

SUMMARY
Cerebral cavernous malformations (CCM) are vascular malformations of the brain that lead to cerebral hemorrhages. In 20% of CCM patients, this results from an autosomal dominant condition caused by loss-of-function mutations in one of the three CCM genes. High expression levels of the CCM genes in the neuroepithelium indicate that CCM lesions might be caused by a loss of function of these genes in neural cells rather than in vascular cells. However, their in vivo function, particularly during cerebral angiogenesis, is totally unknown. We developed mice with constitutive and tissue-specific CCM2 deletions to investigate CCM2 function in vivo. Constitutive deletion of CCM2 leads to early embryonic death. Deletion of CCM2 from neuroglial precursor cells does not lead to cerebrovascular defects, whereas CCM2 is required in endothelial cells for proper vascular development. Deletion of CCM2 from endothelial cells severely affects angiogenesis, leading to morphogenic defects in the major arterial and venous blood vessels and in the heart, and results in embryonic lethality at mid-gestation. These findings establish the essential role of endothelial CCM2 for proper vascular development and strongly suggest that the endothelial cell is the primary target in the cascade of events leading from CCM2 mutations to CCM cerebrovascular lesions.
INTRODUCTION
Cerebral cavernous malformations (CCM) are slow-flow vascular anomalies characterized by densely packed vascular sinusoids embedded in a collagen matrix without intervening neural tissue (Russell and Rubinstein, 1989). These clusters of vascular sinusoids (also called caverns) are lined by a thin endothelium and by rare subendothelial cells (Clatterbuck et al., 2001). An absence of tight junctions between endothelial cells (ECs) has been reported in CCM lesions upon ultrastuctural examination. Most of these lesions are located within the central nervous system (CNS) but they may also affect the retina (for a review, see Labauge et al., 2007). Clinical onset is generally around 20–30 years of age but symptoms can start in early infancy and in old age. The most common manifestations include headaches, seizures and focal neurological deficits caused by cerebral hemorrhages.
From large series studies, the prevalence of CCM in the general population has been estimated to be close to 0.1–0.5% and CCM bleeding is involved in more than 10% of young patients who show intracerebral hemorrhage (Otten et al., 1989; Moussa et al., 2006). Both sporadic and familial autosomal dominant forms of the disorder have been identified (Rigamonti et al., 1988; Labauge et al., 1998; Labauge et al., 2007). Familial CCM (FCCM) cases are characterized by the presence of multiple lesions upon cerebral magnetic resonance imaging (MRI) and the major risk endured by FCCM patients is the recurrence of cerebral hemorrhages.
Our group and others have identified three CCM genes namely CCM1/KRIT1, CCM2/MGC4607 (also called malcavernin) and CCM3/PDCD10 (Craig et al., 1998; Laberge Le Couteulx et al., 1999; Liquori et al., 2003; Denier et al., 2004; Bergametti et al., 2005). The mutations detected in CCM patients are loss-of-function mutations. It has been suggested that a Knudson two-hit mechanism is likely to be involved in CCM pathophysiology, as reported previously in some other vascular conditions (Brouillard et al., 2002). This is based on the observed autosomal dominant pattern of CCM inheritance and the presence of multiple lesions in FCCM, contrasting with the detection of a single lesion in nonhereditary cavernous angiomas. In addition, recent data showing biallelic mutations in CCM genes (germline and somatic) have been reported (Gault et al., 2005; Akers et al., 2008; Pagenstecher et al., 2008).
The CCM1 gene encodes KRIT1, a 736-amino acid protein containing three ankyrin domains and a FERM (protein 4.1, ezrin, radixin, moesin) domain. CCM2/MGC4607 encodes a protein containing a phosphotyrosine-binding domain (PTB) and CCM3/PDCD10 encodes a protein, without a known conserved functional domain, which was shown recently to interact with STK serine threonine kinases (Ma et al., 2007; Voss et al., 2007). Ccm genes have highly similar patterns of expression in both the embryo and adult mouse. Within the CNS, Ccm transcripts are detected mostly in neuronal cell layers (Petit et al., 2006; Plummer et al., 2006). By embryonic day (E)14.5, moderate labeling of all three transcripts can be observed in the large arterial and venous blood vessels and in the heart. Ccm1 mRNA is not detected in small cerebral vessels at any stage of development. Ccm2 and Ccm3 mRNAs are weakly, and only transiently, detected within meningeal and parenchymal cortical vessels at P8. Regarding CCM protein expression in tissue sections, controversial results have been published that are most likely explained by a lack of antibody specificity. Inter-relations between neural and vascular cells are crucial for proper cerebrovascular development and, therefore, the mostly neuronal expression pattern of CCM genes raises the question of which cell type, neuronal or vascular, underlies the vascular lesions observed in CCM patients (Proctor et al., 2005).
Recent in vitro data suggest strongly that CCM proteins are scaffold proteins that exist as a complex in cells (Zhang et al., 2001; Zawistowski et al., 2002; Zawistowski et al., 2005; Hilder et al., 2007). CCM1 has been shown to interact with (1) the PTB domain of CCM2 protein, (2) RAP1A, a small Ras-like GTPase (Serebriiski et al., 1997; Glading et al., 2007), and (3) ICAP1, a protein that binds to the cytoplasmic tail of integrin β1 (Zhang et al., 2001; Zawistowski et al., 2002). The yeast ortholog of CCM2, OSM (osmosensing scaffold for MEKK3), has been identified as a scaffold protein that interacts with kinases involved in the p38 mitogen-activated kinase pathway in response to osmolarity stress (Uhlik et al., 2003). It also interacts with CCM1 and CCM3, as well as with MEKK3, RAC1, RIN2 (a protein shown to regulate E-cadherin internalization), actin and tubulins. In addition, CCM2 and CCM3 bind to phosphatidyl insositol phospholipids (Hilder et al., 2007). Altogether, these data suggest that the three CCM proteins are members of a larger signaling complex that is involved in cell-cell junction homeostasis and cytoskeleton remodeling.
These biochemical data have provided useful insights into CCM protein function. However, their in vivo function, particularly during cerebral angiogenesis, remains obscure. In the zebrafish embryo, Ccm1 and Ccm2 are expressed mostly in the brain, the notochord and the posterior cardinal vein. Their inactivation leads to an early death with massive dilation of both the heart and large venous vessels (Mably et al., 2006; Hogan et al., 2008). Constitutive deletion of CCM1 in the mouse leads to abnormal arterial morphogenesis including both enlargement and narrowing of large trunk arteries and ultimately to mid-gestation death (Whitehead et al., 2004).
