

Abstract
Bone morphogenetic proteins (BMPs) are required for normal postnatal bone formation and osteoblast differentiation. There is evidence from recent studies that BMP signaling in osteoblasts is controlled by an ubiquitin-proteasome regulatory mechanism involving a cascade of enzymatic reactions. The specificity of protein ubiquitination is determined by E3 ubiquitin ligases, which play a crucial role in defining substrate specificity and subsequent protein degradation by 26S proteasomes. We have examined the role of the E3 ubiquitin ligase Smad ubiquitin regulatory factor 1 (Smurf1), a member of the Hect domain family of E3 ubiquitin ligases in osteoblast function. Smurf1 has been found to interact with BMP-activated Smad1 and -5 and to mediate degradation of these Smad proteins. Recently we have found that Smurf1 mediates the protein degradation of the osteoblast-specific transcription factor Runx2/Cbfa1. To determine the role of Smurf1 in osteoblast differentiation, in the present studies we transfected a Smurf1 expression plasmid into 2T3 osteoblast precursor cells and found that Smurf1 overexpression inhibits BMP signaling and osteoblast differentiation. To further investigate the role of Smurf1 in bone formation in vivo, we generated transgenic mice in which expression of the epitope-tagged Smurf1 transgene was targeted to osteoblasts using the murine 2.3-kb osteoblast-specific type I collagen promoter. In these transgenic mice, bone formation was significantly reduced during postnatal life. Our results demonstrate for the first time that Smurf1 plays a specific role in osteoblast differentiation and bone formation in vivo.
Bone morphogenetic proteins (BMPs)1 play a specific role in osteoblast differentiation in vitro and bone formation in vivo (1, 2). The role of BMPs in postnatal bone formation and maintenance of bone mass has recently been demonstrated in several animal models. In previous studies, we have shown that selective blockage of BMP receptor signaling by over-expression of a dominant-negative type I BMP receptor in osteoblasts inhibits postnatal bone growth and bone formation (3). Noggin and sclerostin, two glycoproteins, which are produced in bone cells, demonstrate high affinity binding to BMP ligands and prevent the interaction between BMP ligands and their cognate receptors (4). Noggin has high affinity to bind BMP-2, -4, and -7 (5)2 and sclerostin has high affinity to bind BMP-5, -6, and -7 (6, 7). Recently it has been shown that transgenic mice overexpressing noggin or sclerostin transgene in osteoblasts develop an osteopenic/osteoporotic phenotype (8, 6). However, the mechanism(s), that regulate BMP signaling during osteoblast differentiation are not fully understood.
One of the important regulatory mechanisms by which the activity of BMP signaling proteins is modulated involves ubquitin-mediated proteasomal degradation. The ubiquitin-proteasome proteolytic pathway is essential for various important biological processes including cell cycle progression, gene transcription, and signal transduction (9, 10). The formation of ubiquitin-protein conjugates requires three enzymes that participate in a cascade of ubiquitin transfer reactions: ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3). The specificity of protein ubiquitination is determined by E3 enzymes, and proteins polyubiquitinated by these enzymes are targeted to undergo degradation primarily by the 26S proteasome (11, 12).
Smurf1 is a member of the Hect domain family of E3 ubiquitin ligases and has been found to interact not only with BMP signaling proteins Smad1 and 5 but also the bone-specific transcription factor Runx2/Cbfa1 and type I BMP receptors and to mediate the degradation of these proteins (13-15). Recent studies suggest that Smurf1 may also play an important role in osteoblast differentiation (14, 16). The related E3 ubiquitin ligase Smurf2 interacts with Smad2 and mediates degradation of Smad2 and proteins interacting with Smad2, such as type I TGF-β receptor and SnoN (17-19). It has also been reported that Smurf2 mediates Smad1 degradation (20). The role of Smurf2 in osteoblast differentiation is currently unknown.
