

Abstract
Tandem mass spectrometry combined with database searching allows high throughput identification of peptides in shotgun proteomics. However, validating database search results, a problem with a lot of solutions proposed, is still advancing in some aspects, such as the sensitivity, specificity, and generalizability of the validation algorithms. Here a Bayesian nonparametric (BNP) model for the validation of database search results was developed that incorporates several popular techniques in statistical learning, including the compression of feature space with a linear discriminant function, the flexible nonparametric probability density function estimation for the variable probability structure in complex problem, and the Bayesian method to calculate the posterior probability. Importantly the BNP model is compatible with the popular target-decoy database search strategy naturally. We tested the BNP model on standard proteins and real, complex sample data sets from multiple MS platforms and compared it with PeptideProphet, the cutoff-based method, and a simple nonparametric method (proposed by us previously). The performance of the BNP model was shown to be superior for all data sets searched on sensitivity and generalizability. Some high quality matches that had been filtered out by other methods were detected and assigned with high probability by the BNP model. Thus, the BNP model could be able to validate the database search results effectively and extract more information from MS/MS data.
Proteomics has become one of the most active areas of life science research in the postgenomics era. MS is an analytical technique widely used in proteomics research and provides information on protein identification, characterization, and quantification (1). MS/MS can analyze protein mixtures in a high throughput manner and provide sequence information for peptides and proteins (2, 3). Currently MS/MS data are usually processed by the so-called database search method or de novo sequencing (4, 5). Automated database search software, such as SEQUEST (6), Mascot (7), Phenyx (8), and X!Tandem (9), can assign mass spectra to peptides from a protein sequence database quickly and provide scores to measure the quality of these matches. Generally the search engines select the best matches according to their scoring models but do not guarantee the accuracy of the matches. Consequently validation of database search results has been the focus of much attention (10–13). Recently the challenge of simultaneously improving the specificity and the sensitivity of the quality of database search results was addressed by Domon and Aebersold (11). Nesvizhskii et al. (12) also addressed this issue in their review on MS/MS data processing.
The research on the database search result validation focused on finding the new features to distinguish the correct and incorrect matches; improving the sensitivity, specificity, and generalizability is the problem of main concern. As a robust search engine, SEQUEST is commonly used in many researches, and many algorithms, scoring models, and statistical models have been developed to validate SEQUEST database search results (14–17). Among them, the target-decoy database search method is more favored in the practice data processing because it is simple to apply and is robust to the effects of database size, sample quality, experimental conditions, and instrument type (12, 18, 19). Nowadays probability frameworks, which can incorporate the decoy database searching and the multiple result validation features, are touched upon (20, 21), but more comprehensive discussions are needed, such as the estimation of false discovery rate (FDR)1 of the data set as a whole and the correct probability assignment for each match (19).
In this study, we propose a Bayesian nonparametric (BNP) model to incorporate a probability framework into the randomized database searching method. A similar idea was also proposed by Nesvizhskii et al. (12). The BNP model integrates an extended set of features to validate database search results; these features were selected from the literature and cover many characters of the spectrum, including SEQUEST scores, empirical parameters, peptide fragmentation knowledge, and chemical or physical properties of peptides. To compress the feature space and reduce the computational burden, an LDF was constructed based on the “typical labeled data set” from decoy database matches. Then a set of component Gaussian density functions (GDFs) was used to fit the LDF score distribution of random matches; the LDF score distribution of correct matches was fitted with GDFs estimated from the normal database matches, which were based on observations of correct and random results. In the latter step, the contribution of the incorrect matches remained unchanged. Thus, we call our approach “restricted nonparametric probability density function (PDF) estimation.” Finally the correct probability of each assignment was calculated using a Bayesian formula, and the error rates for different cutoff values of the probability score were estimated. This method can also estimate the total number of correct matches and the false negative rate of the filtered data set.
The basic idea behind the BNP model is that, based on the decoy database matches, a degraded filtering model can be used to initialize an iterative process to refine the model and improve the sensitivity. The principle underlying our model is that what constitutes a high quality spectrum can be learned from the analyzed data itself (22). In this way, the BNP model automatically develops a statistical classifier for each data set. By using a nonparametric approach, our model can flexibly adapt to variable score distributions, which are a frequent occurrence in database search result validation in proteomics. Based on randomized database searching, the model is sufficiently robust to analyze data sets derived from different samples, experimental conditions, and mass spectrometry platforms.
The BNP model was evaluated using three MS/MS spectra data sets from standard proteins, and the results indicate that our model performs well for peptide identification validation. We also demonstrate that the new model is suited to different MS instruments and databases, and it identifies more confident peptides than three other commonly used algorithms. Furthermore we applied the BNP model to data sets derived from real, complex samples analyzed by LCQ, LTQ, and LTQ/FT mass spectrometers and obtained results consistent with those from control data sets. When the confidence level is fixed, the BNP model can increase the number of confirmed identified peptides, including those with ambiguous mass difference. Importantly the calculated probability detects some high quality matches that other algorithms may filter out. In summary, the BNP model provides a tool with the potential to extract more information from MS/MS data.
EXPERIMENTAL PROCEDURES
Experimental Data Sets
To conduct a comprehensive evaluation of the Bayesian nonparametric model, we applied our method to three control sample data sets (named D1–D3) and five complex sample data sets (named D4–D8) and compared the results with those generated by three other methods. All the data sets were from different samples analyzed with different mass spectrometers in different laboratories. These data sets included most variable factors that have been shown to significantly impact the generalizability of the different database search result validation models. In the complex data sets, D4–D6 have been used in our previous work (23); D7 and D8 were added to prove that the BNP model was not overfitting.
Control Sample Data Sets—
Three control data sets from the LCQ, LTQ, and LTQ/FT instruments were used to investigate the FDR and estimate error rates as well as some other parameters of the BNP model. 1) The LCQ control data set (D1), published by the BIATECH Institute (Bothell, WA) (24), was generated by analyzing a standard mixture of 23 peptides and 12 proteins using an LCQ Deca XPPLUS platform (Thermo Finnigan, San Jose, CA). Additional details about this data set can be found in Purvine et al. (24). 2) The LTQ control data set (D2), published by Proteomics Standards Research Group (sPRG), was derived from six LC-MS/MS runs on the LTQ (Thermo Finnigan) platform. The sample was designed to contain 49 purified human proteins, but ∼200 proteins have been shown to be present in the sample as announced by the research poster of sPRG 2006. 3) The LTQ/FT control data set (D3), published by the Institute for Systems Biology (25), was generated by analyzing the peptides in a tryptic digest of a mixture of 18 proteins, the “ISB standard protein mix,” on the LTQ/FT platform (raw data of Mixture 4).
