable channel, providing a proton leak pathway in the lysosomal membrane (Soyombo et al., 2006). Therefore, loss of TRPML1 leads to over-acidification of the lysosomal lumen in tissue culture cells (Miedel et al., 2008). As an indirect measure of intra-lysosomal pH, we relied on the intensities of LysoTracker staining and found that lysosomes of relatively the same size were significantly brighter in trpml1 photoreceptor cells (Figure S8, arbitrary units: wild-type, 17.3±2.0; trpml1, 46±3.5; p≤10−5). This result may indicate over-acidification of lysosomes in the trpml mutant.
Autophagic and proteosomal pathways are coupled such that incomplete autophagy can cause macromolecules that are normally degraded by autophagy to enter the proteosomal pathway, thereby increasing the levels of polyubiquitinated proteins (Settembre et al., 2007). We found that trpml1 brains (Figures 3Q–3R) and head extracts showed significantly elevated anti-ubiquitin staining (Figures 3S–3T; 4.6±1.6-fold higher).
Inhibition of autophagy can lead to the accumulation of damaged mitochondria and oxidative stress (Terman and Brunk, 2004). Mitochondria undergo cycles of fusion and fission, which sometimes gives rise to uneven daughters, one of which has a disrupted trans-membrane potential (ΔΨ) and is targeted for degradation via autophagy (Twig et al., 2008). Therefore, disruption of autophagy can lead to accumulation of damaged mitochondria. We found that photoreceptor cell bodies from 21 day-old trpml1 displayed a small but significant increase in the number of mitochondria (Figure 3U; wild-type, 4.3±0.4; trpml1, 6.0±0.5; p≤0.05). Although the number of mitochondria increased, the intensity of Mitotracker-orange CM-H2TMRos staining in trpml1 ommatidia was reduced four-fold relative to wild-type (Figures 3V–3X; arbitrary fluorescence units: wild-type, 29±1.8; trpml1, 7.8±1.0; p≤5×10−6). Since mitochondrial uptake of Mitotracker-orange CM-H2TMRos is dependent upon functional ΔΨ (Deshmukh et al., 2000), the accumulation of mitochondria with dissipated ΔΨ in trpml1 neurons is indicative of a disruption of autophagic clearance of dysfunctional mitochondria.
One indication of oxidative stress in trpml1 was the increase in lipofuscin. To evaluate oxidative stress in trpml1 further, we measured the levels of the reactive oxygen species, H2O2 and found that the mutant flies showed ~40% higher H2O2 levels compared to controls (Figure 3Y; p≤5×10−6). This elevation is physiologically significant, since 30–60% increases in H2O2 can induce abnormally high apoptosis and neurodegeneration (Li et al., 2000; St-Pierre et al., 2006).
Loss of trpml enhances polyglutamine mediated neurotoxicity
Introduction of a polyglutamine repeats in the Drosophila eye results in macromolecular aggregation and cytotoxicity (Warrick et al., 1999). Upregulation of autophagy promotes the clearance of these aggregates thereby suppressing polyglutamine mediated toxicity (Ravikumar et al., 2004). To test whether defective autophagy in trpml1 enhanced polyglutamine mediated toxicity, we expressed a 120 polyglutamine repeat protein in trpml1 photoreceptor cells (gmr-HDQ120) (Warrick et al., 1999) and used an assay on intact eyes to test for loss of rhabdomeres (Figure 4A). Most newly eclosed trpml+ flies expressing gmr-HDQ120/+ had a full set of seven rhabdomeres, but displayed rapid retinal degeneration during the first 3 days post-eclosion (Figures 4A–4B). In combination with the trpml1 mutation, the time-course of degeneration was enhanced significantly, starting from the day of eclosion (Figures 4A–4B). In the absence of gmr-HDQ120, the trpml1 mutation did not cause rhabdomere loss over this time-span (Figure 4B). Since autophagy protects against polyglutamine induced toxicity, the result that trpml1 enhances HDQ120-mediated neurotoxicity is consistent with disrupted autophagy in trpml1 neurons.
FIGURE 4. Toxic aggregation of macromolecules in trpml.
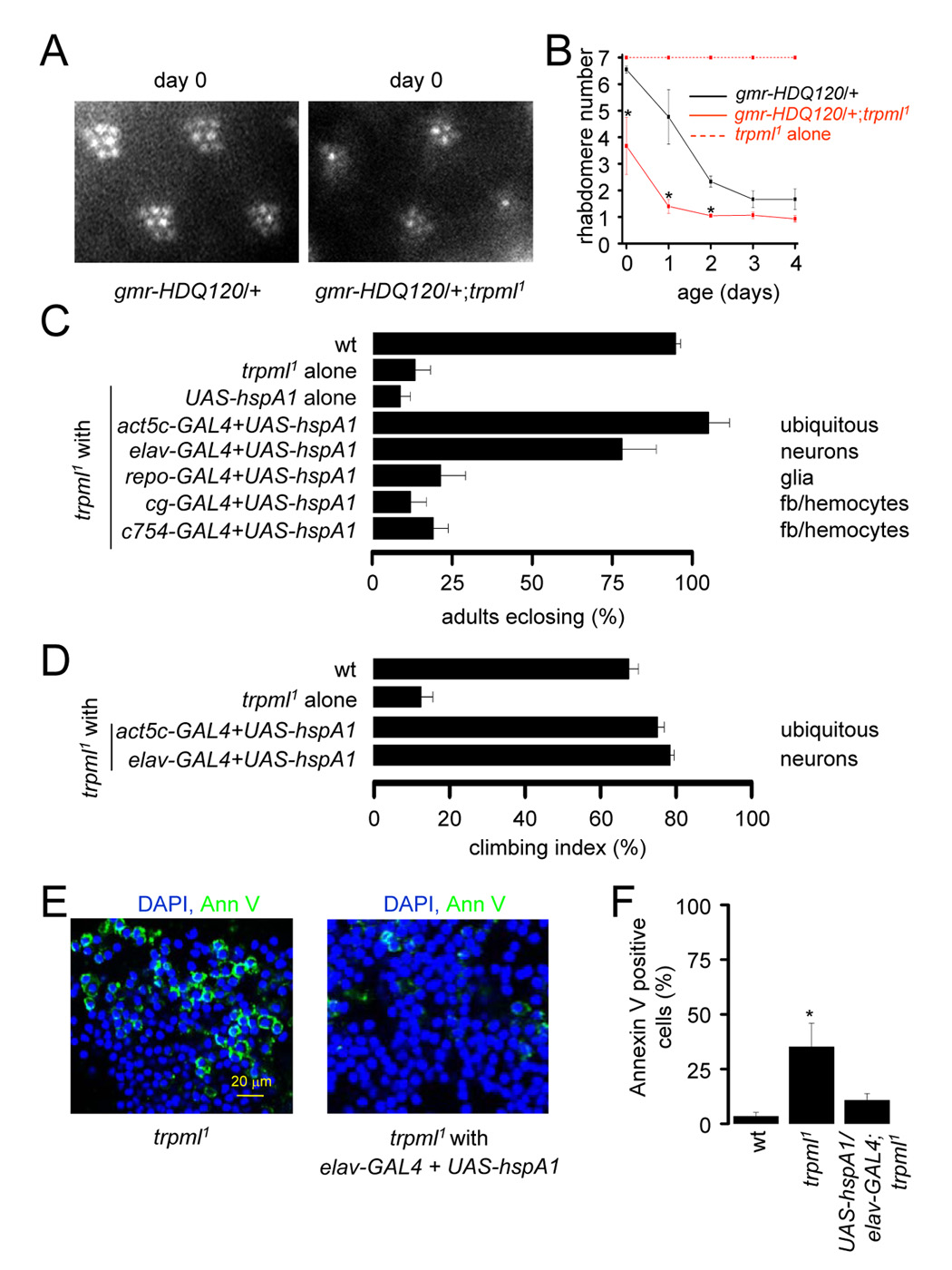
(A) Ommatidia viewed by the optical neutralization technique ≤24 h post-eclosion.
