

Abstract
Cellular function and adaptive behavior is often driven by signals generated in response to the local tissue microenvironment. Cell surface receptors that detect changes in extracellular matrix composition and modifications to extracellular matrix components, are ideally positioned to provide highly responsive sensors of changes in the microenvironment and mediate changes in cellular function required to maintain tissue integrity. Receptors can act as “on/off” switches, but ligand/receptor complexes that provide “rheostatic” control may be more sensitive, provide a more rapid mechanism of control and allow for fine-tuning of cellular responses to the microenvironment. Herein, we review evidence that transitions in the physiochemical properties of the extracellular glycosaminoglycan hyaluronan and in the function of its major receptor, CD44, differentially regulate ERK and Rac signal transduction pathways to provide critical rheostatic control of mesenchymal cell proliferation.
Introduction
Mesenchymal cells were historically considered a scaffold that maintained the structure of tissues and supported other cell types responsible for tissue function. However, the paradigm has shifted so that mesenchymal cells such as smooth muscle cells, fibroblasts and myofibroblasts are now appreciated for the essential roles they play in virtually every tissue. Our growing appreciation of their contribution to organ function stems from our expanded knowledge of mesenchymal cell biology including their dynamic nature, the diversity of their origins, their phenotypic heterogeneity, their plasticity and their functional capacities. As for a number of other cell types, many of the fascinating features of mesenchymal cells were revealed by comparing their structural, genetic, phenotypic and functional parameters under steady state or homeostatic conditions and under stress conditions imposed by tissue injury or tissue malfunction. It is becoming increasingly evident that mesenchymal cells are endowed with a remarkable capacity to sense changes in the local milieu when tissue integrity is compromised.
Receptors expressed on mesenchymal cells act as sensors that alert the cells to changes in the structure and composition of their environment including the surrounding extracellular matrix (ECM). Interestingly, this cadre of sensors overlaps significantly with those expressed by innate immune cells. On innate immune cells, these receptors, such as Toll-like receptors (TLRs), scavenger receptors, and others, sense exogenous threats against host integrity by recognizing pathogens and pathogen-derived molecules that bind and mediate the signals that activate phagocytosis and inflammatory host defense mechanisms [1]. However, recent studies have revealed that these same receptors also bind to endogenous (or “self”) components modified as the result of oxidative stress, inflammation, injury and tissue malfunction, apoptosis or necrosis [1], thus expanding the role of innate immune cells to protection against internal as well as external threats to host integrity. By analogy, a primary function of mesenchymal cells may be to serve as sensors of tissue integrity and as first responders to disruption of tissue integrity due to injury or organ malfunction. Consistent with this proposed function, it has become clear that mesenchymal cells are critical not only to matrix remodeling but are also active participants in the inflammatory response through, for example, the expression of adhesion molecules, cytokines, chemokines, lipid mediators, and reactive oxygen (ROS) and reactive nitrogen (RNS) species.
Here, we review the evidence that the cell surface receptor CD44 on mesenchymal cells acts as an important sensor of changes in the microenvironment by differentially regulating cytoskeletal dynamics and signal transduction in response to native and modified forms of its major ligand, the ECM glycosaminoglycan hyaluronan (HA). We describe work indicating that the functions of HA and CD44 change in normal homeostatic versus inflammatory conditions, and that this change in function allows HA and CD44 to act as a biological rheostat. The rheostatic control through CD44-HA interactions discussed here also illustrates the intimate relationship between inflammation and the function of mesenchymal cells in biological processes ranging from wound healing to cardiovascular disease.