To determine the role of Smurf1 in osteoblast differentiation and bone formation, in the present studies we have utilized a Smurf1 expression plasmid stably transfected into osteoblast precursor 2T3 cells. We also generated transgenic mice in which expression of a Smurf1 transgene is targeted to osteoblasts using the osteoblast-specific type I collagen promoter (21). We found that expression of Smurf1 inhibits osteoblast differentiation in 2T3 cells. In addition, overexpression of Smurf1 in osteoblasts in vivo causes a significant reduction in trabecular bone volume and bone formation rates. Moreover, both osteoblast proliferation and differentiation are inhibited in Smurf1 transgenic mice. Our results demonstrate that Smurf1 plays a specific role in regulating BMP signaling, osteoblast differentiation, and bone formation.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
2T3 osteoblast precursor cells were cultured in α minimal essential medium (αMEM) supplemented with 10% fetal calf serum (FCS) (22). FLAG-tagged Smurf1 expression plasmid and pcDNA3 empty vector (1 μg/well) were stably transfected into 2T3 cells in 6-well culture plates using LipofectAMINE plus reagents (Invitrogen). After transfection (48 h), cells were treated with G418 (300 μg/ml) for 3 weeks. Pooled G418-resistant cells were then cloned using a limiting dilution method and expression of Smurf1 mRNA and protein in selected subclones screened. Four Smurf1-positive subclones (2T3/Smurf1) were analyzed for alkaline phosphatase activity (ALP), expression of osteoblast marker genes and mineralized bone nodule formation, and compared with control subclones (2T3/vector).
Generation of Col1a1-Smurf1 Transgenic Mice
To generate the Col1a1(2.3)-Smurf1 transgene, the truncated type IB BMP receptor (trBMPR-IB) cDNA at the BamHI site of a Col1a1-trBMPR-IB vector (3) was replaced with a FLAG-tagged full-length Smurf1 cDNA. The transgene was released by KpnI/NarI digestion. Approximately 1 to 2 μl of a solution of purified DNA at a concentration of 2 μg/ml was microinjected into the pronuclei of fertilized one-cell mouse embryos derived from C57BL/6 mice. The injected embryos were reimplanted into B6D2F1 (C57BL/6 x DBA/2) pseudopregnant females. The presence of the transgene in the resulting pups was determined by preparing genomic DNA from the tail tissue and assaying by Southern blot analysis and PCR.
Alkaline Phosphatase Activity Assay
Empty vector or Smurf1 expressing-2T3 subclones or primary osteoblasts isolated from wild-type or Col1a1-Smurf1 transgenic mice were plated into 96-well culture plates and grown to ∼60% confluence. The medium was then changed into αMEM with 2% FCS. 48 h after incubation, cells were washed with phosphate-buffered saline twice, and cell lysates were extracted with 0.05% Triton X-100. ALP activity in cell lysates was measured using a Sigma ALP assay kit (Sigma) and normalized by the protein content.
Mineralized Bone Nodule Formation Assay
2T3/vector and 2T3/Smurf1 cells and primary osteoblasts isolated from the calvariae of neonatal wild-type and Smurf1 transgenic mice were used for mineralized bone nodule formation assays as described previously (3, 22). The cells were plated in 24-well culture plates at a density of 2 × 104 cells/well and cultured with αMEM supplemented with 10% FCS. When the cells reached confluence (day 0), the medium was changed to αMEM containing 5% FCS, 100 μg/ml ascorbic acid, and 5 mm β-glycerol phosphate with or without 50 ng/ml BMP-2. The medium was changed every other day and fresh reagents were added. After 10-day incubation, von Kossa staining was performed to analyze the formation of mineralized bone nodules.
Luciferase Assay
The BMP and TGF-β signaling reporter constructs, 12×SBE-OC-Luc (14) and p3TP-Lux (23), were transfected into 2T3/vector and 2T3/Smurf1 cells. Cells were incubated for 24 h in the presence or absence of 50 ng/ml BMP-2 or 2 ng/ml TGF-β (R&D Systems, Minneapolis, MN). Cell lysates were extracted 48 h after transfection, and luciferase activity was measured using a Promega luciferase assay kit and normalized by β-galactosidase activity.