(B) Time course of photoreceptor degeneration viewed by optical neutralization. n≥3 flies, ≥50 ommatidia/fly; *, difference at each time point, p≥0.05.
(C) Percentage of pharate adults without the TM3 balancer in wt, trpml1 and trpml1 with the indicated transgenes, n=7–11. GAL4 expression is indicated (fb, fat bodies).
(D) Climbing indices. n=3–8, 10–20 flies each.
(E) Merged confocal images of brains from 21 day-old flies viewed at 310 nm to detect DAPI and 488 nm to detect Annexin V-FITC.
(F) Percentages of adult brain cells labeled with Annexin V-FITC. n≥3 brains; *, difference from wt, p≤0.05.
All statistical analyses, t-test.
Targeted expression of hspA1L in neurons restores viability and motor function
A factor contributing to the cell death from protein aggregation is the irreversible association of molecular chaperones with these aggregates, thereby lowering the level of chaperones available for newly synthesized proteins (Brignull et al., 2007). Increasing expression of the molecular chaperone, hspA1L (human homolog of HSP70), protects against polyglutamine repeat neuronal toxicity in the Drosophila eye (Warrick et al., 1999).
We assessed the effectiveness of suppressing the pupal semi-lethality and motor deficits in trpml1 by introduction of hspA1L. Expression of UAS-hspA1L using either ubiquitous or neuronal GAL4 drivers rescued the pupal semi-lethality (Figure 4C), while expression in glia and fat-bodies/hemocytes did not (Figure 4C). Introduction of hspA1L in neurons also restored normal motor activity in trpml1 flies (Figure 4D). Thus, it appeared that the primary defect leading to the increased lethality and motor deficits in trpml1 animals occurred in neurons. The rescue of the locomotor activity may be attributed to a suppression of the elevated cell death. Therefore, we assayed Annexin V-FITC labeling in hspA1L expressing neurons and found significant rescue of the cell death (Figures 4E–4F; Annexin V labeled cells: wild-type, 3.4±1.8%; trpml1, 35.1±10.9%; hspA1L and trpml1, 10.7±3.0%, not significantly different from wild-type).
Disruption of synaptic transmission in the absence of TRPML
We noticed that the trpml1 flies remained immobilized longer than wild-type after exposure to CO2 anesthesia. After four minutes, 90.7±9.3% of wild-type recovered mobility, in contrast to only 6.7±3.3% of trpml1. The return of activity of all trpml1 flies required 9 minutes, twice as long as for wild-type (Figure 5A). Since CO2 immobilizes flies through inhibition of synaptic transmission at the neuromuscular junction (NMJ) (Badre et al., 2005), we tested whether there was a decrease in synaptic transmission in trpml1. This possibility seemed plausible given that increased oxidative stress correlates with reduced NMJ synaptic transmission (Giniatullin et al., 2006).
FIGURE 5. Impairment of synaptic transmission in the trpml.
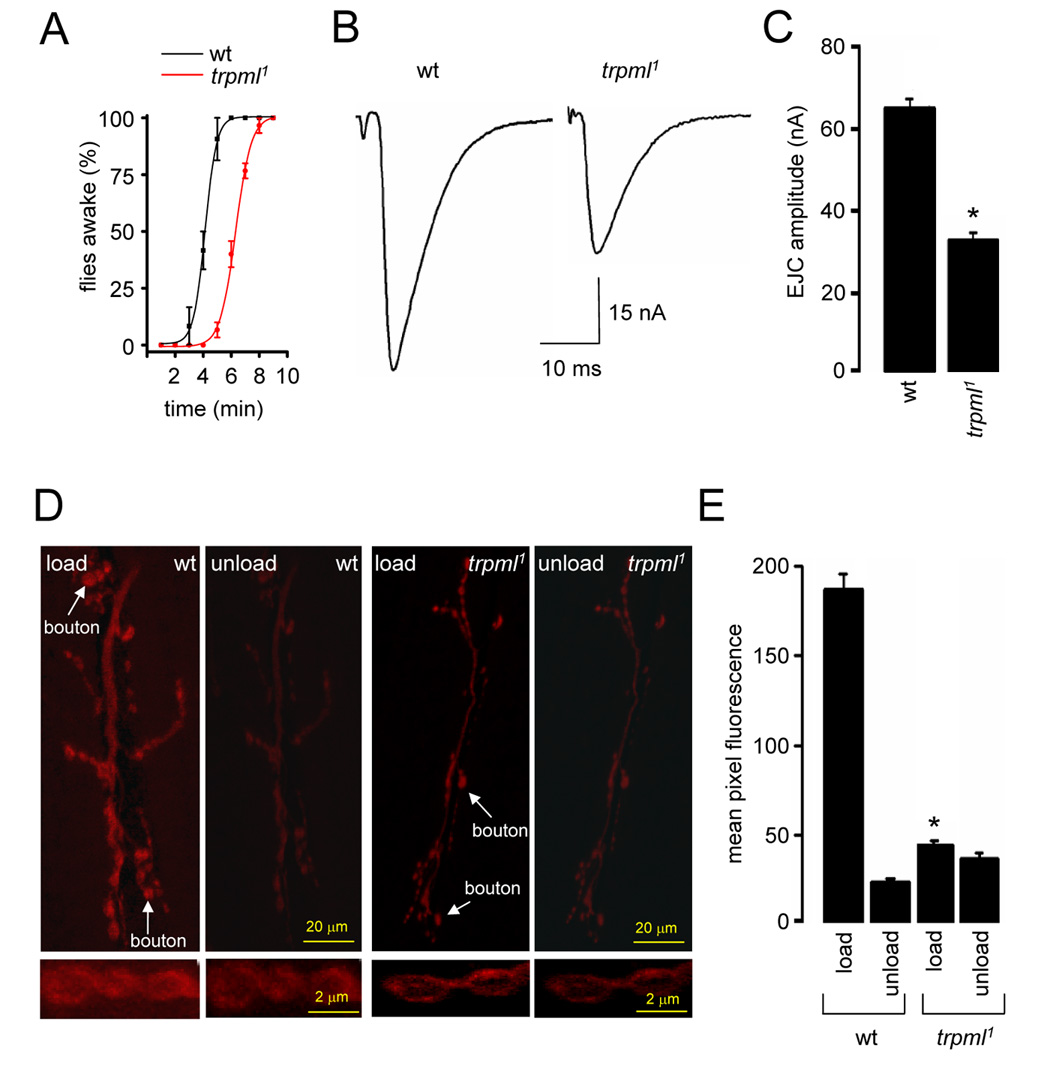
(A) Time-course of flies with restored mobility following a 3 min exposure to CO2. n=3, 10 flies per experiment.
