

Abstract
Representing receptors as ensembles of protein conformations during docking is a powerful method to approximate protein flexibility and increase the accuracy of the resulting ranked list of compounds. Unfortunately, docking compounds against a large number of ensemble members can increase computational cost and time investment. In this manuscript, we present an efficient method to evaluate and select the most contributive ensemble members prior to docking for targets with a conserved core of residues that bind a ligand moiety. We observed that ensemble members that preserve the geometry of the active site core are most likely to place ligands in the active site with a conserved orientation, generally rank ligands correctly and increase interactions with the receptor. A relative distance approach is used to quantify the preservation of the three-dimensional interatomic distances of the conserved ligand-binding atoms and prune large ensembles quickly. In this study, we investigate dihydrofolate reductase as an example of a protein with a conserved core; however, this method for accurately selecting relevant ensemble members a priori can be applied to any system with a conserved ligand-binding core, including HIV-1 protease, kinases and acetylcholinesterase. Representing a drug target as a pruned ensemble during in silico screening should increase the accuracy and efficiency of high throughput analyses of lead analogs.
Keywords: docking, DHFR, crystal structure, solution structure, homology models, ensembles, protein flexibility
Introduction
Virtual screening, a process in which ligands are docked to the structure of a target receptor and ranked according to their interactions with the receptor, has become an integral part of identifying lead compounds in modern drug discovery. Accurately ranking closely related cogeners is essential to successfully guide the design of optimized leads; incorporating the flexibility of the target receptor and the ligand while considering their conformations is critical to achieving accurate ranking results. It is well established that representing the receptor as an ensemble of structures can substantially improve accuracy in ranking1-4. Ensembles can be generated by compiling experimentally determined structures5 or stochastically determined conformations via molecular dynamics (MD). MD is well established as a viable method of studying protein motion and generating realistic conformations of apo and ligand-bound protein complexes.6-12 During docking, the ensembles may better represent protein flexibility9,13,14, overcome energetic barriers and thermodynamic issues15,16 or present alternate binding conformations over the ligand-tuned version offered by crystal structures determined with a ligand.17-20
In previous work1, we docked a lead series of known compounds against structures of a species of dihydrofolate reductase (DHFR) derived from X-ray crystallography and NMR solution data as well as homology models. We showed that the docking scores of the compounds fell into the correct generalized ranking order when the crystal structure was used as the target receptor and that the solution and homology modeled structures did not serve as good representations of the target. We then created ensembles of the target receptors and docked the same series of compounds against the ensemble members. When ensembles of the solution and homology modeled structures were used as target receptors, the accuracy of docking and ranking increased to the same level as the crystal structure.
In the work described above1, as is typical in studies that have employed ensembles of protein conformations, the entire ensemble with several tens of members is used as the target. While this is the most thorough approach, there is a significant computational cost and time loss. A more efficient approach would be to evaluate and select the most contributive ensemble members prior to docking, therefore reducing the ensemble size and saving substantial computational time as well as increasing the reliability of the ensemble. A limited number of studies have focused on finding a method to filter or compress the ensemble to the contributive members. As a related example, the unified receptor approach attempts to combine ensemble members to a single representative member.12,21
A conserved core approach has been used by others and proven to be an effective means of accurately identifying correct ligand-receptor interactions. The “Structural Interaction Fingerprints” algorithm reduces ligand-protein interactions to a group of strings based on ligand binding to conserved residues. These strings are then used to filter a virtual library for known hits and cluster poses of a ligand in a receptor.22 The multiple protein structure (MPS) method also uses conserved binding regions of a target to map a pharmacophore.3,11,23
Here we report a method that uses the geometry of conserved core residues to select, a priori, ensemble members with a strong positive contribution to accurate ligand orientation, ranking, and docking. An MD simulation allows the residues to explore conformational space and a relative distance approach is used to consider the preservation of the three-dimensional interatomic distances of the relevant ligand-binding atoms. The relative difference method is capable of culling ensemble sets to their most contributive members by up to 75 %, substantially reducing computational time while also dramatically increasing the predictive ability and docking metrics of the pruned ensemble.
In this study, we have chosen DHFR as an example of a protein with a conserved core, since the reported ligands contain a 2,4-diaminopyrimidine or analogous moiety that binds to conserved residues.24-28 With knowledge of the conserved binding of the pyrimidine ring, we can judge the correct placement of ligands within the active site whose orientation has not been proven via experimental methods. However, this method for accurately selecting contributive ensemble members a priori is not restricted to DHFR. Rather, it has potential for any system with a conserved active site core and related ligand-binding moiety. Such families include common drug targets such as the CDK family29,30, HIV-1 nef31 and protease29, many acetylcholinesterases6, p3822, many protein kinase families22, as well as DHFR. Additionally, improving ensembles with the relative distance approach would be especially useful for drug targets that currently have limited or no experimentally determined structures when a suitable homology model scaffold exists. Homology models have traditionally been of questionable utility for drug design, but we describe a method for substantially increasing their ability to not only correctly dock ligands, but also to accurately rank groups of ligands in relation to one another.
Methods
All ligands were drawn in Sybyl32 and brought to their local energy minima as discussed previously.1 Experimentally determined structures were obtained from the PDB and prepared by adding hydrogens, checking geometries and calculating formal charges. Individual ensemble members from NMR data were selected: 1LUD_1 is the representative structure and 1LUD_av is the average structure. Homology models were created using the L. casei DHFR (LcDHFR) sequence from 3DFR25 and various modeling servers (EsyPred3D33, Geno3D34, or Swiss-Model35) using a ClustalW36 sequence alignment. Models were based on three scaffolds: the DHFR structure from B. stearothermophilus that has the highest DHFR sequence identity (PDB ID 1ZDR37, 36% sequence identity), the DHFR structure from T. maritime that has the lowest DHFR sequence identity (PDB ID 1CZ338, 31% sequence identity), or using a multiple-template approach with several DHFR scaffolds (PDB IDs 1ZDR, 1DYH39, 1RF740, 2INQ41). The multiple-template models were selected based on a range of sequence identities resulting from a BlastP42 search of the 3DFR sequence. Ligands and the co-factor were added to these models and were minimized with one of two methods: a 3.5 Å radius around the added ligands or the entire construct. The resulting homology models are named based on the modeling server, the scaffold, and the minimization technique. For example, the model generated by the Geno3D server using a 1ZDR scaffold and a whole-structure minimization is referred to as Geno3D 1ZDR Wholemin.
MD simulations were performed upon the starting structure with a 300 K or 1000 K temperature for 10000 fs or 50000 fs, as indicated in the text. Motion was confined to a specifically defined active site and the NADPH cofactor was held rigid. Conformational snapshots were obtained at 500 fs increments, and resulting structures were minimized using a Amber force field for 1000 iterations and a terminal energy change of 0 kcal/mol. Receptors were checked for correct geometry and Φ/ψ angles. Structures are labeled with their time course snapshot (i.e. R1500 indicates the conformational snapshot at 1500 fs), with RS indicating the starting structure.
A specially defined ensemble set was created based on the backbone flexibility observed in two loop regions of two DHFR NMR solution structure ensemble sets, one from LcDHFR (PDB ID 1LUD43) and the second from human (hDHFR, PDB ID 1YHO44). Using an MD simulation that included atoms within a 3.5 Å radius around the ligand as well as the near-neighbors of the two flexible loops resulted in ensembles in which residues 44-55, 97, 13-20, 29-30, and 123-126 (numbering as per LcDHFR) were allowed to move.
Docking
Docking was carried out as previously discussed. 1 An in-house script was used to automatically determine the top-scoring pose with the conserved orientation of the 2,4-diaminopyrimidine moiety, which was defined as the N1H atom located within 2.75 Å of the conserved acidic residue oxygen atoms. To ensure that the ligand was relatively planar in the active site, a second constraint was used in which Hb (Figure 1) on the amino substituent at C2 must fall within 4.2 Å of the conserved Thr residue oxygen.