Complex Sample Data Sets—
We applied our model to three biological sample data sets analyzed by the LCQ, LTQ, and LTQ/FT mass spectrometers. 1) The LCQ-shotgun data set (D4) was generated from the K562 cell line sample and was downloaded from the Open Proteomics Database. This data set had been used by Resing et al. (26) to illustrate the use of multisource information to improve reproducibility and sensitivity in identifying human proteins by shotgun proteomics. 2) The LTQ-shotgun data set (D5) was generated from MS/MS analyses of a human liver tissue sample (27). Peptides generated by tryptic digestion were analyzed by reversed-phase LC-MS/MS using a Thermo Finnigan linear ion trap mass spectrometer (LTQ) with an ESI source. 3) The LTQ/FT-shotgun data set (D6), was also generated from the human liver tissue sample. Strong cation exchange chromatography was performed on the treated protein mixtures, and each of 43 fractions collected was analyzed by the LTQ/FT platform. This data set was produced by the Beijing Proteome Research Center and was described previously (23). 4) The LTQ-shotgun data set (D7) was generated from yeast proteins analyzed by nano-LC-MS/MS using a nanoflow HPLC system connected to a linear ion trap mass spectrometer (LTQ) (28), and the raw data were downloaded from the PeptideAtlas (PAe000324). 5) The LTQ/FT-shotgun data set (D8)2 was produced by the Beijing Proteome Research Center by 10 reduplicated MS/MS analyses on yeast samples.
Protein Sequence Database
All data searching was performed using SEQUEST against the modified randomized sequence database by BioWorks 3.2 (29). For control data sets the standard sequences, which included the purified proteins and peptides as well as possible contaminants provided with the data sets, were combined with the protein sequences from Methanosarcina acetivorans C2A (4520 sequences in total; downloaded from the National Center for Biotechnology Information (NCBI)) to construct the target database. The human International Protein Index (IPI) version 3.19 database, containing 60,397 protein sequences, was the searched database of complex data sets D4, D5, and D6. The Saccharomyces cerevisiae ORF protein sequence (download from the Saccharomyces Genome Database (SGD) at Stanford University) was used as the searched database for D7 and D8. Simultaneously the random permutation amino acid sequence of digested peptide (RSDP) method (30) was proposed to construct a randomized database for each normal data set, and then the normal and randomized data sets were combined and used for database search.
Database Searching
The raw files were searched against the combined database using a local Turbo SEQUEST v.27 server using the same database search parameters for all data sets. The SEQUEST parameters were as follows. The monoisotopic mass was used for both peptide and fragment ions with fixed modification (Carbamidomethyl, +57 Da) on Cys and variable modification (oxidation, +16Da) on Met. The mass tolerance for precursors of all data sets was 3.0 Da, and the fragment ion mass tolerance was 1.0 Da for D7 and D8 and 0.6 Da for the others. Tryptic cleavage at only Lys or Arg was selected, and up to two missed cleavage sites were allowed. Only b and y fragment ions were taken into account. The peptide mass ranged from 400 to 5000 Da when creating *.dta files, and the threshold of the total ion intensity for the LTQ and the LCQ was 100 and 10,000, respectively.
BNP Model Work Flow
The work flow of the BNP model is shown in Fig. 1 and contains three main steps. After MS/MS spectra were searched against the combined database, the first step begins with the construction of two typical labeled subsets. The first subset includes all decoy matches, which were taken as negatives and designated y = 0. The second subset consisted of matches validated (positives) by the cutoff-based method with FDREst = 0.01 and designated y = 1. Based on these subsets, the coefficients of the LDF score (Equation 1) could be estimated by multivariate linear regression. We were then able to calculate the LDF score of all matches.
Fig. 1.

Work flow for constructing and applying the BNP model. This procedure can be divided into three essential steps. 1, an LDF is conducted on two typical labeled subsets, and the LDF score is calculated for each match. 2, the BNP model is constructed, and a probability is computed for every assignment. 3, the PScore cutoff value is determined for a given FDR.
In the second step, the LDF score distribution of decoy matches was fitted by a nonparametric PDF with the maximum likelihood parameter estimation. By restricting the decoy matches as a constant part and applying the expectation-maximization (EM) algorithm, the LDF score PDF of correct matches can be estimated from the normal database matches, which consisted of the combined observations of correct and incorrect assignments. Consequently the correct probability of each assignment can be calculated using the Bayesian formula and the conditional distributions of correct and incorrect matches.
At the last step, we were able to make a decision according to the cost function, which is presented here as the FDR; the FDR can be replaced by estimated error rates in the probability framework. The percentages of correct and incorrect matches were also estimated at this step. Therefore, we can calculate the total number of correct assignments and provide the model specificity at each probability score (PScore) cutoff value. When applied to large data sets, the BNP model can reduce the computational burden by randomly resampling 30,000 observations of the whole data set for the model-building process.
Features Involved in the BNP Model and LDF
Many parameters (referred to as “features” in this study) have been used to validate SEQUEST database search results; these include 1) database search scores, including Xcorr, ΔCn, Sp, RSp, and Ions; 2) physical and chemical properties of the peptide and the basic properties of the experimental MS/MS spectra, such as peptide length (PLen), predicted peptide chromatographic retention time (tRT), peptide molecular weight, and number of peaks in the MS/MS spectrum (PNum); and 3) the empirical parameters used in previous studies, such as RScore (17), Cont (16), and SimScore (31). We used a total of 28 features to improve the discriminant power of the BNP model because a large amount of information was being extracted from the MS data; these are listed, along with the corresponding transformations, in Table I, and additional details are briefly summarized in supplemental File S1. Table I lists the transformation of these features to reduce the variance and improve their discriminant power (32).
Table I.
Features used in the BNP model
Three main classes and 28 features in total are introduced into the BNP model to measure the characteristics of MS/MS spectra and database search assignment.
| Feature class and feature | Notea/Ref. | Transformb |
|---|---|---|
| Database search scores | ||
| Xcorr | 1/6 | — |
| ΔCn | 2/6 | — |
| Sp | 3/6 | Log |
| RSp | 3/6 | Reciprocal |
| Ions | 4/32 | Absolute |
| MS/MS spectrum or peptide properties | ||
| dm | 5/26 | Absolute |
| (M + H)+ | The molecular weight of peptide/42 | /100 |
| TIC | The total ion current of MS/MS spectrum/42 | Log |
| PLen | Peptide length/42 | — |
| PNum | Peak number in the MS/MS spectrum/43 | Log |
| tRT | Predicted retention time/44 | — |
| ScanNum | MS/MS spectrum scan number/1000 | /1000 |
| Ch | Charge state/45 | — |
| Ch_MS_Idx | 6/46 | — |
| Ch_Seq_Idx | 6/47 | — |
| Empirical parameters | ||
| Max_Tag_Len | 7/48 | — |
| Cont | 7/16 | — |
| Pts | 7/49 | — |
| Fdm_Avg | Average mass error of matched fragment ions | — |
| Fdm_std | Standard deviation of mass error of matched fragment ions | — |
| HPM | 8/50 | — |
| iIons | 9/42 | — |
| nIons | 9/16 | — |
| XIII | 9/51 | — |
| MPF | 10/45 | — |
| SimScore | 11/31 | — |
| VEMS | 12/52 | — |
| RScore | 13/17 | — |
The number denotes the order of description in the supplemental file S1.
