

Abstract
Variola virus, the causative agent of smallpox, is a potential bio-weapon. The development of new antiviral compounds for smallpox prophylaxis and treatment is critical, especially since the virus can acquire resistance to the drugs that are currently available. We have identified novel small chemical inhibitors that target DNA synthesis of vaccinia, the prototypical poxvirus. Robotic high-throughput screening of 49,663 compounds and follow-up studies identified very potent inhibitors of vaccinia DNA synthesis, with IC50 values as low as 0.5 ∞M. Cell-based assays showed that 16 inhibitors effectively blocked vaccinia infection with minimal cytotoxicity. Three inhibitors had selectivity indexes that approximate that of cidofovir. These new non-nucleoside inhibitors are expected to interfere with components of the vaccinia DNA synthesis apparatus that are distinct from cidofovir. Based on the high sequence similarity between the proteins of vaccinia and variola viruses, these new inhibitors are anticipated to be equally effective against smallpox.
Keywords: Smallpox, variola, vaccinia, DNA synthesis, DNA polymerase, rapid plate assay, high-throughput screen, inhibitors, antivirals
Introduction
Poxviruses are large, double stranded DNA viruses that replicate solely within the cell cytoplasm.1 The most famous member of the poxvirus family is variola virus, the causative agent of smallpox. Our knowledge of poxvirus biology has been largely obtained through studying the vaccinia virus. This prototypical poxvirus, which shares over 97% amino acid sequence identity with variola,2, 3 was used as the vaccine in the global immunization campaign that successfully eradicated smallpox in 1980.4
While smallpox no longer occurs in the natural world, reintroduction of the virus as a bio-weapon could trigger a pandemic, as more than half of the world’s current population is not vaccinated. In the event of widespread exposure, potent antiviral therapeutics could serve as the first line of defense. The drugs currently recommended for short-term prophylaxis and emergency treatment against smallpox are cidofovir (CDV) and 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide (30).
CDV, (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine,5, 6 is a phosphonate analogue of cytosine that targets viral DNA polymerase by acting as a chain terminator of nucleotide incorporation.7 CDV has a broad-spectrum antiviral activity, proving to be effective against many poxviruses and herpesviruses. However, the oral bioavailability of CDV is low and intravenous administration leads to nephrotoxicity.8 CDV’s poor cellular absorption is due to the presence of two negative charges on the phosphonate group.9 Neutralization of these negative charges by esterification with lipophilic groups has resulted in prodrugs with significantly enhanced oral bioavailability.10-12
A drug more recently approved for the prevention and treatment of smallpox is 30 (ST-246, SIGA Technologies), that targets the P37 protein of vaccinia, a membrane component required for the formation of extracellular viral particles.13 Compound 30 was shown to inhibit the growth of multiple orthopoxviruses in cell cultures, including two strains of variola virus.14 Compound 30 was effective in protecting against viral spread when administered orally to mice challenged with lethal doses of cowpox, vaccinia or ectromelia virus.13, 15
However, as often the case, the use of single therapeutics can lead to the emergence of resistant strains. Indeed, poxviruses resistant to CDV or 30 have been generated in cell culture.13, 16, 17 This raises the concern that poxvirus strains can be easily produced by genetic manipulation to become unresponsive to the therapeutics currently available. One means of circumventing this problem is to have in place several therapeutics directed against different viral targets. Viable new viral targets include the replicating proteins of vaccinia, comprising DNA polymerase (E9),18 processivity factor (A20),19 uracil DNA glycosylase (D4),20, 21 DNA-independent nucleoside triphosphatase (D5),22 and serine/threonine protein kinase (B1).23, 24 Since the amino acid sequences of the viral DNA polymerase and its accessory proteins are highly conserved amongst poxviruses, drugs that target vaccinia DNA synthesis are predicted to inhibit variola DNA synthesis.
Previously we developed a rapid plate assay to screen for inhibitors of viral DNA synthesis.25 This in vitro assay was used to identified chemical inhibitors of Kaposi’s sarcoma-associated herpesvirus processive DNA synthesis.26 Recently, we modified this assay to select inhibitors that block vaccinia virus DNA synthesis.27 Using this modified plate assay, we screened a small library of 2,000 compounds, representing a wide spectrum of chemical structures, and identified inhibitors that were able to block vaccinia virus infection. In this study, we adapted the rapid plate assay for robotic high-throughput screening (HTS) and tested 49,663 compounds. We present 16 compounds that block viral infection with low cytotoxicity. In particular, three of the compounds had selectivity indexes that approximate that of the nucleoside derivative CDV. However, unlike CDV, which is a nucleoside analog, all of our newly identified inhibitors are non-nucleosides, serving to extend the range and diversity of poxvirus inhibitors and lessen the potential of generating drug-resistant mutants.
Results and Discussions
Identification of small chemical inhibitors of vaccinia virus DNA synthesis with a rapid plate assay designed for high-throughput screening
We recently developed a rapid plate assay to identify inhibitors of vaccinia virus DNA synthesis that target the viral DNA polymerase and its associated factors required for processivity.25, 27 Briefly, a 5′-biotinylated DNA template annealed to an oligonucleotide primer at its 3′-end is immobilized onto streptavidin-coated wells. In the presence of the viral polymerase and its associated factors, dNTP’s and digoxigenin-labeled 2′-deoxy-uridine-5′-triphosphate (DIG-11-dUTP) are incorporated into the newly synthesized DNA strand. DNA synthesis activity is measured by the detection of digoxigenin using an enzyme-linked antibody that generates a colorimetric reaction.