Hyaluronan and CD44
The ECM is comprised of matrix proteins (such as collagen, fibronectin and vitronectin) and several non-proteinaceous components. Hyaluronan is a glycosaminoglycan (GAG) that is one of the major non-proteinaceous ECM components. It is distinct from other GAGs in that it is non-sulfated and relatively simple non-branching polymer of repeating dissacharides of D-glucuronic acid and D-N-acetylglucosamine linked via alternating β-1,4 and β-1,3 glycosidic bonds. HA is especially enriched in pericellular matrices surrounding migrating and proliferating cells during embryonic development, tissue repair, inflammation and tumorigenesis [2]. HA influences proliferation, migration and adhesion of cells via interactions at the cell surface where it binds to receptors such as CD44, RHAMM and layilin [3-6], or remains associated with the cell surface as the result of retention of newly synthesized HA by HA synthase, of which there are three isoform, HAS-1, HAS-2 and HAS-3. The most widely-distributed form of HA in normal tissues is a high-molecular weight extra- and peri-cellular polysaccharide, usually of several million Daltons (HMW-HA or native HA). HMW-HA forms a highly viscous network important for molecular exclusion, flow resistance, tissue osmosis, lubrication and hydration. The structure of ECM also depends on the interactions of HA with other extracellular macromolecules referred to as HA-binding proteins or hyaladherins, such as versican, aggrecan and others. Importantly, at sites of inflammation, there is an accumulation of lower molecular weight forms of HA (LMW-HA) that are synthesized de novo or generated by enzymatic degradation through the activity of hyaluronidases and oxidative hydrolysis of HMW-HA [7]. The HMW- and LMW- forms of HA have different physiochemical properties and may differ in their interactions with hyaladherins. In addition, the receptor-mediated effects of HMW-HA and LMW-HA on cell behavior are clearly distinct. Most importantly, LMW-HA has multiple proinflammatory effects not observed for HMW-HA. In fact, HMW-HA can block the proinflammatory effects of LMW-HA [8]. As reviewed herein, the differences in biologic activity of HMW- and LMW-HA are due to differential receptor-mediated signaling.
CD44 is a type I transmembrane glycoprotein encoded by a single gene and expressed as multiple isoforms. The structural diversity of CD44 is generated by alternative RNA splicing as well as differential posttranslational modifications including glycosylation and the attachment of glycosaminoglycans [9-11]. The HA-binding domain is present in all isoforms of CD44. The gene for CD44 contains 21 exons of which 7 encode the extracellular domain of the most common form of CD44, referred to as the “standard” or “hematopoietic” form. The multitude of variant CD44 isoforms are generated by the insertion of up to 11 additional alternatively spliced exons (V1-V11) into a single site within the membrane-proximal portion of the extracellular domain [12].
CD44 is expressed by virtually all cells of neurectodermal origin. Under normal conditions the majority of primary cells, except epithelial cells which express CD44E, express the standard form of CD44, though cellular activation leads to expression of additional variant forms in many cell types. Malignant derivatives of many cell types express variant isoforms although recent evidence indicates that expression of CD44 is repressed with progression to metastatic disease [13]. Interestingly, many primary cells, for example leukcocytes, do not bind to HA (in spite of expressing ample amounts of CD44) until activated, while CD44 on other cells, such as fibroblasts and smooth muscle cells, may be constitutively active as is the case on most primary tumor cells. The transition to high affinity HA binding has been associated with increased expression and receptor clustering of the standard form of CD44 or induction of CD44v expression, although neither is required for HA binding, and with changes in posttranslational modifications including phosphorylation, sulfation and glycosylation of the receptor [14-20]. Thus, regulation of the CD44 binding affinity is one mechanism by which CD44/HA interactions are controlled.
The function of CD44 depends on its cellular context. Some of the best documented functions of CD44 are its role in leukocyte homing to sites of inflammation and its role in HA turnover by mesenchymal cells. CD44 can also regulate cell proliferation and apoptosis in different contexts and in a variety of cell types. Structural variations in CD44 might provide a mechanism for modulating CD44 function: alternatively spliced variants or post-translationally modified forms preferentially serve as docking/binding sites for MMPs and ECM proteins [21-24]. It is not yet clear whether differential splicing of CD44 results in modification of receptor signaling.
We previously showed that the predominant form of CD44 expressed in vascular smooth muscle cells (VSMCs) is the standard form of CD44 that includes none of the variant exons [25]. Hyaluronan binds to CD44 and, as discussed below, can be coupled to the actin cytoskeleton to mediate signal transduction. In addition to direct signaling, oligomeric degradation products of HA transduce their inflammatory signal through Toll-like receptor 2 (TLR2), TLR4, or both, in innate immune cells (macrophages and dendritic cells) [26,27]. HA binding to CD44 may also modulate growth factor receptor signaling [28,29]. Although investigating the higher order interactions involved in initiating or modulating signals provoked by HA binding to CD44 is a priority, we focus here on the downstream signaling induced in mesenchymal cells by CD44-HA interactions independent of these added complexities.
Proliferation-associated effects of HA and CD44 on the actin cytoskeleton and Rho family GTPases
The signaling pathways coupled to CD44 are not fully defined, but it is well established that CD44 provides an important connection between the plasma membrane and the cytoskeleton and that reorganization of the cytoskeleton regulates both the ligand binding capacity of CD44 and CD44-mediated signal transduction. A linchpin of this connection appears to be the interaction between CD44, ERM (ezrin/radixin/moesin) proteins and merlin [30,31]. CD44 is also reported to bind to ankyrin in some cell types [32]. ERM proteins connect the cytoskeleton to the plasma membrane by binding to actin and several transmembrane proteins. At least in some cell types, the predominant species co-precipitated with ERM proteins is CD44 [33].
