

Abstract
Mutations in genes encoding desmosomal proteins have been implicated in the pathogenesis of heart and skin diseases. This has led to the hypothesis that defective cell-cell adhesion is the underlying cause of injury in tissues that repeatedly bear high mechanical loads. In this study, we examined the effects of two different mutations in plakoglobin on cell migration, stiffness, and adhesion. One is a C-terminal mutation causing Naxos disease, a recessive syndrome of arrhythmogenic right ventricular cardiomyopathy (ARVC) and abnormal skin and hair. The other is an N-terminal mutation causing dominant inheritance of ARVC without cutaneous abnormalities. To assess the effects of plakoglobin mutations on a broad range of cell mechanical behavior, we characterized a model system consisting of stably transfected HEK cells which are particularly well suited for analyses of cell migration and adhesion. Both mutations increased the speed of wound healing which appeared to be related to increased cell motility rather than increased cell proliferation. However, the C-terminal mutation led to dramatically decreased cell-cell adhesion, whereas the N-terminal mutation caused a decrease in cell stiffness. These results indicate that different mutations in plakoglobin have markedly disparate effects on cell mechanical behavior, suggesting complex biomechanical roles for this protein.
Keywords: cell mechanics, cell adhesion, junctional proteins
Introduction
Cell mechanical properties are known to be governed by several factors including the cytoskeleton and cytoskeletal-regulating proteins. Additionally, growth factors and cytokines are known to induce changes in cell migration and wound healing. Recently, however, attention has been directed towards the regulation of cell mechanical properties by junctional molecules. For example, it has been shown that mutations in the gap junction protein connexin26 alter wound healing in skin cells (Djalilian et al. 2006). Other studies have demonstrated that junctional-adhesion molecule-A regulates cell migration, microtubule stability and cell-substrate adhesion in endothelial cells (Bazzoni et al. 2005; Huang et al. 2006). Genetically engineered alterations in plakoglobin have been shown to alter cell migration and adhesion in keratinocytes. (Yin et al. 2005a; Yin et al. 2005b) Because naturally occurring mutations in plakoglobin have been implicated in the pathogenesis of several skin and cardiac diseases, we performed the present study to determine how these disease-causing mutations affect cell mechanical properties.
Desmosomal proteins including plakoglobin form plaques that are responsible for cell-cell adhesion (Green and Gaudry 2000). Mutations in many of these proteins have been linked to various cutaneous diseases including striate palmoplantal keratoderma and skin fragility/wooly hair syndrome (Armstrong et al. 1999; Whittock et al. 2002). Moreover, defects in desmosomal cadherins are thought to contribute to a number of skin diseases including pemphigus foliaceous, pemphigus vulgaris, fogo selvagem and staphylococcal scalded skin syndrome (Kljuic et al. 2003; Whittock and Bower 2003; Wu et al. 2000). Plakoglobin has also been implicated in the regulation of epithelial cell apoptosis (Charpentier et al. 2000). Finally, changes in plakoglobin expression levels have been suggested to play a role in tumor cell invasion and motility (Ben-Ze'ev and Geiger 1998; Canes et al. 2005; Pan et al. 2007).
Two distinct mutations in plakoglobin have been implicated in the pathogenesis of heart and skin diseases. One is a recessive mutation that introduces a premature stop codon leading to truncation of the C-terminus. This mutation causes Naxos disease, a cardiocutaneous syndrome characterized by woolly hair, palmoplantar keratoderma and the typical cardiac features of arrhythmogenic right ventricular cardiomyopathy (ARVC) (McKoy et al. 2000; Protonotarios et al. 1986). The other is a dominant mutation predicted to lead to insertion of an extra serine residue in the N-terminal region of plakoglobin (insS) (Asimaki et al. 2007). This mutation causes autosomal dominant inheritance of ARVC but without apparent hair or skin abnormalities. While the genetic bases for these conditions have been identified, the mechanisms by which these mutations alter cell behavior are poorly understood. One hypothesis is that impaired mechanical integrity causes injury in tissues bearing high mechanical loads, in which desmosomes play a major supporting role. In this study, we tested the hypothesis that these mutations in plakoglobin affect cell mechanics. Specifically, we asked whether these different plakoglobin mutations caused similar or distinct alterations in overall cell mechanical properties such as cell stiffness, intercellular adhesion and rates of single cell migration and wound healing.
Because primary cultures of adult cardiac myocytes are difficult to maintain and study in terms of different aspects of cell mechanics, and because mechanical behavior in various cell types is likely regulated by similar basic mechanisms, we created a model using HEK293 cells. These cells make typical desmosomes (Asimaki et al. 2007) but unlike cardiac myocytes, they divide and migrate in vitro thus providing the opportunity to examine a broad range of cellular biomechanical behavior that may be relevant to diverse disease phenotypes including those in patients with these mutations. Moreover, an epithelial cell model allows us to test the hypothesis that expression of a disease phenotype in a specific tissue is related, at least in part, to mechanical stresses experienced by that tissue. If this hypothesis is true, then we would expect epithelial cells to exhibit altered cell mechanics in ways that tend to weaken or disperse cells.
We found that different mutations in plakoglobin lead to dramatically different effects on cellular biomechanics. Our observations suggest that changes in cell mechanics caused by plakoglobin mutations have complex underlying biomechanical mechanisms which can affect a range of cell types, despite the phenotypical localization in heart and skin tissues. Thus, we propose that one reason some tissues do not exhibit disease-related changes in vivo is that they do not experience sufficiently high mechanical stresses.
Materials and Methods
Cell Culture and Generation of Stable Cell Lines
Generation, growth and characterization of HEK293 cells expressing wildtype or insS forms of plakoglobin have been described previously (Asimaki et al. 2007). Vectors containing plakoglobin harboring the Naxos disease mutation were generated using site-directed mutagenesis and confirmed by sequencing. HEK293 cells were stably transfected to express this mutant form of plakoglobin using the protocol described for the insS cell line (Asimaki et al. 2007).