RNA Analysis
Total RNA was isolated from 2T3/vector and 2T3/Smurf1 cells using RNAzol B solution (Tel-Test Inc., Friendswood, TX). Expression of osteoblast-specific genes was analyzed by Northern analysis (22). Expression of Smurf1 transgene in 2T3/Smurf1 cells and primary osteoblasts isolated from Col1a1-Smurf1 transgenic mice was analyzed by reverse transcriptase-PCR. For reverse transcriptase-PCR analysis, DNase I-treated total RNA was reverse transcribed using oligo(dT). cDNA were amplified using primers specific for the FLAG-Smurf1 transgene.
Western Blot Analysis
To determine expression of Smurf1 and to examine Smad1 and Runx2 protein levels in 2T3/Smurf1 cells, cell lysates were extracted from 2T3/vector and 2T3/Smurf1 cells or from osteoblasts isolated from wild-type and transgenic mice, mixed with sample buffer, and run on SDS-PAGE gels (Mini-PROTEIN II Ready gels, Bio-Rad). The proteins were transblotted onto a polyvinylidene difluoride membrane (Bio-Rad) in transblotting buffer (20 mm Tris, 150 mm glycine, 20% methanol, pH 8.0; TBS-T) at 4 °C, 100v for 1 h. The membrane was blocked with 5% dry milk in TBS-T for 1 h at room temperature and incubated with anti-FLAG monoclonal antibody in TBS-T for 2 h at room temperature. Incubation with horseradish peroxidase-conjugated anti-mouse IgG (Amersham Biosciences) was performed at room temperature for 1 h. The membrane was then washed five times with TBS-T for 5 min. Immunostaining was detected using an enhanced chemiluminescence (ECL) system (Amersham Biosciences, Backinghamshire, UK).
Histological and Histomorphometric Analyses
Histomorphometric analyses were performed on proximal tibial metaphyses in 3-month-old transgenic and wild-type mice that had been injected intraperitoneal with calcein 8 and 2 days before animals were sacrificed (3). Half of the bone samples from either group were decalcified in 14% EDTA, embedded in paraffin, and sections stained with hematoxylin and eosin or for tartrate-resistant acid phosphatase. Histological changes in trabecular bones were analyzed and changes in osteoblast and osteoclast numbers quantitated as described previously (3). The other half of the bone samples were embedded un-decalcified in methylmethacrylate and sections used for measurements of bone formation rate (BFR) or stained with von Kossa to determine changes in the cancellous bone mineralization and changes in bone volume. Trabecular bone volume was quantitated in the entire bone marrow cavity 1-3 mm from the growth plates and expressed as a percentage of total tissue volume. Values were expressed as the means ± S.E. calculated from three non-consecutive sections per mouse from each of 10 transgenic mice and 10 wild-type littermates. The calcein-labeled bone surface or mineralizing surface and the distance between two fluorescent labels was measured, and mineral appositional rates (μm/day) and bone formation rates (BFR/bone surface, μm3/μm2/day) were calculated (3).
BrdUrd Labeling
5-Bromo-2′-deoxyuridine (BrdUrd) labeling was performed using Zymed Laboratories Inc., South San Francisco, CA BrdUrd labeling reagent and BrdUrd staining kit according to the manufacture’s protocols (Zymed Laboratories Inc.). Briefly, BrdUrd was injected intraperitonally into mice 24 h before sacrifice. Bone tissues were fixed in 10% neutral buffered formalin and embedded in paraffin. 3-4 μm thick sections were cut and placed on polylysine-coated slides. Slides were dried in a 60 °C oven overnight, deparaffinized in two changes of xylene for 5 min each, and then rehydrated in a graded series of alcohol. Slides were stained for BrdUrd according to manufacture’s protocols. Streptavidin-peroxidase was used as a signal generator for the BrdUrd system, and deaminobenzidine in the presence of hydrogen peroxide was used as a chromogen, staining BrdUrd-incorporated nuclei dark brown (24).