(B) Excitatory junctional current (EJC) from the 3rd instar NMJ.
(C) Quantification of the EJC amplitudes in wt and trpml1. n=5 animals; 10 NMJs for each genotype; *, p≤0.001.
(D) NMJ synapses following FM1-43 loading and unloading. Arrows indicate synaptic boutons. Panels below show enlarged magnification of synaptic boutons.
(E) Quantification of FM1-43 fluorescence intensity in NMJ boutons following dye loading and unloading. n=5 animals,10 NMJs for each genotype; *, difference from wt, p≤0.001.
All statistical analyses, t-test.
To assess synaptic function, we made evoked excitatory junction current (EJC) recordings at the NMJ synapse of 3rd instar larvae, using the two-electrode voltage-clamp (TEVC) configuration. To reveal changes in basal synaptic function, we performed assays in a bath solution containing 0.5 mM extracellular [Ca2+] (Rohrbough et al., 1999; Trotta et al., 2004). Control animals exhibited a mean EJC amplitude of 64.1±5.2 nA, while trpml1 showed a ~50% decrease in transmission strength (32.3±4.6 nA; Figures 5B–5C).
To investigate whether the reduction in synaptic transmission was due to an alteration in the size or cycling of the endo-exo synaptic vesicle (SV) pool, we used the lipophilic fluorescent dye FM1-43 to assay SV endocytosis and exocytosis (Kuromi and Kidokoro, 2000; Fergestad and Broadie, 2001; Trotta et al., 2004). We exposed NMJ preparations to FM1-43 in the presence of 90 mM [K+] saline, which depolarized the nerve terminal and induced vesicular cycling and loading of FM1-43. We then depolarized the preparations in the absence of FM1-43 to assess dye unloading by SV exocytosis. Based on comparison of the initial mean fluorescence values, trpml mutant synaptic boutons displayed a four-fold decrease in loading (Figures 5D–E; wild-type, 187.3±9.3; trpml1, 48.2±3.2). This lower level of loading was sufficient to assess whether the loaded boutons in trpml1 unloaded properly. Following the second depolarization, the wild-type boutons displayed a >90% reduction in fluorescence (unloading), whereas the trpml1 mutants showed little decrease (Figures 5D–5E; wild-type, 29.2±3.5; trpml1, 39.3±3.6). These data indicate that the diminished synaptic transmission in trpml1 was due to presynaptic impairment of SV cycling.
Expression of trpml+ in neurons, hemocytes or glia rescues trpml phenotypes
To determine the cellular requirements for TRPML, we expressed wild-type UAS-trpml under the control of promoter-GAL4 transgenes that were expressed in different cell types. Introduction of the UAS-trpml transgene alone in trpml1 had no impact on pupal semi-lethality, the behavioral climbing index and synaptic transmission amplitude. However, these deficits were rescued fully upon expression of UAS-trpml under the control of ubiquitously expressed (act5c-GAL4) or pan-neuronally expressed (elav-GAL4) GAL4 drivers (Figures 6A–6D).
FIGURE 6. TRPML expression in either hemocytes/fat bodies or glia rescues the trpml pupal semi-lethality, locomotor defects and synaptic transmission impairment.
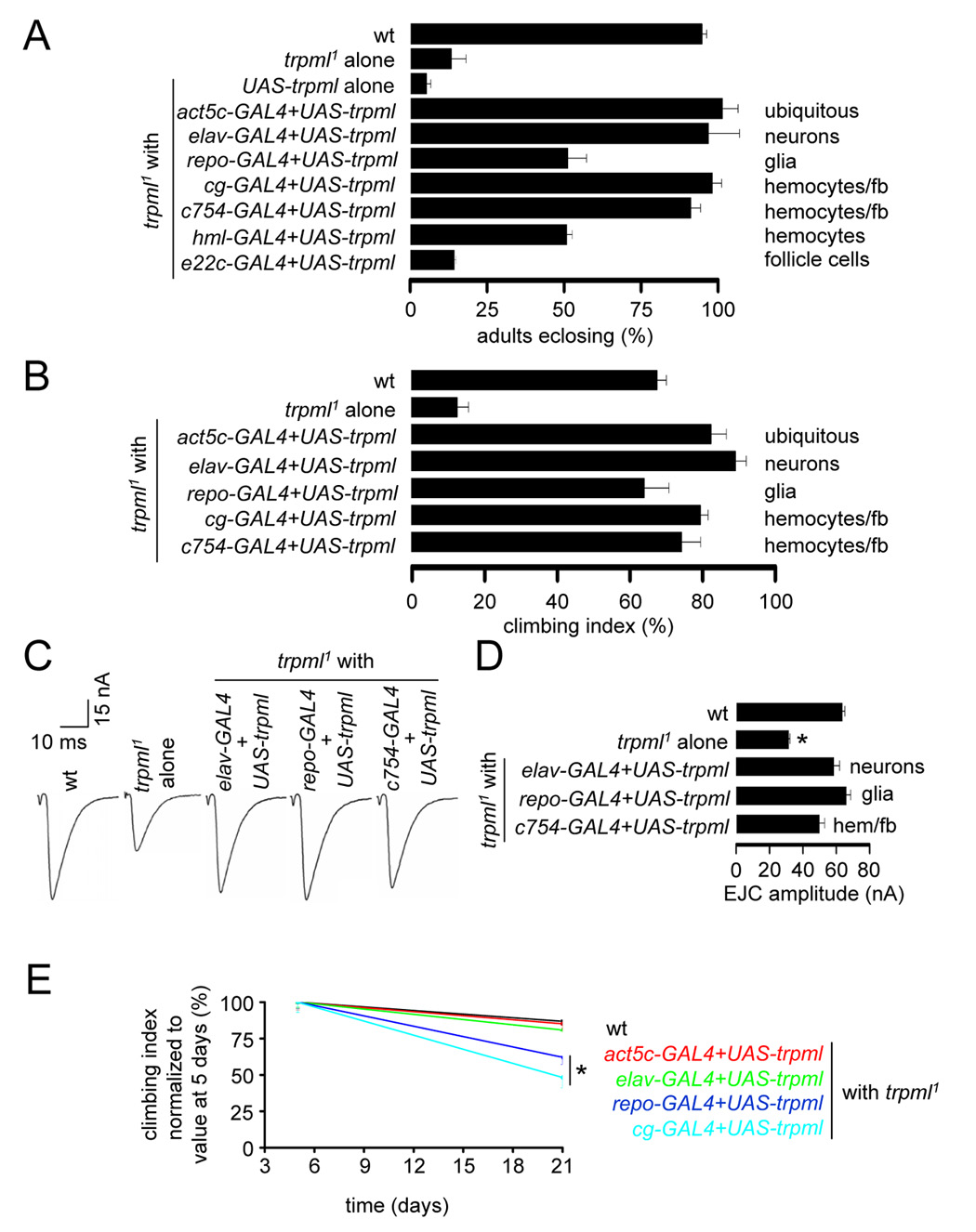
(A) Percentage of pharate adults without the TM3 balancer. n=7–11. GAL4 expression patterns are indicated.