Some transformations, e.g. log, are implemented on the features where indicated, and — means no transformation has been done to the corresponding feature.
Therefore, the LDF score can be defined as
 |
(Eq. 1) |
where a0–a26 are the coefficients that can be derived by regression from the “typical labeled data sets.” We constructed the LDF model for each charge state (Ch) individually and used peptide length PLen ≥6 as a prefilter in applying the model.
BNP Model and EM Algorithm
Based on the theory that the random matches and the correct matches can be grouped into subclasses and that the LDF score of each subclass should have a simple distribution (e.g. normal distribution; some detailed discussion can be found in the supplemental File S1), we used the Gaussian component distributions to simulate the mixture distribution of the observations. The format of the hypothesis mixture PDF is
 |
(Eq. 2) |
where
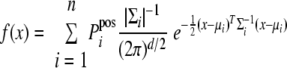 |
(Eq. 3) |
and
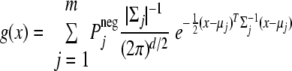 |
(Eq. 4) |
and the μj, j = 1, 2, … m, and μi, i = 1, 2, … n, are means of the component Gaussian distributions. Σj, j = 1, 2, … m, and Σi, i = 1, 2, …, n, are their covariance matrices. These parameters satisfy Σj = 1mPjneg = 1 and Σi = 1nPipos = 1; Ppos ≥ 0, Pneg ≥ 0; Pipos ≥ 0, Pjneg ≥ 0, and Ppos + Pneg = 1.
First the negative component contributing to random matches can be estimated from the decoy matches by the fully nonparametric probability density function estimate procedure proposed by Archambeau and Verleysen (33) and Duda et al. (34) that was implemented by the maximum likelihood estimation with the EM algorithm. Then the positive component contributing to correct matches can be estimated from the mixture observations of the normal database matches by a restricted fully nonparametric probability density function estimate. This iterative EM procedure can be read as described previously (23) with keeping Pjneg, j = 1, 2, …, n, unchanged when updating the parameters in the M-step. Here x is the LDF score, a scalar.
By trial and error, we found that five component GDFs can provide an accurate PDF fitting. We initialized the parameters in the EM procedure by partitioning the observations into five intervals on the LDF score axis and keeping the number of observations in each interval equal.
After estimation of the conditional PDF, the correct probability of a match with LDF score x can be given as follows.
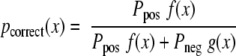 |
(Eq. 5) |
The estimated number of correct matches is Npos = KPpos and the number of incorrect matches is Nneg = KPneg where K is the total number of observations. The FDR and false negative rate (FNR) under different LDF score cutoff values can be estimated by the conditional distribution and the prior probability as follows.
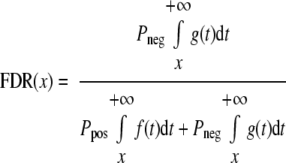 |
(Eq. 6) |
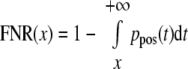 |
(Eq. 7) |
Assuming the expected FDR is α, we can determine the filtration threshold of the LDF score xα according to Equation 6. At the same time, the estimated sensitivity and discriminating power can be estimated as follows.
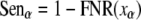 |
(Eq. 8) |
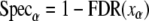 |
(Eq. 9) |
Finally we can calculate the estimated error rate (Err) under different PScore thresholds based on the correct probability of every identified peptide using
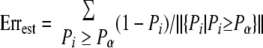 |
(Eq. 10) |
where ‖{Pi|Pi ≥ Pα}‖ denotes the number of the elements in aggregate {Pi|Pi ≥ Pα}. In practice, we found that ErrEst was close to the actual FPR. So in the following sections, ErrEst was used as the estimation of FDR for the BNP model.
RESULTS
Estimation of the FDR—
The control data sets (D1–D3) were used to verify the accuracy of the estimated FDR of the BNP model. When the PScore cutoff is small, the FDR (Err) estimated by the BNP model is larger than the actual FPR. For high quality filtration, Err is close to the actual FPR (Fig. 2). Table II compares the performance of the BNP model (M3), the cutoff-based method (M1), PeptideProphet (M2; contained in the Trans-Proteomic Pipeline version 4.0.1), and our previously published nonparametric model (M4) (23) under two typical FDRs. In the cutoff-based method, an exhaustive search procedure was used to identify the optimal threshold value of the Xcorr/ΔCn pair by maximizing the number of validated normal database matches and keeping the estimated FDR estimated lower than the expected FDR. PeptideProphet provides an estimated error rate for different probability score cutoffs. For all three data sets, the sensitivity of the BNP model surpassed that of the three other filtered methods when the estimated errors/FDRs were the same. The traditional cutoff method produced high quality results with quite a low actual FPR at a cost of the loss of some sensitivity. Thus, the total correct numbers validated by the cutoff method were much lower than those validated by the BNP model.
Fig. 2.

Accuracy of the FDR estimated by the BNP model. The solid lines are the actual FPRs for different PScore cutoff values, and the dashed lines are the estimated FDR calculated by BNP model under the same criterion. The FDR (Err) estimated by the BNP model is larger than the actual FPR for lower PScore values, and for the filtered results with high quality (larger PScore), Err is close to the actual FPR.
Table II.
Performance of the BNP model on three control data sets
| Data seta and methodb | Expected FDR = 5%
|
Expected FDR = 1%
|
||||
|---|---|---|---|---|---|---|
| Actual FPR | Total/correct | Sensitivity | Actual FPR | Total/correct | Sensitivity | |
| % | % | % | % | |||
| D1 | ||||||
| M1 | 2.23 | 719/703 | 78.29 | 0.53 | 567/564 | 62.81 |
| M2 | 2.59 | 733/714 | 79.51 | 0.89 | 674/668 | 74.39 |
| M3 | 2.20 | 820/802 | 89.31 | 0.40 | 758/755 | 84.08 |
| M4 | 2.72 | 810/788 | 87.75 | 1.39 | 722/712 | 79.29 |
| D2 | ||||||
| M1 | 1.92 | 5,875/5,762 | 68.20 | 0.36 | 4,964/4,946 | 58.54 |
| M2 | 2.17 | 6,775/6,628 | 78.45 | 0.51 | 5,895/5,865 | 69.42 |
| M3 | 3.16 | 7,426/7,191 | 85.11 | 1.04 | 6,754/6,684 | 79.11 |
| M4 | 1.91 | 7,001/6,867 | 81.28 | 0.55 | 6,333/6,298 | 74.54 |
| D3 | ||||||
| M1 | 0.13 | 10,284/10,271 | 74.80 | 0.03 | 9,182/9,179 | 83.70 |
| M2 | 0.42 | 11,477/11,429 | 93.14 | 0.17 | 10,699/10,681 | 87.04 |
| M3 | 0.50 | 11,983/11,923 | 97.16 | 0.09 | 11,388/11,378 | 92.72 |
| M4 | 0.32 | 10,885/10,850 | 88.42 | 0.16 | 10,117/10,101 | 82.32 |
D1, LCQ control data set; D2, LTQ control data set; D3, LTQ/FT control data set. For details see “Experimental Data Sets.”
