

Abstract
Cholecystokinin [CCK] is a peptide released as a hormone by the proximal gut in response to the presence of peptones and fatty acid in the gut. Considerable evidence suggests that CCK inhibits feeding behavior and gastric function by acting as a paracrine modulator of vagal afferents in the periphery, especially in the duodenum. CCK is also widely distributed throughout the mammalian brain and appears to function as a neurotransmitter and neuromodulator. More recent studies have suggested that CCK may act directly within the CNS to activate central vagal afferent terminal inputs to the solitary nucleus. We have developed an in vitro calcium imaging method that reveals, for the first time, the direct effects of this peptide on vagal terminals in the solitary nucleus. In vitro imaging reveals that CCK provokes increases in intracellular calcium in vagal afferent terminals as a consequence of a complex interaction between protein kinase A [PKA] and phospholipase C [PLC] transduction mechanisms that open L-type calcium channels and causes endoplasmic reticular [ER] calcium release. The subsequent activation of PKC may be responsible for initiating calcium spiking which is dependent on a TTX-sensitive mechanism. Thus, imaging of the isolated but spatially intact hindbrain slice has allowed a more complete appreciation of the interdependent transduction mechanisms used by CCK to excite identified central vagal afferent fibers and varicosities.
Keywords: calcium imaging, satiety, hindbrain
Introduction
Cholecystokinin [CCK] is a peptide released from gut endocrine cells in response to the presence of peptones and fatty acids in the gut [19]. CCK regulates pancreatic enzyme secretion, gallbladder contractions, gastrointestinal motility, and is a potent satiety factor. CCK-A receptors are abundant on peripheral vagal afferent fibers and on the cell bodies of origin in the nodose ganglion [10, 15, 36, 57]. Peripheral vagal afferent responsiveness to CCK has been demonstrated using electrophysiological recordings of teased vagal afferent fibers and from nodose ganglion cell bodies [18, 36, 53, 57].
While there is little doubt that CCK activates vagal afferents, there is some question concerning where and how this activation takes place. Previous studies have shown that the effects of CCK to promote satiety, reduce gastric motility, and provoke pancreatic exocrine secretion are a consequence of the peptides action on vagal afferents [[37, 40, 47], respectively]. Many of these effects appear to be a consequence of paracrine-like effects of CCK on the peripheral vagus [56]. However, recent reports suggest that CCK may also have direct CNS effects to control digestion and feeding behavior. For example, in vitro electrophysiological studies have shown that solitary nucleus neurons [NST] and efferent neurons in the dorsal motor nucleus of the vagus [DMN] are activated by CCK [3, 5, 42, 43, 57]. cFOS activation of solitary nucleus neurons on both sides of the neuraxis still occurs after peripheral administration of CCK to unilateral, cervically vagotomized animals [4]. These points suggest a parallel sensitivity for CCK shared both by peripheral vagal afferents and by vagal afferent circuit elements within the dorsal medulla. This redundancy is supported by behavioral observations where CCK antagonists that can penetrate the blood brain barrier are more effective in suppressing feeding and gastrointestinal effects of CCK than those antagonists that do not have central actions [44, 46].
Recordings made from cultured nodose ganglion neurons [36, 57] suggest that CCK action, mediated through CCK-A receptors, causes a significant elevation in intracellular calcium. These studies suggest that the principal means of activation of this calcium flux in nodose neurons may be extracellular calcium entry through membrane channels. However, work on non-neural (i.e., pancreatic and gastrointestinal) cell types suggests that CCK may also augment calcium release from endoplasmic reticular [ER] stores [63]. Other studies of the CCK-A receptor suggest that the intracellular domain can activate both Gq- and Gs-initiated transduction pathways; Gq pathways activate ER calcium release by activating a phospholipase C [PLC] mechanism while the Gs-protein kinase A [PKA] path can regulate the sensitivity of cation channels [21, 23, 25]. Our in vitro calcium imaging method allows us to directly examine how CCK activates vagal afferent terminals in the NST [48, 49].