(B) Climbing indices of adult flies. n=3–8 experiments, 10–20 flies each.
(C) Representative EJC traces of larvae.
(D) Quantification of the EJC amplitudes. n=5 animals, 10 NMJs for each genotype; *, p≤10−4, ANOVA. hem, hemocytes.
(E) Age-progressive decline in climbing index of wt (black) and trpml1 flies expressing UAS-trpml and the indicated GAL4s. Indices are normalized with 5 day values as 100%. n≥3, 10–20 flies per experiment; *, difference from wt, p≤0.05, ANOVA.
As controls, we expressed UAS-trpml using GAL4s that directed expression in follicle cells (e22c-GAL4), hemocytes/fat-bodies (cg-GAL4, c754-GAL4 and hml-GAL4) and glia (repo-GAL4). As expected, the combination of the e22c-GAL4 with UAS-trpml did not improve adult viability (Figure 6A). To our surprise, the hemocyte/fat body and glial drivers restored normal survival, climbing activity and synaptic transmission (Figures 6A–6D). While the onset of motor deficits following the expression of trpml+ in glia or hemocytes/fat-bodies was delayed significantly, there was some subsequent age-dependent decline in climbing activity (Figure 6E). Nevertheless, the significant rescue of the trpml1 impairments by expression in hemocytes/fat bodies or glia was unanticipated, since we expected that the trpml1 phenotypes were due to loss of function solely in neurons.
Accumulation of late apoptotic cells suppressed by expression of trpml+ in hematopoietic cells
In Drosophila, both hemocytes and glia are involved in the clearance of apoptotic cells during development and in the adult nervous system (Wood and Jacinto, 2007). Clearance of dead cells is particularly important during developmental stages in which there is pronounced apoptosis, such as during pupal to adult metamorphosis and late neural development when many surplus cells are pruned (Lee and Baehrecke, 2001; Awasaki et al., 2006). Rapid clearance of early apoptotic neurons minimizes neuroinflammation, which otherwise occurs through the release of antigenic material by late apoptotic cells with compromised plasma membranes (Franc, 2002). Late apoptotic and necrotic cells release cytotoxic agents, which can induce cell death in neighboring cells, thereby leading to widespread neurodegeneration. Several aspects of the trpml1 phenotype raised the possibility that there was a buildup of late apoptotic and necrotic cells, indicative of a defect in clearance. These include widespread neurodegeneration and rescue of these phenotypes by reintroduction of trpml+ in hemocytes and glia.
To address whether there was accumulation of late apoptotic and necrotic cells in trpml1, we stained brains with Annexin V-FITC or propidium iodide (PI). PI is a membrane impermeant nuclear dye that only stains cells that have lost plasma membrane integrity due to cell death (Franc et al., 1999). Annexin V-FITC staining without PI staining is indicative of early-apoptotic cells with an intact plasma membrane. Late-apoptotic/necrotic cells have compromised membrane integrity. Therefore, staining with both Annexin V-FITC and PI is indicative of an accumulation of late-apoptotic/ necrotic cells (Franc et al., 1999). We performed staining with Annexin V-FITC on unfixed tissue, while we used fixed tissue for PI staining since the duration of the procedure without fixation caused necrosis in intact adult brains, possibly due to acute hypoxia. 21 day-old trpml1 brains showed significant increases in the percentage of cells with Annexin V-FITC membrane staining (Figures 7A–7B; wild-type, 3.4±1.8%; trpml1, 35.1±10.9%; p≤0.05) and PI positive nuclei (Figures 7A–7B; wild-type, 0.1±0.1%; trpml1, 35.5±11%; p≤10−5). These data demonstrate that there was a significant elevation in the accumulation of late apoptotic/necrotic cells in trpml brains, indicating a deficit in the clearance of dying cells.
FIGURE 7. Reduced clearance of late-apoptotic cells in trpml mutants Images are by confocal microscopy.