M1, cutoff-based method; M2, PeptideProphet; M3, BNP model; M4, nonparametric model.
Although we used a relatively large parent ion mass tolerance setting (3.0 Da) for the FT/LTQ database search, the actual mass accuracy of the FT mass spectrometer is in the range of a few ppm. Mass accuracy filtering was proposed for this high accuracy data (35, 36). As the statistical mass error for D3 ranged from −2 to 6 ppm (23), validated results with a mass error larger than 10 ppm were taken as false positives and were excluded from the output lists of all filter methods. The peptide assignment lists of three control data sets are provide in Supplemental Tables S1_LCQ, S1_LTQ, and S1_FT, and the corresponding filter criteria can be found in supplemental File S2.
Searching a Larger Database—
It is generally acknowledged that search algorithms lose sensitivity as the search space is increased because more peptides are queried (37). Larger databases increase the number of candidate peptides for each MS/MS spectrum, and the probability of randomized matches increases as well. We constructed a large combined database containing 13,936 protein sequences from four different Archaea species (M. acetivorans C2A, Archaeoglobus fulgidus DSM 4304, Methanosarcina barkeri strain fusaro chromosome 1, and Methanosarcina mazei Go1; all downloaded from NCBI) and repeated the database search to test the performance of the BNP model on different searched databases. As the search space was expanded, fewer matches were identified. When the estimated FDR was set at 0.05 and 0.01, the BNP model confirmed 804 and 708 matches, respectively (supplemental Table S1_LCQ). The actual FPR was 2.86 and 0.71%, and the sensitivity was 91.24 and 82.13%, respectively. These values are nearly identical to those observed when searching a smaller database. The results indicate that the BNP model is reliable and accurate on different searched databases.
Among the validated matches of the large and small database search results, 778 matches were the same. Only 15 of the 40 matches validated by large database searching alone were from the control sequences. On the other hand, 43 MS/MS spectra matched with the control sequence were confirmed only in the small database search. These matches may possibly be false positives, the MS/MS spectra of which would be matched with a more appropriate peptide in a different database. These observations indicate that not all matches assigned to control sequences are correct because some spectra matched with different peptides in the large and small search spaces, and some were randomly matched with control peptides. Thus, we used four empirical rules to refine these possible correct matches for sensitivity calculation: 1) Rsp ≤ 50, 2) PLen ≥ 6; 3) PNum ≥ 20, and 4) Max_Tag_Len ≥ 4.
Quality of the Results Confirmed by the BNP Model—
We also validated the confirmed matches identified by the BNP model (M3) in the real, complex sample data sets using the empirical rules (Table III). These empirical rules came from different sources in the literature (16, 26, 38). In Table V, MTL is the abbreviation for Max_Tag_Len, and other parameters are introduced under “Features Involved in the BNP Model and LDF.” Most of the matches confirmed by the BNP model are of high quality in view of these empirical rules, and the quality of the results improves as the accuracy of the data increases. As a comparison, we calculated these percentages for results obtained with the cutoff-based method (M1; without the rule of Rsp ≤ 50). In some cases, the cutoff-based method seemed to generate slightly better results, but the difference was negligible, especially on the LTQ/FT data set (D6).
Table III.
Validating the quality of confirmed matches on the real sample data sets
| Empirical rulea and methodb | Data setc
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Expected FDR = 5%
|
Expected FDR = 1%
|
|||||||||
| D4 | D5 | D6 | D7 | D8 | D4 | D5 | D6 | D7 | D8 | |
| MIT ≥ 4 (%) | ||||||||||
| M1 | 93.21 | 98.94 | 99.55 | 98.81 | 99.97 | 94.51 | 99.55 | 99.85 | 99.36 | 99.99 |
| M3 | 92.12 | 96.06 | 99.45 | 97.70 | 99.86 | 93.62 | 97.84 | 99.79 | 98.70 | 99.94 |
| Ions ≥ 0.2 (%) | ||||||||||
| M1 | 99.59 | 99.97 | 100.00 | 99.95 | 99.92 | 99.77 | 100.00 | 100.00 | 99.98 | 99.99 |
| M3 | 99.60 | 99.94 | 99.26 | 99.92 | 99.92 | 99.70 | 99.96 | 99.56 | 99.96 | 99.98 |
| RSp ≥ 10 (%) | ||||||||||
| M1 | 96.88 | 91.96 | 90.33 | 89.88 | 98.45 | 98.99 | 96.05 | 95.50 | 94.67 | 99.74 |
| M3 | 93.68 | 89.90 | 99.01 | 87.52 | 97.76 | 96.47 | 93.55 | 99.66 | 92.61 | 99.86 |
| Cont ≥ 0.2 (%) | ||||||||||
| M1 | 84.06 | 94.93 | 98.43 | 91.63 | 99.84 | 85.34 | 96.17 | 99.13 | 92.68 | 99.93 |
| M3 | 84.62 | 95.16 | 99.15 | 90.70 | 99.64 | 85.43 | 95.96 | 99.54 | 91.75 | 99.76 |
| iIons ≥ 0.25 (%) | ||||||||||
| M1 | 97.71 | 93.71 | 96.23 | 98.77 | 95.83 | 98.41 | 95.21 | 97.85 | 99.15 | 95.81 |
| M3 | 97.54 | 90.57 | 98.10 | 98.29 | 95.76 | 98.13 | 93.25 | 98.60 | 98.85 | 95.94 |
| nIons ≥ 0.2 (%) | ||||||||||
| M1 | 96.88 | 98.96 | 99.90 | 99.88 | 100.00 | 97.61 | 99.47 | 99.98 | 99.94 | 100.00 |
| M3 | 96.07 | 95.29 | 99.94 | 99.83 | 99.99 | 96.97 | 97.16 | 99.97 | 99.88 | 100.00 |
MTL, Max_Tag_Len; other parameters are described under “Features Involved in the BNP Model and LDF.”
M1, cutoff-based method; M3, BNP model.
For details of the complex data sets D4–D8 see “Experimental Data Sets.” D4, LCQ real sample data set; D5, LTQ real sample data set; D6, LTQ/FT real sample data set.
Table V.