Methods
Long-Evans rats [200–400 g; both males and females], obtained from the breeding colony located at Pennington Biomedical Research Center, were used in these studies. All animals were maintained in a room with a 12:12 hour light-dark cycle with constant temperature and humidity, and given food and water ad libitum. All experimental protocols were performed according to the guidelines set forth by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committees at the Pennington Biomedical Research Center.
Afferent prelabeling
Techniques for performing calcium imaging studies of identified vagal afferent fibers and varicosities have been described in detail [48, 49]; the methods are abstracted here. Prior to surgery, glass microinjection pipettes are pulled from 1.8 mm OD starbore capillary tubing [Radnoti Glass Technologies, Inc, Monrovia, CA] using Narishige Model 1D puller [Tokyo, Japan]. Tips are beveled at ~20° angle on a rotating diamond-coated glass disk [Sutter BV-10]; final tip diameter ~50microns. Micropipettes were filled with 20% CalciumGreen 1-dextran 3000MW conjugate [CGD; Invitrogen-Molecular Probes; Carlsbad, CA] in a solution of 1% Triton-X 100 and distilled water.
Rats are anesthetized with pentobarbital [Nembutal; Abbott Laboratories, Chicago, IL; 50mg/kg, ip]. Using aseptic technique, the nodose ganglion is exposed at the jugular foramen. Access is made through a midline longitudinal incision over the tracheal area. The sternohyoidus and omohyoidus muscles are slightly separated to expose the common carotid artery and the attached cervical vagus nerve. The vagus is carefully dissected from the carotid in the rostral direction. The nodose appears as a pink translucent swelling of the vagus at the posterior lacerated foramen [22]. The micropipette containing CGD is connected to a source of pulsed air pressure [Picospritzer, General Valve Corp., Fairfield NJ] and the tip of the pipette is guided by hand through the sheath of the nodose ganglion. Pressure pulses [~2–10 PSI] are applied to the pipette; dye is injected to fill the ganglion. Total injected volume is approximately 200nL. The pipette is withdrawn; the cervical wound is closed with 4-0 nylon suture. Upon recovery from the anesthesia, animals are returned to their home cage for 2–5 days to allow anterograde transport of CGD to central vagal varicosities.
Brainstem Slice Preparation
Animals are re-anesthetized with urethane [ethyl carbamate; Sigma-Aldrich, St. Louis, MO; 1.5g/kg i.p.]; this anesthesia readily washes out of tissue and does not have long-term effects on the activity of neurons in these slices [24]. After decapitation and swift removal of the brainstem to cold, carbogenated, cutting solution [~4°C; 95% O2; 5% CO2; see recipe below], 300micron thick slices are cut coronally through the medulla with a sapphire knife on a Vibratome [model 1500; St. Louis, MO]. Slices are then transferred to a scintillation vial containing carbogenated normal Krebs [recipe below] at 29°C for approximately 1 hr prior to conducting experiments. Brain slices are transferred using a Pasteur pipette whose tip is cut off and fire-polished, leaving a bore of ~ 6mm.
In vitro perfusion solutions
Cutting Krebs solution contained (in mM): 110 choline chloride, 25 NaHCO3, 2.5 KCl, 7 MgSO4- 7H2O, 1.25 NaH2PO4; 10 glucose; 0.5 CaCl2-2H2O; bubbled with 95% O2/ 5% CO2 during the entire cutting process.
Normal Krebs contained (in mM): 124 NaCl, 25 NaHCO3, 3.0 KCl, 1 MgSO4-7H2O, 1.5 NaH2PO4; 10 glucose; 1.5 CaCl2-2H2O; bubbled with 95% O2/ 5% CO2, continuously; osmolality was 300 ± 5 mOsm; pH = 7.3.