Replication of poxviruses occurs solely within the cytoplasm.1 Cytoplasmic extracts from infected BS-C-1 cells were used as the source of vaccinia proteins for the rapid plate assay. These cytoplasmic extracts were confirmed to be completely free of nuclear proteins by western blot with antibody specific for the nuclear marker retinoblastoma protein Rb1 (Figure 1A). Moreover, cytoplasmic extracts of uninfected cells had little to no DNA synthesis activity in the rapid plate assay, indicating that the DNA synthesis activity of infected cell extracts was exclusively due to vaccinia cytoplasmic proteins and not to nuclear enzymes involved in cellular DNA synthesis (Figure 1B).
Figure 1.

DNA synthesis activity of the vaccinia virus-infected cell cytoplasmic extract. A. Western blot analysis showing separation of cytoplasmic and nuclear extracts of vaccinia virus-infected BS-C-1 cells. Nuclear (N) and cytoplasmic (C) fractions were analyzed by western blot probed with a retinoblastoma monoclonal antibody (Rb1 antibody ab24, Abcam, Cambridge, MA, USA). Retinoblastoma, which served as a nuclear marker, is indicated by the arrow. The cytoplasmic fraction was free of nuclear contaminants as the Rb1 protein could not be observed even after gel overloading and long exposure times. B. DNA synthesis activity of the infected cytoplasmic extract. Shown are rapid plate assay reactions catalyzed by uninfected cytoplasmic extract (lane 1); vaccinia-infected cytoplasmic extract (lane 2); vaccinia-infected cytoplasmic extract in the presence of EDTA inhibitor (lane 3).
Before proceeding with the HTS, the rapid plate assay was adapted for use with the Epson compound transfer robot (Epson Robots, Carson, CA). DNA synthesis reactions were optimized without inhibitors in streptavidin-coated 384-well plates. This robotic procedure exhibited plate uniformity and no signal variability when tested in multiple plates on different days. No drift or edge effects were observed and the calculated values for signal-to-background, signal-to-noise, coefficient of variation, and screening window coefficient (Z’-factor) validated the assay as suitable for HTS. The robotic plate assay was reproducible and the Z’-factor had an excellent value of 0.95.
Next, the optimized assay was validated by testing 1,520 chemical compounds with known pharmacological activities (Supporting Information Table S1). These known bioactive compounds are compiled in two libraries and include more than half of the drugs currently approved by the Food and Drug Administration. Out of the 58 compounds that blocked the colorimetric reaction of the rapid plate assay, 39 are known to bind DNA or inhibit DNA or RNA polymerases, which confirmed that the assay was indeed capable of detecting inhibitors of DNA synthesis. Interestingly, with respect to the remaining 19 known compounds, this is the first report to reveal that they are capable of inhibiting DNA synthesis.
The optimized robotic plate assay was then used to identify novel vaccinia DNA synthesis inhibitors by performing a HTS of 45,832 small synthetic compounds (MW<500) and 2,311 partially purified natural extracts (Supporting Information Table S1). In this primary screen, we identified 383 natural extracts and 446 synthetic compounds that inhibited vaccinia virus DNA synthesis, representing a hit rate of 1.6%. An extensive survey of the current literature showed that the majority of these hit compounds were never reported to function as inhibitors of DNA or RNA synthesis. Of the total 829 hit compounds, 178 were ranked as strong (greater than 70% inhibition), 271 were ranked as intermediate (50-70% inhibition), and 380 were ranked as weak (30-50% inhibition), (Supporting Information Table S1), based on the colorimetric intensity of the read-out signal relative to the control signal obtained with no inhibitor (DMSO alone). Interestingly, the percentage of natural extracts that inhibited DNA synthesis was greater than that of the synthetic compounds (Supporting Information Table S1). Likely, the DNA synthesis reaction is more vulnerable to the multitude of chemical entities present in natural extracts than the singly purified compounds from synthetic libraries. It is noted that the group of synthetic compounds with known biological activities also had a greater hit rate than the synthetic compounds from combinatorial libraries. This is not surprising, since many of these known compounds have been shown to modulate activities that utilize DNA (e.g. topoisomerization).
At this point, we decided to pursue only the synthetic hit compounds since they have defined structures and are available as purified chemicals. These synthetic hit compounds represent a wide range of structures, which we were able to classify into 59 different chemical families. All of the strong and most of the intermediate inhibitors from the combinatorial synthetic libraries were confirmed to block vaccinia DNA synthesis upon manual re-testing in the 96-well rapid plate assay (data not shown). Follow-up studies were conducted on 93 of the synthetic inhibitors, from which all of the structural families are represented. These compounds were evaluated for potency in blocking vaccinia DNA synthesis in vitro, protecting cultured cells from infection with vaccinia virus, as well as cellular toxicity.
Potency of hit compounds in blocking vaccinia virus DNA synthesis in vitro
We determined the inhibitory concentrations (IC50) for each of the 93 synthetic compounds. The assay was performed in 96-well plates using conditions similar to those used for the primary HTS. Each compound, dissolved in DMSO, was tested in triplicate over a range of concentrations for inhibition of DNA synthesis catalyzed by vaccinia proteins. The percentage inhibition values were fitted on sigmoidal dose-response curves from which the IC50 was determined. Three representative dose-response curves are presented in Figure 2A. The IC50 values for 89 compounds ranged from 0.5 ∞M to 400 ∞M, while 4 compounds showed less than 50% inhibition at concentrations of 400 ∞M or higher (Tables 1 and 2, Supporting Information Tables S2 and S3).
Figure 2.