Figure 1.

Atoms used in the relative difference calculation of LcDHFR, showing the conserved orientation of the 2,4-diaminopyrimidine ring within the active site, with hydrogen bonding defined by crystallographic data.
Docking results are presented as a combination of metrics. The docking score is expressed in −log10(Kd) units with higher scores indicating a stronger affinity, as a sum of the hydrophobic, polar, electrostatic, repulsive, entropic, and solvation terms.45,46 The docking scores are averaged across ensembles, as all members are considered to have contributed equally. A grouped ranking score represents the receptor's ability to accurately rank ligands by placing them in the correct ligand-affinity bins. These are calculated using a near-neighbor technique in which a ligand is considered correctly binned if it falls into the matched affinity bin or its neighbor's bin. This creates a soft edge between adjacent bins and alleviates the problem of numerical ranking when docking scores are very close or identical.1 The grouped ranking score is represented as a percentage of ligands that are correctly grouped and ranked, with 100 % equaling perfect order. Grouped ranking scores must be greater than random in order to be statistically relevant. The random cutoff is determined for each ligand set.
DHFR has a highly conserved 2,4-diaminopyrimidine binding orientation, allowing the orientation of novel ligands to be assessed in the absence of structural data. The improper orientation rate represents the percentage of ligands that were docked with an incorrect orientation for the 2,4-diaminopyrimidine moiety as the top-scoring pose. In such cases, the top-scoring correct pose is reported and used in the calculation but the ligand is flagged as having originally been improperly oriented. An improper orientation rate (represented as IO %) of 0 indicates that all ligands were correctly oriented by the docking program.
A relative distance scoring metric is used to compare the spatial geometries of relevant conserved residues within the active sites of different models. This function is the averaged total of similarity scores of distances between the relevant atoms of two models, as defined by Equation 147.
where c = comparative model
r = reference model
nmeasures = number of compared measures
Vki = measure k of molecule i
Equation 1: The relative difference score comparing the similarity of measures between two models.
The atoms used in the relative difference calculation are those that can be involved in hydrogen bonds with the 2,4-diaminopyrimidine ring (Figure 1): both oxygens on the sidechain of Asp 26 (numbering is defined by 3DFR), the hydroxy oxygen of Thr 116, the backbone carbonyl oxygen of Trp 5, the backbone carbonyl oxygen of Leu 4, and the backbone carbonyl of Ala 97. While the backbone carbonyl of Trp 5 and the hydroxyl oxygen of Thr 116 do not appear to form hydrogen bonds in the crystal structure, they played an important role in ligand orientation in docking. For hDHFR, the sidechain oxygen of Tyr 121 can also interact with the ligand and is used in the calculation. The conserved acidic residue Asp 26 carries additional weight (two atoms from this residue are used in the calculation) because of its importance to the correct orientation of the 2,4-diaminopyrimidine ring.
Results and Discussion
We have previously shown that docking compounds in a lead series to ensembles of the receptor structure with different conformations can improve the resulting grouped ranking of the docked compounds. However, it is evident that not all ensemble members are positive contributors to the final scores. Here, we first briefly explore various definitions of the active site and the resultant motion across an MD time course to determine if a wider range of motion impacts the predictive ability of the receptor. We then investigate whether maintaining a conserved core orientation as defined by the relative interatomic measures of the corresponding binding residues provides a metric that can assess the predictive worth of an individual ensemble member a priori, effectively allowing ensembles to be substantially trimmed to their positively contributive members.
Exploring parameters of the MD simulation
To begin this study, it was important to determine whether the parameters used in the MD simulation to create the target receptor ensemble affected the degree to which the ensembles improved docking scores, grouped ranking scores and improper orientation rates. We therefore explored two different temperatures (300 K and 1000 K) during the simulation and several different definitions of the active site. The same receptors were used as in our previous study: a crystal structure of LcDHFR (PDB ID 3DFR25) and two crystal structures of hDHFR (PDB ID 1KMV48 and 1OHJ49), the representative member of the ensemble of the NMR solution structure of LcDHFR (1LUD43) as well as the averaged NMR structure and several different LcDHFR homology models created with different templates and different modeling algorithms. Ensembles were created via conformational snapshots across an MD simulation. Motion was constrained to various active site definitions in which all residues with at least one atom falling within a specific sphere around the co-crystallized ligand were included. These radii were 3.5 Å, 6 Å, the ‘special set’ described in Methods, or an MD simulation in which all residues were allowed to move across the time course (“full set”).
The special set was created to model DHFR-specific protein flexibility, as determined by the variance in backbone orientation with NMR solution structure ensembles. The average3d.py50 Pymol script was used to create an average structure as well as a sausage-structure of backbone flexibility with increased radius at higher variance from the average structure alpha carbons, as shown in Figure 2. This averaged structure indicated substantial mobility in the undefined loops above and in front of the active site and minimal flexibility along the alpha-helices and beta-sheets of the active site. This mobility is in agreement with E. coli and P. carinii DHFR studies that showed similar flexibility across the ligand binding event.40,51,52
Figure 2.

Representative structures of backbone location variation as determined by tube radius and color (blue to red) with the script average3d.py for 1LUD (left) and 1YHO (right).
Two families of monocylic 2,4-diaminopyrimidine LcDHFR analogs were selected based on their wide range of inhibition constants (0.05 nM – 1949.8 nM) and structural variety (shown in Supplementary Information)53-55. These 40 ligands were split into six groups based on logical breaks in Ki value and equivalent bin size. The random cutoff for grouped ranking score was determined to be 31 %. These ligands exhibit drug-like properties with molecular weights ranging from 201.2 to 382.5, logP values ranging from 0.12 to 4.55, three to six rotatable bonds per ligand (with three outliers of seven, nine and twelve rotatable bonds) and conformational energies ranging from 2.54 kcal/mol to 23.17 kcal/mol.
The results of the docking exercises for which the MD parameters were varied are presented in Tables I-III. It is important to consider all three metrics: docking score, grouped ranking score and improper orientation rate in order to evaluate whether a receptor or an ensemble accurately docks the lead series. Docking scores reflect the number of ligand-receptor interactions and should be considered within their relative sets. Grouped ranking scores reflect the percentage of ligands that fall in the correct affinity bin and improper orientation rates reflect the percentage of ligands with the conserved orientation of the pyrimidine ring. While there is no statistical relationship between the three metrics, they are often interdependent.
Table I.
LcDHFR experimental structures and their ensembles. Structures are labeled based on their active site radius size. MD simulations are at 300K unless otherwise indicated. The Cluster column indicates the clustering behavior of the relative difference ensembles (Yes, No and O for rocky slope or small groups of clusters), and are color coded as follows: the titles are colored based on their clustering pattern (green: clusters, yellow: rocky slope or small groups of clusters, red: does not cluster). Since relative difference was not used for full ensembles (22 members) and cannot be used for single receptor cases, this column is blank. The improvement of metrics with a relative difference ensemble over the original ensemble is shown in the Improvement column (Yes, No, or = for equivalent metrics), colored based on the improvement (green: overall metrics improve; yellow: overall metrics equivalent).