Here, we used constitutive and tissue-specific inactivation of CCM2 to investigate its role in mouse vascular development. We showed that (1) neuroepithelial expression of CCM2 is dispensable for proper vascular development and (2) endothelium-specific inactivation of CCM2 results in embryonic lethality at mid-gestation, abnormal angiogenesis and massive heart and blood vessel defects.
RESULTS
Targeting Ccm2 in mice
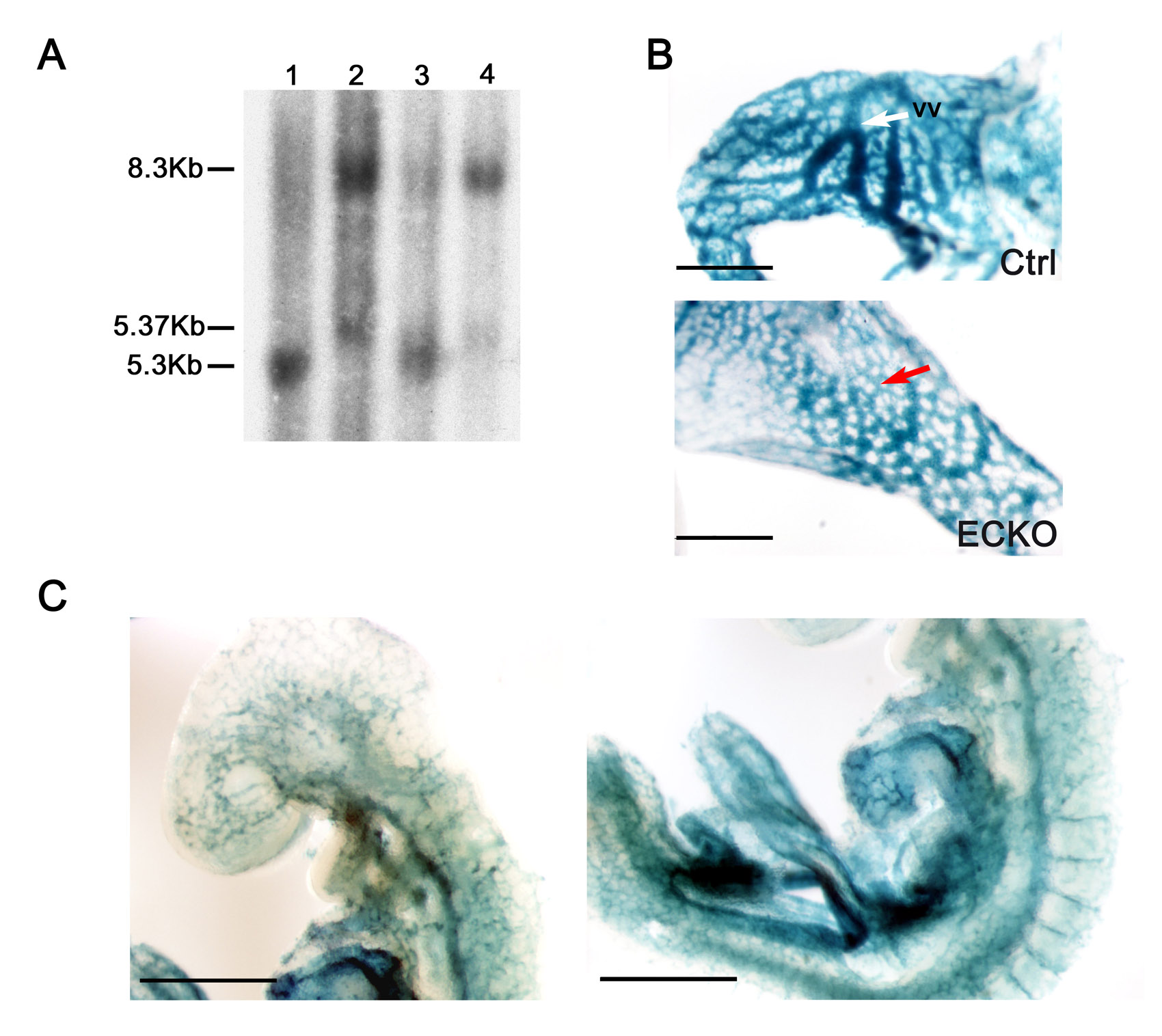
We targeted the Ccm2 locus in embryonic stem (ES) cells by using a linearized construct containing exons 3 and 4 of the Ccm2 gene, which encode most of the PTB (Fig. 1A and Methods). Mutations leading to transcripts with these two exons deleted have been detected in several CCM patients and result in a frameshift and a premature stop codon, leading to partial mRNA decay (Denier et al., 2004) (supplementary material Fig. S1). Ccm2 gene targeting was confirmed by Southern blot on ES cells and mice, and by northern blot and western blot analysis (Fig. 1B–D; supplementary material Fig. S2).
Fig. 1.

Targeted disruption of the mouse Ccm2 gene. (A) Schematic drawing of the Ccm2 gene targeting strategy. The Ccm2 locus was targeted by homologous recombination using a construct containing three loxP sites (open triangles) flanking a hygromycin resistance cassette and Ccm2 exons 3 and 4 (black boxes). The relevant restriction sites (X and H), primers for genotyping (F, forward; R, reverse) and probes for Southern blotting (P1 and P2) are indicated. (B,C) Validation of Ccm2 targeting by Southern blot analysis. 10 μg of genomic DNA from cells, mouse tissues or embryos (as indicated) was digested with XbaI and hybridized with the 5′ external probe, P1. (B) Identification of the ES clone and mice (M1 and M2) containing the targeted Ccm2 allele (indicated as the trilox allele). The 5.3 kb and the 8.3 kb DNA fragments represent the wild-type Ccm2 and targeted alleles, respectively. (C) Southern blot analysis for Ccm2 in either wild-type animals (WT), or wild-type embryos (+/+), heterozygous Ccm2+/– embryos (+/–) or homozygous deleted embryos (null). The 5.3 kb and the 8.2 kb DNA fragments represent the wild-type Ccm2 and the deleted alleles, respectively. (D) Western blot analysis of CCM2 protein expression using 15 μg of total protein lysates from E9.5 embryos (Ccm2+/+ and Ccm2-null embryos). Protein lysate from HEK cells transfected with the Ccm2 cDNA was used as a positive control (200 ng). Immunoblotting for α-tubulin on the same blot was performed as a control for the amount of protein loaded. F, forward primer; H, HindIII; Hyg, hygromycine; P, probe; R, reverse primer; X, XbaI.