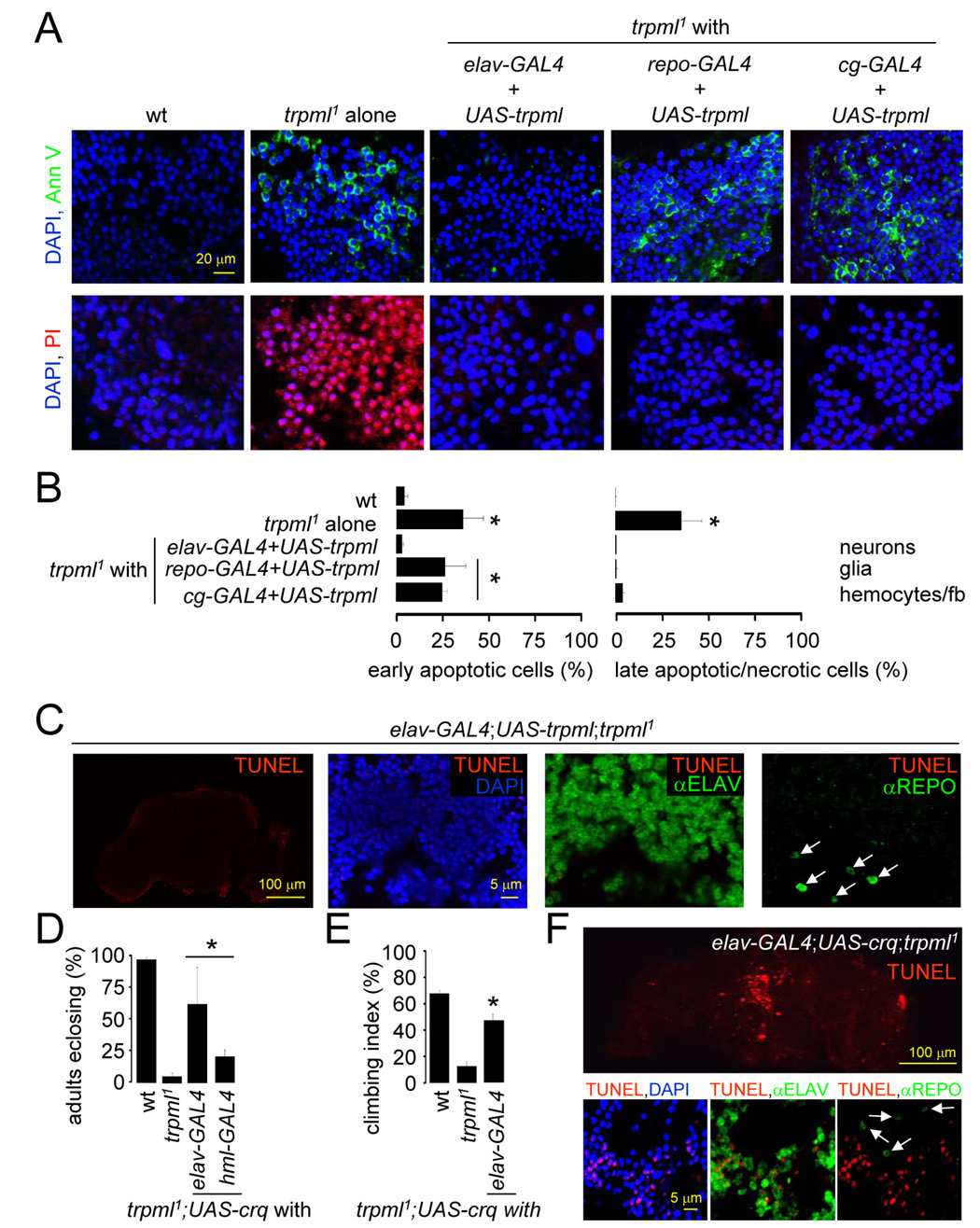
(A) DAPI/Annexin V-FITC and DAPI/PI stained brains from 21 day-old flies.
(B) Percentage of early apoptotic and late apoptotic/necrotic cells in brains from 21 day-old flies. n≥3; *, difference from wt, p≤0.01, ANOVA.
(C) Brains from 21 day-old flies viewed at 310 nm to detect DAPI, 488 nm to detect anti-REPO, 568 nm to detect TUNEL and 633 nm to detect anti-ELAV. Arrows indicate glia.
(D) Percentage of pharate adults without the TM3 balancer. n=3–11; *, difference from trpml1, p≤0.05, ANOVA.
(E) Climbing indices of adult flies. n≥5; *, difference from trpml1, p≤5×10−5, t-test.
(F) Brains from 21 day-old flies viewed at 310 nm to detect DAPI, 488 nm to detect anti-REPO, 568 nm to detect TUNEL and 633 nm to detect ELAV. Arrows indicate glia.
To determine whether expression of trpml in hematopoietic and glial cells reduced the accumulation of late apoptotic/necrotic cells, we expressed UAS-trpml under the control of the GAL4 lines described above. Upon expression of UAS-trpml using the pan-neuronal GAL4 (elav-GAL4), there was virtually no staining with Annexin V-FITC or PI, similar to wild-type (Figures 7A–7B). When we expressed UAS-trpml in hemocytes/fat bodies (cg-GAL4) or glia (repo-GAL4), the number of late-apoptotic cells again returned to the very low levels typical of wild type, although early apoptotic cells were still detected (Figures 7A–7B). Since only pan-neuronal expression of trpml fully suppresses cell death, these results suggest that the initial cell death occurs in neurons and that trpml expression in hemocytes or glia contributes to clearance of dying neurons. Consistent with this observation, trpml expression in neurons abolished all cell death in the adult brain (Figure 7C). The glial cell death that occurred in trpml1 brains (Figures 2M–2O) was also rescued by expression of trpml in neurons (Figure 7C). Although, we cannot exclude that elav-GAL4 is expressed in some glia, our results indicate that glial cell death occurs in a non-cell-autonomous manner and is a secondary consequence of a defect in the clearance of adjoining apoptotic neurons. Since necrotic cells also display DNA fragmentation (Pang and Geddes, 1997), the increased TUNEL staining of neurons and glia in trpml1 brains could indicate pronounced secondary necrosis due a defect in the clearance of apoptotic neurons.
The croquemort (crq) gene encodes a CD36-related scavenger receptor, which is involved in the clearance of cells undergoing both apoptotic and autophagic cell death (Franc et al., 1999; Lee and Baehrecke, 2001). Although predominantly expressed in macrophage populations, exogenous expression of crq in COS cells confers them with the ability to phagocytize apoptotic cells (Franc et al., 1996). Therefore, we tested whether expression of crq in trpml1 neurons enabled them to phagocytize nearby apoptotic cells, thereby preventing the accumulation of late-apoptotic/necrotic cells and the locomotor defects. Furthermore, since autophagic cell death promotes the removal of superfluous tissues during pupal metamorphosis (Franc et al., 1999; Lee and Baehrecke, 2001), the pupal semi-lethality in trpml1 might also be due to a defect in autophagic cell clearance in the developing nervous system and be suppressed by increasing expression of crq in trpml1 neurons.
To test whether exogenous over-expression of crq rescues the defects associated with trpml, we drove expression of UAS-crq using either pan-neuronal or hemocyte GAL4s (elav-GAL4 or hml-GAL4 respectively). Introduction of the UAS-crq transgene alone in trpml1 had no impact, whereas expression of crq in neurons or hemocytes rescued the pupal semi-lethality (Figure 7D; p≤0.05). Furthermore, the motor defect was suppressed significantly in 5 day-old trpml1 by neuronal expression of crq (Figure 7E; p≤5×10−5). Expression of crq in brain neurons also decreased TUNEL labeling and the residual staining was only in neurons (Figure 7F). This further supports the finding that primary cell death occurs in neurons and reduced clearance of apoptotic neurons induces secondary death in adjoining cells, including glia.
Discussion
Requirement for TRPML for lysosomal function and completion of autophagy
Impairments in autophagy are implicated in the pathophysiology of several neurodegenerative diseases (Klionsky, 2007). MLIV may also involve a perturbation in autophagy, as suggested by a pharmacological study using a tissue culture model (Jennings et al., 2006). We found that trpml mutant cells displayed impaired autophagy in vivo. However, in contrast to other models of LSDs (Settembre et al., 2007), there does not appear to be a block in fusion between the lysosomes and autophagosomes in trpml cells. Rather, there appeared to be reduced macromolecular degradation in autolysosomes following fusion.