| Grouped Ranking |
% IO | Score Range |
Ensemble Members |
|||||
|---|---|---|---|---|---|---|---|---|
| Cluster | Improvement | % | (% A.R.) | High | Low | |||
| 3DFR RS | 80% | 49% | 0% | 8.79 | 4.07 | - | ||
| 3DFR 3.5Å | 75% | 44% | 3.6% | 8.06 | 3.91 | 22 | ||
| 3DFR 6Å | 78% | 47% | 41.3% | 7.17 | 3.08 | 22 | ||
| 3DFR 6Å Rel Dif | Y | Y | 80% | 49% | 31% | 7.28 | 2.19 | 5 |
| 3DFR 6Å 1000K | 80% | 49% | 39.8% | 7.14 | 2.73 | 22 | ||
| 3DFR 6Å 1000K Rel Dif | Y | Y | 80% | 49% | 38% | 7.39 | 2.69 | 10 |
| 3DFR SS | 78% | 47% | 7% | 7.74 | 2.95 | 22 | ||
| 3DFR SS Rel Dif | O | = | 75% | 44% | 5.6% | 7.81 | 2.91 | 20 |
| 3DFR Full | 85% | 54% | 31% | 6.51 | 2.97 | 22 | ||
| 3DFR Full Rel Dif | O | = | 80% | 49% | 25.5% | 6.67 | 3.2 | 11 |
| 1LUD_1 RS | 30% | −1% | 0% | 10.5 | 3.31 | - | ||
| 1LUD 1 3.5Å Ens | 80% | 49% | 10.3% | 7.82 | 3.39 | 22 | ||
| 1LUD_1 3.5Å Ens Rel Dif | O | Y | 80% | 49% | 6.5% | 8.35 | 3 | 10 |
| 1LUD_1 SS | 75% | 44% | 13.4% | 7.75 | 4.04 | 22 | ||
| 1LUD_1 SS Rel Dif | N | N | 38% | 7% | 7.5% | 9.12 | 4.56 | 3 |
| 1LUD_av | 40% | 9% | 20% | 10.41 | 1.82 | - | ||
| 1LUD_av3.5Å Ens | 80% | 49% | 7.3% | 8.66 | 3.31 | 22 | ||
| 1LUD_av3.5Å Ens Rel Dif | N | N | 75% | 44% | 7.5% | 8.72 | 3.35 | 10 |
| 1LUD_av SS | 83% | 52% | 13.4% | 8.64 | 3.58 | 22 | ||
| 1LUD_av SS Rel Dif | N | = | 80% | 49% | 11.5% | 9.05 | 3.46 | 10 |
Table III.
LcDHFR homology models and their ensembles. Structures are labeled based on their modeling server engine, the scaffold structure, the minimization technique applied after the ligand and co-factor were added, and the radius of the active site definition. All simulations were performed at 300 K. The relative difference ensembles are labeled based on the length of MD simulation used. Rows are colored as per Table I.
| Grouped Ranking |
% IO | Score Range |
Ensemble Members | |||||
|---|---|---|---|---|---|---|---|---|
| Cluster | Improvement | % | (% A.R.) | High | Low | |||
| 1CZ3 | 58% | 27% | 2.50% | 9.83 | 5.78 | - | ||
| 1ZDR | 68% | 37% | 15% | 8.81 | 4.61 | - | ||
| Esypred 1CZ3 Wholemin RS | 75% | 44% | 58.0% | 6.94 | 0.2 | - | ||
| Esypred 1CZ3 Wholemin 3.5Å Ens | 75% | 44% | 48.0% | 7.2 | 0.15 | 22 | ||
| Esypred 1CZ3 Wholemin 6Å Ens | 83% | 52% | 49.2% | 7.09 | 1.49 | 22 | ||
| Esypred 1CZ3 Wholemin 3.5 Å Ens 10000fs Rel Dif | Y | Y | 75% | 44% | 45.8% | 7.27 | 0.32 | 6 |
| Esypred 1CZ3 Wholemin 3.5 Å Ens 50000fs Rel Dif | Y | Y | 83% | 52% | 40.7% | 7.9 | 0.8 | 7 |
| Geno 1ZDR Wholemin RS | 80% | 49% | 80% | 6.25 | 1.88 | - | ||
| Geno 1ZDR Wholemin 3.5Å Ens | 78% | 47% | 78% | 5.98 | 1.69 | 22 | ||
| Geno 1ZDR Wholemin 3.5 Å Ens 10000fs Rel Dif | Y | Y | 80% | 49% | 80% | 6.02 | 1.85 | 7 |
| Geno 1ZDR Wholemin 3.5 Å Ens 50000fs Rel Dif | N | = | 78% | 47% | 78% | 6.01 | 1.63 | 2 |
| Multiple-template 3.5 Å min RS | 53% | 22% | 5.0% | 8.73 | 5.11 | - | ||
| Multiple-template 3.5 Å min 3.5 Å Ens | 53% | 22% | 10.5% | 8.38 | 5.04 | 22 | ||
| Multiple-template 3.5 Å min 6 Å Ens | 80% | 49% | 16.8% | 6.76 | 4.09 | 22 | ||
| Multiple-template 3.5 Å min 3.5 Å 10000fs Rel Dif | O | Y | 58% | 27% | 3.1% | 8.87 | 5.81 | 4 |
| Multiple-template 3.5 Å min 3.5 Å 50000fs Rel Dif | O | Y | 55% | 24% | 1.6% | 9.15 | 5.91 | 8 |
| Swiss 1CZ3 3.5 Å min RS | 40% | 9% | 73.0% | 8.41 | 2.88 | - | ||
| Swiss 1CZ3 3.5 Å min 3.5Å Ens | 48% | 17% | 67.3% | 6.7 | 0.24 | 22 | ||
| Swiss 1CZ3 3.5 Å min 6 Å Ens | 65% | 34% | 29.2% | 8.08 | 5.11 | 22 | ||
| Swiss 1CZ3 3.5 Å min 3.5 Å Ens 10000fs Rel Dif | Y | Y | 53% | 22% | 63.0% | 6.63 | 0 | 5 |
| Swiss 1CZ3 3.5 Å min 3.5 Å Ens 50000fs Rel Dif | Y | Y | 48% | 17% | 66.9% | 6.41 | 0.61 | 9 |
| Swiss 1ZDR Wholemin RS | 63% | 32% | 10.0% | 8.38 | 5.04 | - | ||
| Swiss 1ZDR Wholemin 3.5 Å Ens | 83% | 52% | 5.1% | 8.14 | 4.55 | 22 | ||
| Swiss 1ZDR Wholemin 6 Å Ens | 65% | 34% | 33.3% | 8 | 3.81 | 22 | ||
| Swiss 1ZDR Wholemin 3.5 Å 10000fs Ens Rel Dif | N | N | 63% | 32% | 22.5% | 7.91 | 3.99 | 11 |
Data from Tables I and II show that ensembles based on crystal structures do not improve the metrics of docking and ranking. In fact, larger allowed radii of motion during the simulation yield worse metrics. For instance, the metrics for 3DFR decrease as the radius increases from 3.5 Å to the special set to 6 Å to the full MD set. While the grouped ranking score increases by a few percentage points with each iteration, the IO rates steadily increase and the docking scores decrease, indicating that potential ligand-receptor binding interactions are lost. The crystal structure of hDHFR, 1KMV, also demonstrates a decrease in metrics when the radius is increased from 3.5 Å to 6 Å. Another crystal structure of hDHFR, 1OHJ, exhibits better docking metrics with a 6 Å radius compared to a 3.5 Å radius but both ensembles exhibit high IO rates and large decreases in docking score as compared to the single crystal structure receptor. If the crystal structure is considered to be the optimal docking conformation, then larger motions move farther away from these orientations. When higher temperature MD simulations (1000 K as opposed to 300 K) were performed on the crystal structures, the results were inconsistent. For 3DFR, a 1000 K simulation offered slight improvement over 300 K, while for 1KMV and 1OHJ, the higher temperature simulation further degenerated the metrics of the ensemble by nearly doubling the IO rate.