ERM proteins exist in a dormant conformation that requires an activating phosphorylation at T558 (based on the moesin sequence). This activating phosphorylation can be catalyzed by growth factor receptors, PKC, or other kinases. Activated ezrin monomers or head-to-tail oligomers associate directly with F-actin through a domain in its C-terminus, and with the membrane through its N-terminal domain [33]. The association of ezrin with transmembrane proteins can be direct, as in the case of CD44 (via a motif rich in basic amino acid residues in the juxtamembrane region of CD44 [34]) or indirect through EBP50 [31].
Interestingly, the ERM-CD44 complex can affect signaling by Rho family GTPases. Rho GDP dissociation inhibitor (Rho GDI) directly interacts with activated ERM proteins, resulting in enhanced Rho activity [31]. In turn, Rho activity has been closely linked to organization of the f-actin cytoskeleton [35,36]. The ERM-mediated interaction of CD44 with the actin cytoskeleton and its stimulation of Rho activity can have important implications for integrin signaling because Rho activity and an organized actin cytoskeleton is important for ligand-induced integrin clustering and focal adhesion formation [37]. Disruption of the actin cytoskeleton inhibits the integrin-dependent autophosphorylation of focal adhesion kinase (FAK) at Y397, and consequently, the recruitment of Src to focal adhesion complexes [38,39]. Thus, both integrin and Src signaling can be affected by changes in CD44-dependent organization of the actin cytoskeleton. Hyaluronan and CD44 also regulate Rac activity. We recently showed that the HMW form of HA efficiently inhibits Rac GTP-loading in VSMCs while lower molecular weight forms of HA synergize with mitogens to stimulate Rac GTP-loading [40]. In addition to CD44, Rho and Rac activities are regulated by growth factor receptors and integrins. Recent studies have begun to identify synergistic effects of HA-CD44 and the PDGF receptor [41,28]. There has been continuing speculation about crosstalk between CD44 and integrins, though this has yet to be directly demonstrated.
The effects of CD44 on Rho and Rac activity have important implications for the regulation of cell proliferation. Rho-dependent organization of the actin cytoskeleton is associated with the sustained activation of ERK, and sustained ERK activity has been closely linked to the induction of cyclin D1 mRNA [42]. Rac signaling is also required for cyclin D1 gene expression [43] and the translation of cyclin D1 mRNA, at least in endothelial cells [44]. Thus, several pro-mitogenic signaling pathways, especially those targeting cyclin D1 (viz., ERK, Rho and Rac) can be regulated by CD44. Understanding the receptor proximal signaling pathways by which HA binding to CD44 coordinately regulates these pathways and cellular proliferation are major issues for future study.
Regulation of CD44 signaling by merlin phosphorylation
The ERM-related protein, merlin (also called NF2), interacts with CD44 in a phosphorylation-dependent manner (Figure 1). The binding of hypo-phosphorylated merlin to CD44 is growth inhibitory, presumably because merlin binding to CD44 displaces associated ERM proteins [45]. Since merlin lacks the F-actin binding motif found in ERM proteins [30,31], its binding to CD44 disrupts the linkage between CD44 and the cortical actin cytoskeleton. The CD44-merlin complex antagonizes Ras-dependent activation of ERK as well as the GTP-loading of Rac [46, 47] (Figure 1). Hyper-phosphorylation of CD44-associated merlin leads to recruitment of activated ERM proteins to the complex to form a growth permissive signaling complex [45]. The details of merlin phosphorylation and dephosphorylation are not fully understood, but PAK can phosphorylate merlin at S518 [48], and merlin dephosphorylation may be catalyzed by a CD44-recruited phosphatase [47].
Figure 1.