Perfusion chamber
Individual brain slices are transferred to the temperature regulated perfusion chamber [Delta T4 culture dish, Bioptechs, Inc, Butler, PA]. The 25mm diameter chamber dishes have integral heating elements and a clear cover-slip bottom that allows trans-illumination with an ordinary white light source. Thermocontrol is assisted through the use of an objective heater for the microscope. Temperature of the slice chamber and objective are maintained at 33–34°C. The inside, bottom of the chamber is coated with a thin film of silicone elastomer [Sylgard®, Dow Corning, Midland, MI] to help hold the tissue slice to the bottom of the chamber. Slices placed in the chamber are held fast with a harp-type pressor foot made from 22 gauge gold wire and nylon hosiery fibers.
The recording chamber is continuously perfused at a rate of 2 ml/min with carbogenated Krebs [or experimental solutions] warmed to 33°C. Perfusion solutions are carbogenated in individual plastic cups; solenoid valves are used to select perfusates to be directed to the slice. Perfusates are delivered through a manifold to a roller pump to switch between the normal bathing solution and the challenge or experimental solutions without interruption to perfusion of the experimental slice. A period of at least 10min elapses between positioning the slice in the chamber and beginning of the imaging trial to assure that the tissue and harp have settled in the chamber so as to avoid focus shifts while imaging.
Imaging instrumentation and data processing
Time-lapse laser confocal calcium imaging is carried out with a "Ultraview" Nipkow disk imager [model CSU21; PerkinElmer, Boston, MA] combined with a digital camera [Hammamatsu-Orca-ER; Japan] on a Nikon E600FN fixed stage upright microscope equipped with 10X and 40X water immersion objectives. CGD is excited by the 488nm laser line and the 509nm confocal fluorescent signal is processed by the Perkin Elmer Ultraview software.
In vitro Calcium Signal Analysis
Relative changes in cytoplasmic calcium in response to agonist solutions are expressed as changes in fluorescence: (ΔF/F)%, where F is the fluorescence intensity within an area of interest [ROI; e.g., the outline of a vagal afferent fibers or varicosities] prior to stimulation and ΔF is the change from this value during neuronal activity [26]. Background fluorescence [i.e., non-involved areas adjacent to the area of interest] was subtracted from both ΔF and F.
Experimental procedures
Basic calcium response characteristics of CG-labeled vagal afferent varicosities to CCK and concentration-dependent responses
The effects of repeated CCK application on vagal terminal responses were established to determine whether or not these receptors would show rapid adaptation. In this case, the slice was perfused with CCK [100nM for 60sec] in normal Krebs followed by a 10min washout in normal Krebs. This CCK concentration was chosen as a search and pharmacologic test dose because it has been established as the minimum dose necessary to produce the maximum excitatory post-synaptic current [EPSC] effects on solitary nucleus neurons [5]. Thus, this dose provided a robust initial response against which the effects of receptor and transduction antagonists could be compared. In all experiments, CCK-responsive varicosities were identified as those that produced a minimum change of 20% [(ΔF/F)%] in relative calcium flux on first exposure to the peptide. The CCK application was then repeated in normal Krebs, providing a control for both repetitive application and time interval. This “time control” data indicated that repetitive exposure to CCK did not result in adaptation and allowed us to use each set of ROI as its own control to make comparisons for drug [e.g., signal transduction blockers] effects on CCK-induced calcium changes. As described above, changes in calcium flux are recorded as a change in the magnitude of fluorescence within the ROI.
Next, the concentration dependence of the vagal terminal calcium response to CCK was examined. As before, varicosities were initially identified as CCK-responsive by applying a 60sec pulse of 100nM CCK to the bath and identifying those with a minimum 20% increase in calcium green fluorescence. After a 10min rest interval with normal Krebs perfusion, the vagal afferent varicosity response to CCK was retested at concentrations between 1pM and 100nM. Statistical differences between calcium responses at different concentrations were established by one-way analysis of variance followed by the construction of 99% confidence limits around the individual means
Dependence of CCK-evoked vagal afferent calcium response on CCK-A receptors
The principal effect of CCK to activate peripheral vagal afferent processes has been attributed to effects on the CCK-A receptor [36, 40, 57]. This possibility was examined in vagal afferent varicosities in the brainstem by applying CCK [100nM] to the slice in normal Krebs, followed by a 10min exposure to the specific CCK-A receptor antagonist, lorglumide [1uM; Sigma-Aldrich, St Louis; [57]]; CCK [100nM] was then reapplied to the bath. Unsulfated CCK, a selective CCK-B agonist, was also applied to the preparation in preliminary studies and was found to have no effect on vagal terminal calcium levels. No further experiments were performed with this agonist.