Table II.
hDHFR crystal structures and their ensembles. Structures are labeled based on their active site radius size. MD simulations were performed at 300K unless otherwise indicated. The relative difference rows are color coded as per Table I.
| Grouped Ranking |
% IO | Score Range |
Ensemble Members |
|||||
|---|---|---|---|---|---|---|---|---|
| Cluster | Improvement | % | (% A.R.) | High | Low | |||
| 1KMV RS | 84% | 23% | 0% | 11.34 | 6.16 | - | ||
| 1KMV 3.5Å Ens | 68% | 7% | 4.3% | 10.47 | 3.74 | 22 | ||
| 1KMV 3.5Å Ens Rel Dif | O | = | 63% | 2% | 3.4% | 10.59 | 4.02 | 20 |
| 1KMV 6Å Ens | 79% | 18% | 16.7% | 9.06 | 2.65 | 22 | ||
| 1KMV 6Å Rel Dif | Y | Y | 74% | 13% | 7.2% | 10.1 | 4.03 | 11 |
| 1KMV 6Å 1000K | 79% | 18% | 28.9% | 8.59 | 3.76 | 22 | ||
| 1KMV 6Å l000K Rel Dif | Y | Y | 74% | 13% | 10.5% | 10.7 | 4.46 | 4 |
| 1OHJ RS | 90% | 29% | 0% | 10.49 | 3.48 | - | ||
| 1OHJ 3.5Å Ens | 63% | 2% | 35.6% | 7.81 | 1.62 | 22 | ||
| 1OHJ 3.5Å Ens Rel Dif | Y | Y | 84% | 23% | 11.8% | 9.39 | 2.4 | 8 |
| 1OHJ 6Å Ens | 79% | 18% | 20.5% | 7.51 | 0.2 | 22 | ||
| 1OHJ 6Å Ens Rel Dif | O | Y | 79% | 18% | 20.6% | 7.79 | 0.22 | 20 |
| 1OHJ 6Å l000K Ens | 90% | 29% | 35% | 6.93 | 1.8 | 22 | ||
| 1OHJ 6Å l000K Rel Dif | O | Y | 90% | 29% | 17% | 8.15 | 2.41 | 9 |
In the case of 1LUD, the LcDHFR NMR solution structure, an MD ensemble based on individual members dramatically improves the docking and ranking metrics (Table I). However, larger radii degenerate the ensembles. Ensembles based on 1LUD_1, the representative structure, and 1LUD_av, the averaged structure, both have the best combination of metrics when a 3.5 Å radius is used (a 3.5 Å radius is roughly equivalent to the special set) and then decrease for both the 6 Å and full set. The full-motion MD data are not shown in the table as the majority of the ligands were not correctly placed within 200 poses, making it impossible to calculate a grouped ranking score. It is likely that for experimentally observed structures only small motions are needed to ‘fine-tune’ the receptor orientations when they are not initially optimal, as in the case of NMR solution structures as compared to crystal structures. Large motions likely explore far too much conformational space and rapidly escape their optimal orientations. In conclusion, after exploring different temperatures and radii for the MD simulation, stand-alone crystal structures still remain the best receptor model, followed by small radii MD ensembles of NMR solution structures.
The homology models present a converse case to the experimentally determined models (Table III). In four out of five cases, an ensemble based on a 6 Å radius MD simulation presents a better combination of metrics over a 3.5 Å radius. In most cases this is an improvement of both the grouped ranking score and improper orientation rate. In the case of the multiple-template model, the 6 Å radius is still considered optimal despite the 6.3 % increase in improper orientation and drop in docking score because of the substantial 27 % increase in grouped ranking score. Given the wide-ranging structural variability of homology models, it is possible that the larger motions are needed to place the side-chains into better orientations. However, the varied nature of the improvements makes absolute statements in these cases difficult.
In summary, we have previously determined that docking and ranking metrics for solution and homology modeled structures are improved by the use of MD ensembles with a 3.5 Å radius of allowed motion. With larger ranges of motion across the MD timecourse and increased temperatures, our findings were consistent in that no type of explored ensemble improved the metrics of a crystal structure. While NMR and homology models are improved by ensembles, the solution structure members are best served by limited radii of motion and homology models are best served with larger radii of motions. However, overall, varying the active site definition yields minimal and varied improvement, leading us to the conclusion that the parameters in the MD simulation are less important factors to consider.
Relative Difference
Here, we investigated the hypothesis that ensemble members that maintain the geometry of the conserved core will be the most positive contributors to the docking metrics. In order to find a descriptor that measured whether the geometry of the conserved core was maintained relative to a reference structure, we first explored using a simple root mean squared deviation (RMSD) measurement of selected atoms. We observed that RMSD has no correlation to the preservation of the conserved core because RMSD measures the differences in the location of the residues themselves as opposed to what they should be representing: the ability to correctly orient a conserved ligand-binding moiety. It is the interatomic distances of the relevant atoms of these residues that must be conserved as opposed to the location of the entire residue itself. Relative difference as explored here identifies receptors which preserve these interatomic distances, and therefore, receptors that will orient the ligands correctly. We have found no statistical relationship between relative difference and RMSD, or RMSD and the resulting metrics, while there is a direct and substantial relationship between relative difference and scoring metrics.
Eight relative distances are used in the calculation for LcDHFR, with most forming a triangle that maintains order in 3-dimensional space, as shown in Figure 3. Six of the eight measures involve the location of the conserved acidic residue (Asp 26 in LcDHFR). In the case of hDHFR, Tyr 121 is an additional residue that can be involved in orienting the 2,4-diaminopyrimidine moiety. By using this residue in the calculation, there are a total of 12 distances involved, shown in Figure 3. Six of these measures involve the conserved acidic residue Glu 30. In the case of DHFR, this is well warranted as the conserved residue has been shown to play a critical role in both the docking and orientation of ligands. If the N1 atom is oriented to make the relevant hydrogen bonds with this conserved residue, the other residues are oriented at certain distances in order to form hydrogen bonds to the 2,4 diaminopyrimidine moiety at the other locations.
Figure 3.

The relative distances used in the LcDHFR (left) and hDHFR (right) calculation, with residue numbering as per 3DFR (LcDHFR) and 1KMV (hDHFR).
The relative difference calculation uses a reference structure that has been experimentally determined with the ligand docked into the conserved core to demonstrate optimal distances. In the case of MD ensembles based on crystal or NMR solution structures, the reference structure is the original structure. The calculation assigns a score to each of the indicated measures based on the similarity of the same measure in the reference structure. Each score is then averaged to provide each ensemble member with a single ‘relative difference score’.
For homology models, the original scaffold structure is used as the reference structure. A DHFR homology model should preserve the critical conserved geometries. If the resulting LcDHFR homology model has a low relative difference to the conserved core of the scaffold model, it should also be similar to the conserved core of 3DFR. In fact, there is a strong correlation (R2=0.94) between the relative difference of the homology models to 3DFR and the relative difference of the homology models to the scaffold model. The relationship between the scaffolding structure and resulting homology model at the conserved core can be used to identify homology models that do not maintain the conserved geometry, and therefore are not likely to reflect the actual structure.
The resulting relative difference scores are sorted and graphically analyzed for clustering patterns, obvious groups of more than two that occur because the conformations cluster in locations of least steric strain (see Figure 4 for examples). Several patterns can emerge in the graphed scores: clear clustering (Fig. 4a), a ‘rocky slope’ with small groups of distinct clusters (Fig. 4b) or no clustering (Fig. 4c and 4d). For the first two types of patterns, the ensemble members in the cluster with the least relative difference are separated and ligands are docked to the pruned ensemble. A comparison of the docking and ranking metrics from the pruned and full ensembles shows that the pruned ensemble returns metrics equivalent to or better than the full set (Tables I-III). These scores for the pruned ensembles led us to the conclusion that the relative difference calculation is the primary descriptor that allows selection of contributive ensemble members prior to docking.
Figure 4.

A comparison of clustering patterns using a relative difference calculation on an ensemble set. A) Geno3D 1ZDR Wholemin shows a clear clustering pattern, b) the Multiple Template calculation shows small regions of defined clusters (RS is very low scoring, and then an obvious cluster of three is seen). The data points corresponding to selected relative difference clusters are shown as open circles. C) The 1LUD_av 3.5Å radius ensemble calculation shows no apparent clustering, with a steady increase across the ensemble. D) Swiss 1ZDR also shows no clustering. RS is low scoring, followed by a smooth gradient of all other ensemble members.