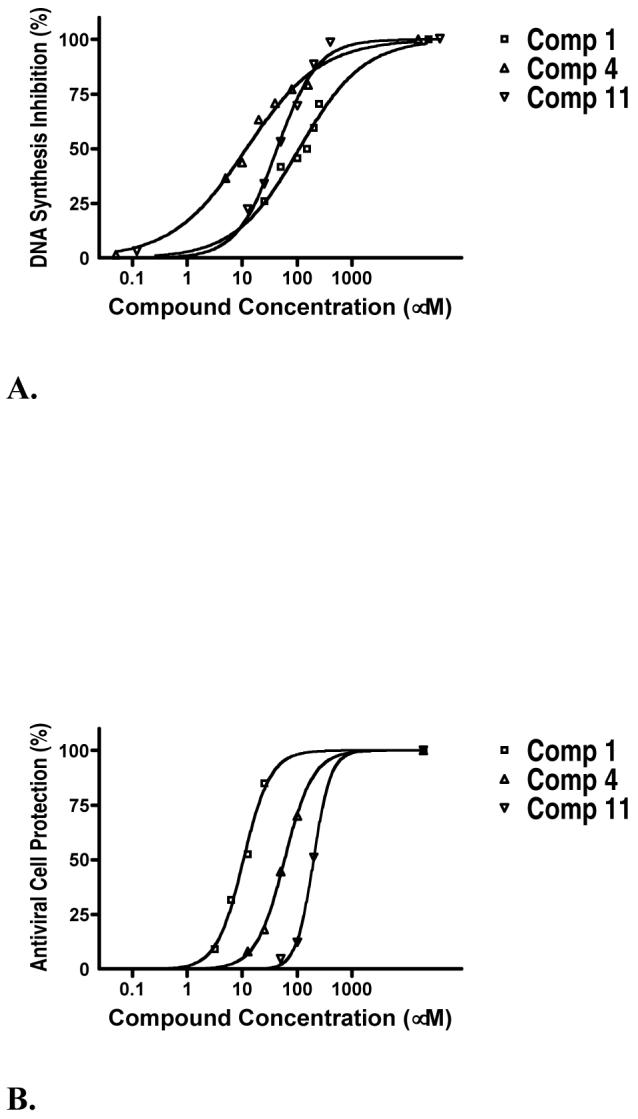
Inhibition of vaccinia virus DNA synthesis and antiviral activity of 3 typical hit compounds. A. Inhibition of vaccinia virus DNA synthesis by each compound was tested in triplicate over a range of concentrations in the DNA synthesis rapid plate assay. Percent inhibition was calculated relative to the uninhibited negative control (DMSO) and the completely inhibited positive control (EDTA). The Prism software was used to plot the inhibitory dose-response and to calculate the IC50 values. B. Antiviral activity measured in the cell protection assay. The ability of compounds to block vaccinia virus infection in BS-C-1 cells was measured over a wide range of concentrations. Twenty hours after infection, cells were fixed with formaldehyde, stained with crystal violet and the extent of antiviral protection was quantified by measuring absorbance at 570 nm. Every compound was tested in triplicate and the percent protection was calculated relative to the DMSO and uninfected controls. The Prism software was used to plot the protection dose-response and to calculate the EC50 values.
Table 1.
Summary of activities for 16 synthetic hit compounds
| compda | DNA Synth IC50 (∞M) | Cell Prot EC50 (∞M) | Plaque Red EC50 (∞M) | Tox 20 h TC50 (∞M) | Tox 72 h TC50 (∞M) | Tox 120 h TC50 (∞M) | Selectivity Indexb | Trapped by dsDNA |
|---|---|---|---|---|---|---|---|---|
| 1 | 108 | 9.6 | 10 | 127 | 20 | 13.2 | No | |
| 2 | 374 | 30 | 25 | 159 | 5.3 | No | ||
| 3 | 131 | 50 | 50 | >200 | 36 | 3 | >4.0 | No |
| 4 | 12 | 50 | 15 | 157 | 40 | 3.1 | No | |
| 5 | 129 | 45 | 15 | 115 | 5 | 2.6 | No | |
| 6 | 30 | 95 | 75 | >200 | 50 | 20 | >2.1 | No |
| 7 | 26 | 100 | 75 | >200 | 38 | 20 | >2 | No |
| 8 | 52 | 100 | 75 | >100 | >100 | >100 | >2 | No |
| 9 | 26 | 104 | 75 | >200 | 40 | 20 | >1.9 | No |
| 10 | 188 | 90 | 25 | 166 | 1.8 | No | ||
| 11 | 43 | 130 | 75 | >200 | 174 | 120 | >1.5 | No |
| 12 | 32 | 80 | 75 | >100 | >100 | 50 | >1.2 | No |
| 13 | 46 | 100 | 50 | >100 | 13 | 30 | >1 | No |
| 14 | 75 | 188 | 150 | >200 | >200 | 50 | >1 | No |
| 15 | 111 | 236 | 100 | >200 | >200 | 75 | >1 | No |
| 16 | 239 | 100 | 50 | >100 | >100 | 50 | >1 | No |
| CDV | 132 | 50 | >500 | >500 | 300 | >3.8 | No |
Refer to Chart 1 for the structures of compounds 1 to 16.
The Selectivity Index was obtained as the ratio between TC50 in the 20 h cytotoxicity assay and EC50 in the 20 h cell protection assay.
Table 2.