Comparison among the three methods on the protein level
| Data seta and methodb | Expected FDR = 5%
|
Expected FDR = 1%
|
||
|---|---|---|---|---|
| Protein numberc | Proteins with at least 2/3 peptide hits | Protein number | Proteins with at least 2/3 peptide hits | |
| % | % | |||
| D4 | ||||
| M1 | 1,894 | 51.9/35.7 | 1,630 | 54.0/36.0 |
| M2 | 2,237 | 47.7/30.9 | 1,761 | 54.2/35.7 |
| M3 | 2,362 | 50.8/35.1 | 1,916 | 55.9/39.3 |
| M4 | 2,025 | 53.1/37.5 | 1,704 | 56.4/38.9 |
| D5 | ||||
| M1 | 3,363 | 54.6/37.6 | 2,733 | 58.3/39.3 |
| M2 | 4,573 | 49.1/33.4 | 3,175 | 59.1/41.5 |
| M3 | 4,412 | 51.2/35.4 | 3,272 | 59.0/42.2 |
| M4 | 3,511 | 57.1/40.7 | 2,810 | 61.6/42.8 |
| D6 | ||||
| M1 | 2,723 | 42.0/23.0 | 2,193 | 42.5/22.8 |
| M2 | 2,150 | 44.4/24.1 | 1,938 | 42.8/22.6 |
| M3 | 3,714 | 41.1/22.5 | 2,861 | 45.2/25.9 |
| M4 | 2,844 | 44.2/25.1 | 2,466 | 45.2/25.0 |
| D7 | ||||
| M1 | 2,295 | 49.6/33.2 | 1,273 | 58.6/45.7 |
| M2 | 3,071 | 55.0/35.1 | 1,815 | 51.8/38.5 |
| M3 | 3,124 | 56.3/35.5 | 1,797 | 52.1/38.7 |
| M4 | 2,242 | 51.4/34.1 | 1,240 | 61.1/48.1 |
| D8 | ||||
| M1 | 518 | 33.4/23.4 | 246 | 49.6/39.4 |
| M2 | 224 | 50.4/41.1 | 161 | 57.1/44.1 |
| M3 | 895 | 27.4/15.6 | 418 | 37.8/27.3 |
| M4 | 565 | 31.7/21.9 | 305 | 47.9/36.4 |
For details of the complex data sets D4–D8 see “Experimental Data Sets.”
M1, cutoff-based method; M2, PeptideProphet; M3, BNP model; M4, nonparametric model.
The minimal protein lists including “protein group.”
Comparison among Different Methods on Complex Data Sets—
We compared the performance of the BNP model (M3) with the cutoff method and PeptideProphet on complex data sets (D4–D8; Table IV). The BNP model confirmed about 14–39% more results, both total and unique peptides, than the cutoff-based method. PeptideProphet appeared to be influenced by data quantity and quality. For complex data sets D5 and D7 containing more than 106 matches we had to separate the *.pep.xml files (60 files in total) into several runs because PeptideProphet requires too much memory, generates too large (greater than 4 gigabytes) a temporary file to be accommodated by Windows, and requires an unacceptable amount of time to complete the modeling. While conducting the search on complex data set D6 (data from 46 LC runs in total), the MS/MS data quality of some LC runs was so poor that PeptideProphet was not able to finish the modeling and validated very few peptides (only 18,446 correct with 1% FDR). Therefore, we used the results of D6 from an earlier version (PeptideProphet 1.9) that were superior to those derived using Trans-Proteomic Pipeline version 4.0.1.
Table IV.
Comparison of three filtering methods on large data sets
| Data seta and methodb | Expected FDR = 5%
|
Expected FDR = 1%
|
||
|---|---|---|---|---|
| Confirmed matches | Non-redundant peptides | Confirmed matches | Non-redundant peptides | |
| D4 | ||||
| M1 | 13,632 | 5,378 | 11,512 | 4,555 |
| M2 | 13,776 | 5,769 | 11,928 | 4,878 |
| M3 | 18,151 | 6,942 | 15,897 | 5,941 |
| M4 | 16,276 | 6,101 | 13,543 | 5,120 |
| D5 | ||||
| M1 | 45,153 | 10,767 | 36,855 | 8,851 |
| M2 | 57,009 | 13,593 | 47,304 | 10,737 |
| M3 | 60,565 | 13,888 | 52,145 | 11,479 |
| M4 | 53,561 | 12,048 | 44,602 | 9,940 |
| D6 | ||||
| M1 | 40,746 | 5,540 | 34,185 | 4,458 |
| M2 | 31,328 | 4,477 | 26,964 | 3,899 |
| M3c | 52,181 | 7,562 | 46,923 | 6,224 |
| M4 | 45,470 | 6,049 | 40,047 | 5,249 |
| D7 | ||||
| M1 | 99,952 | 9,010 | 80,222 | 6,313 |
| M2 | 111,075 | 11,314 | 95,390 | 7,865 |
| M3 | 123,499 | 12,177 | 104,598 | 8,131 |
| M4 | 113,140 | 9,403 | 92,951 | 6,761 |
| D8 | ||||
| M1 | 32,230 | 1,522 | 28,251 | 1,047 |
| M2 | 16,486 | 897 | 12,430 | 648 |
| M3d | 36,709 | 2,099 | 33,217 | 1,436 |
| M4 | 34,912 | 1,651 | 33,060 | 1,297 |
For details of the complex data sets D4–D8 see “Experimental Data Sets.”
M1, cutoff-based method; M2, PeptideProphet; M3, BNP model; M4, nonparametric model.
The PeptideProphet results for D6 were generated using version 1.9.
The decoy hits were not used to pin down the negative distribution in the D8 processing because there were too few decoy hits in the database search result of D8 to meet the need of the modeling process of PeptideProphet.
The list of peptide assignments of real, complex sample data sets by M1, M2, M3, and M4 and the corresponding proteins are provided in supplemental Tables S2_D4, S2_D5, S2_D6, and S3.
The Venn diagram in Fig. 3 shows the classification of confirmed peptides using these four methods. The peptides confirmed by the BNP model represented more than 92% of the merged results of the cutoff-based method and PeptideProphet (M1 ∪ M2) and represented more than 91% of the nonparametric model; the BNP model confirmed many additional results, indicating that the sensitivity of the BNP model is much higher than that of the other two methods. By manually checking the records that were discarded by the BNP model but confirmed by the other two methods, we found that some records had relative large Xcorr and ΔCn; careful inspection of these records showed that some other feature scores, such as Ions, iIons, Cont, and nIons were small, indicating that they were potential incorrect matches.
Fig. 3.

Overlap of peptides identified by the three methods. M1 denotes the cutoff-based method without the rule RSp ≤ 50, M2 denotes PeptideProphet, and M3 denotes the BNP model. Taking 0.01 as the FDR, the peptides identified by the BNP model covered more than 92% of the results of the cutoff-based method and PeptideProphet. The BNP model can also confirm significantly more results.
Conversion to Protein Identifications—
In analyzing complex samples, the most important criterion is the number of proteins identified with confidence as output. The number of unique protein counts and high confidence protein identifications (more than two or three peptide hits) for an FDR of 1 and 5% in each experimental data set are shown in Table V. The minimal protein lists were assembled according to the parsimony principle applied by the DBParser algorithm (39), and an in-house software written in C++ was developed to support our file format. It appears that the percentages of proteins with two or three peptide hits provided by the four methods are close. However, the BNP model can generate a large protein list with a greater number of high confidence proteins. It is interesting that the percentage of high confidence proteins cannot be improved by improving the confidence level of resulting matches if only one method (M1, M2, M3, or M4) is used.