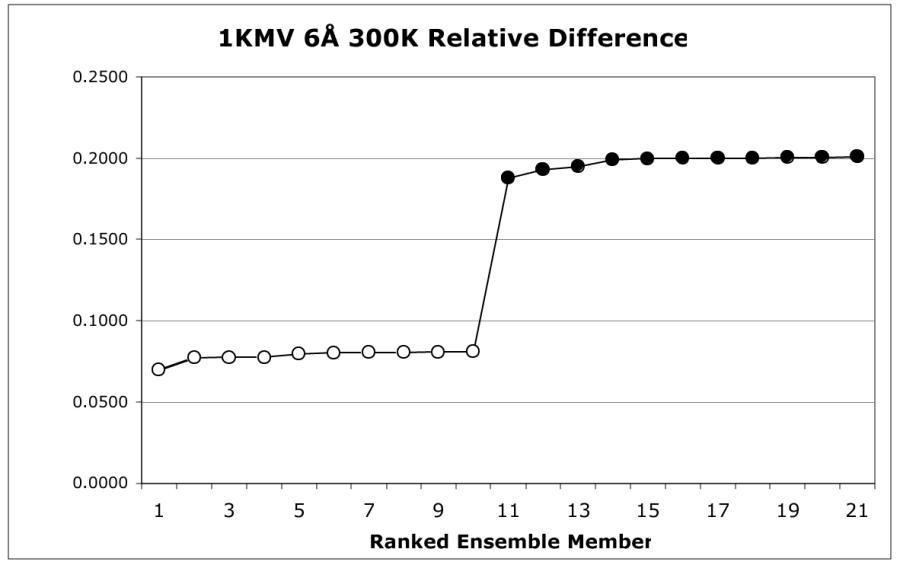
A detailed example using 1KMV 6 Å 300 K illustrates both the process and the interpretation of results. First, using MD, an ensemble is created based on 1KMV as the reference structure with a 6 Å sphere defining the active site at 300 K. Relative differences are calculated for the conserved core using the ensemble members and 1KMV as the reference structure; these differences are then sorted and result in a clear clustering pattern as shown in Table IV.
Table IV.
The relative difference scores and clustering of 1KMV 6Å 300K. Relative difference scores are shown on the left sorted ascendingly, with those in the lower cluster highlighted.
|
Ensemble Member |
Relative Difference |
|
|||||
| 1KMV_6_RO | 0.0698 | ||||||
| 1KMV_6_R8500 | 0.0772 | ||||||
| 1KMV_6_R5500 | 0.0776 | ||||||
| 1KMV_6_R500 | 0.0777 | ||||||
| 1KMV_6_R8000 | 0.0796 | ||||||
| 1KMV_6_R7000 | 0.0804 | ||||||
| 1KMV_6_R7500 | 0.0804 | ||||||
| 1KMV_6_R9000 | 0.0806 | ||||||
| 1KMV_6_R6500 | 0.0807 | ||||||
| 1KMV_6_R5000 | 0.0811 | ||||||
| 1KMV_6_R3000 | 0.1878 | ||||||
| 1KMV_6_R4500 | 0.1930 | ||||||
| 1KMV_6_R6000 | 0.1950 | ||||||
| 1KMV_6_R2000 | 0.1990 | ||||||
| 1KMV_6_R2500 | 0.1995 | ||||||
| 1KMV_6_R3500 | 0.1998 | ||||||
| 1KMV_6_R10000 | 0.1998 | ||||||
| 1KMV_6_R1500 | 0.1998 | ||||||
| 1KMV_6_R1000 | 0.2005 | ||||||
| 1KMV_6_R4000 | 0.2006 | ||||||
| 1KMV_6_R9500 |
0.2009 |
||||||
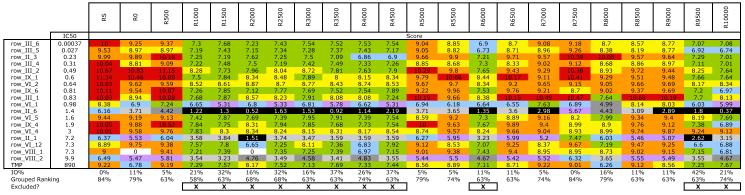
The complete docking score matrix for the ensemble is shown in Table V. The excluded members, belonging to the cluster with substantially different conserved core orientations and therefore, higher relative difference scores, are indicated with an X in the last row of the table. A quick visual inspection shows that the excluded ensemble members are those with much lower docking scores, as indicated by the collection of cool colors. These members are also generally those with higher improper orientation rates and most also have lower grouped ranking scores. However, there is no statistical correlation between relative difference and improper orientation rate (R2 = 0.601) or grouped ranking score (R2 = 0.205). In this example, there is a correlation between maximum docking score and relative difference (R2 = 0.907), but this is not consistent across all ensembles created during this study. While several of the excluded members have high grouped ranking scores, they also have high improper orientation rates.
Table V.
1KMV 6Å 300K ensemble docking scores and grouped ranking scores. X indicates ensemble members that are excluded based on relative difference clustering. Docking scores are colored in a heat map scheme, moving from warm (red, 10+; orange, 9; yellow, 8) to cool (green, 7; blue, 6; purple, 5) to monochrome (dark grey, 4; light grey, 3; black, <2).
|
Pruning the ensemble to select members in the best-scoring cluster results in improved docking and ranking metrics relative to the full ensemble (Table II). The grouped ranking scores are roughly equivalent for the small number of ligands used in the hDHFR set, but the improper orientation substantially decreases with the relative difference set, from 16.7 % to 7.2 %, showing that more compounds maintained the conserved orientation in the active site. The docking score also increases by greater than one point (an increase of 11 %), indicating increased interactions with the receptor. More importantly, the ensemble set has been decreased by half: from 22 members to 11 members. This cuts the processing time in half and means that more ligands can be investigated, with more accurate results, in the same amount of time.
As shown in Tables I-III, there is a consistent trend in the application of relative difference, especially for ensembles based on solution structures and homology models. If the graphed relative difference scores cluster, the resulting best-scoring cluster always improves some combination of docking, ranking or orientation metrics (usually all three) while substantially reducing the number of ensemble members. If there is a rocky slope, or small groups of clusters, then the resulting metrics may be equivalent or improved. In these cases, however, there is still a substantial reduction in the number of ensemble members and therefore a significant improvement in computational time over the original ensemble. If the calculation results in no clustering pattern, then no ensemble members are better than others, they cannot be judged a priori and the ensemble cannot be pruned. In the case of a non-clustering smooth gradient, selection of the lower-scoring members may actually decrease the docking score metrics.
It should be noted in the reported scores for Geno 1ZDR Wholemin 50K and Multiple-template 3.5Å 50K in Figure 4a and 4b and Table VI that the members in the low-scoring cluster are not sequential, nor are they necessarily found within one region of the MD simulation. This indicates that as conformational space is explored across the time course that the residues move in and out of an orientation that conserves the core structure. A superposition of the selected ensemble members shows that conformational changes, both inside and outside the conserved core, are clearly evident.
Table VI.