Heterozygous Ccm2+/– and Ccm2+/flox mice and homozygous floxed (Ccm2flox/flox) mice were viable, fertile and indistinguishable from control littermates. A similar level of CCM2 protein was detected in wild-type (WT) and floxed mice (data not shown).
Ubiquitous CCM2 ablation is lethal in embryonic mice
When Ccm2+/– mice were interbred, we did not obtain any Ccm2-null pups (from a total of 96 offspring) (Table 1), strongly suggesting in utero lethality. During embryogenesis, all of the different genotypes were recovered at a Mendelian ratio (Table 1). The absence of the CCM2 protein in null embryos was confirmed by western blots (Fig. 1D). No phenotypic difference between embryos was detected upon dissection at E8.5 (supplementary material Fig. S3). At E9.5, mutant mice were recognized because their very pale yolk sacs (YSs) showed a wrinkled appearance (supplementary material Fig. S3). Homozygous null embryos showed a developmental delay with failure to complete turning and signs of resorption. Most Ccm2-null embryos had a massive pericardial edema at E9.5 and their hearts, which were sometimes still beating, were showing a retarded S-shape. No Ccm2-null embryos were found alive at E10.5; all of them were already in resorption (supplementary material Fig. S3). Altogether, our results show that constitutive deletion of Ccm2 leads to cardiovascular failure and mid-gestation embryonic death.
Table 1.
Genotype of the progeny from Ccm2+/– intercrosses
|
Ccm2 genotype* |
||||
|---|---|---|---|---|
| Age | Total | +/+ | +/– | Null |
| E8.5 | 71 | 13 (19) | 40 (56) | 18 (25) |
| E9.5 | 60 | 18 (30) | 28 (47) | 14 (23) |
| E10.5 | 33 | 8 (24) | 16 (49) | 9† (27) |
| P21 | 96 | 36 (37) | 60 (63) | 0 |
Numbers are given with percentage in brackets;
all 9 null embryos were already in resorption.
Cerebrovascular development does not require expression of CCM2 in neuroepithelial precursor cells
To assess whether CCM2 expression in neuroepithelial cells is required for proper vascular development in the mouse brain, we specifically ablated CCM2 in these cells by using a nestin-Cre line that expresses Cre under the control of the neural enhancer element of the nestin promoter (Tronche et al., 1999). This transgene drives Cre-mediated recombination in neural precursor cells giving rise to both neurons and glia, but does not drive recombination in ECs (Graus-Porta et al., 2001). Briefly, we sequentially crossed the transgenic nestin-Cre line with Ccm2+/–and Ccm2 floxed mice to generate NPKO mice (nestin-Cre; Ccm2–/flox). We obtained NPKO mice with the expected Mendelian ratio at weaning (27% from a total of 176 offspring). NPKO mice were fertile and indistinguishable from their control littermates at up to 1 year of age. They did not have any significant weight difference or detectable neurological defect.
The neural-specific recombination of the floxed allele and the absence of the CCM2 protein within the brain of NPKO mice were confirmed at P8 (Fig. 2A,B). In addition, X-Gal staining performed on E12.5 embryos, obtained from crosses between nestin-Cre; Ccm2 floxed mice with a Rosa26R reporter line (Soriano, 1999) (Fig. 2C), showed specific expression of Cre recombinase in neural cells, in agreement with previously published data (Graus-Porta et al., 2001; Proctor et al., 2005).
Fig. 2.

Conditional deletion of CCM2 from neuroglial precursors does not lead to major cerebrovascular defects. (A-C) Analysis of the specific inactivation of CCM2 in the neuroglial compartment. (A) Southern blot analysis using 12 μg of genomic DNA extracted from tissues at P8. DNA was digested by HindIII and hybridized with the external radiolabeled 3′ probe P2. The floxed allele (8.2 kb) in NPKO was recombined specifically within the brain of NPKO mice. A Ccm2–/flox brain from a littermate was used as a control for the absence of recombination (second lane from the left). The 6.3 kb and the 8.2 kb DNA fragments represent the Ccm2-deleted and floxed alleles, respectively. Note that the DNA fragment in the first lane is slightly lower than the floxed fragment and corresponds to the 8.13 kb wild-type allele. (B) Western blot analysis of CCM2 protein expression within the brain (100 μg protein lysates) and the lung (70 μg protein lysates) from NPKO mice and a control littermate at P8. CCM2 protein was not detected in NPKO brain lysates. Protein lysate from Ccm2-transfected HEK cells was used as a positive control (200 ng). Note that the second lane of the blot is free of sample. Immunoblotting for α-tubulin on the same blot was performed as a loading control. (C) β-galactosidase expression analysis on a E12.5 embryo obtained after crossing a nestin-Cre; Ccm2 floxed animal with a Rosa26R reporter line (genotype of the embryo: nestin-Cre; Rosa26R; Ccm2+/flox). Note that the blood vessels (red arrow) are not blue, demonstrating an absence of recombination in the ECs. (D,E) Analysis of the brains from NPKO and control mice. (D) Analysis of 2 mm-thick brain coronal sections from 2-month-old NPKO and control animals under a dissecting microscope, showing slices of the cerebrum (left panels) and the cerebellum (right panels). (E) Hematoxylin and eosin (H&E) staining on 10 μm paraffin-embedded brain sections from NPKO or control mice at P19. B, brain; C, control; cc, corpus callosum; co, cortex; cpu, caudate putamen; Fb, forebrain; fd, fascia dentata; H, heart; Hb, hindbrain; hi, hippocampus; K, kidney; Li, liver; Lu, lung; Mb, midbrain; Nt, neural tube; ob, olfactory bulb; S, spleen; T, toe; v, ventricle.
Analysis of NPKO mice at P8 (n=8), P19 (n=7), 2 months of age (n=11) and 6 months of age (n=8) did not reveal any macroscopic anomaly in major organs. Analysis of 2 mm coronal slices of brain under a dissecting microscope did not show any cerebral hemorrhage in NPKO mutants (Fig. 2D). Moreover, histological and immunostaining analyses of the vasculature did not show any gross brain structure anomaly or any obvious vascular defect within the NPKO brain (Fig. 2E and data not shown).