DISCUSSION
Proteomics research has generated vast amounts of MS/MS data. SEQUEST is a robust algorithm that is appropriate for processing low accuracy ion trap MS/MS data. Using external tools to separate correct from incorrect SEQUEST database search results has been the focus of much attention. We developed BNP to filter the false-positive matches in shotgun proteomics database searching. This strategy is based on a randomized database method and nonparametric density distribution functions. By applying this model to control protein data sets and complex data sets from real samples, we demonstrate that the BNP model has greater power to discriminate between correct and incorrect assignments and can effectively control the false-positive ratio of peptide identifications. Furthermore the BNP model can greatly increase the number of confirmed peptides and proteins, and it is suited for use with several MS platforms.
BNP Model Versus PeptideProphet—
Recently Choi et al. (21) presented a variable component mixture model and a semiparametric mixture model to remove the restrictive parametric assumptions in the mixture modeling approach of PeptideProphet. The most recent version of PeptideProphet provides an option to use nonparametric modeling with target-decoy database searches. The process works well on D3, which was the test data set in Ref. 19. The number of validated peptides in D1 increased (964 for 5% FDR and 798 for 1% FDR), but the actual FPR increased as well (10.06 and 3.26% respectively); the numbers of identified peptides (7080 for 5% FDR and 6072 for 1% FDR) as well as the actual FPRs (5.25 and 1.15%, respectively) do not improve on the LTQ control data set.
BNP Model Versus Nonparametric Model—
Both the BNP model and our previously published nonparametric model (23) are based on the target-decoy database search strategy. The nonparametric model, using the nonparametric density estimation technique, aims to estimate the multivariate PDF of the database search scores directly and takes the contour lines as the candidate discriminant functions to filter out false-positive results. Based on the hypothesis that what constitutes a high quality match can be learned from the treated data itself (22), the BNP model was able to model the probability structure from the target-decoy search results and then automatically classify the results. The nonparametric PDF estimation in the BNP model provided a flexible framework for the probability structure.
The primary parameters in the nonparametric model are three commonly used database scores: Xcorr, ΔCn, and SimScore; incorporation of an additional feature dictates an additional dimension of the feature space, and the complexity of the model increases accordingly. The BNP model incorporates 28 features into a linear discriminant function and permits convenient incorporation of more features as required. Furthermore the BNP model can provide a correct probability of each assignment that facilitates subsequent processes, such as application of the EBP (Empirical Bayes Protein identifier) model for protein inference (40).
Extension of the BNP Model to a Higher Dimension—
In this study all 28 features were integrated by an LDF. This process reduced the computational complexity but may result in the loss of some information contained in the raw features. From the view of principal component analysis, only the first main principal component was used by the LDF model. It is possible to use more principal components and extend the BNP model to a higher dimension space. The model building procedure will not require modification, but new techniques will be needed to compress the feature space. Partial least squares regression (41) can utilize the target classification information and complete the principal component analysis and regression at the same time; this is a useful tool to use the typical labeled data set to compress the dimension of the feature space. But when more principal components are taken, the initial procedure of the EM algorithm will have to be adjusted, and more computational time will be required.
Why Did the BNP Model Validate More Matches?—
The BNP model is able to identify more high confident proteins (with at least two peptide hits) from an MS/MS data set under the same estimated FDR compared with PeptideProphet and the cutoff-based method. Within 1% peptide FDR in the D4 data set, more than 90% of the proteins with two or more peptide hits that were identified using PeptideProphet and the cutoff-based method were also identified by the BNP model.
Confirming a higher number of confident peptides is the greatest strength of the BNP model; thus, the BNP model could offer a larger high confidence protein list under the established peptide identification FDR, and it can provide more information for downstream biological analysis. The capacity of the BNP model to confirm more peptides is due to its ability to detect high quality matches that other algorithms might filter out based on only a few features of these matches. There are some conditions that would result in high quality assignments being filtered out by other methods. The masses of some amino acid pairs (e.g. Lys/Gln and Leu/Ile) as well as several amino acid combinations (Table VI) are indistinguishable when the resolution of the instrument is low. Those may cause the ΔCn score to be small and, in some cases, as low as zero. There are also some conditions for which the theoretical spectra of rank 1 and rank 2 identified peptides are similar in SEQUEST outputs, which would also make the ΔCn score smaller than the commonly acceptable value of other methods.
Table VI.
Examples of high quality matches detected by the BNP model
| PScore | Rank | Peptide sequencea | Xcorr | ΔCn |
|---|---|---|---|---|
| 1 | 1 | GVVDSEDIPLNLSR | 4.7929 | 0 |
| 1 | GVVDSEDLPLNISR | 4.7929 | ||
| 0.981 | 1 | SETAPAAPAAAPPAEK | 3.5665 | 0.0286 |
| 2 | SETAPAAPAAPAPAEK | 3.5016 | ||
| 0.973 | 1 | IEDLSQEAQLAAAEK | 5.4036 | |
| 2 | IEDLSEQAQLAAAEK | 5.3374 | ||
| 0.992 | 1 | AQIHDLVLVGGSTR | 4.5778 | 0 |
| 2 | AKIHDIVLVGGSTR | 4.5778 | ||
| 1 | 1 | NPQQHLNAQPQVTMQQPAVHVQGQEPLTASMLASAPPQEQK | 6.1680 | 0.0282 |
| 2 | NPQQHLNAQPQVTMQQPAVHVQGQEPLTASMLASAPPQEEK | 5.9939 | ||
| 1 | 1 | RMEELHNQEVQK | 3.7482 | 0.0271 |
| 2 | PEIKLESLKEDIK | 3.3659 |
Indistinguishable amino acids and amino acid combinations are indicated in bold.
We investigated the LCQ complex data set and found 118 undistinguished assignment cases whose ΔCn was less than 0.05. Some examples are listed in Table VI. The BNP model assigned high confidence probabilities (PScore) for those matches filtered by both PeptideProphet and the cutoff-based method. Practically we might not be able to confirm which was the true hit when we do not know the existing proteins at all. To some extent, the BNP model may provide a more objective judgment. In its present form the BNP model cannot accommodate the similarity of theoretical spectra systematically, and introduction of a new parameter to measure this characteristic would improve the performance of the model in the future. The BNP model algorithm tool as well as other scripts used for the SEQUEST search process will be made publicly available.
Acknowledgments
We thank Dr. Jianqi Li at the Beijing Proteome Research Center for thoughtful discussion. The LCQ control data set was kindly provided by the BIATECH Institute. Dr. Zhongqi Zhang at Amgen Inc. kindly provided a program for predicting the MS/MS spectrum.
Footnotes
Published, MCP Papers in Press, November 12, 2008, DOI 10.1074/mcp.M700558-MCP200
The abbreviations used are: FDR, false discovery rate; LDF, linear discriminant function; GDF, Gaussian density function; PDF, probability density function; EM, expectation-maximization; Err, estimated error rate; BNP, Bayesian nonparametric; FPR, false-positive rate; PScore, probability score; PLen, peptide length; PNum, number of peaks in the MS/MS spectrum; VEMS, Virtual Expert Mass Spectrometrist; MPF, mobile proton factor; HPM, hypergeometric probability model.