RESULTS
Smurf1 Inhibits Osteoblast Differentiation in Vitro
To determine the effect of Smurf1 on osteoblast differentiation, subclones of the osteoblast precursor 2T3 cells stably overexpressing wild-type Smurf1 were generated. Four individual clones expressing high levels of Smurf1 mRNA (Fig. 1a) and Smurf1 protein (Fig. 1b) were selected and further characterized. Four clones transfected with empty vector were used as controls. The effects of Smurf1 on ALP activity, expression of osteoblast-specific genes, and mineralized bone nodule formation in these cells were examined. In 2T3/Smurf1 clones, there was an over 80% reduction in ALP activity compared with 2T3/vector clones (Fig. 1c). Expression of several osteoblast-specific genes, osterix, Runx2, type I collagen, and osteocalcin, were also markedly decreased in 2T3/Smurf1 cells (Fig. 1d). To determine the effect of Smurf1 on osteoblast terminal differentiation, we examined the effect of Smurf1 on formation of mineralized bone nodules. By day 10 in the presence of ascorbic acid, β-glycerol phosphate and BMP-2 (50 ng/ml), 2T3/vector subclones had formed bone nodules. In contrast, in cultures of 2T3/Smurf1 cells, there were no bone nodules formed even in the presence of the same amount of BMP-2 (Fig. 1e), demonstrating that Smurf1 plays a specific role in BMP signaling and osteoblast differentiation.
Fig.1. Smurf1 inhibits osteoblast differentiation in 2T3 cells.
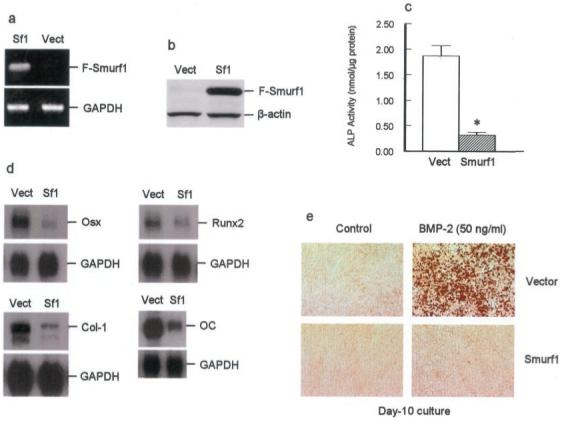
a and b, mRNA and protein expression of Smurf1 in 2T3 cells. FLAG-Smurf1 expression plasmid was transfected into 2T3 cells, and four stably transfected clones were selected and characterized. Expression of transfected FLAG-tagged Smurf1 mRNA was detected by reverse transcriptase-PCR using primers encoding FLAG sequence (upper primer) and Smurf1 cDNA (lower primer) (a). Expression of FLAG-Smurf1 protein was detected by Western blot analysis using an anti-FLAG monoclonal M2 antibody (b). c, Smurf1 inhibits ALP activity. ALP activity from four clones of 2T3/vector and four clones of 2T3/Smurf1 was measured. Smurf1 overexpression inhibited ALP activity. d, Smurf1 inhibits expression of osteoblast-specific genes. mRNA expression of osterix (Osx), Runx2, type I collagen (Col-I), and osteocalcin (OC) was analyzed by Northern blot analysis. Following phosphorimaging, the expression levels of these osteoblast-specific genes were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Smurf1 overexpression inhibited mRNA expression of Osx, Runx2, Col-I, and OC in 2T3 cells. e, Smurf1 inhibits mineralized bone nodule formation. 2T3/vector and 2T3/Smurf1 cells were cultured for 10 days in the absence or presence of BMP-2 (50 ng/ml). Transfection of Smurf1 inhibited BMP-2-induced mineralized bone nodule formation.