Altogether, these data strongly suggest that, in spite of Ccm2 being expressed essentially in neuronal layers within the CNS, ablation of CCM2 from neuroglial precursor cells does not lead to major cerebrovascular defects or obvious cerebral anomalies.
Endothelial-specific ablation of CCM2 leads to mid-gestation embryonic death
In order to determine whether the disruption of CCM2 in ECs leads to vascular defects, we generated mice with an endothelium-restricted deletion of CCM2, by using a previously well-characterized Tie2-Cre transgenic mouse (Kisanuki et al., 2001). Intercrosses between Tie2-Cre; Ccm2+/– and Ccm2 floxed mice did not produce any Tie2-Cre; Ccm2–/flox pups (ECKO) at weaning (from a total of 77 offspring) (Fig. 3A). During embryogenesis, all the genotypes were obtained at the expected Mendelian ratios at the different stages tested.
Fig. 3.
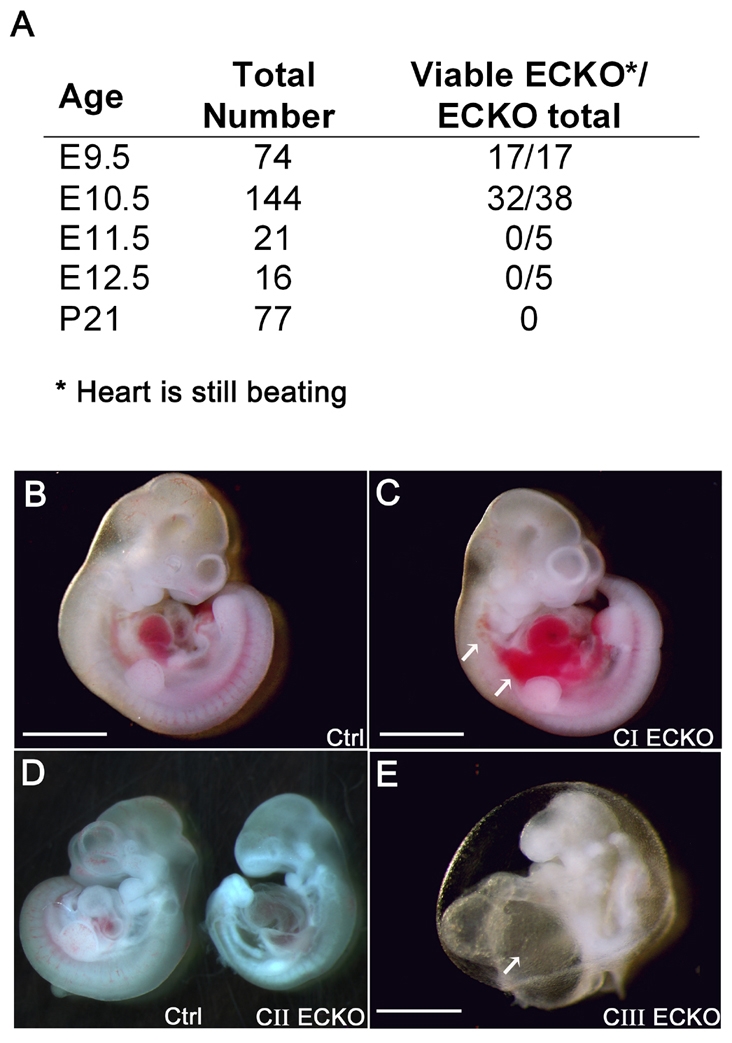
Conditional deletion of CCM2 from endothelial cells leads to a variable phenotype at E10.5 and finally to death. (A) ECKO progeny resulting from intercrosses between Tie2-Cre; Ccm2+/– and homozygous Ccm2 floxed mice. Viability was assessed by the presence of heartbeats. At E11.5 and E12.5, no ECKO embryos were found alive (genotyping was performed on embryos under resorption). (B-E) ECKO embryo classes reflecting the severity of the phenotype at E10.5. (B) Control embryo. (C) Class I ECKO embryo (CI) without developmental delay. Note the hemorrhage in the trunk (white arrows). (D) Class II ECKO embryo (CII, right) with a control littermate (left). (E) Class III ECKO embryo (CIII) showing failure to complete turning and an enormous heart with a massively enlarged atrium (white arrow). Note that this embryo shows signs of resorption. Bars, 1 mm (B,C); 500 μm (E).
EC-specific Cre expression was confirmed in the YS and embryo vasculature at E9.5, and Cre-mediated recombination of the Ccm2 floxed allele was observed in the endothelium (supplementary material Fig. S4 and S5).
At E8.5, ECKO embryos could not be distinguished from controls (data not shown). At E9.5, most of the ECKO embryos were comparable in size to the control embryos and did not show any obvious anomaly. However, 10% of them were retarded and failed to complete turning, showing a phenotype similar to the one described in the constitutive null embryos at this stage (supplementary material Fig. S3 and data not shown). At E10.5, ECKO embryos exhibited phenotypes of variable severity, which we assigned to three classes (Fig. 3B–E). Class I included 19% of ECKO embryos dissected at this stage and corresponded to ECKO embryos showing no significant size difference compared with controls (Fig. 3C). A slight developmental delay restricted to the craniofacial part of the embryos was noticed in several ECKO embryos of this group. Most ECKO embryos were assigned to class II (58%); they were characterized by a marked general developmental delay (Fig. 3D). Class III included the remaining 23% of ECKO embryos, which showed general growth arrest, a failure to complete turning and/or signs of resorption; the heart was still beating in most of these embryos (Fig. 3E). At E10.5, most ECKO embryos had a pericardial edema that was more severe in class II and III embryos. Half of the mutants had signs of hemorrhage in the pericardial cavity and in the trunk. Interestingly, we never observed any hemorrhage within the upper part of the embryo and the head. No ECKO embryos were found alive at E11.5 and E12.5.
Abnormal remodeling of the extra-embryonic vasculature in ECKO embryos
The blood islands of the YS are the first sites of vasculogenesis and their fusion gives rise to the primitive vascular plexus. In wild-type embryos at around E9, the initial honeycomb-like pattern of the YS vasculature is remodeled into a more mature and branched pattern of vitelline vessels (Risau, 1997). YSs of ECKO embryos were wrinkled, similar to those of Ccm2-null embryos, allowing the identification of mutant embryos during dissection. YS vessels remained in the honeycomb pattern and failed to develop into large vitelline vessels (Fig. 4B; supplementary material Fig. S4B). Endodermal and mesodermal layers were less frequently connected compared with in control embryos and almost no blood cells were detected in the immature vessels (Fig. 4C,D). These data suggest that vasculogenesis occurred normally in ECKO YSs but that angiogenesis was impaired, with severe remodeling defects.