Structures and IC50 values for 19 synthetic compounds that are strong inhibitors of vaccinia DNA synthesis in vitroa
| Librarya | Compound Structure | DNA Synthesis IC50 (∞M) |
|---|---|---|
| 1 | 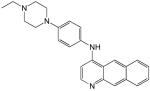 |
0.5 |
| 5 | 1.3 | |
| 1 | 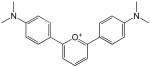 |
3.0 |
| 1 | 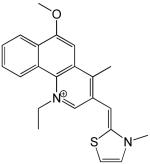 |
3.5 |
| 3 |  |
3.8 |
| 1 | 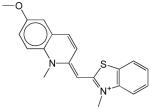 |
4.0 |
| 1 | 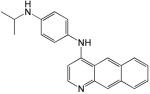 |
4.1 |
| 4 |  |
5.0 |
| 4 |  |
5.3 |
| 1 | 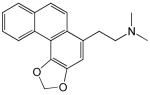 |
5.4 |
| 1 | 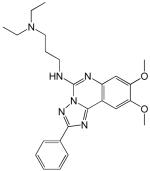 |
5.9 |
| 1 | 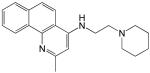 |
6.0 |
| 3 | 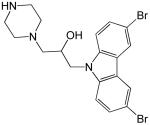 |
6.3 |
| 1 | 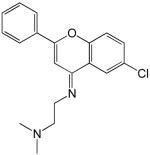 |
6.7 |
| 1 | 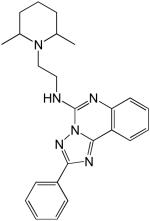 |
7.3 |
| 1 | 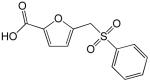 |
7.4 |
| 4 |  |
7.4 |
| 1 | 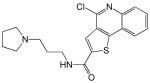 |
10 |
| 1 | 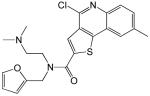 |
10 |
These compounds do not have favorable antiviral activity in cell-based assays since they are either highly toxic or failed to block viral infection.
The synthetic compounds belong to the following libraries: ChemDiv 3 (library 1); Maybridge 4 (library 3); ActiMol TimTec (library 4); and Bionet 2 (library 5).
Potency of hit compounds in blocking vaccinia virus infection in vitro
The 93 DNA synthesis inhibitors were first tested in a plaque reduction assay to determine if they could prevent the formation of vaccinia virus plaques on BS-C-1 cell monolayers. For the plaque reduction assay, cells were infected at a low multiplicity of infection (moi) to generate approximately 50 vaccinia virus plaques in each well of a 48-well plate. Each compound was tested in triplicate over a range of concentrations and the results were used to estimate the EC50, the effective concentration of compound required to reduce the number of viral plaques by 50%. The EC50 values for 75 compounds ranged from 5 ∞M to 200 ∞M, while 18 compounds had no effect on the number of plaques (Tables 1 and 2, Supporting Information Tables S2 and S3).
Next, the 93 compounds were tested in a cell protection assay. This assay was performed with amounts of virus capable of lysing every cell in the culture during the 20-hour incubation period. Compounds with antiviral properties protect the cell monolayer from infection, as determined by the cellular mass, stained for absorbance at 570 nm. Each compound was tested in triplicate over a range of concentrations up to 200 ∞M in two-fold serial dilution increments. Dose-dependent plots were generated by fitting the results on four-parameter sigmoidal response curves. These plots were used to calculate for each compound the concentration at which 50% antiviral protection is obtained relative to the unprotected DMSO control (Figure 2B). Twenty-nine of the 75 compounds that were active in the plaque assay also protected cells from viral infection with EC50 values as low as 9.6 ∞M (Table 1 and Supporting Information Table S2).
While both the plaque reduction and the cell protection assays measure the antiviral activity of compounds, they differ dramatically by the amount of virus used to infect the cell monolayer. Nevertheless, as seen in Table 1, there is an overall concordance between the EC50 values obtained with these two assays, confirming the efficacy of these hit compounds in blocking viral infection.
Cellular toxicity of hit compounds
The cell protection assay presented above, indirectly measures cytotoxicity, in that compounds that protect against viral infection must, a priori, perform this function without disrupting the cells. However, in order to directly assess the cytotoxicity of each of the 93 compounds, we employed a cytotoxicity assay that measures the metabolic activity of cells exposed to chemicals in the absence of virus. After culturing cells in the presence of inhibitors, cell viability was determined by spectrophotometrically measuring the reduction of a water-soluble tetrazolium salt to a dark-red formazan, which is catalyzed by a mitochondrial activity.28 The percent cytotoxicity relative to the DMSO control was fitted onto four-parameter sigmoidal dose-response curves and the 50% toxic concentration values (TC50) were determined. After 20 hours, 15 compounds had negligible cytotoxicity even at the highest working concentration of 200 ∞M. Not surprisingly, longer incubation periods of 72 and 120 hours lead to increased cytotoxicity (Table 1 and Supporting Information Table S2).
Based on the results of cell culture assays, the 93 compounds fell into 3 groups: group I consisted of 18 compounds that had no effect on cells (Table 2 and Supporting Information Table S3); group II consisted of 46 compounds that inhibited viral infection but were cytotoxic at similar concentrations (Table 2 and Supporting Information Table S3); group III consisted of 29 compounds that inhibited viral infection at concentrations lower than the cytotoxic concentration (Chart 1, Table 1 and Supporting Information Chart S1, Table S2).
Chart 1.

Structures of the 16 most promising synthetic hit compounds that inhibit vaccinia virus infection.
Compounds 7, 11, 14, 15 belong to ChemDiv 3 library; compounds 4, 5, 6, 8, 9 belong to Maybridge 4 library; compounds 12, 16 belong to ActiMol TimTec library; compound 3 belongs to Enamine 1 library; compounds 1, 2, 10, 13 belong to I.F. Lab 1 library.