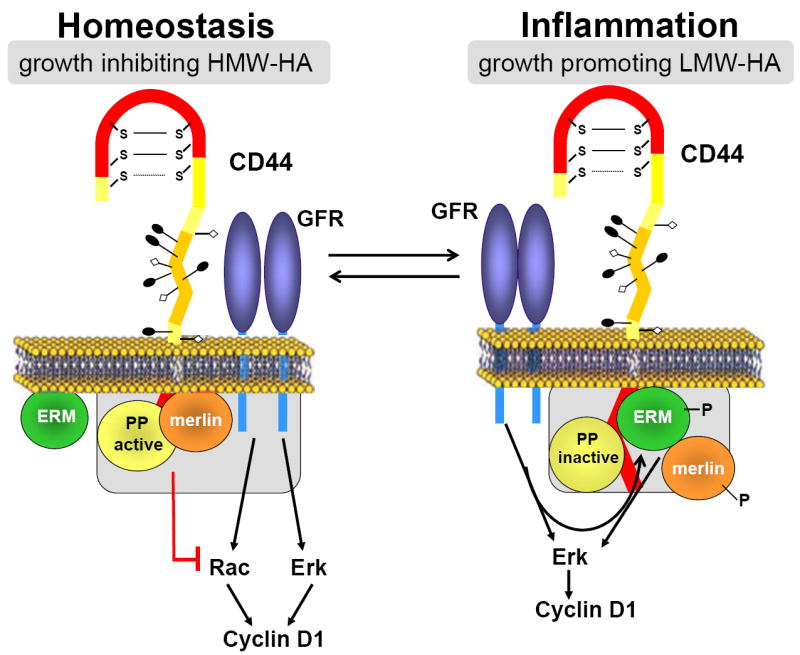
Working model showing how inflammation-associated changes in the molecular weight of HA may regulate the phosphorylation of merlin, the association of ERM proteins, and the ERK and Rac signaling pathways to cyclin D1, leading to rheostatic control of cell proliferation.
Regulation of CD44 signaling by modulating the size of hyaluronan
A particularly intriguing way of regulating CD44 signaling is by controlling the size of its major ligand, hyaluronan (see above). In primary VSMCs and fibroblasts, the binding of HMW-HA to CD44 inhibits S phase entry in response to a strong mitogenic stimulus whereas the binding of LMW-HA to CD44 stimulates progressions phase entry in response to a weak mitogenic stimulus [49]. Moreover, as determined using both CD44-null cells and neutralizing antibodies, the opposing effects of HMW- and LWM-HA on mitogenesis both require CD44. Since inflammation is an important determinant of the size of hyaluronan, the bifunctionality of CD44 has implications for several diseases (see below).
Our most recent studies show that the opposing cell cycle effects of HMW-HA and LMW-HA result from differential regulation of CD44-regulated signaling pathways to cyclin D1 [40,50]. HMW-HA binding to CD44 selectively inhibits the GTP-loading of Rac and Rac-dependent signaling to the cyclin D1 gene while LMW-HA binding to CD44 selectively stimulates ERK activation and ERK-dependent cyclin D1 gene expression (Figure 1). These results describe a novel mechanism of growth control in which a ligand-receptor system generates opposing effects on mitogenesis by differentially regulating signaling pathways to a common cell cycle target. They also emphasize how a seemingly subtle change in matrix composition, viz. the size of HA, can result in diametrically opposed effects on cell cycle progression and proliferation.
A working model integrating the effects of CD44 on cytoskeletal dynamics and cell proliferation
As summarized above, ERM proteins bind to CD44 and, at least in part, mediate the effects of CD44 on organization of the actin cytoskeleton [31,30]. Hypo-phosphorylated merlin binds to CD44 to displace ERM proteins from CD44, and this change in binding partners is thought to mediate growth inhibition in response to HA (Figure 1). Although initial studies focused on the ability of CD44-bound merlin to inhibit Ras signaling to ERK [45]), merlin also inhibits Rac activation and signaling to PAK [47,51,52]. PAK has been reported to regulate cyclin D1 gene expression [53-55], and ectopic expression of merlin can inhibit the induction of cyclin D1 [56]. Thus, the phosphorylation-dependent interaction of merlin with CD44 may explain the anti-mitogenic effects of CD44 in VSMCs treated with HMW-HA (Figure 1). ERM binding to CD44 can be restored by hyper-phosphorylation of merlin at S518, and this event may be involved in the pro-mitogenic effect of LMW-HA (Figure 1).
Thus, based on the work of Morrison et al. [45], we hypothesize that hypo-phosphorylated merlin is associated with CD44 in serum-starved VSMCs and that LMW-HA binding to CD44 leads to hyper-phosphorylation of merlin and the association of ERM proteins with CD44. Based on the model in Figure 1, we predict that the addition of ERM to the complex would eliminate the inhibitory effect of CD44-associated merlin on Rac activation. Moreover, the consequent binding of ERM proteins to CD44 may be required for ERK activation [45-47]. Together, the ability to activate Rac and ERK would lead to optimal mitogenic signaling. It is easy to envision how interactions between CD44 and RTKs or integrins could also contribute to ERK activity in this pro-mitogenic environment.