Fig. 4.

Vascular defects in extra-embyonic tissues in ECKO embryos at E10.5. (A,B) Newly dissected E10.5 ECKO YSs are easily distinguishable from control ones owing to their wrinkled surface and their immature vasculature, which remains in a honeycomb pattern. (C,D) H&E staining of E10.5 YS cross sections. Compared with the control YSs (C), the endodermal (e) and mesodermal (m) layers are rarely connected in the ECKO YSs [red arrows in panel (D)]. (E,F) Placenta histology in control (E) or ECKO (F) embryos at E10.5. Note the rare embryonic nucleated red blood cells (arrowheads) in the labyrinthine layer in the ECKO placenta, and the presence of maternal red blood cells (arrows). Cp, chorionic plate; Gc, giant cells; Lb, labyrinthine trophoblaste; Md, maternal decidua; Sp, spongiotrophoblast.
Vascular defects were also observed in the placental labyrinth. At E10.5, the different layers of the placenta were difficult to define and appeared poorly organized in ECKO embryos (Fig. 4F). Further, embryonic blood vessels did not invade the labyrinthine layer to the same extent as seen in control placentas.
Embryonic vasculature in ECKO embryos is abnormal and shows impaired recruitment of vascular smooth muscle cells
Visualization of the embryonic vasculature at E9.5, using whole-mount platelet endothelial cell adhesion molecule (PECAM) staining, revealed the presence of all major blood vessels, paired dorsal aortae (DA), branchial arch arteries, cardinal veins and intersomitic vessels in ECKO embryos, suggesting that vasculogenesis occurred normally.
However, in E9.5 ECKO embryos, the DA already had an irregular appearance and were thinner than in controls (Fig. 5B). At E10.5, the vascular phenotype was even more severe; the DA were now highly irregular in appearance and had narrow lumens (Fig. 5F,J). In the caudal region, DA failed to fuse in most ECKO embryos (except in a few class I embryos) and remained present as two stenotic DA (Fig. 5H,L). Interestingly, we never observed DA dilations or arterio-venous malformations. With regard to venous vessels, we observed an important dilation of the large vessels connecting to the heart, including the common cardinal veins and the sinus venosus (Fig. 5F,J). In the head, the cephalic plexus failed to remodel into a hierarchical branched network and ECKO embryos exhibited a rather coarse vascular network with poorly organized vessels, making it difficult to distinguish the internal carotid artery or primary head veins, which seemed to narrow in some ECKO embryos (Fig. 5D and data not shown). However, vessel sprouting into the neural tube did occur in ECKO embryos (data not shown). Intersomitic vessels were also less organized and their limits were poorly defined compared with control embryos.
Fig. 5.

Conditional deletion of CCM2 from endothelial cells results in major vascular defects in ECKO embryos. (A-H) Whole-mount PECAM staining of control (left panels) and ECKO embryos (right panels) at E9.5 (A,B) and E10.5 (C-H). (I-L) E10.5 PECAM-stained embryo sections after counterstaining with eosin. The presence of PECAM-positive DA, somitic vessels and branchial arch arteries (numbered in red) at E9.5 demonstrates the occurrence of vasculogenesis (A,B). However, at E10.5 (C-H), the arterial and venous blood vessels are abnormal in ECKO embryos and the internal carotid artery is difficult to distinguish [red arrow (D)]. Further, DA are highly irregular in appearance and common cardinal veins are enlarged (F,J). In some animals, the caudal part of the DA fails to fuse (red arrows (H,L)]. (M-P) Double staining for PECAM [red (M,N)] and SMA [green (O,P)] on E10.5 DA sections from the trunk. Note the impaired recruitment of vascular smooth muscle cells/pericytes in the DA of ECKO embryos (P). CCV, common cardinal vein; DA, dorsal aorta; ICA, internal carotid artery; LB, limb bud; NT, neural tube; S, somite; Bars, 500 μm (A,B); 50 μm (M-P).
To further explore the angiogenic defects of ECKO embryos, we used smooth muscle actin immunostaining to search for the presence of vascular smooth muscle cells/pericytes. In control embryos, mural cell recruitment could be detected at E9.5 and DA sections were clearly SMA positive at E10.5 in both the trunk and the caudal region (Fig. 5O and data not shown). In contrast, mural cells were hardly detectable in the DA of ECKO embryos although SMA staining was detected in the somites and in the heart (Fig. 5P; Fig. 6L and data not shown). Only the class I ECKO embryos showed some rare mural cell recruitment in the DA (data not shown).
Fig. 6.

Cardiac defects in ECKO embryos. (A-D) Hearts from control (A) and ECKO embryos (B-D) after whole-mount PECAM staining at E10.5. (E-J) Heart sections from PECAM-stained control (E) and ECKO (F) embryos, counterstained with eosin, showing the common atrial chamber (a) and the common ventricular chamber (v). The black and red boxes in (E,F) are shown enlarged in (G,H) and (I,J), respectively. (G,H) A reduction of cells was observed in the atrioventricular canal in the ECKO heart. (I,J) Ventricular trabeculations are strongly reduced in the ECKO heart (J). (K,L) Double staining for PECAM (red) and SMA (green) showing the atrial wall from a control (K) and an ECKO (L) embryo. Sections were counterstained with DAPI. Bars, 100 μm (E,F); 25 μm (G-J); 20 μm (K,L).
In order to explore whether the Ccm2 deletion affects arterial identity, we examined the arterial expression of an early arterial-specific molecular EC marker, the transmembrane ligand ephrin B2 (Efnb2), prior to establishment of aortal blood flow at E8.5, using an Efnb2-tau-lacZ reporter transgenic mouse (Wang et al., 1998) and did not detect any difference between control and Ccm2-null embryos (supplementary material Fig. S6).
Altogether, our results suggest that EC-specific ablation of CCM2 does not affect vasculogenesis but severely hampers angiogenesis and the remodeling of capillary, arterial and venous vessels, and that it is associated with a defect in mural cell recruitment.