Expression of wildtype and mutant plakoglobin was assessed by immunofluorescence microscopy and western blotting. Cells were prepared for confocal immunofluorescence microscopy as described in previous studies (Saffitz et al. 2000; Yamada et al. 2005; Zhuang et al. 2000). Anti-plakoglobin antibodies that recognized either N-terminal (Sigma, St. Louis, MO) or C-terminal (RDI, Concord, MA) epitopes were used. In preparation for immunoblotting, cells were disrupted in lysis buffer (20 mM tris-HCl, 1% triton-X-100, 0.1 % SDS, 0.15 M NaCl, 1 mM EDTA) and protein (7 μg/lane) was loaded on 1% polyacrylamide gels. After electrophoresis, proteins were transferred to nitrocellulose membranes which were washed and incubated with the same antibodies used in immunohistochemistry. The membranes were then incubated with anti-mouse IgG secondary antibody (1:10000 dilution, GE Healthcare UK Limited, UK), washed, treated with Immobilon (Millipore, Billerica, MA) and exposed to film.
Magnetic Micromanipulation Experiments
Receptor-dependent cell adhesiveness and cell stiffness were measured using a previously described magnetic micromanipulator (Huang et al. 2001; Huang et al. 2005; Lammerding et al. 2003a). Briefly, this apparatus consists of a core of CMI-C metal rod which was machined to a sharp chisel tip, annealed and coated with a thin gold film to minimize corrosion, and wrapped with 300 turns of 18 gauge copper wire. Magnetic beads, 4.5 μm in diameter (M-450, Dynal, Invitrogen) were coated with antibodies against beta-1 integrins (HACD29, Fitzgerald, Concord, MA) according to the manufacturer's protocol. A limited number of beads were added to dishes of confluent cells such that an individual cell had no more than one bead on its surface. After incubation for 30 min at 37°C, the cultures were rinsed to remove unattached beads and placed on a temperature-controlled stage (Warner Instruments, Hamden, CT) maintained at 37°C on an inverted microscope (Olympus IX-71, Center Valley, PA). The tip of the magnetic micromanipulator was brought into close proximity of a bead identified on the surface of a cell. Magnetic force was applied in 5 increasing steps to a maximum of 5 nN. Bead detachment rates were recorded, along with the maximum displacement of the beads during the step-wise application of magnetic force using custom Matlab tracking algorithms (Huang et al. 2005; Lammerding et al. 2003b). Magnetic micromanipulation experiments were done pair-wise due to time constraints. More than fifty beads were analyzed in each condition.
To measure the amount of beta-1 integrins expressed on the surface of HEK cell lines expressing wildtype or mutant forms of plakoglobin, we used flow cytometry as previously reported (Huang et al. 2006). Cells were resuspended in 1 ml Hank's balanced salt solution (HBSS) and split into 2 aliquots. One was incubated with a FITC-conjugated anti-rat CD29 (beta-1 integrin) antibody and the other was incubated with an Alexa488-conjugated anti-rat IgG antibody to control for non-specific binding. Cells were incubated on ice for 1 hr, centrifuged and resuspended in 1 ml HBSS, and analyzed using a Becton-Dickinson flow cytometer.
Cell-Matrix Adhesion Assay
Cells expressing wildtype plakoglobin or either of the mutant forms of plakoglobin were dispersed into single cell suspensions using trypsin. The number of cells in each suspension was counted and the volumes adjusted to equalize densities (cells/ml culture medium) for each cell type. The cells were then loaded with calcein (Vybrant Cell Adhesion kit, Invitrogen) according to the manufacturer's instructions and ∼40,000 cells were seeded into individual fibronectin-coated wells in two separate plates (8 wells in each plate for each cell type) where they were allowed to settle and attach for 1 hr at 37°C. Then, the wells in one plate were washed 4 times with HBSS and refilled with culture medium. Both washed and unwashed plates were analyzed using a fluorometer. Readings from the washed wells were normalized to the unwashed wells as a measure of the proportion of total cells that remained attached to the fibronectin-coated surface.
Wound Healing Experiments
Cells were plated on 35mm fibronectin-coated plastic dishes and grown to confluence. A linear wound approximately 1 mm in width was created by scraping the tip of a P-1000 pipette across each dish. The extent to which cells had filled in the wound was determined at 2 and 24 hr post-wounding by imaging at the same location at 4x magnification. To determine whether cells expressing mutant forms of plakoglobin exhibited different rates of cell division in response to wounding, cells were plated on fibronectin-coated glass coverslips (to permit use of a high-NA water immersion objective for fluorescence microscopy), grown to confluence and wounded as described above. At 24 hr post-wounding, cells were loaded with BrdU (Sigma) for 30 min, and then fixed in 4% paraformaldehyde (Sigma) for 15 min at room temperature. Cells were permeabilized in 0.1% triton-X100 for 5 min at room temperature and then incubated with G3G4, an antibody against BrdU (Developmental Studies Hybridoma Bank)(Hakuno et al. 2005) in PBS containing 1% BSA for 1 hr at 37°C. The cells were then incubated with a fluorescent secondary antibody, washed several times and loaded with propridium iodide for 5 min at room temperature to label all nuclei. The cells were examined by fluorescence microscopy under the green channel to identify dividing cells (BrdU-positive) and red channel to identify all nuclei (propridium iodide staining). The percentage of BrdU-positive nuclei in cells near the wound was then calculated. Four representative fields were analyzed in each of 3 separate dishes.
Cell Migration Assay
To assess the speed of migration of isolated cells, cells were plated in low density (∼20,000 cells/cm2) on fibronectin-coated plastic dishes and allowed to attach overnight. The dishes were placed on the temperature-controlled microscope stage and images of individual cells were acquired every 10 min at 10x for 2.5 hr. Displacements between successive cell centroids were measured using a custom Matlab program and averaged to yield a measure of cell migration speed.
Cell-cell Adhesion Assay Using a Deform-Drag Method
Cells were plated on fibronectin-coated glass coverslips and grown to confluence. Cell nuclei were labeled by incubating them in a 1:4000 dilution of Hoechst. The dishes were placed on a temperature-controlled microscope stage and imaged at 10x magnification. A 1 mm diameter glass rod in a motorized micromanipulator (Eppendorf) was positioned so that the tip was just touching the glass surface, through the cell monolayer. This location was determined by a lowering the rod until a small wound was created at the touch-down location. Thus, when the rod was moved horizontally, the cells would be scraped off. The rod was then moved by the manipulator at a constant rate of 100 μm/sec across the field of view. Images were acquired using a CoolsnapHQ at 2.5 frames/sec throughout the entire interval of rod motion and for several seconds thereafter. The detachment and dragging of cells in the rod's path resulted in deformation of the cell monolayer adjacent to the cells being dragged. The extent of this deformation is an indicator of the relative strength of cell-cell adhesion and cell-matrix adhesion. To quantify this parameter, the frames acquired during each experiment were merged into a single image and the vertical extent of nuclear smearing was used as a quantitative measure of cell-cell adhesion. The location of measurement was chosen to maximize the degree of smearing, without letting the smearing exit the field of view, and was in the same horizontal location relative to the initial location of the glass rod for each set of experiments.