Relative proportion of vagal varicosities responding to CCK
In a subset of experiments [17 slices obtained from 7 animals], we sought to estimate the proportion of identified vagal profiles that respond to CCK. We functionally-identified vagal afferent varicosities with capsaicin [100nM; a VR1 receptor agonist and selective activator of C-type afferent fibers] or ATP [100uM; a P2×3 agonist and selective activator of A-type fibers [30]]. The effects of CCK [100nM] on these identified fibers were examined 10 minutes after exposing the slice to capsaicin or ATP.
Transduction mechanisms responsible for CCK evoked increases in vagal afferent calcium responses
Previous studies [36, 57] have further suggested that CCK activates vagal afferent cell bodies by increasing transmembrane cation fluxes. Studies performed in non-neuronal cell types [63, 64] suggest that CCK can also activate intracellular calcium release from the endoplasmic reticulum [ER]. The CCK-A receptor has the potential to activate either or both mechanisms, in parallel, because the intracellular domain of this receptor possesses activation domains for Gs [65] and Gq [55, 67]. One downstream effect of Gs is protein kinase A [PKA] activation and this event, in turn, is causally linked to the opening of L-type calcium channels [23, 31]. Gq activation leads to activation of phospholipase C [PLC] and, secondarily, protein kinase C [PKC]. These events are linked with intracellular calcium release and subsequent depolarization of excitable cell membranes [6]. With these possible outcomes in mind, we examined the transduction mechanism(s) of CCK activation of vagal afferent terminals by employing the same basic protocol as described, above, with lorglumide.
After the initial exposure of the slice to CCK in normal Krebs, the slice was then exposed to one of the following during the 10min resting period:
nitrendipine [10uM; selective L-type calcium channel blocker; Sigma-Aldrich],
U73122 [10uM; selective phospholipase C antagonist; Tocris, Ellisville, MO],
combination of U73122 and nitrendipine [each at 10uM],
H89 [1uM; selective PKA antagonist; Sigma-Aldrich],
chelerythrine [10uM; selective PKC antagonist; Sigma-Aldrich],
cadmium chloride [100uM; general blocker of membrane calcium channels; Sigma-Aldrich], OR
TTX [1uM; selective sodium channel antagonist; Sigma-Aldrich].
At the end of this 10min exposure, the slice was again perfused with CCK [100nM]. As before, a minimum change of 20% in calcium flux [on first exposure to CCK] as evidenced by a change in magnitude of fluorescence was required for the varicosity to be included for additional study of underlying signaling mechanisms. For ease of comparison, data are expressed as a percentage of the initial calcium response to 100nM CCK equal to 100%. Thus, each varicosity served as its own control, minimizing variability. These normalized changes in calcium flux, [ΔF/F]%, to the second CCK exposure [i.e., post treatment] were compared across all groups via one-way ANOVA followed by a Dunnett’s post hoc test for significance vs. CCK time control.
Results
Response characteristics of CG-labeled vagal afferent varicosities to CCK
A total of 510 terminals were identified in 110 slices [N = 40 rats] that responded to an initial application of 100nM CCK at the minimum criterion [i.e., 20% (ΔF/F)]. Individual vagal afferent varicosities responding to CCK typically produce an erratic spiking calcium oscillation [Figure 1]. When viewing a whole field of varicosities in the NST in the brainstem slice, perfusion with CCK produces an effect on the labeled varicosities that could be described as “sparkling” in appearance. One of the advantages of using this terminal imaging method is that it is possible to monitor a large field of vagal afferent varicosities and quickly determine which of the labeled processes is viable and responsive to the peptide.