Heart defects in ECKO embryos
Although most ECKO embryo hearts were still beating at E10.5 (the hearts of a few class III embryos were not), they were enlarged and showed edematous pericardial swelling and bleeding, a hallmark of cardiac failure (Fig. 3C–E). In class I and some class II ECKO embryos, the size of the heart, assessed on an atrioventricular axis, relative to the size of the embryo from the hindbrain/midbrain boundary to the rostral part of the limbud, was about 17% larger than in control embryos. Chamber specification appeared normal and, at this stage, ECKO embryos showed both a common atrial and a common ventricular chamber (Fig. 6F).
Several embryos had an enlarged outflow tract and almost all of them had an enlarged atrium (Fig. 6A–D). In most severe cases, the massively enlarged atrium and sinus venosus led to distortion of the embryo. However, the atrial wall of ECKO embryos was comparable to control embryos, with a single layer of endocardial cells surrounded by a few layers of cardiomyocytes, including a monolayer of SMA-positive cells (Fig. 6F,L). Interestingly, the dilations that were seen in the atrial chamber of the heart, as well as in the venous system, could not be correlated to an increase in the proliferative rate of ECs, as determined by double labeling with antibodies for PECAM and phosphorylated histone 3 (data not shown).
Ventricular trabeculations were strongly reduced with, in some extreme cases, detachment of the endocardial cells from the myocardium (Fig. 6J and data not shown). This much thinner ventricular wall could be responsible for blood cells leaking into the pericardial cavity, which was observed in some ECKO embryos. In addition, there was a paucity of cells in the cushions in the atrioventricular canal (Fig. 6H), suggesting a reduction in the ability of endocardial cells to invade the cardiac jelly and undergo endocardial-mesenchymal transformation (Conway et al., 2003).
DISCUSSION
Loss-of-function mutations in the CCM2 gene in humans lead to cerebrovascular malformations, causing recurrent brain hemorrhages. Here, we demonstrate that, in spite of CCM2 being predominantly expressed in the neuronal layers within the CNS, deletion of CCM2 from neuroglial precursor cells in mice does not have a major phenotypic effect. In contrast, deletion of CCM2 from ECs leads to a very severe vascular phenotype. Vasculogenesis appears normal in ECKO embryos, contrasting with marked angiogenesis remodeling abnormalities in the extra-embryonic and embryonic vasculature that lead to major heart defects and both arterial and venous defects. These data establish the role of endothelial CCM2 in angiogenesis.
In constitutive null embryos, lethality occurred one embryonic day earlier than in ECKO embryos. Further, whilst all null embryos are already in resorption at E10.5, only 15% of ECKO embryos are dead at this stage. This delayed lethality might be explained by the incomplete and progressive endothelial-specific recombination induced by the Tie2-Cre cell line. Kisanuki et al. showed that EC-specific recombination induced by this Tie2-Cre line starts at E7.5 in some blood islands but is still not complete at E9.5. However, we cannot exclude a role for CCM2 in other cell types, such as in vascular smooth muscle cells, which might also explain the more severe phenotype of constitutive null embryos.
The neural expression of Ccm2 and other Ccm genes suggested that CCM vascular lesions could be secondary to neural defects. However, NPKO mice did not show neurological symptoms and we did not observe any vascular lesion within the brain of NPKO mice, up to 6 months of age. Interestingly, constitutive ablation of CCM1 did not lead to neural lesions in Ccm1–/– embryos (Whitehead et al., 2004). Further analysis of NPKO mice would be required to search for subtle neuropathological lesions that might affect the cerebral cortex and to investigate the functional consequences of a complete loss of CCM2 in neurons.
Some of the vascular defects described here in ECKO embryos are reminiscent of those reported in embryos with constitutive CCM1 inactivation (Whitehead et al., 2004). In Ccm1–/– embryos, vasculogenesis seems intact but angiogenesis is severely compromised. Ccm1–/– embryos die at E10.5 and show multiple arterial morphogenesis defects including a variable narrowing of both branchial arch arteries and the proximal dorsal aorta, as well as heart defects including atrial enlargement and signs of cardiovascular failure. In addition, Ccm1–/– embryos show a defect in mural cell recruitment, similar to that observed in our ECKO embryos. However, the main phenotype reported in Ccm1–/–embryos is an extensive vascular dilation of the dorsal and caudal aortae, and of the cranial and intersomitic vessels, which was never observed in our ECKO embryos. These defects have been associated with an increase in the proliferative rate of ECs of the dilated dorsal aortae and a downregulation of arterial-specific markers including Efnb2, neither of which was observed in ECKO embryos. Interestingly, no dilation of the venous compartment was reported in Ccm1–/– embryos, contrasting with the dilation of the common cardinal vein and sinus venosus in CCM2 ECKO embryos and dilation of the major large venous vessels in ccm1–/– zebrafish embryos (Hogan et al., 2008).
Recessive loss-of-function mutations in santa and valentine, the respective orthologs of CCM1 and CCM2 in zebrafish, lead to similar and major cardiac and vascular defects (Mably et al., 2006; Hogan et al., 2008). The cardiac phenotype observed in these mutants is also highly similar to that observed in heart of glass mutants (Mably et al., 2003) and is characterized by a massive dilation of the heart chambers, which show only one layer of cardiomyocytes instead of the two or three layers present in wild-type embryos. Enlargement of the atrium is one of the main phenotypic features in ECKO embryos; however, we did not observe any anomaly in the number of cell layers composing the atrial wall of these embryos. The vascular phenotype of Ccm1–/–embryos, and to a lesser degree ccm2–/– zebrafish embryos, is characterized by a progressive and massive dilation of venous vessels including the caudal vein and posterior cardinal vein, contrasting with the normal development of dorsal aorta and intersomitic vessels (Hogan et al., 2008). This venous defect is associated with a progressive thinning and spreading of ECs, which is EC cell-autonomous, strongly suggesting that CCM1 is involved in the control of EC shape. Interestingly, the authors did not observe an increase in EC proliferation rate or any modification in the expression of the arterial specification markers.