Cell-cell Adhesion Assay Using the Dispase Dissociation Method
To validate the novel deform-drag method and to provide additional independent evidence of the effects of mutant plakoglobin on the strength of cell-cell adhesion, we also performed a dispase dissociation assay similar to that described by Yin et al (Yin et al. 2005b). Cells were plated in 10cm2 cell culture dishes and grown to confluence. Cultures were washed with HBSS without calcium or magnesium, and then incubated in 2.4U/ml dispase (Roche) for 15 min until the monolayer lifted from the culture dish as an intact sheet of cells. The monolayers were rinsed in HBSS, transferred into a 15 ml tube and then poured into a 30 cm2 dish. The dishes were examined at a magnification of 4x and 5 images were acquired from each dish. The number of clusters composed of >3 cells was counted in each image. A greater number of cell clusters indicated more extensive disruption of the monolayer, thus providing evidence of reduced strength of cell-cell adhesion independent of cell-matrix interactions.
Effects of Stretch on Expression of Junctional Proteins
HEK293 cells were grown to confluence on silicone membranes and then subjected to linear pulsatile stretch at a frequency of 3 Hz and a strain of 10% using a custom-designed apparatus as previously described (Zhuang et al. 2000). After being stretched for 1 or 4 hr, cells were prepared for quantitative confocal immunofluorescence microscopy to measure the amount of immunoreactive signal for plakoglobin, N-cadherin (Sigma, UK) and Cx43 (Zymed, UK) at cell-cell junctions. Detailed methods for immunostaining and quantitative confocal microscopy have been validated and implemented in numerous previous studies (Saffitz et al. 2000; Yamada et al. 2005; Zhuang et al. 2000).
Statistics
Unless otherwise indicated, data are expressed as mean ± SD. Wound healing rates, cell migration speed, dispase dissociation, Vybrant cell adhesion readouts and deform-drag results were compared using one-way ANOVA and Dunn's post test. Cell adhesion and cell division rates were compared using contingency tables. Cell stiffness was compared using the Mann-Whitney test because these experiments were performed in a pair-wise fashion, and based on past experience, may not always exhibit normal distribution. Stretch responses by confocal analysis were compared using t-tests. Significance in all studies was defined as p<0.05.
Results
Effects of Plakoglobin Mutations on Cell Growth in Vitro
HEK293 cells transfected to stably express wildtype plakoglobin or mutant forms of plakoglobin (insS or Naxos) were passaged under standard in vitro conditions. No obvious differences in individual cell morphology or size were observed in lines expressing the different forms of plakoglobin. However, under sub-confluent conditions, it was readily apparent that cells expressing the Naxos mutation showed a lower degree of clustering than cells expressing either wildtype or insS plakoglobin (Figure 1A). This observation provided a first hint that cells expressing the Naxos mutation exhibited different cell-cell interactions compared to wild-type-expressing cells.
Figure 1.

(A) Differences in the degree of cell aggregation seen in sub-confluent cultures of cells expressing the Naxos mutant compared with cells expressing wildtype (wt) or the insS mutant form of plakoglobin. The Naxos cells are more dispersed (cluster less) than either the wildtype or insS mutant cells. Magnification is 4x; image digitally magnified 2x, scale bars = 200 μm. (B) Western blot showing roughly equal amounts of wildtype and mutant plakoglobin expressed in transfected cell lines. The Naxos protein migrates with a lower molecular weight, consistent with the premature termination. Numbers on the left represent molecular weights of a standard ladder. (C) Western blot using a C-terminal-region antibody against plakoglobin showing that the Naxos plakoglobin band is absent, consistent with C-terminal truncation, but also showing a faint band in the Naxos cell lane which presumably represents endogenous (wildtype) plakoglobin. (D) Confocal images of cells stained with an antibody recognizing a C-terminal epitope of plakoglobin. There is abundant junctional localization of plakoglobin in the wildtype and insS mutant cells but not in the Naxos cells. Scale bar = 10 μm. (E) Confocal images of cells stained with an antibody recognizing an N-terminal epitope of plakoglobin. Again, there is abundant junctional localization of plakoglobin in the wildtype and insS mutant cells whereas in the Naxos cells there is nuclear localization. Scale bar = 10 μm.
Expression of wildtype or mutant forms of plakoglobin by the stably transfected cells was determined by western blotting and confocal immunofluorescence microscopy. Immunoblotting showed that the 3 cells lines expressed plakoglobin in roughly equal amounts (Figure 1B). As expected, a truncated species of plakoglobin was observed in cells expressing the Naxos mutant, whereas the wildtype and insS forms migrated with the expected molecular weight of ∼82kDa (McKoy et al. 2000). Repeat blotting using an antibody specific for the C-terminal region of plakoglobin showed little signal for the cells expressing Naxos plakoglobin, whereas plakoglobin bands remained strong in cells expressing the wildtype or insS mutant versions of plakoglobin (Figure 1C). Confocal microscopy images of the 3 cell lines labeled with an antibody against a C-terminal epitope demonstrated strong junctional signal in cells expressing wildtype and insS plakoglobin but virtually none in cells expressing Naxos plakoglobin (Figure 1D). Interestingly, staining with an antibody against the N-terminus showed the expected junctional staining in wt and insS cells but marked nuclear labeling in Naxos cells (Figure 1E). This indicates that the Naxos mutation acts in a dominant negative fashion to prevent normal formation of junctional plakoglobin while also leading to marked nuclear accumulation of mutant plakoglobin.