K. Liu, J. Zhang, J. Wang, L. Zhao, X. Peng, W. Jia, W. Ying, Y. Zhu, H. Xie, F. He, and X. Qian, Anal.Chem., in press.
This work was supported by the Chinese Ministry of Science and Technology (Grants 2006CB910803, 2006CB910700, 2006AA02A312, and 2006AA02Z334), National Natural Science Foundation of China (Grant 30621063), and Beijing Municipal Science and Technology Project (Grant H030230280590).
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Hernandez, P., Muller, M., and Appel, R. D. ( 2006) Automated protein identification by tandem mass spectrometry: issues and strategies. Mass Spectrom. Rev. 25, 235–254 [DOI] [PubMed] [Google Scholar]
- 2.Aebersold, R., and Mann, M. ( 2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 3.Cañas, B., López-Ferrer, D., Ramos-Fernández, A., Camafeita, E., and Calvo, E. ( 2006) Mass spectrometry technologies for proteomics. Brief. Funct. Genomics Proteomics 4, 295–320 [DOI] [PubMed] [Google Scholar]
- 4.Sadygov, R. G., Cociorva, D., and Yates, J. R. ( 2004) Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat. Methods 1, 195–202 [DOI] [PubMed] [Google Scholar]
- 5.Xu, C., and Ma, B. ( 2006) Complexity and scoring function of MS/MS peptide de novo sequencing. Comput. Syst. Bioinformatics Conf. 2006, 5, 361–369 [PubMed] [Google Scholar]
- 6.Eng, J. K., McCormack, A. L., and Yates, J. R. ( 1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 7.Perkins, D. N., Pappin, D. J., Creasy, D. M., and Cottrell, J. S. ( 1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 8.Colinge, J., Masselot, A., Giron, M., Dessingy, T., and Magnin, J. ( 2003) OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 3, 1454–1463 [DOI] [PubMed] [Google Scholar]
- 9.Craig, R., and Beavis, R. C. ( 2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 10.Chamrad, D., and Meyer, H. E. ( 2005) Valid data from large-scale proteomics studies. Nat. Methods 2, 647–648 [DOI] [PubMed] [Google Scholar]
- 11.Domon, B., and Aebersold, R. ( 2006) Challenges and opportunities in proteomic data analysis. Mol. Cell. Proteomics 5, 1921–1926 [DOI] [PubMed] [Google Scholar]
- 12.Nesvizhskii, A. I., Vitek, O., and Aebersold, R. ( 2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 4, 787–797 [DOI] [PubMed] [Google Scholar]
- 13.Carr, S., Aebersold, R., Baldwin, M., Burlingame, A., Clauser, K., and Nesvizhskii, A. ( 2004) The need for guidelines in publication of peptide and protein identification data: Working Group on Publication Guidelines for Peptide and Protein Identification Data. Mol. Cell. Proteomics 3, 531–533 [DOI] [PubMed] [Google Scholar]
- 14.Tabb, D. L., McDonald, W. H., and Yates, J. R. ( 2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian, W. J., Liu, T., Monroe, M. E., Strittmatter, E. F., Jacobs, J. M., Kangas, L. J., Petritis, K., Camp, D. G., and Smith, R. D. ( 2005) Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: the human proteome. J. Proteome Res. 4, 53–62 [DOI] [PubMed] [Google Scholar]
- 16.Sun, W., Li, F., Wang, J., Zheng, D., and Gao, Y. ( 2004) AMASS: software for automatically validating the quality of MS/MS spectrum from SEQUEST results. Mol. Cell. Proteomics 3, 1194–1199 [DOI] [PubMed] [Google Scholar]
- 17.Li, F., Sun, W., Gao, Y., and Wang, J. ( 2004) RScore: a peptide randomicity score for evaluating tandem mass spectra. Rapid Commun. Mass Spectrom. 18, 1655–1659 [DOI] [PubMed] [Google Scholar]
- 18.Higdon, R., Hogan, J. M., Van Belle, G., and Kolker, E. ( 2005) Randomized sequence databases for tandem mass spectrometry peptide and protein identification. OMICS 9, 364–379 [DOI] [PubMed] [Google Scholar]
- 19.Elias, J. E., and Gygi, S. P. ( 2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 20.Choi, H., and Nesvizhskii, A. I. ( 2008) Semisupervised model-based validation of Peptide identifications in mass spectrometry-based proteomics. J. Proteome Res. 7, 254–265 [DOI] [PubMed] [Google Scholar]
- 21.Choi, H., Ghosh, D., and Nesvizhskii, A. I. ( 2008) Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J. Proteome Res. 7, 286–292 [DOI] [PubMed] [Google Scholar]
- 22.Nesvizhskii, A., Roos, F. F., Grossmann, J., Vogelzang, M., Eddes, J. S., Gruissem, W., Baginsky, S., and Aebersold, R. ( 2006) Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data: toward more efficient identification of post-translational modifications, sequence polymorphisms, and novel peptides. Mol. Cell. Proteomics 5, 652–670 [DOI] [PubMed] [Google Scholar]
- 23.Jiyang, Z., Jianqi, L., Xin, L., Hongwei, X., Yunping, Z., and Fuchu, H. ( 2008) A nonparametric model for quality control of database search results in shotgun proteomics. BMC Bioinformatics 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purvine, S., Picone, A. F., and Kolker, E. ( 2004) Standard mixtures for proteome studies. OMICS 8, 79–92 [DOI] [PubMed] [Google Scholar]
- 25.Klimek, J., Eddes, J. S., Hohmann, L., Jackson, J., Peterson, A., Letarte, S., Gafken, P. R., Katz, J. E., Mallick, P., and Lee, H. ( 2008) The Standard Protein Mix Database: a diverse data set to assist in the production of improved peptide and protein identification software tools. J. Proteome Res. 7, 96–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Resing, K. A., Meyer-Arendt, K., Mendoza, A. M., Aveline-Wolf, L. D., Jonscher, K. R., Pierce, K. G., Old, W. M., Cheung, H. T., Russell, S., Wattawa, J. L., Goehle, G. R., Knight, R. D., and Ahn, N. G. ( 2004) Improving reproducibility and sensitivity in identifying human proteins by shotgun proteomics. Anal. Chem. 76, 3556–3568 [DOI] [PubMed] [Google Scholar]
- 27.Chen, M., Ying, W., Song, Y., Liu, X., Yang, B., Wu, S., Jiang, Y., Cai, Y., He, F., and Qian, X. ( 2007) Analysis of human liver proteome using replicate shotgun strategy. Proteomics 7, 2479–2488 [DOI] [PubMed] [Google Scholar]
- 28.Piening, B. D., Wang, P., Bangur, C. S., Whiteaker, J., Zhang, H., Feng, L. C., Keane, J. F., Eng, J. K., Tang, H., and Prakash, A. ( 2006) Quality control metrics for LC-MS feature detection tools demonstrated on Saccharomyces cerevisiae proteomic profiles. J. Proteome Res. 5, 1527–1534 [DOI] [PubMed] [Google Scholar]