With regard to the anti-mitogenic effect of HMW-HA, we hypothesize that HMW-HA could antagonize mitogen-dependent cyclin D1 expression by stimulating merlin dephosphorylation (presumably through protein phosphatase (PP) recruitment [45], thereby enforcing merlin association with CD44, dissociation of ERM proteins and the growth-inhibitory state (Figure 1). We have not detected an inhibitory effect of HMW-HA on ERK activity, but HMW-HA inhibits FBS-stimulated Rac-activation, and Rac signaling regulates cyclin D1 mRNA levels in VSMCs. In these ways, the CD44-merlin complex may serve as a molecular switch that controls proliferation in conjunction with inflammation-associated changes in the size of HA.
CD44 as a rheostat for cell proliferation
Under homeostatic conditions, HA exists in its native, high molecular weight form (Figure 2), but lower molecular weights forms can arise from the action of specific hyaluronidases or from oxidative degradation (Figure 2). The degradation of HMW-HA to LMW-HA occurs at sites of inflammation (as seen in atherosclerosis, rheumatoid arthritis and tumorigenesis), and we propose that this dynamic balance allows HA and CD44 to act as a proliferation rheostat (Figure 2). First, decreases in the levels of HMW-HA would reduce the anti-mitogenic effect of CD44. Second, the consequent increase in the levels of LMW-HA would enhance the pro-mitogenic effect of CD44. The rheostat would work, at least in part, by differentially regulating the ERK and Rac signaling pathways to cyclin D1 as described above. Interestingly, ERK and Rac also regulate cell migration, and deletion of CD44 has profound affects on actin stress fiber formation and directional migration [57]. Thus, differential regulation of ERK and Rac signaling by CD44 may affect cellular functions well beyond proliferation.
Figure 2.
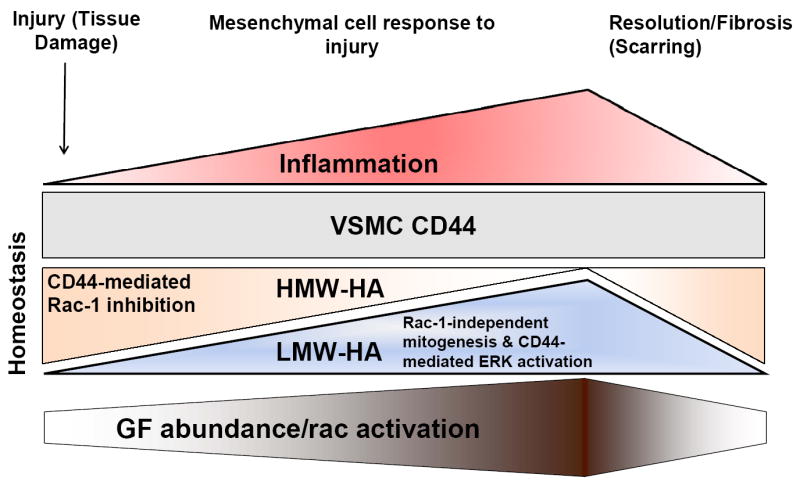
Working model of the interplay between inflammation, HA, CD44 and mitogenic signaling in response to tissue damage or injury.
Projected biological relevance of a CD44 rheostat
Under homeostatic conditions, parenchymal cells reside in tissues in a relatively quiescent state, dividing only as required to compensate for normal cellular turnover. Quiescence is often thought of as a default state that reflects the absence of pro-mitogenic signals required to induce proliferation. We have proposed, instead, that quiescence of VSMCs requires active signaling by negative regulators of cell cycle progression. Key among these is the HMW-form of HA that is present under homeostatic conditions and can counter-act the mitogenic effects of serum-derived growth factors. Moreover, in association with vascular injury or malfunction, the ensuing inflammatory response results in a shift in the prevalence of HMW-HA towards LMW-HA that, in contrast, provides pro-mitogenic signals to VSMCs. The simultaneous loss of HA-mediated anti-mitogenic signals and induction of HA-mediated pro-mitogenic signals provides for rheostatic control that should be more sensitive and efficient than would be expected of an “on/off switch” type of growth control. As recruitment of mesenchymal cell progenitors and migration of resident mesenchymal cells also contribute to the expansion of local stromal cell content following tissue injury, it is interesting to note that the HMW- and LMW-forms of HA have been reported to differentially affect VSMC migration which may amplify the differential impact of HMW- and LMW-HA on the response to tissue injury. It will be of considerable interest to determine the degree to which these regulatory mechanisms are conserved in other mesenchymal-derived stromal cells, such as fibroblasts which undergo activation in response to injury during wound healing, fibrosis and cancer. If this is the case, then targeting the signaling mechanisms that shift the balance of CD44-HA mediated pro- and anti-mitogenic effects may provide a means to regulate the extent and duration of mesenchymal cell proliferation following injury, thereby optimizing tissue repair and minimizing fibrosis or scarring in a variety of pathophysiologic settings.