Smurf1 Specifically Inhibits BMP Signaling
We then determined the effects of Smurf1 on steady-state levels of Smad1 and Runx2. In 2T3/Smurf1 cells, Smad1 and Runx2 levels were significantly reduced compared with 2T3/vector cells (Fig. 2, a and b). Since Runx2 expression is regulated by the BMP/Smad1 signaling pathway, the decrease in Runx2 protein could be due to a combination of reduced mRNA expression and increased protein degradation. It has been reported that Smurf1 forms a complex with Smad7, which is then exported from the nucleus and targeted to the type I TGF-β receptor for degradation (25, 26). To determine the effect of Smurf1 on BMP and TGF-β signaling, we transfected a BMP signaling reporter 12×SBE-OC-Luc construct (14) and the TGF-β signaling reporter p3TP-Lux construct (23) into 2T3/vector and 2T3/Smurf1 cells and treated cells with BMP-2 or TGF-β. In 2T3/Smurf1 cells, BMP signaling was significantly reduced (Fig. 2c). In contrast, TGF-β signaling remained intact in 2T3/Smurf1 cells (Fig. 2d), suggesting that Smurf1 specifically inhibits BMP signaling in osteoblast precursor cells. Transfection of Smad6 into 2T3/Smurf1 cells further inhibited BMP signaling (Fig. 2c), suggesting that Smad6 and Smurf1 inhibit BMP signaling by independent pathways.
Fig.2. Smurf1 inhibits BMP signaling in 2T3 cells.

a and b, reductions in Smad1 and Runx2 protein levels in 2T3/Smurf1 cells. Expression of Smad1 (a) and Runx2 (b) protein was detected by Western blot analysis. In 2T3/Smurf1 cells, Smad1 and Runx2 protein levels were decreased. c, inhibition of BMP signaling in 2T3/Smurf1 cells. 2T3/vector and 2T3/Smurf1 cells were transfected with BMP signaling reporter construct, 12×SBE-OC-Luc, and treated with 50 ng/ml BMP-2. Expression of Smurf1 inhibited BMP-2-stimulated luciferase activity. Transfection of Smad6 further inhibited BMP-2-stimulated luciferase activity. *, p < 0.05; #, p < 0.05, unpaired t test, compared with the same treatments in 2T3/vector group. d, the effect of Smurf1 on TGF-β signaling in 2T3 cells. 2T3/vector and 2T3/Smurf1 cells were transfected with TGF-β signaling reporter construct, p3TP-Lux, and treated with 2 ng/ml TGF-β. Transfection of Smurf1 had no significant effect on TGF-β signaling in 2T3 cells.
Smurf1 Inhibits Bone Formation in Vivo
To further determine whether Smurf1 plays an essential role in bone formation in vivo, we generated transgenic mice (Col1a1-Smurf1) in which expression of a FLAG-tagged Smurf1 transgene was targeted to osteoblasts using the 2.3-kb type I collagen promoter (21). The Col1a1-Smurf1 transgenic mice were viable and fertile and survived into adulthood and expression of the FLAG-Smurf1 transgene in osteoblasts isolated from the transgenic mice was confirmed (Fig. 3a). Histological analyses of two independent lines of 3-month-old transgenic mice revealed a reduction of about 33% in average trabecular bone volume (BV/TV) (Fig. 3, b and c). Dynamic histomorphometric analyses showed a significant decrease of about 40% in bone formation rates in transgenic mice compared with their wild-type littermates (Fig. 3, d and e). In addition, ALP staining in trabecular bone was also significantly decreased in Smurf1 transgenic mice (Fig. 3f).
Fig.3. Smurf1 inhibits bone formation in Col1a1-Smurf1 transgenic mice.