Of note, the 18 synthetic compounds from group I did not have any effect in cell-based assays when tested at concentrations up to 200 ∞M: did not inhibit viral plaque formation, did not protect cells from viral infection, and did not show any cytotoxicity. All of these 18 compounds had tested positive in the rapid plate assay, some of them being very potent inhibitors of vaccinia DNA synthesis in vitro, with IC50 values as low as 1.3 ∞M (Table 2). This lack of antiviral activity and cytotoxicity is most probably due to the inability of these compounds to permeate the cell membrane. Chemical modifications that increase lypophilicity and cell permeability may confer antiviral activity to these in vitro DNA synthesis inhibitors and convert them into attractive lead compounds.
Selectivity index of hit compounds
The selectivity index (SI) for each compound was calculated as the ratio between TC50 in the 20-hour cytotoxicity assay and EC50 in the cell protection assay. Six compounds had a SI of 4 or higher with the most effective compound having a SI of 13, reflecting the ability to block vaccinia virus infection at concentrations significantly lower than those producing cytotoxicity (Table 1 and Supporting Information Table S2). Of note, the SI for many compounds is a conservative estimate, since 50% cytotoxicity was not attained at the highest tested concentration of 200 ∞M (e.g. compound 3 in Table 1). As reference for antiviral activity, we tested the well-known anti-poxvirus drug CDV, which functions as a chain terminator to inhibit viral DNA synthesis. In our hands, CDV had a SI of greater than 4, which is in accord with the values previously reported.16, 29 Our compounds have potencies comparable to CDV, suggesting their significance as potential poxvirus therapeutics.
Sixteen hit compounds retain their antiviral activity in the presence of a DNA trap
The compounds identified in our primary HTS were selected by their ability to block vaccinia virus DNA synthesis. Although many hit compounds are expected to inhibit DNA synthesis by interfering with a specific mechanistic step (e.g. the catalytic incorporation of dNTP’s by DNA polymerase), we were concerned that some hit compounds bind to DNA (e.g. by intercalation), in which case they would non-specifically inhibit DNA polymerases from incorporating nucleotides. To eliminate such non-specific inhibitors, we tested their antiviral activities in the presence of large amounts of genomic double-stranded DNA added to the media of vaccinia-infected cells.30 Since cells do not take up exogenous DNA under normal culturing conditions, this DNA serves to trap non-specific DNA binding compounds and disable their cellular uptake. Compounds that were affected by the exogenous DNA were identified by the loss of antiviral activity. As shown in Table 1, a total of 16 compounds retained their antiviral activity in the presence of the extracellular DNA trap, indicating that these compounds were relevant inhibitors that block a mechanistic step in DNA synthesis.
When tested in 20h assays, 3 relevant hit compounds had a SI of 4 or higher with the most effective compound having a SI of 13 (Table 1). We also tested these 16 compounds for cell protection and cytotoxicity over longer times (48h and 72h). At these longer times, the compounds showed diminishing antiviral activity and increasing cytotoxicity, as compared to the overnight assays. The antiviral activities of 4 compounds were high enough to allow calculation of EC50 values, but 12 compounds showed less than 50% protection in the 72h assays. When calculable, the SI for each compound was lower in the 72h assay than in the overnight assays. For example, compound 1 has an SI of 13.2 over 20h, and an SI of 3.2 over 72h.
We also verified the solubility of the 16 compounds in cell growth medium over the range of concentrations used in the activity assays. Light scattering measurements indicated that 11 compounds were completely soluble, while 5 compounds (8, 10, 12, 13, 16) were just slightly insoluble at the EC50.
Conclusions
We have discovered 16 inhibitors of vaccinia DNA synthesis that have antiviral activity. These 16 inhibitors were identified by the HTS of 49,663 compounds using an in vitro DNA synthesis rapid plate assay. All of the inhibitors effectively block viral infection with minimal toxicity to the cells and could not be trapped outside the cells by exogenous DNA, indicating that their antiviral activity is mediated by the disruption of an essential step in the mechanism of viral DNA synthesis. Of particular interest, three of the inhibitors had selectivity indexes that approximate that of CDV, the well-known poxvirus DNA synthesis inhibitor. Since these new inhibitors are not nucleoside analogs, they are expected to block vaccinia DNA synthesis through a mechanism that is distinct from that of CDV, a nucleoside analog. Further development of these 16 synthetic compounds could lead to useful pox antiviral compounds that will complement the inhibitory activity of CDV, and thus, reduce the emergence of drug resistant mutants. Based on the high sequence similarity between the proteins of vaccinia and variola viruses that are responsible for DNA synthesis, these new inhibitors are anticipated to be equally effective against smallpox. Future enzymatic and virological studies will identify the specific DNA synthesis proteins targeted by these poxvirus inhibitors.