Recent in vitro studies suggest that mammalian TRPML1 is a proton-permeable channel that provides a proton leak pathway in the lysosomal membrane (Soyombo et al., 2006; Miedel et al., 2008). Therefore, in the absence of the channel, we propose that over-acidification of the lysosomal lumen impairs normal degradation in autolysosomes, since lysosomal proteases are intimately dependent on the normal lysosomal pH.
Mitochondrial dysfunction and oxidative stress in trpml mutant
In addition to playing a cytoprotective role by promoting clearance of toxic macromolecules, autophagy is important for the turnover of entire organelles, including damaged mitochondrial with disrupted trans-membrane potential (ΔΨ) (Twig et al., 2008). trpml neurons accumulated mitochondria with dissipated ΔΨ, indicating disruption of autophagic clearance of dysfunctional mitochondria. Moreover, trpml cells accumulated lipofuscin, indicative of oxidative stress, and the mutant animals displayed a significant increase in H2O2 in a range that leads to elevated apoptosis and neurodegeneration (Li et al., 2000; St-Pierre et al., 2006). Thus, oxidative stress appears to be a key factor contributing to the neurodegeneration in the trpml mutant.
The molecular chaperone, HSP70, suppresses the trpml phenotype
To cope with oxidative stress and toxic aggregation of macromolecules, cells increase expression of molecular chaperones such as heat-shock proteins (Menoret et al., 2002). Exogenous expression of the human homolog of HSP70 (HspA1L) can suppress the toxicity associated with HttQ120 (Warrick et al., 1999). Similarly, introduction of HspA1L into trpml neurons, but not in other cell types, rescued the mutant phenotypes. This finding indicates that the impairments in trpml arose primarily in neurons and possibly due to an accumulation of macromolecules.
Model for motor defects due to loss of TRPML
Loss of TRPML causes a decrease in lysosomal degradation, resulting in an increase in dysfunctional mitochondrial, aggregation of toxic macromolecules and oxidative stress. Lipofuscin, which forms under oxidative stress (Terman and Brunk, 2004), can cause a further decrease in lysosomal function. We propose that in trpml mutants there is an amplifying cycle of increased oxidative stress and defective autophagy and lysosomal function, which leads to progressive neurodegeneration and motor impairment. We suggest that elevated oxidative stress also underlies the reduced NMJ synaptic transmission in trpml animals, which in turn contributes to the deficit in behavioral motor function. Consistent with this model, increased oxidative stress is linked to inhibition of synaptic transmission (Giniatullin et al., 2006). Our results raise the possibility that a combination of neurodegeneration and loss of NMJ synaptic transmission accounts for the diminished motor activity in MLIV patients. Although we cannot rule out that the impaired synaptic transmission in trpml is an indirect consequence of unhealthy neurons, it still provides an explanation for the impaired motor function that precedes neurodegeneration in MLIV.
The trpml phenotype is reminiscent of the spinster/benchwarmer Drosophila mutant. Similarities include accumulation of lipofuscin-loaded effete lysosomes, loss of NMJ synaptic function and neurodegeneration (Nakano et al., 2001; Sweeney and Davis, 2002; Dermaut et al., 2005). The Spinster/Benchwarmer protein also resides in a presynaptic lysosomal compartment and is implicated in efficient synaptic vesicle recycling (Sweeney and Davis, 2002; Dermaut et al., 2005).
TRPML is required in hematopoietic cells and glia to mediate clearance of apoptotic cells and to prevent widespread neurodegeneration
Initially as a control, we introduced the wild-type trpml+ transgene in glia and fat-bodies/hemocytes and to our surprise expression of trpml+ in these cells rescued the trpml mutant phenotypes. Thus, the issue arises as to why expression of TRPML in hematopoietic cells or glia rescues the trpml defects. Clarifying this mechanism may have relevance to other LSD models of progressive neurodegeneration, since re-introduction of Drosophila NPC1 in glia reduces adult lethality associated with the dnpc1a mutation (Phillips et al., 2008).
Neural tissue in trpml animals accumulates early- and late-apoptotic/necrotic cells. Normally, early apoptotic cells are rapidly cleared by hemocytes and glia (Wood and Jacinto, 2007). If dead cells are not cleared rapidly, they lose plasma membrane integrity and release antigenic and cytotoxic material, which induce neuroinflammation and secondary necrosis in neighboring bystander cells (Franc, 2002). Loss of trpml function in neurons induces cell-autonomous apoptosis and defective clearance of these cells induces secondary cell death in nearby cells. Consistent with this proposal, introduction of trpml+ in neurons prevents cell death of both neurons and glia, indicating that trpml functions cell-autonomously for neuronal viability and non-autonomously for the viability of adjacent cells such as glia. When trpml was introduced in hemocytes or glia there was no accumulation of late-apoptotic cells or necrotic cells, although mutant cells still underwent early apoptosis.
We suggest that expression of trpml+ in hemocytes and glia promotes the clearance of early apoptotic cells before their membrane integrity is compromised. In the absence of this function, there is an accumulation of late apoptotic/necrotic cells, leading to widespread neuroinflammation and progressive cell death in adjoining cells that would otherwise remain unaffected.
Since both late apoptotic/necrotic neurons and oxidative stress are mediators of neuroinflammation (Franc, 2002), our results raise the possibility that neuroinflammation may be a hitherto unexplored mediator of MLIV associated neurodegeneration. This is the first link between a TRP channel and either neuroinflammation or clearance of apoptotic cells.
Development of therapeutic strategies
The finding that expression of wild-type trpml+ in hematopoietic cells is sufficient to delay the onset of the trpml mutant phenotypes raises the exciting possibility that bone marrow transplantation (BMT) in patients with MLIV might delay disease progression. In favor of this proposal, several reports and case studies describe the successful use of BMT to ameliorate other LSDs in patients and in murine models (Bruni et al., 2007). With the recent development of TRPML1 knockout mice (Venugopal et al., 2007), the feasibility of this approach can now be tested in a mammalian animal model. Our results also raise the possibility that one or more of the approved drugs that stimulate either autophagy or HSP1AL may also suppress MLIV, especially in combination with BMT. Thus, this Drosophila model for MLIV provides the framework for developing strategies to treat MLIV.
Experimental Procedures
Generation of transgenic and mutant trpml strains
trpml1 and trpml2 were generated by imprecise excision of the P-element (GE22279). The deletions in trpml1 and trpml2 removed −456 - +641 and −234 - +860 base-pairs respectively relative to the translation start site. P[trpml] was generated by cloning the region between the genes Gyc76C and CG32209 into pCaSpeR. The UAS-trpml transgenic flies were generated by subcloning the trpml cDNA into pUAST. Other strains are described in the Supplemental Experimental Procedures.
Locomotor activity assays
10–20 flies were placed in a 50 ml graduated cylinder. The climbing index was the fraction of flies that climbed to the 25 ml mark within 15 seconds after being tapped down. Total daily activity was determined by placing individual flies in a DAMS actometer (Trikinetics) for 24 h. Animals were not exposed to CO2 for ≤24 h prior to performing the assays.