Acknowledgments
Supported by grants from the NIH, the Pennsylvania Department of Health and the Ludwig Institute for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Curtiss LK, Tobias PS. The Toll of Toll-Like Receptors, Especially Toll-Like Receptor 2, on Murine Atherosclerosis. Current Drug Targets. 2008;8:1230–1238. doi: 10.2174/138945007783220605. [DOI] [PubMed] [Google Scholar]
- 2.Fraser JR, Laurent TC, Laurent UB. Hyaluronan: its nature, distribution, functions and turnover. J Intern Med. 1997;242:27–33. doi: 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- 3.Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 4.Culty M, Miyake K, Kincade PW, Sikorski E, Butcher EC, Underhill C, Silorski E. The hyaluronate receptor is a member of the CD44 (H-CAM) family of cell surface glycoproteins. J Cell Biol. 1990;111:2765–2774. doi: 10.1083/jcb.111.6.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardwick C, Hoare K, Owens R, Hohn HP, Hook M, Moore D, Cripps V, Austen L, Nance DM, Turley EA. Molecular cloning of a novel hyaluronan receptor that mediates tumor cell motility. J Cell Biol. 1992;117:1343–1350. doi: 10.1083/jcb.117.6.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bono P, Rubin K, Higgins JM, Hynes RO. Layilin, a novel integral membrane protein, is a hyaluronan receptor. Mol Biol Cell. 2001;12:891–900. doi: 10.1091/mbc.12.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawkins CL, Davies MJ. Degradation of hyaluronic acid, poly-and monosaccharides, and model compounds by hypochlorite: evidence for radical intermediates and fragmentation. Free Rad Biol Med. 1998;24:1396–1410. doi: 10.1016/s0891-5849(98)00009-4. [DOI] [PubMed] [Google Scholar]
- 8.Hodge-Dufour J, Marino MW, Horton MR, Burdick MD, Strieter RM, Noble PW, Hunter CA, Puré E. Inhibition of IFN-gamma induced IL-12 production: a potential mechanism for the anti-inflammatory activities of TNF. Proc Natl Acad Sci. 1998;95:13806–13811. doi: 10.1073/pnas.95.23.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenfield B, Wang W-C, Marquardt H, Piepkorn M, Wolff EA, Aruffo A, Bennett KL. Characterization of the heparan sulfate and chondroitin sulfate assembly sites in CD44. J Biol Chem. 1999;274:2511–2517. doi: 10.1074/jbc.274.4.2511. [DOI] [PubMed] [Google Scholar]
- 10.Brown TA, Bouchard T, St John T, Wayner E, Carter WG. Human keratinocytes express a new CD44 core protein (CD44E) as a heparan-sulfate intrinsic membrane proteoglycan with additional exons. J Cell Biol. 1991;113:207–221. doi: 10.1083/jcb.113.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stamenkovic I, Aruffo A, Amiot M, Seed B. The hematopoietic and epithelial forms of CD44 are distinct polypeptides with different adhesion potentials for hyaluronate-bearing cells. Embo J. 1991;10:343–348. doi: 10.1002/j.1460-2075.1991.tb07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241–319. doi: 10.1016/s0065-230x(08)60101-3. [DOI] [PubMed] [Google Scholar]
- 13.Huang Q, Gumireddy K, Schrier M, le Sage C, Nagel R, Nair S, Egan DA, LI A, Huang G, Klein-Szanto AJ, et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008;10:202–210. doi: 10.1038/ncb1681. [DOI] [PubMed] [Google Scholar]
- 14.Perschl A, Lesley J, English N, Trowbridge I, Hyman R. Role of CD44 cytoplasmic domain in hyaluronan binding. Eur J Immunol. 1995;25:495–501. doi: 10.1002/eji.1830250228. [DOI] [PubMed] [Google Scholar]
- 15.Puré E, Camp RL, Peritt D, Panettieri RA, Jr, Lazaar AL, Nayak S. Defective phosphorylation and hyaluronate binding of CD44 with point mutations in the cytoplasmic domain. J Exp Med. 1995;181:55–62. doi: 10.1084/jem.181.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown KL, Maiti A, Johnson P. Role of sulfation in CD44-mediated hyaluronan binding induced by inflammatory mediators in human CD14(+) peripheral blood monocytes. J Immunol. 2001;167:5367–5374. doi: 10.4049/jimmunol.167.9.5367. [DOI] [PubMed] [Google Scholar]
- 17.Maiti A, Maki G, Johnson P. TNF-alpha induction of CD44-mediated leukocyte adhesion by sulfation. Science. 1998;282:941–3. doi: 10.1126/science.282.5390.941. [DOI] [PubMed] [Google Scholar]