Magnetic Micromanipulation Experiments
To determine whether expression of mutant plakoglobin affects cell-matrix adhesiveness or cell stiffness, we used magnetic micromanipulation. Measurements were made on confluent cells which had been incubated with magnetic beads coated with anti-beta-1 integrin antibodies, which bound to abundant cell surface receptors that link common matrix components to the actin cytoskeleton. We first assessed the strength of cell-matrix adhesion by measuring the rate of bead detachment. The tendency for beads to detach from the cell surface is a reasonable surrogate for the strength of adhesion of the cell to the fibronectin-coated surface of the culture dish. Bead detachment rates were similar for all 3 cell lines (28 of 90 beads detached for insS versus 25/88 for wildtype; 24/90 for Naxos versus 29/90 for wildtype, p=NS for both). To determine whether there were any differences in the amount of beta-1 integrins on the surface of cells expressing wildtype or mutant plakoglobin, we employed flow cytometry of cells labeled with fluorescently-conjugated antibodies against beta-1 integrins. No differences were seen (data not shown). Taken together, the bead detachment and flow cytometry data indicate that the strength of cell-matrix adhesion was roughly similar in cells expressing wildtype or mutant forms of plakoglobin. This conclusion was independently confirmed using the Vybrant cell adhesion assay. No differences were observed in the proportion of cells remaining attached to a fibronectin-coated surface (Figure 2).
Figure 2.

Cell adhesion assay showing no significant differences in adhesion to a fibronectin-coated surface of cells expressing wildtype (wt) or mutant plakoglobin (p=NS).
Next, we assessed the stiffness of cells expressing wildtype or mutant plakoglobin by measuring displacements of the magnetic beads on the cell monolayer in response to step-wise increases in magnetic force. Only firmly attached beads were analyzed. Any beads that detached or moved more than one bead diameter (indicating loose attachment) were excluded from analysis. Greater bead displacements indicated lower cell stiffness (greater compliance). As shown in Figure 3, cells expressing the insS mutation were significantly less stiff than cells expressing wildtype plakoglobin (p<0.05), whereas cells expressing the Naxos mutation were not significantly different than wildtype cells with respect to stiffness.
Figure 3.
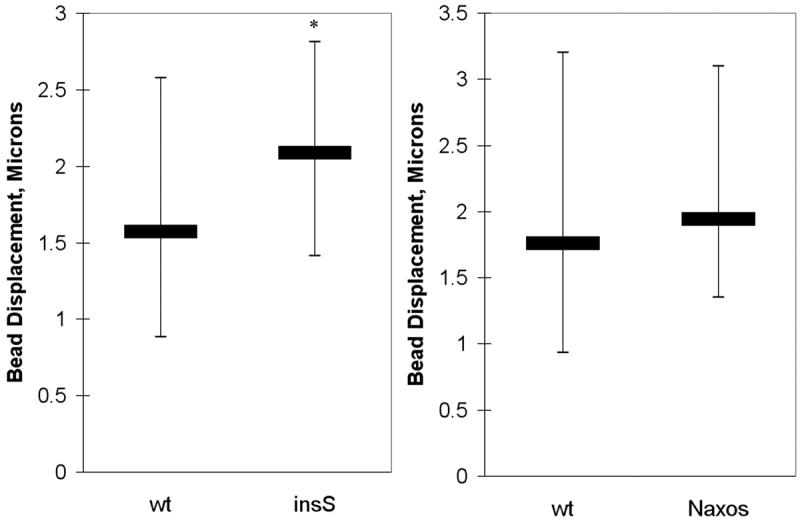
Magnetic micromanipulation experiments reveal differences in cell stiffness as measured by bead displacement. Cells expressing the insS mutant cells are significantly less stiff (exhibit greater bead displacement) than cells expressing wildtype (wt) plakoglobin, whereas cells expressing the Naxos mutant exhibit similar stiffness to cells expressing wildtype plakoglobin (the thick horizontal bars show the median value and the thin verticle bars show the range in values between the 25th and 75th percentiles; n>60 for each group; * p<0.05 compared to wildtype).
Effects of Plakoglobin Mutations on Wound Healing and Migration
To determine whether disease-causing mutations in plakoglobin alter cell motility, we employed two assays, a wound healing assay and a single-cell migration assay, to delineate the rate of cell motion and the dependence of such motility on cell-cell contact. Cultures grown to confluence were wounded with a pipette tip and allowed to heal. The wound healing rate was measured by determining the distance of closure between the two leading edges of the wound. As shown in Figure 4, wound healing occurred more rapidly during the first 24 hr in cells expressing either mutant form of plakoglobin compared with cells expressing wildtype plakoglobin (p<0.01 for both).
Figure 4.

Wound healing during the first 24 hr is significantly accelerated in cells expressing mutant plakoglobin, but cell division rates are not increased. (A) Representative images of each cell type 2 and 24 hr after wounding (4x magnification). (B) Group data showing the extent of wound closure (n >7 for each group; * p <0.01). (C) Group data showing the proportion of BrdU-positive nuclei in the vicinity of the wound (n = 12 fields for each group; p = NS).
To determine whether increased wound healing was related to greater rates of cell division in cells expressing mutant plakoglobin, we incubated cultures with BrdU during active wound healing and counted the number of labeled nuclei. Cell division rates were equivalent in all cell lines (Figure 4). Similar results have been reported for cells expressing the insS mutation during the first 24 hours of growth in vitro (Asimaki et al. 2007). Thus, more rapid wound healing does not appear to be attributable to increased proliferation in cells expressing mutant plakoglobin.
To determine whether changes in the speed of cell migration contributed to increased wound healing, the movements of cells grown in low density were tracked over 2 hr using a custom program described previously (Huang et al. 2006). As shown in Figure 5, cells expressing either the insS or Naxos forms of mutant plakoglobin exhibited increased speed of migration compared with cells expressing wildtype plakoglobin (p<0.05). This result suggests that the increased wound healing rates are related, at least in part, to enhanced cell mobility caused by expression of mutant plakoglobin.
Figure 5.
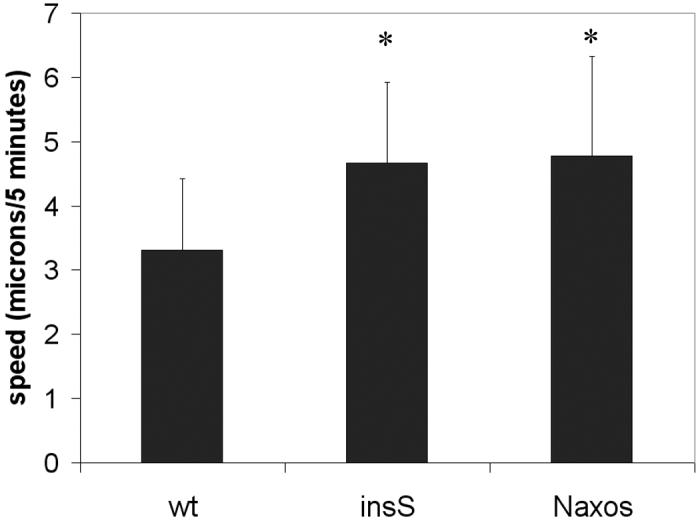
The speed of migration of individual cells in low density cultures is significantly increased in cells expression either mutant form of plakoglobin compared with cells expressing wildtype (wt) plakoglobin (n >11 for each group; * p<0.05 compared to wildtype).