Assays for apoptosis
Brains were dissected from flies immediately after dissection to prevent necrosis due to hypoxia. TUNEL staining was performed as described (Roche). PI staining (1:3000 dilution in PBS) was performed for 10 min. To determine the types of cells dying, brains were dissected, stained with antibodies that label neurons or glia, followed by TUNEL staining as described (Supplemental Experimental Procedures). Images were viewed by confocal microscopy.
Light microscopy and transmission EM
Heads were dissected from flies reared under a 12 h light/dark cycle or constant darkness and embedded as described (Porter et al., 1992), except that 0.1 M sodium phosphate (pH 7.4) was the buffer. For light microscopy, 1 µm sections were prepared (50–100 µm depth), stained with 1% toluidine blue and viewed by light microscopy. For transmission EM, 85 nm sections were prepared at a depth of 30 µm.
Cytoplasmic membrane inclusions and autophagosomes were evaluated in photoreceptor cell bodies. Vesicles were characterized as multilamellar or multivesicular bodies based on established morphological criteria (Dermaut et al., 2005). The numbers of cytoplasmic inclusions per photoreceptor cell body were counted in ≥50 ommatidia per fly (n≥3 flies).
Detection of GFP-ATG8
Wild-type and trpml1 flies containing the hsGFP-ATG8 transgene were heat-shocked for 1 h at 37°C 48 h prior to performing experiments. Brains were dissected and imaged as described above. For the quantification of GFP-ATG8 positive vesicles, ommatidia were dissected as described above from a set of 6 flies per experiment (n≥3). 1 µm sections spanning the entire ommatidia (thickness of 10–12 µm on the cover-slips) were obtained by confocal microscopy and projected into a single image. The total numbers of GFP-positive vesicles per ommatidia were determined for the quantification of the GFP-ATG8 puncta.
Detection of Rh1 degradation
Flies were exposed to either ambient or blue (480 nm) light for 6 h before dissecting heads under a photographic safelight. Heads were homogenization in SDS-sample buffer. Proteins were fractionated by SDS-PAGE, transferred to nitrocellulose, probed with mouse anti-Rh1 and rabbit anti-TRP antibodies and with anti-rabbit IRDye 800 CW and anti-mouse IRDye 680 conjugates (Odyssey). Signals were detected and quantified using a LICOR imaging system (Odyssey).
MitoTracker staining
Ommatidia were dissected, loaded with MitoTracker-orange CM-H2TMRos (100 nM; Invitrogen) for 30 min, fixed for 15 min in 4% PFA, washed, mounted and imaged as described above.
Two Electrode Voltage Clamp (TEVC) recordings and FM1-43 Imaging
Recordings were performed on 3rd instar NMJ using two electrode voltage-clamp (TEVC) techniques as described (Rohrbough et al., 1999). Optical FM1-43 dye imagings were performed at the NMJ as described (Trotta et al., 2004) (Supplemental Experimental Procedures).
Measurement of H2O2 levels
10 flies were homogenized in PBS, incubated at 4°C for 15 min, the extracts were centrifuged and the supernatants retained. H2O2 levels were measured using the Amplex-red H2O2 kit (Invitrogen) using a Fluorstar Optima fluorescent plate reader (BMG). Samples without fly extracts were used to determine backgrounds.
Optical neutralization technique
To assay the time-course of photoreceptor cell degeneration, the numbers of rhabdomeres/ommatidium were determined as described (Xu et al., 2004). Each data point was based on ≥50 ommatidia per fly (n≥3 flies).
Statistics
All statistical values indicate means±SEMs. We used ANOVA to make multi-group comparisons and Student’s t-tests to compare two sets of values.
Supplementary Material
Acknowledgments
We thank the Bloomington Stock Center for fly stocks, T. Neufeld for the hsGFP-ATG8 flies, N. Franc for the UAS-crq flies, J. Corden, J. Lorsch, R. Rao and S. Muend for use of equipment and M. Sepanski for preparing sections of fly heads. We thank S. J. Moon, Y. Jiao and X. Wang for technical advice and M. Köttgen, Y. Kwon and D. Wasserman for helpful discussions. K.V. was supported in part by a MLIV Foundation postdoctoral fellowship. The work in the K.B. laboratory was supported by the NINDS (NS41740) and the research in the C.M. laboratory was supported by grants from the March of Dimes and the NEI (EY08117).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest.
References
- Awasaki T, Tatsumi R, Takahashi K, Arai K, Nakanishi Y, Ueda R, Ito K. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–867. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Bach G. Mucolipin 1: endocytosis and cation channel-a review. Pflugers Arch. 2005 doi: 10.1007/s00424-004-1361-7. [DOI] [PubMed] [Google Scholar]
- Badre NH, Martin ME, Cooper RL. The physiological and behavioral effects of carbon dioxide on Drosophila melanogaster larvae. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005;140:363–376. doi: 10.1016/j.cbpb.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D, Bach G. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 2000;26:118–123. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am. J. Hum. Genet. 2000;67:1110–1120. doi: 10.1016/s0002-9297(07)62941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignull HR, Morley JF, Morimoto RI. The stress of misfolded proteins: C. elegans models for neurodegenerative disease and aging. Adv. Exp. Med. Biol. 2007;594:167–189. doi: 10.1007/978-0-387-39975-1_15. [DOI] [PubMed] [Google Scholar]
- Bruni S, Loschi L, Incerti C, Gabrielli O, Coppa GV. Update on treatment of lysosomal storage diseases. Acta. Myol. 2007;26:87–92. [PMC free article] [PubMed] [Google Scholar]
- Cooper JD. Progress towards understanding the neurobiology of Batten disease or neuronal ceroid lipofuscinosis. Curr. Opin. Neurol. 2003;16:121–128. doi: 10.1097/01.wco.0000063762.15877.9b. [DOI] [PubMed] [Google Scholar]
- Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y, Callaerts P, Bellen HJ. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J. Cell Biol. 2005;170:127–139. doi: 10.1083/jcb.200412001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Kuida K, Johnson EM., Jr Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares H, Greenwald I. Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat. Genet. 2001;28:64–68. doi: 10.1038/ng0501-64. [DOI] [PubMed] [Google Scholar]
- Fergestad T, Broadie K. Interaction of stoned and synaptotagmin in synaptic vesicle endocytosis. J. Neurosci. 2001;21:1218–1227. doi: 10.1523/JNEUROSCI.21-04-01218.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher BL, Dillard CJ, Tappel AL. Measurement of fluorescent lipid peroxidation products in biological systems and tissues. Anal. Biochem. 1973;52:1–9. doi: 10.1016/0003-2697(73)90327-8. [DOI] [PubMed] [Google Scholar]