Relative difference scores for ensemble members belonging to simulations from Geno3D 1ZDR Wholemin 50K and Multiple-template 3.5Å 50K. Selected relative difference cluster members are shown in grey.
| Ensemble Member | Relative Difference | Ensemble Member | Relative Difference | |
|---|---|---|---|---|
| Geno 1ZDR W 50K R7500 | 0.1922 | Multiple-template 3.5Å 50K RS | 0.0958 | |
| Geno 1ZDR W 50K R6500 | 0.1938 | Multiple-template 3.5Å 50K R10000 | 0.1923 | |
| Geno 1ZDR W 50K R9500 | 0.1949 | Multiple-template 3.5Å 50K R9500 | 0.1929 | |
| Geno 1ZDR W 50K R6000 | 0.1953 | Multiple-template 3.5Å 50K R6500 | 0.1929 | |
| Geno 1ZDR W 50K R7000 | 0.1956 | Multiple-template 3.5Å 50K R0 | 0.2003 | |
| Geno 1ZDR W 50K R10000 | 0.1956 | Multiple-template 3.5Å 50K R1500 | 0.2029 | |
| Geno 1ZDR W 50K RS | 0.1980 | Multiple-template 3.5Å 50K R3000 | 0.2029 | |
| Geno 1ZDR W 50K R1500 | 0.3120 | Multiple-template 3.5Å 50K R7500 | 0.2031 | |
| Geno 1ZDR W 50K R8500 | 0.3123 | Multiple-template 3.5Å 50K R6000 | 0.2031 | |
| Geno 1ZDR W 50K R3000 | 0.3126 | Multiple-template 3.5Å 50K R1000 | 0.2033 | |
| Geno 1ZDR W 50K R8000 | 0.3131 | Multiple-template 3.5Å 50K R8500 | 0.2033 | |
| Geno 1ZDR W 50K R5000 | 0.3132 | Multiple-template 3.5Å 50K R2500 | 0.2035 | |
| Geno 1ZDR W 50K R2000 | 0.3137 | Multiple-template 3.5Å 50K R2000 | 0.2040 | |
| Geno 1ZDR W 50K R9000 | 0.3141 | Multiple-template 3.5Å 50K R9000 | 0.2041 | |
| Geno 1ZDR W 50K R2500 | 0.3143 | Multiple-template 3.5Å 50K R5500 | 0.2042 | |
| Geno 1ZDR W 50K R0 | 0.3152 | Multiple-template 3.5Å 50K R8000 | 0.2044 | |
| Geno 1ZDR W 50K R5500 | 0.3153 | Multiple-template 3.5Å 50K R7000 | 0.2051 | |
| Geno 1ZDR W 50K R3500 | 0.3158 | Multiple-template 3.5Å 50K R4500 | 0.2051 | |
| Geno 1ZDR W 50K R500 | 0.3165 | Multiple-template 3.5Å 50K R5000 | 0.2054 | |
| Geno 1ZDR W 50K R4500 | 0.3169 | Multiple-template 3.5Å 50K R3500 | 0.2055 | |
| Geno 1ZDR W 50K R1000 | 0.3169 | Multiple-template 3.5Å 50K R4000 | 0.2058 | |
| Geno 1ZDR W 50K R4000 | 0.3176 | Multiple-template 3.5Å 50K R500 | 0.2147 | |
To investigate whether clustering improved with a larger number of possible ensemble members, MD simulations over 50,000 fs were performed (resulting in 100-member ensembles) using the same constraints of the 10,000 fs simulation and the homology models that showed clustering. The clustering patterns in the relative difference calculations were the same, often with simply more members per cluster, but no new low-scoring clusters emerged that would offer better ligand placement. For example, in Geno 1ZDR Wholemin the same low-scoring relative difference cluster was found in the 50,000 fs run with 35 members as the 10,000 fs run with seven members. As the 35 members do not offer a computational time savings, the 10,000 fs run is sufficient for exploring the space, increasing the docking and ranking metrics and reducing the computational time. A longer time course in the systems investigated here with constrained motion is not likely to explore radically different conformations. This is not surprising as other studies exploring steadily increasing time courses have found no substantial improvement with additional snapshots or a consistent trend with improved metrics.11
Additionally, despite the fact that different MD simulations with the same parameters can generate different conformations across a time course, the resulting relative difference clustering patterns are consistent. Models that show a clustering pattern in the initial simulation show a similar clustering pattern with subsequent simulations. Conversely, if models do not show a clustering pattern with an initial simulation, they typically do not show a clustering pattern within four simulations.
Generally speaking, excluded ensemble members will have lower docking scores, higher improper orientation rates and lower grouped ranking scores or some combination of the three. The trend for the change in metric is not consistent due to the contributive effect of each ensemble member. The grouped ranking score of an ensemble is not an average of the individual members' score, but the grouped ranking score of the averaged docking scores. The final improper orientation rate is the total percentage across the ensemble, not the average. By excluding sets based on the underlying cause for some combination of poor metrics, rather than an individual metric itself, the resulting pruned ensembles are generally improved all around.
While our study focused on species of DHFR, there are no unique structural features of the ligand set or receptor that would make the methodology specific to only DHFR. The ensemble reduction method presented here would be useful with other families of receptors that possess either a conserved core within the active site and/or whose ligands include a conserved moiety that is always placed in the same active site location. For example, HIV-1 protease presents a highly conserved region of ligand-binding residues in the active site56, as demonstrated in the overlay of several crystal structures in Figure 5.
Figure 5.
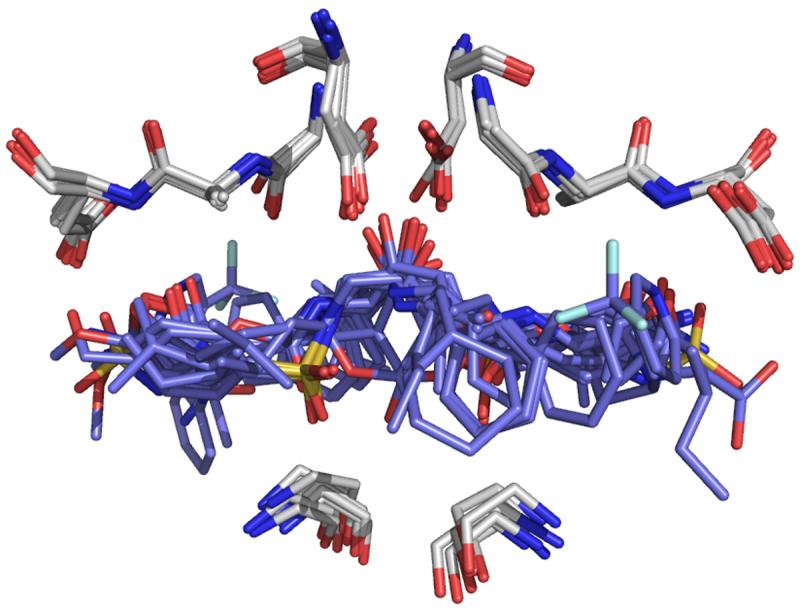
The conserved core of HIV-1 protease. HIV-1 protease is shown with crystal structures and varying ligands (PDB IDs 1T7I57, 2AOC58, 2FGV59, 2HB360, 2HS161, 2I4U62, 2O4L63, 3B7V64) overlaid with the highly conserved residues Asp 25, Gly 27, Ala 28, Glu 29, and Gly 49 shown in white and ligands superposed in blue.
Conclusion
In our previous work we established that the use of an ensemble of structures of the target receptor can substantially improve the docking and ranking metrics of a non-crystallographic structure. In this study we have explored other parameters of the MD simulation to determine that varying the temperature and definition of the active site make only minor further improvements to the ensemble approach. Since the contribution of the individual ensemble members vary, we sought a method to select those with a positive contributive effect a priori.
We have determined a method that can select positively contributing ensemble members prior to any docking experiments providing that the system has a conserved core. This method uses a relative difference calculation based on the preservation of specific interatomic distances of conserved residues that are critical for binding of a conserved ligand moiety. Using distances as opposed to Cartesian coordinates allows the residues to move and explore other possible conformations while preserving the critical distances. Maintenance of the conserved core, as measured by relative differences, has proven to be a reliable predictive measure for pruning ensembles and increasing docking metrics such as correct ligand orientation, grouped ligand ranking, and docking score. The relative difference approach can substantially reduce the size of the ensemble, typically by at least 50 % and up to 75 %. This reduction in ensemble size makes high throughput analysis of lead analogs via an ensemble approach a very reasonable and reliable option for drug development.