Experimental Section
Primary high-throughput screen
For HTS, the rapid plate assay25, 27 was adapted to a robotic platform. The DNA synthesis reactions were performed in 384-well plates coated with streptavidin (SigmaScreen plates, Sigma-Aldrich cat # S8686). In each well, reactions were conducted with 0.5 ∞L vaccinia virus extract containing 0.2 mU DNA polymerase activity in 20 mM Tris-Cl pH7.5, 100 mM ammonium sulfate, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 4% glycerol, 40 ∞g/mL BSA, 5 ∞M of each dATP, dCTP, and dGTP, 4 ∞M dTTP, 0.5 ∞M DIG-11-dUTP, in a total volume of 30 ∞L. Two pmoles of biotinylated primer-template dissolved in 30 ∞L PBS were immobilized on streptavidin-coated wells. The unbound primer-template was removed, the wells were washed twice with 50 ∞L PBS and loaded with 20 ∞L reaction buffer (20 mM Tris-Cl pH7.5). One hundred nL of each compound or natural extract dissolved in DMSO were transferred from the library plates by an Epson transfer robot (Epson Robots, Carson, CA) fitted with a 384-pin array. DNA synthesis was initiated by loading10 ∞L of a 3x reaction mixture. After brief centrifugation, plates were incubated at 37°C for 30 minutes. The DNA synthesis reaction was stopped with 30 ∞L of a solution containing 50 mM EDTA and 2% SDS in 10 mM Tris pH8. Incorporation of DIG-11-dUTP in the newly synthesized DNA strand was detected with peroxidase-conjugated anti-digoxigenin antibody (anti-DIG-POD). Wells were flow-washed with 400 ∞L PBS containing 0.1% Tween-20 at the lowest dispensing speed and loaded with 4.5 mU of anti-DIG-POD in 30 ∞L PBS/blocking solution. After gently rocking at room temperature for 1h, the antibody solution was removed and wells were flow-washed as above. Thirty ∞L of 2, 2′-azino-bis(3-ethylbenzthiazoline)-sulfonate (ABTS) peroxidase substrate dissolved in citrate buffer was added and plates were gently rocked at room temperature for 1h. Color development was stopped with 10 ∞L of 4% SDS and absorbance at 405 nm was measured. Readings from each well were divided by the plate median and the percent inhibition was determined relative to the values of uninhibited reaction controls.
Cytoplasmic extracts of vaccinia-infected BS-C-1 cells
Confluent BS-C-1 cells, infected with vaccinia virus WR at an MOI of 15 in the presence of 10 mM hydroxyurea, were harvested 6 hours post infection. Hydroxyurea inhibits viral DNA replication and transcription of intermediate and late viral mRNA’s, allowing only the expression of early gene products and not the abundant late viral genes.31 Processive DNA synthesis is an early activity of the vaccinia virus and thus, is not affected by hydroxyurea.32 Infected cells were carefully scraped off the plates, washed once with hypotonic buffer (1.5 mM MgCl2, 10 mM KCl, 10 mM Hepes pH 7.5), resuspended in cold hypotonic buffer, incubated on ice for 10 minutes and lysed by gentle dounce homogenization. Nuclei were removed by centrifugation at 1000×g for 10 minutes. The cytoplasmic extracts were filtered twice through 0.2 ∞m and shown to be free of infectious particles as determined by plaque assay. Glycerol was added to 20% final concentration and aliquots were stored at -80°C.
Screened libraries
The 1,520 chemical compounds with known biological activities used for assay validation were from two collections: BIOMOL ICCB Known Bioactives 1 (480 compounds) and NINDS Custom Collection (1,040 compounds). A total of 45,832 synthetic compounds, representing 11 independent libraries: ChemDiv 3 (16,544 compounds), MixCommercial 5 (268 compounds), Maybridge 4 (4,576 compounds), ActiMol TimTec 1 (8,518 compounds), Bionet 2 (1,700 compounds), Enamine 1 (6,004 compounds), I.F. Lab 1 (6,543 compounds), I.F. Lab 2 (292 compounds), Maybridge 2 (704 compounds), MixCommercial 4 (331 compounds), and Peakdale 2 (352 compounds) were tested in duplicate, at a single concentration of 16.7 ∞g/mL, which is equivalent to a molar concentration of 33-167 ∞M, depending on the molecular weight of each individual compound. Natural extracts with inhibitory activity were identified by screening the Starr Foundation Extracts 2 library (1,000 extracts from plants used in traditional Chinese medicine), and two collections of partially purified extracts from endophytic fungi: ICBG Fungal Extracts 1 (851 extracts) and ICBG Fungal Extracts 2 (460 extracts). The natural extracts, resuspended in DMSO at 15 mg/mL, were also tested in duplicate at 50 ∞g/mL final working concentration. The chemical libraries were supplied by the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Disease (NSRB) at the Harvard Institute of Chemistry and Cell Biology (ICCB, Harvard Medical School), where the robotic HTS was performed.
DNA synthesis assay
Milligram quantities of the HTS hit compounds for follow-up studies were purchased from various vendors and suspended in DMSO at a final concentration of 20 mM. Each hit compound was re-tested over a range of concentrations for its ability to inhibit vaccinia virus DNA synthesis. Assays were performed in 96-well plates in conditions similar to those for the HTS. Each compound was tested in triplicate in two-fold serial dilutions and the IC50 was calculated with the Prism software (GraphPad Software, Inc.) for linear regression.
Viral plaque reduction assay
BS-C-1 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), 50 mg/L gentamicin sulfate (Gibco BRL Life Technologies, Gaithersburg, MD) and grown at 37°C in a humidified atmosphere with 5% CO2. For the plaque reduction assay, 6×104 cells/well were seeded in 48-well plates and allowed to attach overnight. Cells were infected with 50 plaque forming units (pfu)/well of vaccinia virus WR in the presence of the HTS hit compounds at various concentrations. After 20 hours, cells were fixed with 5% formaldehyde in PBS and stained with 0.2% crystal violet dissolved in 50% ethanol. Wells were washed, dried and plaques were counted under the microscope. The EC50 values were estimated as the concentration of compound that caused a reduction in the number of plaques by 50%.