Effects of Plakoglobin Mutations on Cell-Cell Adhesion
Because our results showed no apparent differences in the strength of cell-matrix adhesiveness, we turned our attention to the effects of mutant plakoglobin on cell-cell adhesion. Such analysis is of obvious relevance regarding the role of abnormal intercellular adhesion in the pathogenesis of ARVC and cutaneous disease. However, we were unaware of any existing assays designed to directly measure pure cell-cell adhesion forces, although several different approximations have been previously used (van Wetering et al. 2002; Vedula et al. 2005; Yin et al. 2005b).
We developed a new method (the deform-drag method) to assess cell-cell adhesion as a complement to the dispase dissociation assay experiment. By taking images in sequence as a glass rod was dragged across the field of view and merging the images together, neighboring cells that moved significantly could be detected by the smearing of the nuclear label across the field of view. The distance of the deformation, as assessed by this smearing, perpendicular to the direction of the rod's motion, was used as a quantitative measure of the strength of cell-cell adhesiveness in the monolayer (Figure 6A and 6B). This measure of cell-cell adhesion is modified by the strength of the cell-matrix interaction, which we found in experiments described above to be similar for all the cell types. To validate the new deform-drag method, we first analyzed cells expressing wildtype plakoglobin. We then repeated the experiment on additional wildtype cultures to which trypsin had been added for 3-4 min. Brief exposure to trypsin should dissociate both cell-cell and cell-matrix adhesion, but because the deformation is limited primarily by the strength of cell-cell adhesion, we would expect a decrease in deformation with trypsin. Indeed, we observed a substantial decrease in cell-cell adhesion (p<0.005, Figure 6C), thus providing additional evidence that the deform-drag method is a sensitive means of quantifying this complex biomechanical property.
Figure 6.

Deform-drag experiments performed on confluent cell monolayers. (A) Representative images illustrating the principal of the deform-drag method in a culture of cells expressing wildtype wt) plakoglobin (10x magnification). A glass rod was placed near a corner of the field of view (lower left region of the left image), where a small wound was created. The cells had been labeled with Hoechst to visualize nuclei in the live state. The rod was then dragged across the field using a motorized micromanipulator (from left to right), resulting in removal of cells in the path of the rod (see movies in supplemental files). The resulting wound can be seen in the center and right images. During active wounding, cells adjacent to the region of wounding may be pulled (deformed) along as cells in the rod's path are removed. The extent of deformation of adjacent cells is a function of the strength of cell-cell adhesiveness. (B) By merging the frames acquired during dragging, the motion of nuclei in adjacent cells from cultures expressing wildtype or mutant plakoglobin can be visualized and quantified. The extent of motion in the vertical direction (e.g., perpendicular to the direction of the wound) can be used as a measure of the strength of cell-cell adhesiveness, assuming comparable cell-matrix adhesiveness. (C) A validation experiment showing that brief exposure to trypsin of cells expressing wildtype plakoglobin significantly decreases drag-deformation (* p<0.005). (D) Group data showing that cells expressing the Naxos mutation, but not the insS mutation, exhibit markedly diminished cell-cell adhesiveness compared to wildtype cells (n = 6 for each group; * p<0.05 compared to wildtype and insS). (E) A second validation experiment showing that brief dispase treatment significantly increases drag-deformation in cells expressing Naxos plakoglobin (* p<0.005), suggesting that the balance of cell-matrix and cell-cell adhesion has shifted toward the latter.
We next performed deform-drag experiments using cells expressing mutant plakoglobin. We observed that the extent of deformation was equivalent in cells expressing wildtype plakoglobin and the insS mutant plakoglobin (Figure 6D). The significant degree of cells moving together indicates that these cells were strongly connected to one another. In marked contrast, however, cells expressing the Naxos plakoglobin exhibited complete separation immediately adjacent to the wound location, indicating that compared with wildtype and insS cells, these cells had dramatically reduced intercellular adhesion (p<0.05, Figure 6B and 6D; also see supplemental movies of these experiments). To further validate this result, cells expressing Naxos plakoglobin were treated with dispase (2.4U/ml) for 30 min and the deform drag experiment was repeated. Under these conditions, the monolayers remained attached to the dish but the dispase presumably weakened cell-matrix connections without affecting cell-cell adhesion. As shown in Figure 6E, the extent of deformation increased significantly (p<0.005), suggesting that when cell-matrix adhesiveness is diminished while maintaining cell-cell adhesion (via dispase), the deformation can propagate further.
To independently confirm these findings, we performed a dispase dissociation assay on cells expressing wildtype or mutant plakoglobin using a method similar to that previously used to demonstrate altered cell-cell adhesion caused by plakoglobin mutations in keratinocytes (van Wetering et al. 2002; Vedula et al. 2005; Yin et al. 2005b). Intact monolayers lifted with dispase were removed from the culture dishes and subjected to mechanical stress by first pouring them to a 15ml tube and then into a 30cm2 dish. In this assay, diminished cell-cell adhesion leads to greater breakup of the monolayer into separate clumps of cell. As expected based on results of previous deform-drag experiments, monolayers of cells expressing Naxos plakoglobin became much more fragmented indicating markedly diminished cell-cell adhesion compared to cells expressing either the wildtype or insS plakoglobin (p<0.001, Figure 7).
Figure 7.

Dispase dissociation assays confirm the results of deform-drag experiments. (A) Intact sheets of cells expressing Naxos plakoglobin became disrupted into smaller, more numerous fragments after being mixed. (B) Quantitative data showing significantly more fragments per field of view in cells expressing Naxos plakoglobin compared with wildtype (wt) or insS plakoglobin (* p<0.001 for wildtype versus Naxos and for insS versus Naxos; wildtype versus insS was not different).