- 18.Levesque MC, Haynes BF. TNFalpha and IL-4 regulation of hyaluronan binding to monocyte CD44 involves posttranslational modification of CD44. Cell Immunol. 1999;193:209–18. doi: 10.1006/cimm.1999.1456. [DOI] [PubMed] [Google Scholar]
- 19.Katoh S, Zheng Z, Oritani K, Shimozato T, Kincade PW. Glycosylation of CD44 negatively regulates its recognition of hyaluronan. J Exp Med. 1995;182:419–429. doi: 10.1084/jem.182.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesley J, English N, Perschl A, Gregoroff J, Hyman R. Variant cell lines selected for alterations in the function of the hyaluronan receptor CD44 show differences in glycosylation. J Exp Med. 1995;182:431–437. doi: 10.1084/jem.182.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cichy J, Bals R, Potempa J, Mani A, Puré E. Proteinase-mediated release of epithelial cell-associated CD44: Extracellular CD44 complexes with components of cellular matrices. J Biol Chem. 2002;277:44440–44447. doi: 10.1074/jbc.M207437200. [DOI] [PubMed] [Google Scholar]
- 22.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 23.Ehnis T, Dieterich W, Bauer M, Lampe B, Schuppan D. A chondroitin/dermatan sulfate form of CD44 is a receptor for collagen XIV (undulin) Exp Cell Res. 1996;229:388–397. doi: 10.1006/excr.1996.0384. [DOI] [PubMed] [Google Scholar]
- 24.Iczkowski KA, Omara-Opyene AL, Shah GV. The predominant CD44 splice variant in prostate cancer binds fibronectin, and calcitonin stimulates its expression. Anticancer Res. 2006;26:2863–2872. [PubMed] [Google Scholar]
- 25.Zhao L, Hall A, Levenkova N, Lee E, Middleton MK, Zukas AM, Rader DJ, Rux JJ, Puré E. CD44 regulates vascular gene expression in a proatherogenic environment. Arterioscler Thromb Vasc Biol. 2007;27:886–892. doi: 10.1161/01.ATV.0000259362.10882.c5. [DOI] [PubMed] [Google Scholar]
- 26.Liang J, Jiang D, Griffith J, Yu S, Fan J, Zhao X, Bucala R, Noble PW. CD44 is a negative regulator of acute pulmonary inflammation and lipopolysaccharide-TLR signaling in mouse macrophages. J Immunol. 2007;178:2469–2475. doi: 10.4049/jimmunol.178.4.2469. [DOI] [PubMed] [Google Scholar]
- 27.Kawana H, Karaki H, Higashi M, Miyazaki M, Hilberg F, Kitagawa M, Harigaya K. CD44 suppresses TLR-mediated inflammation. J Immunol. 2008;180:4235–4245. doi: 10.4049/jimmunol.180.6.4235. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Asteriou T, Bernert B, Heldin CH, Heldin P. Growth factor regulation of hyaluronan synthesis and degradation in human dermal fibroblasts: importance of hyaluronan for the mitogenic response of PDGF-BB. Biochem J. 2007;404:327–336. doi: 10.1042/BJ20061757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orian-Rousseau V, Chen L, Sleeman JP, Herrlich P, Ponta H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002;16:3074–3086. doi: 10.1101/gad.242602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun CX, Robb VA, Gutmann DH. Protein 4.1 tumor suppressors: getting a FERM grip on growth regulation. J Cell Sci. 2002;115:3991–4000. doi: 10.1242/jcs.00094. [DOI] [PubMed] [Google Scholar]
- 31.Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 32.Kalomiris EL, Bourguignon LYW. Mouse T lymphoma cells contain a transmembrane glycoprotein (GP85) that binds ankyrin. J Cell Biol. 1988;106:319–327. doi: 10.1083/jcb.106.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsukita S, Oishi K, Sato N, Sagara J, Kawai A, Tsukita S. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol. 1994;126:391–401. doi: 10.1083/jcb.126.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yonemura S, Hirao M, Doi Y, Takahashi N, Kondo T, Tsukita S. Ezrin/Radixin/Moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J Cell Biol. 1998;140:885–895. doi: 10.1083/jcb.140.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–99. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 36.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 37.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 39.Jacamo R, Jiang X, Lunn JA, Rozengurt E. FAK phosphorylation at Ser-843 inhibits Tyr-397 phosphorylation, cell spreading and migration. J Cell Physiol. 2007;210:436–444. doi: 10.1002/jcp.20870. [DOI] [PubMed] [Google Scholar]