Supplementary Material
Acknowledgments
The authors thank Jeffrey A. Meunier for his invaluable assistance in generation of the in-house shell script that was used during the Surflex-Dock process as well as NIH (GM067542) and NSF (0133468) for funding.
Footnotes
References
- 1.Bolstad ES, Anderson A. In pursuit of virtual lead optimization: The role of the receptor structure and ensembles in accurate docking. Proteins. 2008 doi: 10.1002/prot.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol. 2002;6(4):447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- 3.Carlson HA, Masukawa KM, Rubins K, Bushman FD, Jorgensen WL, Lins RD, Briggs JM, McCammon JA. Developing a dynamic pharmacophore model for HIV-1 integrase. J Med Chem. 2000;43(11):2100–2114. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 4.Popov VM, Yee WA, Anderson AC. Towards in silico lead optimization: scores from ensembles of protein/ligand conformations reliably correlate with biological activity. Proteins. 2007;66(2):375–387. doi: 10.1002/prot.21201. [DOI] [PubMed] [Google Scholar]
- 5.Rao S, Sanschagrin PC, Greenwood JR, Repasky MP, Sherman W, Farid R. Improving database enrichment through ensemble docking. J Comput Aided Mol Des. 2008 doi: 10.1007/s10822-008-9182-y. [DOI] [PubMed] [Google Scholar]
- 6.Kua J, Zhang Y, McCammon JA. Studying enzyme binding specificity in acetylcholinesterase using a combined molecular dynamics and multiple docking approach. J Am Chem Soc. 2002;124(28):8260–8267. doi: 10.1021/ja020429l. [DOI] [PubMed] [Google Scholar]
- 7.Amaro RE, Baron R, McCammon JA. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comput Aided Mol Des. 2008 doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowman AL, Nikolovska-Coleska Z, Zhong H, Wang S, Carlson HA. Small molecule inhibitors of the MDM2-p53 interaction discovered by ensemble-based receptor models. J Am Chem Soc. 2007;129(42):12809–12814. doi: 10.1021/ja073687x. [DOI] [PubMed] [Google Scholar]
- 9.Carlson HA, Masukawa KM, McCammon JA. Method for including the dynamic fluctuations of a protein in computer-aided drug design. J Phys Chem A. 1999;103:10213–10219. [Google Scholar]
- 10.Lins RD, Briggs JM, Straatsma TP, Carlson HA, Greenwald J, Choe S, McCammon JA. Molecular dynamics studies on the HIV-1 integrase catalytic domain. Biophys J. 1999;76(6):2999–3011. doi: 10.1016/s0006-3495(99)77453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meagher KL, Carlson HA. Incorporating protein flexibility in structure-based drug discovery: using HIV-1 protease as a test case. J Am Chem Soc. 2004;126(41):13276–13281. doi: 10.1021/ja0469378. [DOI] [PubMed] [Google Scholar]
- 12.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49(2):534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 13.Kavraki L. Protein-Ligand Docking, Including Flexible Receptor-Flexible Ligand Docking, Connexions Web site. 2007 [Google Scholar]
- 14.Lerner MG, Bowman AL, Carlson HA. Incorporating Dynamics in E. coli Dihydrofolate Reductase Enhances Structure-Based Drug Discovery. J Chem Inf Model. 2007 doi: 10.1021/ci700167n. [DOI] [PubMed] [Google Scholar]
- 15.Alonso H, Bliznyuk AA, Gready JE. Combining docking and molecular dynamic simulations in drug design. Med Res Rev. 2006;26(5):531–568. doi: 10.1002/med.20067. [DOI] [PubMed] [Google Scholar]
- 16.Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Freer ST, Rose PW. Complexity and simplicity of ligand-macromolecule interactions: the energy landscape perspective. Curr Opin Struct Biol. 2002;12(2):197–203. doi: 10.1016/s0959-440x(02)00310-x. [DOI] [PubMed] [Google Scholar]
- 17.Berndt KD, Guntert P, Wuthrich K. Conformational sampling by NMR solution structures calculated with the program DIANA evaluated by comparison with long-time molecular dynamics calculations in explicit water. Proteins. 1996;24(3):304–313. doi: 10.1002/(SICI)1097-0134(199603)24:3<304::AID-PROT3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 18.Czerminski R, Elber R. Computational studies of ligand diffusion in globins: I. Leghemoglobin. Proteins. 1991;10(1):70–80. doi: 10.1002/prot.340100107. [DOI] [PubMed] [Google Scholar]
- 19.Murray CW, Baxter CA, Frenkel AD. The sensitivity of the results of molecular docking to induced fit effects: application to thrombin, thermolysin and neuraminidase. J Comput Aided Mol Des. 1999;13(6):547–562. doi: 10.1023/a:1008015827877. [DOI] [PubMed] [Google Scholar]
- 20.Sugita Y, Okamtot Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 21.Knegtel RM, Kuntz ID, Oshiro CM. Molecular docking to ensembles of protein structures. J Mol Biol. 1997;266(2):424–440. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 22.Deng Z, Chuaqui C, Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J Med Chem. 2004;47(2):337–344. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 23.Bowman AL, Lerner MG, Carlson HA. Protein Flexibility and Species Specificity in Structure-Based Drug Discovery: Dihydrofolate Reductase as a Test System. J Am Chem Soc. 2007 doi: 10.1021/ja068256d. [DOI] [PubMed] [Google Scholar]
- 24.Anderson AC. Two crystal structures of dihydrofolate reductase-thymidylate synthase from Cryptosporidium hominis reveal protein-ligand interactions including a structural basis for observed antifolate resistance. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2005;61(Pt 3):258–262. doi: 10.1107/S1744309105002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolin JT, Filman DJ, Matthews DA, Hamlin RC, Kraut J. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate. J Biol Chem. 1982;257(22):13650–13662. [PubMed] [Google Scholar]
- 26.Champness JN, Achari A, Ballantine SP, Bryant PK, Delves CJ, Stammers DK. The structure of Pneumocystis carinii dihydrofolate reductase to 1.9 A resolution. Structure. 1994;2(10):915–924. doi: 10.1016/s0969-2126(94)00093-x. [DOI] [PubMed] [Google Scholar]
- 27.Knighton DR, Kan CC, Howland E, Janson CA, Hostomska Z, Welsh KM, Matthews DA. Structure of and kinetic channelling in bifunctional dihydrofolate reductase-thymidylate synthase. Nat Struct Biol. 1994;1(3):186–194. doi: 10.1038/nsb0394-186. [DOI] [PubMed] [Google Scholar]
- 28.Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, Vanichtanankul J, Sirawaraporn W, Taylor P, Walkinshaw MD, Yuthavong Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol. 2003;10(5):357–365. doi: 10.1038/nsb921. [DOI] [PubMed] [Google Scholar]
- 29.Renner S, Derksen S, Radestock S, Morchen F. Maximum Common Binding Modes (MCBM): Consensus Docking Scoring Using Multiple Ligand Information and Interaction Fingerprints. J Chem Inf Model. 2008;48(2):319–332. doi: 10.1021/ci7003626. [DOI] [PubMed] [Google Scholar]
- 30.Sabo A, Lusic M, Cereseto A, Giacca M. Acetylation of conserved lysines in the catalytic core of cyclin-dependent kinase 9 inhibits kinase activity and regulates transcription. Mol Cell Biol. 2008;28(7):2201–2212. doi: 10.1128/MCB.01557-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee CH, Saksela K, Mirza UA, Chait BT, Kuriyan J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell. 1996;85(6):931–942. doi: 10.1016/s0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- 32.SYBYL 7.3. Tripos Inc; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 33.Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002;18(9):1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 34.Combet C, Jambon M, Deleage G, Geourjon C. Geno3D: automatic comparative molecular modelling of protein. Bioinformatics. 2002;18(1):213–214. doi: 10.1093/bioinformatics/18.1.213. [DOI] [PubMed] [Google Scholar]