Effects of Mutant Plakoglobin on Cellular Responses to Stretch
To determine whether expression of mutant plakoglobin alters cellular responses to mechanical perturbation, we subjected cells grown on deformable silicone membranes to linear, pulsatile stretch for 1 or 4 hr and measured the amount of plakoglobin, N-cadherin and Cx43 specifically localized to cell-cell junctions using confocal microscopy. N-cadherin and Cx43 were included in the analysis because previous studies in cardiac myocytes have shown that cyclical stretch not only increases the amounts of desmosomal proteins at intercellular junctions but these non-desmosomal proteins as well (Yamada et al. 2005). Thus, we sought to determine the effects of mutations in plakoglobin on stretch-induced changes in both mechanical and electrical intercellular junctions. As previously observed in cardiac myocytes (Yamada et al. 2005), the amount of signal for plakoglobin, N-cadherin and Cx43 at cell junctions increased significantly following stretch in 293 cells expressing wildtype plakoglobin (Figure 8). In contrast, cells expressing mutant forms of plakoglobin showed blunted responses to stretch. Increases in junctional plakoglobin, N-cadherin and Cx43 were all markedly diminished (Figure 8), although it was not possible to assess changes in cell surface plakoglobin in Naxos cells because of the absence of junctional signal in these cells. Interestingly, the amount of Cx43 was significantly reduced under basal (non-stretched) conditions in cells expressing mutant plakoglobin. This observation is consistent with previous studies showing reduced expression of myocardial Cx43 in patients with Naxos disease (Kaplan et al. 2004). Taken together, these results suggest that mutant plakoglobin interferes with the ability of cells to respond normally to mechanical load by increasing expression of cell-cell junction proteins at the cell surface. Furthermore, the fact that Cx43 expression is reduced in both 293 cells expressing mutant plakoglobin and in the heart of patients with Naxos disease adds credence to the use of 293 cells as a model of altered cellular biomechanics.
Figure 8.

Membrane stretch experiments demonstrate the altered expression and response of plakoglobin, N-cadherin and Cx43 to mechanical stimuli. (A,B) Confocal microscopy images of wildtype (wt) cells under control conditions (A) or after 4 hr of pulstile stretch (B) immunostained to show the amount of junctional N-cadherin expression. Increased levels of N-cadherin at cell borders can be seen in response to stretch. Scale bars = 20 μm. The number of pixels occupied by bright immunoreactive signal was quantified and expressed as a proportion of total pixels occupied by cells to measure the amount of signal for each protein at cell-cell junctions. These measurements are shown in the bar graphs depicted in this figure. (C) The amount of plakoglobin was similar in wt and insS cells at baseline. Stretch-induced up-regulation of plakoglobin was suppressed in insS cells compared to wt cells. Naxos cells could not be analyzed because of the limited amount of junctional plakoglobin. (D,F) Expression of N-cadherin followed a similar pattern as for plakoglobin. While cells expressing wildtype (wt) or mutant plakoglobin had similar N-cadherin expression levels at baseline, up-regulation in response to 1 and 4 hr of stretch was diminished for in cells expressing mutant plakoglobin. (E,G) Expression of connexin43 (Cx43) was diminished under basal (non-stretched) conditions in cells expressing mutant plakoglobin. Stretch-induced up-regulation of Cx43 was also appeared to be diminished compared to wt cells. However, due to the lower baseline, the lower stretch response cannot be attributed exclusively to decreased upregulation. (F, G) Naxos cells exhibited a similar pattern for N-cadherin and Cx43 expression, exhibiting lower expression at one and four hours of stretch, and with decreased expression at baseline for Cx43 only. For each condition in each experiment, n=5. For insS: plakoglobin, p<0.05 at one hour, p< 0.0001 at four hours; for both mutants: for N-cadherin, p<0.01 at one hour and four hours; for Cx43, p<0.05 at baseline, p<0.0001 at one and four hours, each versus wt at the same times.
Discussion
Cell mechanical regulation by junctional proteins such as plakoglobin is not well understood, despite the significant role of plakoglobin mutations in human disease. For example, ARVC affects approximately 1 in 5000 people and, in some parts of the world, is a major cause of sudden death in young individuals (Gemayel et al. 2001; Peters et al. 2004; Thiene et al. 1988). While there has been significant progress in identifying the different mutations that lead to ARVC and various cutaneous diseases, much less is known about the mechanisms by which mutations in desmosomal proteins cause disease. One leading hypothesis is that defects in mechanical linkage between cells cause injury, ultimately leading to degeneration of cardiac myocytes and and/or damage to keratinocytes. Another possibility is that these same mutations induce cell injury by changing molecular signaling pathways (which may or may not be mechanically stimulated) (MacRae et al. 2006). To investigate the ‘mechanical integrity’ hypothesis and explore potential roles of plakoglobin as a determinant of cell mechanics, we produced cell lines expressing two distinct disease-causing mutant forms of plakoglobin and characterized their effects on cellular biomechanical behavior.
While it has been previously established that deletion or truncation mutations of plakoglobin can cause changes in cell properties (Yin et al. 2005a; Yin et al. 2005b), this is, as far as we know, the first study to systematically characterize the biomechanical properties of cells expressing clinically relevant mutations in plakoglobin. It will be of interest in future studies to characterize the effects of these mutations on cardiac myocytes, the target cell in ARVC. However, primary cultures of cardiac myocytes can be challenging to uniformly transfect and rigorous methods to quantitatively characterize biomechanical behavior in cardiac myocytes are rather limited. Furthermore, it is likely that basic disease mechanisms share similarities in various cell types, including keratinocytes and other types of epithelial cells. Thus, to begin to elucidate the effects of desmosomal protein mutations on cell biomechanical behavior, we decided to use a model cell system in which cells could be stably transfected to express wildtype or mutant plakoglobin and in which we could take advantage of more comprehensive readouts and assess changes in cell mechanics over as broad a range of responses as possible.