- 40.Kothapalli D, Flowers J, Xu T, Puré E, Assoian RK. Differential activation of ERK and Rac mediates the proliferative and anti-proliferative effects of hyaluronan and CD44. J Biol Chem. 2008;283:31823–31829. doi: 10.1074/jbc.M802934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L, Heldin CH, Heldin P. Inhibition of platelet-derived growth factor-BB-induced receptor activation and fibroblast migration by hyaluronan activation of CD44. J Biol Chem. 2006;281:26512–26519. doi: 10.1074/jbc.M605607200. [DOI] [PubMed] [Google Scholar]
- 42.Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol. 2001;3:950–957. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- 43.Klein EA, Campbell LE, Kothapalli D, Fournier AK, Assoian RK. Joint requirement for Rac and ERK activities underlies the mid-G1 phase induction of cyclin D1 and S phase entry in both epithelial and mesenchymal cells. J Biol Chem. 2008;283:30911–30918. doi: 10.1074/jbc.M804537200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mettouchi A, Klein S, Guo W, Lopez-Lago M, Lemichez E, Westwick JK, Giancotti FG. Integrin-specific activation of Rac controls progression through the G1 phase of the cell cycle. Mol Cell. 2001;8:115–127. doi: 10.1016/s1097-2765(01)00285-4. [DOI] [PubMed] [Google Scholar]
- 45.Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, Gutmann DH, Ponta H, Herrlich P. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001;15:968–980. doi: 10.1101/gad.189601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jung JR, Kim H, Jeun SS, Lee JY, Koh EJ, Ji C. The phosphorylation status of merlin is important for regulating the Ras-ERK pathway. Mol Cells. 2005;20:196–200. [PubMed] [Google Scholar]
- 47.Morrison H, Sperka T, Manent J, Giovannini M, Ponta H, Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007;67:520–527. doi: 10.1158/0008-5472.CAN-06-1608. [DOI] [PubMed] [Google Scholar]
- 48.Rong R, Surace EI, Haipek CA, Gutmann DH, Ye K. Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene. 2004;23:8447–8454. doi: 10.1038/sj.onc.1207794. [DOI] [PubMed] [Google Scholar]
- 49.Cuff CA, Kothapalli D, Azonobi I, Chun S, Zhang Y, Belkin R, Yeh C, Secreto A, Assoian RK, Rader DJ et al. The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J Clin Invest. 2001;108:1031–1040. doi: 10.1172/JCI12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kothapalli D, Zhao L, Hawthorne EA, Cheng Y, Lee E, Puré E, Assoian RK. Hyaluronan and CD44 antagonize mitogen-dependent cyclin D1 expression in mesenchymal cells. J Cell Biol. 2007;176:535–544. doi: 10.1083/jcb.200611058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, O’Bryan JP, Gupta V, Ratner N, Der CJ, et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev Cell. 2001;1:63–72. doi: 10.1016/s1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 52.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–849. doi: 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 53.Westwick JK, Lambert QT, Clark GJ, Symons M, Van Aelst L, Pestell RG, Der CJ. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nheu T, He H, Hirokawa Y, Walker F, Wood J, Maruta H. PAK is essential for RAS-induced upregulation of cyclin D1 during the G1 to S transition. Cell Cycle. 2004;3:71–74. [PubMed] [Google Scholar]
- 55.Thullberg M, Gad A, Beeser A, Chernoff J, Stromblad S. The kinase-inhibitory domain of p21-activated kinase 1 (PAK1) inhibits cell cycle progression independent of PAK1 kinase activity. Oncogene. 2006;26:1820–1828. doi: 10.1038/sj.onc.1209983. [DOI] [PubMed] [Google Scholar]
- 56.Xiao GH, Gallagher R, Shetler J, Skele K, Altomare DA, Pestell RG, Jhanwar S, Testa JR. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Cell Biol. 2005;25:2384–2394. doi: 10.1128/MCB.25.6.2384-2394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Acharya PS, Majumdar S, Jacob M, Hayden J, Mrass P, Weninger W, Assoian RK, Puré E. Fibroblast migration is mediated by CD44-dependent TGF{beta} activation. J Cell Sci. 2008;121:1393–1402. doi: 10.1242/jcs.021683. [DOI] [PubMed] [Google Scholar]