a, expression of the FLAG-tagged Smurf1 transgene in Col1a1-Smurf1 transgenic mice. Primary osteoblasts were isolated from calvariae of transgenic mice and their wild-type littermates. Expression of FLAG-Smurf1 protein in osteoblasts of Smurf1 transgenic mice was detected by Western blot analysis using the anti-FLAG M2 antibody. b and c, trabecular bone volume (BV) is decreased in Col1a1-Smurf1 transgenic mice. Bone volume was analyzed in two separate lines of transgenic mice in a defined area in proximal tibiae of 3-month-old Smurf1 transgenic mice and wild-type littermates (n = 10) (b). The bone volume was normalized to tissue volume (TV), and in Smurf1 transgenic mice, a 33% decrease in bone volume was observed compared with their wild-type littermates (b, c). *, p < 0.05, unpaired t test. d and e, BFR are decreased in Col1a1-Smurf1 transgenic mice. Bone formation rates were measured and calculated in the same area as bone volume was measured using the Osteomeasure system (Osteometrics Inc., Atlanta, GA). BFR was significantly decreased in Smurf1 transgenic mice compared with littermate control mice. *, p < 0.05, unpaired t test. f, ALP activity is reduced in bones of Col1a1-Smurf1 transgenic mice. Bone samples from Smurf1 transgenic mice and wild-type littermates were fixed in 70% ethanol and embedded in methylmethacrylate without prior decalcification. ALP activity was reduced in transgenic mice compared with their wild-type littermates.
There was a significant decrease (35%) in osteoblast numbers on cancellous bone surfaces (Fig. 4a) but not osteoclast numbers (data not shown) in Smurf1 transgenic mice. To determine whether osteoblast proliferation per se was affected in Smurf1 transgenic mice, we performed BrdUrd labeling experiments and found a significant decrease (38%) in BrdUrd-positive cells on periosteal surfaces of the calvariae of transgenic mice (Fig. 4, b and c). In contrast, osteoblast apoptosis as detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining was not altered in Smurf1 transgenic mice (data not shown). We next isolated primary osteoblasts from 4-day-old neonatal transgenic mice and wild-type littermates and examined ALP activity and mineralized bone nodule formation. Similar to our in vitro findings, ALP activity was significantly reduced (60%) (Fig. 4d), and mineralized bone nodule formation was inhibited in long term cultures of osteoblasts isolated from transgenic mice compared with those from wild-type littermates (Fig. 4e). Taken together, these results strongly suggest that bone formation defects found in Smurf1 transgenic mice are mainly due to decreased osteoblast proliferation and differentiation.
Fig.4. Smurf1 inhibits osteoblast proliferation and differentiation in Col1a1-Smurf1 transgenic mice.

a, osteoblast numbers are decreased in Col1a1-Smurf1 transgenic mice. Osteoblast numbers were counted in the same area as bone volume and bone formation rates were measured. A significant decrease in osteoblast numbers was found in Smurf1 transgenic mice. *, p < 0.05, unpaired t test. b and c, osteoblast proliferation was reduced in Col1a1-Smurf1 transgenic mice. BrdUrd labeling was performed using a BrdUrd assay kit (Zymed Laboratories Inc.). BrdUrd-positive cells on the periosteal surface of calvariae were counted and normalized to the total periosteal cell numbers. A significant decrease in BrdUrd-positive cells was found in Smurf1 transgenic mice. *, p < 0.05, unpaired t test. d, ALP activity is decreased in primary osteoblasts isolated from calvariae of Col1a1-Smurf1 transgenic mice. ALP activity in primary osteoblasts isolated from Smurf1 transgenic mice and their wild-type littermates was measured. In osteoblasts from Smurf1 transgenic mice, ALP activity was significantly decreased. *, p < 0.05, unpaired t test. e, mineralized bone nodule formation was inhibited in primary osteoblasts of Col1a1-Smurf1 transgenic mice. Primary osteoblasts isolated from Col1a1-Smurf1 transgenic mice and their wild-type littermates were cultured for 10 days in the absence or presence of BMP-2 (50 ng/ml). BMP-2-induced mineralized bone nodule formation was inhibited in osteoblasts isolated from Smurf1 transgenic mice.