- 29.Ying, W., Jiang, Y., Guo, L., Hao, Y., Zhang, Y., Wu, S., Zhong, F., Wang, J., Shi, R., Li, D., Wan, P., Li, X., Wei, H., Li, J., Wang, Z., Xue, X., Cai, Y., Zhu, Y., Qian, X., and He, F. ( 2006) A dataset of human fetal liver proteome identified by subcellular fractionation and multiple protein separation and identification technology. Mol. Cell. Proteomics 5, 1703–1707 [DOI] [PubMed] [Google Scholar]
- 30.Zhang, J., Li, J., Xie, H., Zhu, Y., and He, F. ( 2007) A new strategy to filter out false positive identifications of peptides in SEQUEST database search results. Proteomics 7, 4036–4044 [DOI] [PubMed] [Google Scholar]
- 31.Zhang, Z. ( 2004) Prediction of low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 3908–3922 [DOI] [PubMed] [Google Scholar]
- 32.Keller, A., Nesvizhskii, A., Kolker, E., and Aebersold, R. ( 2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 33.Archambeau, C., and Verleysen, M. ( 2003) Fully nonparametric probability density function estimation with finite gaussian mixture models, in 5th International Conference on Advances in Pattern Recognition, Calcutta, India, December 11–13, 2003, pp. 81–84, The International Association for Pattern Recognition
- 34.Duda, R. O., Hart, P. E., and Stork, D. G. ( 2001) Pattern Classification, 2nd Ed., pp. 3–13, John Wiley & Sons, Inc., New Jersey
- 35.Zubarev, R., and Mann, M. ( 2007) On the proper use of mass accuracy in proteomics. Mol. Cell. Proteomics 6, 377–381 [DOI] [PubMed] [Google Scholar]
- 36.Brosch, M., Swamy, S., Hubbard, T., and Choudhary, J. ( 2008) Comparison of Mascot and X!Tandem performance for low and high accuracy mass spectrometry and the development of an adjusted mascot threshold. Mol. Cell. Proteomics 7, 962–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapp, E. A., Schutz, F., Connolly, L. M., Chakel, J. A., Meza, J. E., Miller, C. A., Fenyo, D., Eng, J. K., Adkins, J. N., and Omenn, G. S. ( 2005) An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics 5, 3475–3490 [DOI] [PubMed] [Google Scholar]
- 38.Chen, Y., Kwon, S. W., Kim, S. C., and Zhao, Y. ( 2005) Integrated approach for manual evaluation of peptides identified by searching protein sequence databases with tandem mass spectra. J. Proteome Res. 4, 998–1005 [DOI] [PubMed] [Google Scholar]
- 39.Yang, X., Dondeti, V., Dezube, R., Maynard, D. M., Geer, L. Y., Epstein, J., Chen, X., Markey, S. P., and Kowalak, J. A. ( 2004) DBParser: web-based software for shotgun proteomic data analyses. J. Proteome Res. 3, 1002–1008 [DOI] [PubMed] [Google Scholar]
- 40.Price, T. S., Lucitt, M. B., Wu, W., Austin, D. J., Pizarro, A., Yocum, A. K., Blair, I. A., FitzGerald, G. A., and Grosser, T. ( 2007) EBP, a program for protein identification using multiple tandem mass spectrometry datasets. Mol. Cell. Proteomics 6, 527–536 [DOI] [PubMed] [Google Scholar]
- 41.Put, R., Daszykowski, M., Baczek, T., and Vander Heyden, Y. ( 2006) Retention prediction of peptides based on uninformative variable elimination by partial least squares. J. Proteome Res. 5, 1618–1625 [DOI] [PubMed] [Google Scholar]
- 42.Anderson, D. C., Li, W., Payan, D. G., and Noble, W. S. ( 2003) A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores. J. Proteome Res. 2, 137–146 [DOI] [PubMed] [Google Scholar]
- 43.Baczek, T., Bucinski, A., Ivanov, A. R., and Kaliszan, R. ( 2004) Artificial neural network analysis for evaluation of peptide MS/MS spectra in proteomics. Anal. Chem. 76, 1726–1732 [DOI] [PubMed] [Google Scholar]
- 44.Kaliszan, R., Baczek, T., Cimochowska, A., Juszczyk, P., Wiśniewska, K., and Grzonka, Z. ( 2005) Prediction of high-performance liquid chromatography retention of peptides with the use of quantitative structure-retention relationships. Proteomics 5, 409–415 [DOI] [PubMed] [Google Scholar]
- 45.Ulintz, P. J., Zhu, J., Qin, Z. S., and Andrews, P. C. ( 2006) Improved classification of mass spectrometry database search results using newer machine learning approaches. Mol. Cell. Proteomics 5, 497–509 [DOI] [PubMed] [Google Scholar]
- 46.Hogan, J. M., Higdon, R., Kolker, N., and Kolker, E. ( 2005) Charge state estimation for tandem mass spectrometry proteomics. OMICS 9, 233–250 [DOI] [PubMed] [Google Scholar]
- 47.Huang, Y., Triscari, J. M., Tseng, G. C., Pasa-Tolic, L., Lipton, M. S., Smith, R. D., and Wysocki, V. H. ( 2005) Statistical characterization of the charge state and residue dependence of low-energy CID peptide dissociation patterns. Anal. Chem. 77, 5800–5813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Link, A. J., Eng, J., Schieltz, D. M., Carmack, E., Mize, G. J., Morris, D. R., Garvik, B. M., and Yates, J. R., III ( 1999) Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 17, 676–682 [DOI] [PubMed] [Google Scholar]
- 49.Kristensen, D. B., Brønd, J. C., Nielsen, P. A., Andersen, J. R., Sørensen, O. T., Jørgensen, V., Budin, K., Matthiesen, J., Venø, P., Jespersen, H. M., Ahrens, C. H., Schandorff, S., Ruhoff, P. T., Wisniewski, J. R., Bennett, K. L., and Podtelejnikov, A. V. ( 2004) Experimental Peptide Identification Repository (EPIR): an integrated peptide-centric platform for validation and mining of tandem mass spectrometry data. Mol. Cell. Proteomics 3, 1023–1038 [DOI] [PubMed] [Google Scholar]
- 50.Fridman, T., Razumovskaya, J., Verberkmoes, N., Hurst, G., Protopopescu, V., and Xu, Y. ( 2005) The probability distribution for a random match between an experimental-theoretical spectral pair in tandem mass spectrometry. J. Bioinform. Comput. Biol. 3, 455–476 [DOI] [PubMed] [Google Scholar]
- 51.Fenyö, D., and Beavis, R. C. ( 2003) A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 75, 768–774 [DOI] [PubMed] [Google Scholar]
- 52.Matthiesen, R., Bunkenborg, J., Stensballe, A., Jensen, O. N., Welinder, K. G., and Bauw, G. ( 2004) Database-independent, database-dependent, and extended interpretation of peptide mass spectra in VEMS V2.0. Proteomics 4, 2583–2589 [DOI] [PubMed] [Google Scholar]