To characterize mechanical behavior in cells expressing different plakoglobin mutations, we used some established techniques and also developed a novel deform-drag method to assess the relative strength of cell-cell and cell-matrix interactions. We assumed that the primary interaction between cells and a fibronectin-coated surface is mediated via beta-1 integrins. Because the magnetic micromanipulation studies demonstrated no significant differences in cell-matrix adhesion via beta-1 integrins and because the microplate cell adhesion assay supported this conclusion, our interpretation of the deform-drag experiment is that the dramatic differences observed in cells expressing the Naxos mutation were primarily due to a decrease in cell-cell adhesion. This conclusion is strengthened by the markedly decreased degree of cell clustering seen in sub-confluent cultures of cells expressing the Naxos mutant and in the dispase dissociation assay. It is possible that slight differences in adhesion via beta-1 integrins or differences mediated by other cell-surface receptors could contribute to changes in cell-matrix adhesion overall. Further work to isolate these effects and to elucidate the mechanism of action in regulating cell mechanics is warranted. We note, however, that while complementing the dispase-dissociation assay, the deform-drag assay has the benefit that the cell-cell interaction strength is measured directly on cells without prior treatment (other than with Hoescht for visualization). Dispase dissociation prior to mechanical shearing results in cell-cell contraction and perhaps changes in focal adhesion (as the cell monolayers are lifted) which could alter the biomechanical structure of the cells. Therefore, our conclusion that cell-cell adhesion is altered in cells expressing the Naxos mutant of plakoglobin is strengthened by the concordant observations obtained from both types of experiments.
We observed that disease-causing mutations in plakoglobin lead to significant changes in other cellular mechanical properties. We discovered that cells expressing either plakoglobin mutation (Naxos or insS) exhibit increased wound healing (without increased cell proliferation in the short-term) and increased cell migration speed compared to cells expressing wildtype plakoglobin. While these results are generally consistent with results reported previously by Yin et al.(Yin et al. 2005b) who observed both cell-cell adhesion-dependent and -independent changes in keratinocyte mobility mediated by plakoglobin, this is the first time that clinically relevant mutations have been assessed. Additionally, Yin et al (Yin et al. 2005b) found that expression of an N-terminal truncation variant of plakoglobin, but not a C-terminal truncation form, suppressed cell mobility in keratinocytes lacking wild type plakoglobin. Whereas our Naxos results are consistent with the results of Yin et al (Yin et al. 2005b), we found that the insS mutant plakoglobin which contains an extra N-terminal serine residue but has an intact C-terminus does not suppress cell mobility in a manner similar to wildtype plakoglobin. These data suggest that the presence of an extra serine affects cell mobility in an active manner. We do not know if this serine residue becomes phosphorylated and, if so, whether this affects cell behavior. However, truncation of the N-terminus does not apparently have the same effect as a single amino-acid insertion in the N-terminus.
Experiments in which cells were subjected to pulsatile stretch showed that expression of mutant plakoglobin diminished the normal stretch-induced up-regulation of a variety of junctional proteins. In addition, the presence of mutant plakoglobin reduces the amount of Cx43 at cell-cell junctions even in the absence of mechanical stimulation. Thus, expression of mutant plakoglobin in 293 cells recapitulates at least some features seen in the hearts of patients with ARVC (Kaplan et al. 2004).
Our observations also indicated that the insS mutation in plakoglobin diminishes cell stiffness without affecting cell-cell adhesion, whereas the Naxos mutation markedly diminishes cell-cell adhesion without affecting cell compliance. These disparate effects on cell biomechanical behavior are significant because they underlie potential differences in the pathogenesis of disease. They suggest, for example, that cell injury may arise from loss of cell-cell adhesion in Naxos disease, whereas similar problems could arise from diminished cell stiffness and therefore, persistently increased mechanosensitive responses, in the insS form of ARVC. The fact that “generic” epithelial cells such as HEK293 cells exhibited such responses to mechanical stress suggests that the reason plakoglobin mutations cause disease phenotypes in heart and skin but not elsewhere has to do with the degree to which tissues in the body are subjected to mechanical stresses. At the same time, it should be stressed that the effects of mutant plakoglobin on biomechanical behavior in HEK293 cells cannot necessarily be translated to cardiac myocytes. Future studies will be needed to determine whether these mutations alter the biomechanical properties of ventricular myocytes. Additional insights may also be gained by studying keratinocytes which contain abundant desmosomes and, like cardiac myocytes, are subjected to significant mechanical stress.
We have shown in this study that different mutations in the same protein lead to different biomechanical responses at the cellular level. As a result, there may not be a single common pathway by which diseases such as ARVC develop. There are still many unanswered questions in ARVC including the basis for the disproportionate expression of disease in the right vs the left ventricle, whether mechanical defects per se cause cell injury, and the mechanism by which mutations in non-desmosomal candidate genes may cause disease. Additionally, it is of interest whether non-cardiac and non-keratinocyte cells which express plakoglobin also exhibit alterations in cell mechanical properties in vivo if subjected to high mechanical stresses. It is becoming increasingly clear, however, that the pathogenesis of ARVC cannot be fully characterized without considerations of cell mechanics and mechano-transduction.
Supplementary Material
Acknowledgments and Funding
This study was supported by the NIH EB004646, the Lerner fund, and a grant from the March of Dimes. We thank Dr. Richard Lee for helpful suggestions and support of this project.