DISCUSSION
We have demonstrated for the first time that the E3 ubiquitin ligase Smurf1 plays a specific role in bone formation in vivo and that expression of Smurf1 inhibits osteoblast proliferation and differentiation in vitro and in vivo. Furthermore, our data indicate that these effects of Smurf1 on osteoblast proliferation and differentiation are likely due to its specific inhibition of BMP signaling in osteoblasts. These new findings are consistent with our previous reports that expression of a dominant-negative mutant and catalytically inactive Smurf1 enhanced osteoblast differentiation in BMP-2-responsive osteoblast precursor C2C12 and 2T3 cells (14). This is also consistent with a recent report that expression of Smurf1 in C2C12 cells antagonized BMP-2-induced osteoblast differentiation (16). Mutations in dSmurf in Drosophila also lead to enhanced dpp (a Drosophila homologue of BMP-2/4) signaling and expression of downstream target genes of dpp (27). Taken together, these findings suggest that Smurf1 plays a specific role in regulating BMP signaling and osteoblast differentiation and bone formation. Our current studies were performed using an overexpression approach. The role of Smurf1 in bone formation in vivo needs to be further verified either by overexpressing a dominant-negative mutant Smurf1 in osteoblasts in transgenic mice or by generating Smurf1 null mutant mice, specific to the osteoblast lineage. Nevertheless, our current results argue that Smurf1 plays a specific and physiological role in bone formation in vivo.
Osteoporosis remains a major public health problem in the United States and the Western world and identification and development of anabolic agents for the treatment of osteoporosis remains. Most of the drugs currently used in the treatment of osteoporosis are bone resorption inhibitors including bisphosphonates as well as estrogen and related compounds. Although these types of drugs stabilize bone mass and prevent further bone loss, they have not been shown to increase or restore bone mass. This is a critical shortcoming because patients with established osteoporosis may have lost more than 50% of bone mass at critical sites in the skeleton at time of diagnosis. Therefore, a drug that stimulates formation of a substantial amount of new bone is highly desirable. Recently we have discovered that selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro (28), suggesting that selective inhibition of proteasome-mediated proteolytic degradation could have beneficial effects on bone. Currently available proteasome inhibitors, however, may not be ideal drugs to treat patients with osteoporosis due to their non-selectivity and potential side effects. Our data herein demonstrate that the E3 ligase Smurf1 plays specific roles in recognizing several protein substrates critical for BMP signaling and mediates subsequent proteasomal degradation of these proteins. This now identifies the E3 ligase Smurf1 as a potential molecular target for the development of new anti-osteoporosis drugs.
Acknowledgment
We thank Dr. Gerald Thomsen (Department of Biochemistry and Cell Biology and Institute for Cell and Developmental Biology, State University of New York, Stony Brook, NY) for providing us the FLAG-Smurf1 plasmid.
Footnotes
This work was supported in part by the pilot grant (to D. C.) from the Aging Research and Education Center (Nathan Shock Center) at San Antonio and by Grants AR048920 and AR051189 (to D. C.) from the National Institutes of Health/NIAMS. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- BMP