Viral cell protection assay
The cell protection assay was performed using similar conditions to the plaque reduction assay. BS-C-1 cells were seeded in 96-well plates at 1.5×104 cells/well and grown overnight. The cells were infected with 2000 pfu/well of vaccinia virus WR in the presence of inhibitors. After incubating at 37°C for various times (20, 48, and 72 hours), cells were fixed with formaldehyde and stained with crystal violet. The excess dye was washed away and the cellular mass fixed on the bottom of the wells was allowed to dry overnight. Absorbance at 570 nm was measured. Each compound was tested in triplicate in two-fold serial dilutions and EC50 values were calculated using the Prism software for linear regression.
DNA trap assay
To investigate if the hit compounds retain their antiviral potency in the presence of exogenous DNA, the cell protection assay was performed with 400 ∞g/mL of type III DNA, sodium salt, from salmon testes (Sigma, Saint Louis, MO, USA) added in the growth medium. The final concentration of compound-derived DMSO was 1% (v/v) in all cell-based assays. For the control wells that contained no compound, an appropriate volume of DMSO was added to achieve a final concentration of 1%.
Cytotoxicity assay
Compound cytotoxicity was measured with the Cell Proliferation Reagent WST-1 (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer’s protocol. BS-C-1 cells were seeded in 96-well plates at 5000 cells/well in 50 ∞L growth medium without phenol red. Next day, 50 ∞L/well of growth medium containing the tested compounds were added and cultures were grown for 20 hours at 37°C. Ten ∞L of WST-1 reagent were added to each well, plates were incubated at 37°C for 30 minutes and absorbance at 450 nm was measured. Each compound was tested in triplicate in two-fold serial dilutions and TC50 values were calculated using the Prism software for linear regression. To measure the compound cytotoxicity over longer incubation periods, the number of BS-C-1 cells seeded was adjusted: 1000 cells/well for 3-day cytotoxicity and 200 cells/well for 5-day cytotoxicity. This insured that cells did not grow past confluency by the end of the experiment and that the read-out signal remained in the linear range.
Supplementary Material
Acknowledgment
The authors thank the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Disease (NSRB) at Harvard Institute of Chemistry and Cell Biology (ICCB, Harvard Medical School), for use of the robotic facility and libraries where the HTS was performed. We are especially grateful to Su Chiang, Kyungae Lee, Greg Cuny, Stewart Rudnicki and Sean Johnson of the ICCB for assistance with the screen. A.M.D.S. was supported by an NIH EID T32 training grant. This study was supported by the NIH NIAID Middle Atlantic Regional Center of Excellence grant U54 AI057168NIAID to R.P.R.
Abbreviations
- HTS
high-throughput screening
- CDV
cidofovir
- DMSO
dimethyl sulfoxide
References
- 1.Cairns J. The initiation of vaccinia infection. Virology. 1960;11(3):603–623. doi: 10.1016/0042-6822(60)90103-3. [DOI] [PubMed] [Google Scholar]
- 2.Massung RF, Liu LI, Qi J, Knight JC, Yuran TE, Kerlavage AR, Parsons JM, Venter JC, Esposito JJ. Analysis of the Complete Genome of Smallpox Variola Major Virus-Strain Bangladesh-1975. Virology. 1994;201(2):215–240. doi: 10.1006/viro.1994.1288. [DOI] [PubMed] [Google Scholar]
- 3.Shchelkunov SN, Massung RF, Esposito JJ. Comparison of the Genome DNA-Sequences of Bangladesh 1975 and India 1967 Variola Viruses. Virus Research. 1995;36(1):107–118. doi: 10.1016/0168-1702(94)00113-q. [DOI] [PubMed] [Google Scholar]
- 4.The global eradication of smallpox: final report of the Global Commission for the Certification of Smallpox Eradication. World Health Organization; Geneva: 1980. Report No. 4. [Google Scholar]
- 5.Baker R, Bray M, Huggins JW. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antiviral Research. 2003;57(12):13–23. doi: 10.1016/S0166-3542(02)00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Clercq E. Cidofovir in the treatment of poxvirus infections. Antiviral Research. 2002;55(1):1–13. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magee WC, Hostetler KY, Evans DH. Mechanism of inhibition of vaccinia virus DNA polymerase by cidofovir diphosphate. Antimicrobial Agents and Chemotherapy. 2005;49(8):3153–3162. doi: 10.1128/AAC.49.8.3153-3162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier P, Dautheville-Guibal S, Ronco PM, Rossert J. Cidofovir-induced end-stage renal failure. Nephrology Dialysis Transplantation. 2002;17(1):148–149. doi: 10.1093/ndt/17.1.148. [DOI] [PubMed] [Google Scholar]
- 9.Cundy KC. Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir and adefovir. Clinical Pharmacokinetics. 1999;36(2):127–143. doi: 10.2165/00003088-199936020-00004. [DOI] [PubMed] [Google Scholar]
- 10.Beadle JR, Wan WB, Ciesla SL, Keith KA, Hartline C, Kern ER, Hostetler KY. Synthesis and antiviral evaluation of alkoxyalkyl derivatives of 9-(S)-(3-hydroxy-2-phosphonomethoxypropyl)adenine against cytomegalovirus and orthopoxviruses. Journal of Medicinal Chemistry. 2006;49(6):2010–2015. doi: 10.1021/jm050473m. [DOI] [PubMed] [Google Scholar]