- 35.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31(13):3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HS, Damo SM, Lee SY, Wemmer D, Klinman JP. Structure and hydride transfer mechanism of a moderate thermophilic dihydrofolate reductase from Bacillus stearothermophilus and comparison to its mesophilic and hyperthermophilic homologues. Biochemistry. 2005;44(34):11428–11439. doi: 10.1021/bi050630j. [DOI] [PubMed] [Google Scholar]
- 38.Dams T, Auerbach G, Bader G, Jacob U, Ploom T, Huber R, Jaenicke R. The crystal structure of dihydrofolate reductase from Thermotoga maritima: molecular features of thermostability. J Mol Biol. 2000;297(3):659–672. doi: 10.1006/jmbi.2000.3570. [DOI] [PubMed] [Google Scholar]
- 39.Reyes VM, Sawaya MR, Brown KA, Kraut J. Isomorphous crystal structures of Escherichia coli dihydrofolate reductase complexed with folate, 5-deazafolate, and 5,10-dideazatetrahydrofolate: mechanistic implications. Biochemistry. 1995;34(8):2710–2723. doi: 10.1021/bi00008a039. [DOI] [PubMed] [Google Scholar]
- 40.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36(3):586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 41.Bennett B, Langan P, Coates L, Mustyakimov M, Schoenborn B, Howell EE, Dealwis C. Neutron diffraction studies of Escherichia coli dihydrofolate reductase complexed with methotrexate. Proc Natl Acad Sci U S A. 2006;103(49):18493–18498. doi: 10.1073/pnas.0604977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bethesda BlastP. MD: National Center for Biotechnology Information; Bldg 38A, NIH, 8600 Rockville Pike, Bethesda, MD 20894: [Google Scholar]
- 43.Polshakov VI, Smirnov EG, Birdsall B, Kelly G, Feeney J. NMR-based solution structure of the complex of Lactobacillus casei dihydrofolate reductase with trimethoprim and NADPH. J Biomol NMR. 2002;24(1):67–70. doi: 10.1023/a:1020659713373. [DOI] [PubMed] [Google Scholar]
- 44.Kovalevskaya NV, Smurnyy YD, Polshakov VI, Birdsall B, Bradbury AF, Frenkiel T, Feeney J. Solution structure of human dihydrofolate reductase in its complex with trimethoprim and NADPH. J Biomol NMR. 2005;33(1):69–72. doi: 10.1007/s10858-005-1475-z. [DOI] [PubMed] [Google Scholar]
- 45.Tripos Bookshelf 7.3. Tripos Inc.; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 46.Jain AN. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des. 1996;10(5):427–440. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 47.Wilson PA. University of Montana; Missoula: 2004. A Simple Methodology for the Production of Three-Dimensional Models: Serotonin Transporter as an Example; p. 208. [Google Scholar]
- 48.Klon AE, Heroux A, Ross LJ, Pathak V, Johnson CA, Piper JR, Borhani DW. Atomic structures of human dihydrofolate reductase complexed with NADPH and two lipophilic antifolates at 1.09 a and 1.05 a resolution. J Mol Biol. 2002;320(3):677–693. doi: 10.1016/s0022-2836(02)00469-2. [DOI] [PubMed] [Google Scholar]
- 49.Cody V, Galitsky N, Luft JR, Pangborn W, Rosowsky A, Blakley RL. Comparison of two independent crystal structures of human dihydrofolate reductase ternary complexes reduced with nicotinamide adenine dinucleotide phosphate and the very tight-binding inhibitor PT523. Biochemistry. 1997;36(45):13897–13903. doi: 10.1021/bi971711l. [DOI] [PubMed] [Google Scholar]
- 50.Mura C. The average3d PyMOL module. 2005 [Google Scholar]
- 51.Cody V, Galitsky N, Rak D, Luft JR, Pangborn W, Queener SF. Ligand-induced conformational changes in the crystal structures of Pneumocystis carinii dihydrofolate reductase complexes with folate and NADP+ Biochemistry. 1999;38(14):4303–4312. doi: 10.1021/bi982728m. [DOI] [PubMed] [Google Scholar]
- 52.Rajagopalan PT, Lutz S, Benkovic SJ. Coupling interactions of distal residues enhance dihydrofolate reductase catalysis: mutational effects on hydride transfer rates. Biochemistry. 2002;41(42):12618–12628. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- 53.Hansch C, Hathaway BA, Guo ZR, Selassie CD, Dietrich SW, Blaney JM, Langridge R, Volz KW, Kaufman BT. Crystallography, quantitative structure-activity relationships, and molecular graphics in a comparative analysis of the inhibition of dihydrofolate reductase from chicken liver and Lactobacillus casei by 4,6-diamino-1,2-dihydro-2,2-dimethyl-1-(substituted-phenyl)-s-triazine s. J Med Chem. 1984;27(2):129–143. doi: 10.1021/jm00368a006. [DOI] [PubMed] [Google Scholar]
- 54.Hansch C, Li R, Blaney JM, Langridge R. Comparison of the inhibition of Escherichia coli and Lactobacillus casei dihydrofolate reductase by 2,4-diamino-5-(substituted-benzyl)pyrimidines: quantitative structure-activity relationships, X-ray crystallography, and computer graphics in structure-activity analysis. J Med Chem. 1982;25(7):777–784. doi: 10.1021/jm00349a003. [DOI] [PubMed] [Google Scholar]
- 55.Nelson RG, Rosowsky A. Dicyclic and tricyclic diaminopyrimidine derivatives as potent inhibitors of Cryptosporidium parvum dihydrofolate reductase: structure-activity and structure-selectivity correlations. Antimicrob Agents Chemother. 2001;45(12):3293–3303. doi: 10.1128/AAC.45.12.3293-3303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hou T, McLaughlin WA, Wang W. Evaluating the potency of HIV-1 protease drugs to combat resistance. Proteins. 2008;71:1163–1174. doi: 10.1002/prot.21808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Surleraux DLNG, Tahri A, Verschueren WG, Pille GME, de Kock HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- 58.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 angstroms resolution crystal structures of HIV-1 protease mutants with substrate. FEBS J. 2005;272:5265–5277. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foulkes JE, Prabu-Jeyabalan M, Cooper D, Henderson GJ, Harris J, Swanstrom R, Schiffer CA. Role of invariant Thr80 in human immunodeficiency virus type 1 protease structure, function, and viral infectivity. J Virol. 2006;80:6906–6916. doi: 10.1128/JVI.01900-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-Based Design of Novel HIV-1 Protease Inhibitors To Combat Drug Resistance. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 61.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-high Resolution Crystal Structure of HIV-1 Protease Mutant Reveals Two Binding Sites for Clinical Inhibitor TMC114. J Mol Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cihlar T, He GX, Liu X, Chen JM, Hatada M, Swaminathan S, McDermott MJ, Yang ZY, Mulato AS, Chen X, Leavitt SA, Stray KM, Lee WA. Suppression of HIV-1 Protease Inhibitor Resistance by Phosphonate-mediated Solvent Anchoring. J Mol Biol. 2006;363:635–647. doi: 10.1016/j.jmb.2006.07.073. [DOI] [PubMed] [Google Scholar]
- 63.Muzammil S, Armstrong AA, Kang LW, Jakalian A, B PR, Schmelmer V, Amzel LM, Freire E. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. J Virol. 2007;81:5144–5154. doi: 10.1128/JVI.02706-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovalevsky AY, Chumanevich AA, Liu F, Louis JM, Weber IT. Caught in the Act: The 1.5 A Resolution Crystal Structures of the HIV-1 Protease and the I54V Mutant Reveal a Tetrahedral Reaction. Biochemistry. 2007;46:14854–14864. doi: 10.1021/bi700822g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.