- bone morphogenetic protein
- E3
- ubiquitin ligase
- Smurf1
- Smad ubiquitin regulatory factor 1
- Smad
- Sma/Mother against dpp
- Runx2
- Runt domain transcription factor 2
- Cbfa1
- core-binding factor α1
- TGF-β
- transforming growth factor β
- ALP
- alkaline phosphatase
- SBE
- Smad1 binding element
- OC
- osteocalcin
- dpp
- decapentaplegic
- BFR
- bone formation rates
- BrdUrd
- 5-bro-mo-2′-deoxyuridine
- αMEM
- α minimal essential medium
- FCS
- fetal calf serum
M. Zhao, M. Qiao, G. R. Mundy, and D. Chen, unpublished data.
REFERENCES
- 1.Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz R, Hewick R, Wang EA. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 2.Wozney JM, Rosen V. Clin. Orthop. 1998;346:26–37. [PubMed] [Google Scholar]
- 3.Zhao M, Harris SE, Horn D, Geng Z, Nishimura R, Mundy GR, Chen D. J. Cell Biol. 2002;157:1049–1060. doi: 10.1083/jcb.200109012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, Affolter M, Vale WW, Belmonte JC, Choe S. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- 5.Smith WC, Harland RM. Cell. 1992;70:829–840. doi: 10.1016/0092-8674(92)90316-5. [DOI] [PubMed] [Google Scholar]
- 6.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. EMBO J. 2003;22:6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N. J. Biol. Chem. 2003;278:24113–24117. doi: 10.1074/jbc.M301716200. [DOI] [PubMed] [Google Scholar]
- 8.Devlin RD, Du Z, Pereira RC, Kimble RB, Economides N, Jorgetti V, Canalis E. Endocrinology. 2003;144:1972–1978. doi: 10.1210/en.2002-220918. [DOI] [PubMed] [Google Scholar]
- 9.Weissman AM. Nat. Rev. Mol. Cell. Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 10.Hershko A, Ciechanover A. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 11.Hershko A. Cell. 1983;34:11–12. doi: 10.1016/0092-8674(83)90131-9. [DOI] [PubMed] [Google Scholar]
- 12.Ciechanover A, Orian A, Schwartz AL. BioEssays. 2000;22:442–451. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 13.Zhu H, Kavsak P, Abdollah S, Wrana J, Thomsen GHA. Nature. 1999;400:687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]
- 14.Zhao M, Qiao M, Oyajobi B, Mundy GR, Chen D. J. Biol. Chem. 2003;278:27939–27944. doi: 10.1074/jbc.M304132200. [DOI] [PubMed] [Google Scholar]
- 15.Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T. Mol. Biol. Cell. 2003;14:2809–2817. doi: 10.1091/mbc.E02-07-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ying SX, Hussain ZJ, Zhang YE. J. Biol. Chem. 2003;278:39029–39036. doi: 10.1074/jbc.M301193200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin X, Liang M, Feng XH. J. Biol. Chem. 2000;275:36818–36822. doi: 10.1074/jbc.C000580200. [DOI] [PubMed] [Google Scholar]
- 18.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Mol. Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 19.Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K, Wrana JL. Nat. Cell Biol. 2001;3:587–595. doi: 10.1038/35078562. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Proc. Natl. Acad. Sci. U. S. A. 2001;98:974–979. doi: 10.1073/pnas.98.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossert JA, Eberspaecher H, de Crombrugghe B. J. Cell Biol. 1995;129:1421–1432. doi: 10.1083/jcb.129.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D, Ji X, Harris MA, Feng JQ, Karsenty G, Celeste AJ, Rosen V, Mundy GR, Harris SE. J. Cell Biol. 1998;142:295–305. doi: 10.1083/jcb.142.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carcamo J, Zentella A, Massague J. Mol. Cell. Biol. 1995;15:1573–1581. doi: 10.1128/mcb.15.3.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng JQ, Xing L, Zhang J, Zhao M, Horn D, Chan J, Boyce BF, Harris SE, Mundy GR, Chen D. J. Biol. Chem. 2003;278:29130–29135. doi: 10.1074/jbc.M212296200. [DOI] [PubMed] [Google Scholar]
- 25.Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K. J. Biol. Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki C, Murakami G, Fukuchi M, Shimanuki T, Shikauchi Y, Imamura T, Miyazono K. J. Biol. Chem. 2002;277:39919–39925. doi: 10.1074/jbc.M201901200. [DOI] [PubMed] [Google Scholar]
- 27.Podos SD, Hanson KK, Wang Y, Ferguson EL. Dev. Cell. 2001;1:567–578. doi: 10.1016/s1534-5807(01)00057-0. [DOI] [PubMed] [Google Scholar]
- 28.Garrett IR, Chen D, Gutierrez G, Rossini G, Zhao M, Escobedo A, Kim KB, Hu S, Crews CM, Mundy GR. J. Clin. Invest. 2003;111:1771–1778. doi: 10.1172/JCI16198. [DOI] [PMC free article] [PubMed] [Google Scholar]