References
- Armstrong DK, McKenna KE, Purkis PE, Green KJ, Eady RA, Leigh IM, Hughes AE. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8:143–8. doi: 10.1093/hmg/8.1.143. [DOI] [PubMed] [Google Scholar]
- Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81:964–73. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G, Tonetti P, Manzi L, Cera MR, Balconi G, Dejana E. Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J Cell Sci. 2005;118:623–32. doi: 10.1242/jcs.01661. [DOI] [PubMed] [Google Scholar]
- Ben-Ze'ev A. The dual role of cytoskeletal anchor proteins in cell adhesion and signal transduction. Ann N Y Acad Sci. 1999;886:37–47. doi: 10.1111/j.1749-6632.1999.tb09398.x. [DOI] [PubMed] [Google Scholar]
- Ben-Ze'ev A, Geiger B. Differential molecular interactions of beta-catenin and plakoglobin in adhesion, signaling and cancer. Curr Opin Cell Biol. 1998;10:629–39. doi: 10.1016/s0955-0674(98)80039-2. [DOI] [PubMed] [Google Scholar]
- Canes D, Chiang GJ, Billmeyer BR, Austin CA, Kosakowski M, Rieger-Christ KM, Libertino JA, Summerhayes IC. Histone deacetylase inhibitors upregulate plakoglobin expression in bladder carcinoma cells and display antineoplastic activity in vitro and in vivo. Int J Cancer. 2005;113:841–8. doi: 10.1002/ijc.20634. [DOI] [PubMed] [Google Scholar]
- Charpentier E, Lavker RM, Acquista E, Cowin P. Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J Cell Biol. 2000;149:503–20. doi: 10.1083/jcb.149.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djalilian AR, McGaughey D, Patel S, Seo EY, Yang C, Cheng J, Tomic M, Sinha S, Ishida-Yamamoto A, Segre JA. Connexin 26 regulates epidermal barrier and wound remodeling and promotes psoriasiform response. J Clin Invest. 2006;116:1243–53. doi: 10.1172/JCI27186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemayel C, Pelliccia A, Thompson PD. Arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2001;38:1773–81. doi: 10.1016/s0735-1097(01)01654-0. [DOI] [PubMed] [Google Scholar]
- Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol. 2000;1:208–16. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- Hakuno D, Takahashi T, Lammerding J, Lee RT. Focal adhesion kinase signaling regulates cardiogenesis of embryonic stem cells. J Biol Chem. 2005;280:39534–44. doi: 10.1074/jbc.M505575200. [DOI] [PubMed] [Google Scholar]
- Huang H, Cruz F, Bazzoni G. Junctional adhesion molecule-A regulates cell migration and resistance to shear stress. J Cell Physiol. 2006;209:122–30. doi: 10.1002/jcp.20712. [DOI] [PubMed] [Google Scholar]
- Huang H, Kamm RD, So PT, Lee RT. Receptor-based differences in human aortic smooth muscle cell membrane stiffness. Hypertension. 2001;38:1158–61. doi: 10.1161/hy1101.096456. [DOI] [PubMed] [Google Scholar]
- Huang H, Sylvan J, Jonas M, Barresi R, So PT, Campbell KP, Lee RT. Cell stiffness and receptors: evidence for cytoskeletal subnetworks. Am J Physiol Cell Physiol. 2005;288:C72–80. doi: 10.1152/ajpcell.00056.2004. [DOI] [PubMed] [Google Scholar]
- Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. others. [DOI] [PubMed] [Google Scholar]
- Kljuic A, Gilead L, Martinez-Mir A, Frank J, Christiano AM, Zlotogorski A. A nonsense mutation in the desmoglein 1 gene underlies striate keratoderma. Exp Dermatol. 2003;12:523–7. doi: 10.1034/j.1600-0625.2003.00017.x. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Huang H, So PT, Kamm RD, Lee RT. Quantitative measurements of active and passive mechanical properties of adult cardiac myocytes. IEEE Eng Med Biol Mag. 2003a;22:124–7. doi: 10.1109/memb.2003.1256282. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Kazarov AR, Huang H, Lee RT, Hemler ME. Tetraspanin CD151 regulates alpha6beta1 integrin adhesion strengthening. Proc Natl Acad Sci USA. 2003b;100:7616–21. doi: 10.1073/pnas.1337546100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae CA, Birchmeier W, Thierfelder L. Arrhythmogenic right ventricular cardiomyopathy: moving toward mechanism. J Clin Invest. 2006;116:1825–8. doi: 10.1172/JCI29174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–24. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- Pan H, Gao F, Papageorgis P, Abdolmaleky HM, Faller DV, Thiagalingam S. Aberrant activation of gamma-catenin promotes genomic instability and oncogenic effects during tumor progression. Cancer Biol Ther. 2007;6:1638–43. doi: 10.4161/cbt.6.10.4904. [DOI] [PubMed] [Google Scholar]
- Peters S, Trummel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004;97:499–501. doi: 10.1016/j.ijcard.2003.10.037. [DOI] [PubMed] [Google Scholar]
- Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S, Scampardonis G. Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J. 1986;56:321–6. doi: 10.1136/hrt.56.4.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA. Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol Heart Circ Physiol. 2000;278:H1662–70. doi: 10.1152/ajpheart.2000.278.5.H1662. [DOI] [PubMed] [Google Scholar]
- Salomon D, Sacco PA, Roy SG, Simcha I, Johnson KR, Wheelock MJ, Ben-Ze'ev A. Regulation of beta-catenin levels and localization by overexpression of plakoglobin and inhibition of the ubiquitin-proteasome system. J Cell Biol. 1997;139:1325–35. doi: 10.1083/jcb.139.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–33. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- van Wetering S, van Buul JD, Quik S, Mul FP, Anthony EC, ten Klooster JP, Collard JG, Hordijk PL. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J Cell Sci. 2002;115:1837–46. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- Vedula SR, Lim TS, Kausalya PJ, Hunziker W, Rajagopal G, Lim CT. Biophysical approaches for studying the integrity and function of tight junctions. Mol Cell Biomech. 2005;2:105–23. [PubMed] [Google Scholar]
- Whittock NV, Bower C. Targetting of desmoglein 1 in inherited and acquired skin diseases. Clin Exp Dermatol. 2003;28:410–5. doi: 10.1046/j.1365-2230.2003.01311.x. [DOI] [PubMed] [Google Scholar]
- Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, McLean WH, Pulkkinen L, Uitto J, Christiano AM. Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol. 2002;118:232–8. doi: 10.1046/j.0022-202x.2001.01664.x. others. [DOI] [PubMed] [Google Scholar]
- Wu H, Wang ZH, Yan A, Lyle S, Fakharzadeh S, Wahl JK, Wheelock MJ, Ishikawa H, Uitto J, Amagai M. Protection against pemphigus foliaceus by desmoglein 3 in neonates. N Engl J Med. 2000;343:31–5. doi: 10.1056/NEJM200007063430105. others. [DOI] [PubMed] [Google Scholar]
- Yamada K, Green KG, Samarel AM, Saffitz JE. Distinct pathways regulate expression of cardiac electrical and mechanical junction proteins in response to stretch. Circ Res. 2005;97:346–53. doi: 10.1161/01.RES.0000178788.76568.8a. [DOI] [PubMed] [Google Scholar]
- Yin T, Getsios S, Caldelari R, Godsel LM, Kowalczyk AP, Muller EJ, Green KJ. Mechanisms of plakoglobin-dependent adhesion: desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J Biol Chem. 2005a;280:40355–63. doi: 10.1074/jbc.M506692200. [DOI] [PubMed] [Google Scholar]
- Yin T, Getsios S, Caldelari R, Kowalczyk AP, Muller EJ, Jones JC, Green KJ. Plakoglobin suppresses keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci USA. 2005b;102:5420–5. doi: 10.1073/pnas.0501676102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Yamada KA, Saffitz JE, Kleber AG. Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ Res. 2000;87:316–22. doi: 10.1161/01.res.87.4.316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.