Blood vessel growth and differentiation involves a broad spectrum of genetic and physical signals, such as blood flow; one of the main challenges in the analysis of these various animal models is to differentiate the primary effects of mutations on the vasculature from the secondary effects of altered blood flow (Carmeliet, 2000; Lucitti et al., 2007). Indeed, most of the genes expressed in ECs are expressed in endocardial cells and when mutated, many of them, such as Ccm2, lead to cardiac defects. Altered blood flow may in turn influence extra-embryonic and embryonic vessel morphogenesis and remodeling (le Noble et al., 2004; Jones et al., 2008). Altered expression of arterial-specific markers such as Efbn2 can also be a secondary effect of abnormal blood flow (le Noble et al., 2004). Even very subtle differences in flow patterns may result in remodeling abnormalities (Lucitti et al., 2007). The current generation of mouse models, with their temporally controlled CCM2 deletion from blood vessels that begins at later stages, should help to elucidate this issue and perhaps help to obtain models that more closely recapitulate the human CCM phenotype and thus allow dissection of the relevant cellular and biochemical pathways involved in this condition.
METHODS
Targeting the Ccm2 gene and generation of CCM2 mouse mutants
ES cells (129 background) were electroporated with a linearized 10.1 kb construct, containing three loxP sites flanking a GFP-hygromycin resistance cassette (2.7 kb) and exons 3 and 4 of the Ccm2 gene (Fig. 1A). Hygromycin was used to select a positive ES clone, which was then used to inject C57BL/6 blastocysts. The chimeric tri-lox mice were bred with MeuCre40 mice (Leneuve et al., 2003) to remove the cassette and to obtain mosaic animals; these were then backcrossed with wild-type C57BL/6 mice. Ccm2+/– and Ccm2+/flox mutants, selected to be free of the Cre transgene, were backcrossed with C57BL/6 mice at least seven times to establish a C57BL/6 genetic background.
Southern blot analysis was developed to confirm transmission of the targeted Ccm2 locus. Two different DNA fragments that were external to the targeting construct, one at the 5′ end (245 bp) and one at the 3′ end (282 bp), were radiolabeled before being probed with XbaI- or HindIII-digested genomic DNA.
The absence of the CCM2 protein in mutants was confirmed by western blot. Total protein lysates were prepared from embryos or mouse tissues and lyzed in RIPA buffer that had been supplemented with protease inhibitors. The CCM2 protein (48.8 kDa) was detected using a purified polyclonal antibody, which was raised against a 15-amino acid peptide located at the C-terminal end of the CCM2 protein (peptide sequence: NH2−DDRSAPSEGDEWDRM-COOH; Ab made by Eurogentec). Lysates from full-lengh-CCM2-transfected HEK 293T cells were used as a positive control (200 ng). Western blots for α-tubulin (clone DM 1A, Sigma), performed on the same blots, were used as a control for the amount of protein loaded.
Offspring genotyping was analyzed by PCR on genomic DNA using the following primers (5′ to 3′): wild-type (177 bp) and floxed allele (263 bp), ATGGCACTTTGCTTTTCCAC and TGGCATCGAGAATCTTTCAA; deleted allele (285 bp), ATGGCACTTTGCTTTTCCAC and ACCCTGCTGTCTGAACAAGG; Cre (370 bp), TCCAATTTACTGAGCGTACACC and CGTTTTCTTTTCGGATCC.
Mouse lines
The nestin-Cre mice (Tronche et al., 1999), Tie2-Cre mice (Kisanuki et al., 2001), Rosa26R reporter line (Soriano, 1999) and Efnb2-tau-lacZ reporter transgenic mouse (Wang et al., 1998) have been described previously. Mice were all bred with a C57BL/6 background. The procedure followed in the care and euthanasia of study animals was in accordance with European Community standards on the care and use of laboratory animals (Ministère de l’Agriculture, France).
Histology
Whole brains were fixed by immersion overnight in 4% PFA, before being sectioned in 2 mm coronal slices using an acrylic brain matrix (electron microscopy services) for examination under a dissecting microscope. H&E staining was performed on paraffin-embedded sections.
Placentas and YSs were fixed in 4% PFA and embedded in paraffin. Histological analysis was performed on 7 μm sections using H&E staining.
Immunochemistry and immunofluorescence
The following antibodies were used for immunohistochemistry and/or immunofluorescence: anti-PECAM (1:100; MEC13.3, BD Pharmingen); FITC-conjugated anti-αSMA (1:100; Clone 1A4, Sigma); anti-phosphohistone H3 (1:200; Abcam); peroxidase-conjugated anti-rat IgG (H+L) (1:500; Jackson ImmunoResearch Laboratories); Alexa Fluor 594-conjugated anti-rat IgG (H+L) (1:200; Molecular Probe); FITC-conjugated anti-rabbit IgG (H+L) (1:100; Jackson ImmunoResearch Laboratories).
Immunochemistry on whole embryos was performed as described previously (Nagy et al., 2003) after neutralization of free aldehyde residues in PFA-fixed embryos by incubation with 100 mM glycin for 20 minutes.
X-gal staining on whole embryos was performed as described previously (Moessler et al., 1996). Sections (7 μm) of the paraffin-embedded embryos were counterstained with 1% eosin.
Immunochemistry on frozen sections was performed after fixation in aceton and permeabilization of the tissue. Sections were counterstained with DAPI and mounted in a fluorescent mounting medium (DakoCytomation).