- 11.Kern ER, Hartline C, Harden E, Keith K, Rodriguez N, Beadle JR, Hostetler KY. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrobial Agents and Chemotherapy. 2002;46(4):991–995. doi: 10.1128/AAC.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciesla SL, Trahan J, Wan WB, Beadle JR, Aldern KA, Painter GR, Hostetler KY. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Research. 2003;59(3):163–171. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- 13.Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RLM, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. Journal of Virology. 2005;79(20):13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duraffour S, Snoeck R, de Vos R, van den Oord JJ, Crance JM, Garin D, Hruby DE, Jordan R, De Clercq E, Andrei G. Activity of the anti-orthopoxvirus compound ST-246 against vaccinia, cowpox and camelpox viruses in cell monolayers and organotypic raft cultures. Antiviral Therapy. 2007;12(8):1205–1216. [PubMed] [Google Scholar]
- 15.Quenelle DC, Buller RML, Parker S, Keith KA, Hruby DE, Jordan R, Kern ER. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrobial Agents and Chemotherapy. 2007;51(2):689–695. doi: 10.1128/AAC.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smee DF, Sidwell RW, Kefauver D, Bray M, Huggins JW. Characterization of wild-type and cidofovir-resistant strains of camelpox, cowpox, monkeypox, and vaccinia viruses. Antimicrobial Agents and Chemotherapy. 2002;46(5):1329–1335. doi: 10.1128/AAC.46.5.1329-1335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang G, Harver C, Hruby D, Jordan R. Characterization of virus variants resistant to the antiviral effects of ST-246. Antiviral Research. 2006;70(1):A73–A73. [Google Scholar]
- 18.Challberg MD, Englund PT. Purification and Properties of the Deoxyribonucleic-Acid Polymerase Induced by Vaccinia Virus. Journal of Biological Chemistry. 1979;254(16):7812–7819. [PubMed] [Google Scholar]
- 19.Klemperer N, McDonald W, Boyle K, Unger B, Traktman P. The A20R protein is a stoichiometric component of the processive form of vaccinia virus DNA polymerase. Journal of Virology. 2001;75(24):12298–12307. doi: 10.1128/JVI.75.24.12298-12307.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Upton C, Stuart DT, McFadden G. Identification of a Poxvirus Gene Encoding a Uracil DNA Glycosylase. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(10):4518–4522. doi: 10.1073/pnas.90.10.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuart DT, Upton C, Higman MA, Niles EG, McFadden G. A Poxvirus-Encoded Uracil DNA Glycosylase Is Essential for Virus Viability. Journal of Virology. 1993;67(5):2503–2512. doi: 10.1128/jvi.67.5.2503-2512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez JF, Kahn JS, Esteban M. Molecular-Cloning, Encoding Sequence, and Expression of Vaccinia Virus Nucleic Acid-Dependent Nucleoside Triphosphatase Gene. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(24):9566–9570. doi: 10.1073/pnas.83.24.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rempel RE, Traktman P. Vaccinia Virus-B1 Kinase - Phenotypic Analysis of Temperature-Sensitive Mutants and Enzymatic Characterization of Recombinant Proteins. Journal of Virology. 1992;66(7):4413–4426. doi: 10.1128/jvi.66.7.4413-4426.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin SQ, Wen C, Broyles SS. The Vaccinia Virus B1r Gene-Product Is a Serine Threonine Protein-Kinase. Journal of Virology. 1992;66(5):2717–2723. doi: 10.1128/jvi.66.5.2717-2723.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin K, Ricciardi RP. A rapid plate assay for the screening of inhibitors against herpesvirus DNA polymerases and processivity factors. Journal of Virological Methods. 2000;88(2):219–225. doi: 10.1016/s0166-0934(00)00190-7. [DOI] [PubMed] [Google Scholar]
- 26.Dorjsuren D, Burnette A, Gray GN, Chen XL, Zhu WM, Roberts PE, Currens MJ, Shoemaker RH, Ricciardi RP, Sei S. Chemical library screen for novel inhibitors of Kaposi’s sarcoma-associated herpesvirus processive DNA synthesis. Antiviral Research. 2006;69(1):9–23. doi: 10.1016/j.antiviral.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Silverman JEY, Ciustea M, Shudofsky AMD, Bender F, Shoemaker RH, Ricciardi RP. Identification of polymerase and processivity inhibitors of vaccinia DNA synthesis using a stepwise screening approach. Antiviral Research. 2008 doi: 10.1016/j.antiviral.2008.05.010. doi:10.1016/j.antiviral.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berridge MV, Herst PM, Tan AS, El-Gewely MR. Biotechnology Annual Review. Vol. 11. Elsevier; 2005. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction; pp. 127–152. [DOI] [PubMed] [Google Scholar]
- 29.Kern ER. In vitro activity of potential anti-poxvirus agents. Antiviral Research. 2003;57(12):35–40. doi: 10.1016/S0166-3542(02)00198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirabelli CK, Bartus H, Bartus JOL, Johnson R, Mong SM, Sung CP, Crooke ST. Application of a Tissue-Culture Microtiter Test for the Detection of Cyto-Toxic Agents from Natural-Products. Journal of Antibiotics. 1985;38(6):758–766. doi: 10.7164/antibiotics.38.758. [DOI] [PubMed] [Google Scholar]
- 31.Pogo BGT, Dales S. Biogenesis of Vaccinia - Separation of Early Stages from Maturation by Means of Hydroxyurea. Virology. 1971;43(1):144–151. doi: 10.1016/0042-6822(71)90232-7. [DOI] [PubMed] [Google Scholar]
- 32.McDonald WF, Klemperer N, Traktman P. Characterization of a processive form of the vaccinia virus DNA polymerase. Virology. 1997;234(1):168–175. doi: 10.1006/viro.1997.8639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.