- Franc NC. Phagocytosis of apoptotic cells in mammals, caenorhabditis elegans and Drosophila melanogaster: molecular mechanisms and physiological consequences. Front. Biosci. 2002;7:d1298–d1313. doi: 10.2741/A841. [DOI] [PubMed] [Google Scholar]
- Franc NC, Dimarcq JL, Lagueux M, Hoffmann J, Ezekowitz RA. Croquemort, a novel Drosophila hemocyte/macrophage receptor that recognizes apoptotic cells. Immunity. 1996;4:431–443. doi: 10.1016/s1074-7613(00)80410-0. [DOI] [PubMed] [Google Scholar]
- Franc NC, Heitzler P, Ezekowitz RA, White K. Requirement for croquemort in phagocytosis of apoptotic cells in Drosophila. Science. 1999;284:1991–1994. doi: 10.1126/science.284.5422.1991. [DOI] [PubMed] [Google Scholar]
- Giniatullin AR, Darios F, Shakirzyanova A, Davletov B, Giniatullin R. SNAP25 is a pre-synaptic target for the depressant action of reactive oxygen species on transmitter release. J. Neurochem. 2006;98:1789–1797. doi: 10.1111/j.1471-4159.2006.03997.x. [DOI] [PubMed] [Google Scholar]
- Goldin E, Blanchette-Mackie EJ, Dwyer NK, Pentchev PG, Brady RO. Cultured skin fibroblasts derived from patients with mucolipidosis 4 are auto-fluorescent. Pediatr. Res. 1995;37:687–692. doi: 10.1203/00006450-199506000-00003. [DOI] [PubMed] [Google Scholar]
- Hersh BM, Hartwieg E, Horvitz HR. TheCaenorhabditis elegans mucolipin-like gene cup-5 is essential for viability and regulates lysosomes in multiple cell types. Proc. Natl. Acad. Sci. USA. 2002;99:4355–4360. doi: 10.1073/pnas.062065399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JJ, Jr, Zhu JH, Rbaibi Y, Luo X, Chu CT, Kiselyov K. Mitochondrial aberrations in mucolipidosis Type IV. J. Biol. Chem. 2006;281:39041–39050. doi: 10.1074/jbc.M607982200. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. Journal of Neuroscience. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron. 2000;27:133–143. doi: 10.1016/s0896-6273(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Lee CY, Baehrecke EH. Steroid regulation of autophagic programmed cell death during development. Development. 2001;128:1443–1455. doi: 10.1242/dev.128.8.1443. [DOI] [PubMed] [Google Scholar]
- Li J, Huang CY, Zheng RL, Cui KR, Li JF. Hydrogen peroxide induces apoptosis in human hepatoma cells and alters cell redox status. Cell Biol. Int. 2000;24:9–23. doi: 10.1006/cbir.1999.0438. [DOI] [PubMed] [Google Scholar]
- Manzoni M, Monti E, Bresciani R, Bozzato A, Barlati S, Bassi MT, Borsani G. Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS Lett. 2004;567:219–224. doi: 10.1016/j.febslet.2004.04.080. [DOI] [PubMed] [Google Scholar]
- Menoret A, Chaillot D, Callahan M, Jacquin C. Hsp70, an immunological actor playing with the intracellular self under oxidative stress. Int. J. Hyperthermia. 2002;18:490–505. doi: 10.1080/02656730210146926. [DOI] [PubMed] [Google Scholar]
- Miedel MT, Rbaibi Y, Guerriero CJ, Colletti G, Weixel KM, Weisz OA, Kiselyov K. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J. Exp. Med. 2008;205:1477–1490. doi: 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Fujitani K, Kurihara J, Ragan J, Usui-Aoki K, Shimoda L, Lukacsovich T, Suzuki K, Sezaki M, Sano Y, et al. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol. Cell. Biol. 2001;21:3775–3788. doi: 10.1128/MCB.21.11.3775-3788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z, Geddes JW. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J. Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SE, Woodruff EA, 3rd, Liang P, Patten M, Broadie K. Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J. Neurosci. 2008;28:6569–6582. doi: 10.1523/JNEUROSCI.5529-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JA, Hicks JL, Williams DS, Montell C. Differential localizations of and requirements for the two Drosophila ninaC kinase/myosins in photoreceptor cells. J. Cell. Biol. 1992;116:683–693. doi: 10.1083/jcb.116.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rohrbough J, Pinto S, Mihalek RM, Tully T, Broadie K. latheo, a Drosophila gene involved in learning, regulates functional synaptic plasticity. Neuron. 1999;23:55–70. doi: 10.1016/s0896-6273(00)80753-9. [DOI] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Rubinsztein DC, Ballabio A. Lysosomal storage diseases as disorders of autophagy. Autophagy. 2007;4 doi: 10.4161/auto.5227. [DOI] [PubMed] [Google Scholar]
- Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J. Biol. Chem. 2006;281:7294–7301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS, Jr, Bove C, Kaneski CR, Nagle J, Bromley MC, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000;9:2471–2478. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- Sweeney ST, Davis GW. Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-β-mediated synaptic growth regulation. Neuron. 2002;36:403–416. doi: 10.1016/s0896-6273(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Terman A, Brunk UT. Lipofuscin. Int. J. Biochem. Cell Biol. 2004;36:1400–1404. doi: 10.1016/j.biocel.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Trotta N, Rodesch CK, Fergestad T, Broadie K. Cellular bases of activity-dependent paralysis in Drosophila stress-sensitive mutants. J. Neurobiol. 2004;60:328–347. doi: 10.1002/neu.20017. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Eijnde SM, Boshart L, Baehrecke EH, De Zeeuw CI, Reutelinsperger CP, Vermeij-Keers C. Cell surface exposure of phosphatidylserine during apoptosis is phylogenetically conserved. Apoptosis. 1998;3:9–16. doi: 10.1023/a:1009650917818. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Hofmann T, Montell C. Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J. Biol. Chem. 2006;281:17517–17527. doi: 10.1074/jbc.M600807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal B, Browning MF, Curcio-Morelli C, Varro A, Michaud N, Nanthakumar N, Walkley SU, Pickel J, Slaugenhaupt SA. Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. Am. J. Hum. Genet. 2007;81:1070–1083. doi: 10.1086/521954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Montell C. Phototransduction and retinal degeneration in Drosophila. Pflugers Arch. 2007;454:821–847. doi: 10.1007/s00424-007-0251-1. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Wood W, Jacinto A. Drosophila melanogaster embryonic haemocytes: masters of multitasking. Nat. Rev. Mol. Cell Biol. 2007;8:542–551. doi: 10.1038/nrm2202. [DOI] [PubMed] [Google Scholar]
- Xu H, Lee SJ, Suzuki E, Dugan KD, Stoddard A, Li HS, Chodosh LA, Montell C. A lysosomal tetraspanin associated with retinal degeneration identified via a genome-wide screen. EMBO J. 2004;23:811–282. doi: 10.1038/sj.emboj.7600112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.