Supplementary Material
Acknowledgments
This work was supported by the Agence Nationale pour la recherche grant ANR-07-MRAR-002-01 (to E.T.-L.), the Leducq grant 07 CVD 02 hemorrhagic stroke (to E.T.-L.), INSERM. G.B. has been partly supported by the PHRC grant AOR0301. N.P. was sequentially supported by Lefoulon Delalande and Fédération pour les Maladies Orphelines FMO and ‘Association Cavernomes France’ fellowships. We thank deeply A. Joutel from UMR-S 740, A. Eichmann from INSERM U833 and S.M. Meilhac from CNRS URA 2578 for very helpful discussions, and M. Arnoult for excellent technical help.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/content/2/3-4/168/suppl/DC1
REFERENCES
- Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. (2008). Biallelic somatic and germline mutations in cerebral cavernous malformations (CCM): evidence for a two-hit mechanism of CCM pathogenesis. Hum. Mol. Genet. December 16 [Epub ahead of print] [doi:10.1093/hmg/ddn430]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, et al. (2005). Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 76, 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibe M, Warman ML, Tan OT, Olsen BR, Vikkula M. (2002). Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am. J. Hum. Genet. 70, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. (2000). Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389–395 [DOI] [PubMed] [Google Scholar]
- Clatterbuck RE, Eberhart CG, Crain BJ, Rigamonti D. (2001). Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatr. 71, 188–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. (2003). What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis 35, 1–21 [DOI] [PubMed] [Google Scholar]
- Craig HD, Gunel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK, Ogilvy CS, Berg MJ, Crawford SC, Scott RM, et al. (1998). Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15–13 and 3q25.2–27. Hum. Mol. Genet. 7, 1851–1858 [DOI] [PubMed] [Google Scholar]
- Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, Benabid AL, Comoy J, Frerebeau P, Gilbert B, et al. (2004). Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 74, 326–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J, Shenkar R, Recksiek P, Awad IA. (2005). Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke 36, 872–874 [DOI] [PubMed] [Google Scholar]
- Glading A, Han J, Stockton RA, Ginsberg MH. (2007). KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 179, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. (2001). Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31, 367–379 [DOI] [PubMed] [Google Scholar]
- Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL, Wu CC. (2007). Proteomic identification of the cerebral cavernous malformation signaling complex. J. Proteome Res. 6, 4343–4355 [DOI] [PubMed] [Google Scholar]
- Hogan BM, Bussmann J, Wolburg H, Schulte-Merker S. (2008). ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum Mol Genet 17, 2424–2432 [DOI] [PubMed] [Google Scholar]
- Jones EA, Yuan L, Breant C, Watts RJ, Eichmann A. (2008). Separating genetic and hemodynamic defects in neuropilin 1 knockout embryos. Development 135, 2479–2488 [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230–242 [DOI] [PubMed] [Google Scholar]
- Labauge P, Denier C, Bergametti F, Tournier-Lasserve E. (2007). Genetics of cavernous angiomas. Lancet Neurol. 6, 237–244 [DOI] [PubMed] [Google Scholar]
- Labauge P, Laberge S, Brunereau L, Levy C, Tournier-Lasserve E. (1998). Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Societe Francaise de Neurochirurgie. Lancet 352, 1892–1897 [DOI] [PubMed] [Google Scholar]
- Laberge-le, Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E. (1999). Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat. Genet. 23, 189–193 [DOI] [PubMed] [Google Scholar]
- le Noble F, Moyon D, Pardanaud L, Yuan L, Djonov V, Matthijsen R, Breant C, Fleury V, Eichmann A. (2004). Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development 131, 361–375 [DOI] [PubMed] [Google Scholar]
- Leneuve P, Colnot S, Hamard G, Francis F, Niwa-Kawakita M, Giovannini M, Holzenberger M. (2003). Cre-mediated germline mosaicism: a new transgenic mouse for the selective removal of residual markers from tri-lox conditional alleles. Nucleic Acids Res. 31, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, et al. (2003). Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 73, 1459–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucitti JL, Jones EA, Huang C, Chen J, Fraser SE, Dickinson ME. (2007). Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 134, 3317–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Zhao H, Shan J, Long F, Chen Y, Chen Y, Zhang Y, Han X, Ma D. (2007). PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol. Biol. Cell 18, 1965–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mably JD, Mohideen MA, Burns CG, Chen JN, Fishman MC. (2003). heart of glass regulates the concentric growth of the heart in zebrafish. Curr. Biol. 13, 2138–2147 [DOI] [PubMed] [Google Scholar]
- Mably JD, Chuang LP, Serluca FC, Mohideen MA, Chen JN, Fishman MC. (2006). santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development 133, 3139–3146 [DOI] [PubMed] [Google Scholar]
- Moessler H, Mericskay M, Li Z, Nagl S, Paulin D, Small JV. (1996). The SM 22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development 122, 2415–2425 [DOI] [PubMed] [Google Scholar]
- Moussa R, Harb A, Menassa L, Risk T, Nohra G, Samaha E, Mohasseb G, Okais N, Awad I. (2006). Etiologic spectrum of intracerebral hemorrhage in young patients. Neurochirurgie 52, 105–109 [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. (2003). Manipulating The Mouse Embryo: A Laboratory Manual, pp. 666–669 Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Otten P, Pizzolato GP, Rilliet B, Berney J. (1989). 131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies. Neurochirurgie 35, 82–83; 128–131 [PubMed] [Google Scholar]
- Pagenstecher A, Stahl S, Sure U, Felbor U. (2008). A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum. Mol. Genet. December 16 [Epub ahead of print] [doi:10.1093/hmg/ddn420]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit N, Blecon A, Denier C, Tournier-Lasserve E. (2006). Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development. Gene Expr. Patterns 6, 495–503 [DOI] [PubMed] [Google Scholar]
- Plummer NW, Squire TL, Srinivasan S, Huang E, Zawistowski JS, Matsunami H, Hale LP, Marchuk DA. (2006). Neuronal expression of the Ccm2 gene in a new mouse model of cerebral cavernous malformations. Mamm. Genome 17, 119–128 [DOI] [PubMed] [Google Scholar]
- Proctor JM, Zang K, Wang D, Wang R, Reichardt LF. (2005). Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J. Neurosci. 25, 9940–9948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT, Spetzler RF. (1988). Cerebral cavernous malformations. Incidence and familial occurrence. N. Engl. J. Med. 319, 343–347 [DOI] [PubMed] [Google Scholar]
- Risau W. (1997). Mechanisms of angiogenesis. Nature 386, 671–674 [DOI] [PubMed] [Google Scholar]
- Russell DS, Rubinstein LJ. (1989). Pathology of Tumors of the Nervous System, pp. 730–736 Baltimore, MD: Williams and Wilkins [Google Scholar]
- Serebriiskii I, Estojak J, Sonoda G, Testa JR, Golemis EA. (1997). Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene 15, 1043–1049 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23, 99–103 [DOI] [PubMed] [Google Scholar]
- Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, Horne EA, Dell’Acqua ML, Johnson GL. (2003). Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5, 1104–1110 [DOI] [PubMed] [Google Scholar]
- Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD, Felbor U. (2007). CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8, 249–256 [DOI] [PubMed] [Google Scholar]
- Wang HU, Chen ZF, Anderson DJ. (1998). Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93, 741–753 [DOI] [PubMed] [Google Scholar]
- Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. (2004). Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 131, 1437–1448 [DOI] [PubMed] [Google Scholar]
- Zhang J, Clatterbuck RE, Rigamonti D, Chang DD, Dietz HC. (2001). Interaction between krit1 and icap1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum. Mol. Genet. 10, 2953–2960 [DOI] [PubMed] [Google Scholar]
- Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA. (2002). KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum. Mol. Genet. 11, 389–396 [DOI] [PubMed] [Google Scholar]
- Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA. (2005). CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 14, 2521–2531 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}