Figure 1.

Two characteristic examples of responses by pre-labeled vagal terminal varicosities to CCK in the perfusion media [A–D represent the analysis of one slice; E–H represent another slice]. The complete field of view of the two brainstem slices in the live cell imaging system are shown in (A) and (E) with the dotted boxes indicating the specific regions of interest [ROI] that were highlighted in the remaining panels. In panels (B) and (F), the ROI is shown immediately before the CCK stimulation. In panels (C) and (G), the ROIs are shown at the peak of their activation response. Panels (D) and (H) are displays, over time, of the change in magnitude of fluorescence [(ΔF/F)%] of the ROI indicted in (C)or (G), respectively, in response to 60sec exposure of the slice to CCK perfusion.
Scale bars: A and E = 80micron; B, C, F, and G = 4micron; D and H = 60sec.
In the initial “time control” studies, varicosities were exposed to two 60sec applications of 100nM CCK spaced 10min apart. All varicosities examined [n=21] responded with similar magnitudes of change in fluorescence to both applications of CCK [Average response to 1st exposure: 57.0% + 9.2 (SEM) vs average response to 2nd exposure: 54.0% + 9.3; Figure 2].
Figure 2.

“Time control” studies exposed pre-labeled varicosities to two 60sec applications of 100nM CCK spaced 10min apart. All varicosities responded with similar magnitudes of change in fluorescence to both applications of CCK [Average response to 1st exposure: 57.0% ± 9.2 (SEM) vs average response to 2nd exposure: 54.0% ± 9.3]. These data are also represented in the normalized data in Figure 4 for point of comparison.
Similarly, in the concentration-response studies, the criterion for the initial identification [and inclusion] of a varicosity as being CCK-responsive required a minimum of 20% increase fluorescence [i.e., ΔF/F] to the 100nM concentration of CCK. Varicosities responding with a calcium response of less than 20% to this maximal stimulus were not included for additional study. Once identified CCK-responding varicosities were located [N=164], slices were exposed to a 10min washout period before being challenged by different [lower] concentrations of CCK. The percentage change in fluorescence to these challenges was reported and these may have fallen below the original minimum of 20% in response to the identifying concentration of 100nM CCK. The responses to both 10 and 100pM are small [averaging less than 20%] but they are responsive and the threshold of detection [i.e., ~50% of the CCK-responsive identified varicosities show a response] seems to be at 10pM concentration. These vagal terminals responded to CCK [1pM – 100nM] in a concentration dependent manner; ANOVA F5,158 = 11; p<0.0001 [Figure 3]. One-tailed Dunnett's tests for significance against the 1pM result showed that these incremental doses were statistically different; P < 0.05.
Figure 3.

Pre-labeled afferent varicosities that met initial inclusion criteria for responsiveness to 100nM of CCK demonstrated responses to CCK [1pM – 100nM] in a concentration dependent manner [ANOVA F5,158 = 11; p<0.0001]. These vagal terminal varicosities appear to be minimally sensitive to CCK at concentrations of approximately 10pM [average magnitude of response (ΔF/F)% = 12.6 % ± 3.4 (SEM)]. One-tailed Dunnett's tests for significance against the 1pM result showed that these incremental doses were statistically different; P < 0.05.
Relative proportion of vagal varicosities responding to CCK
In a subset of experiments, we sought to estimate the proportion of identified vagal profiles that respond to CCK. Of 77 varicosities that were responsive to ATP, 24 were also responsive to CCK [32%]. Of 102 varicosities responsive to capsaicin, 37 also responded to CCK [36%]. These values compare well to previous studies [56] that showed 24% of A-type and 38% of C-type vagal afferent cell bodies in the nodose ganglion responded to CCK.
Cellular basis for vagal afferent terminal calcium responses to CCK
In another set of slices, varicosities were first identified as being responsive to CCK [100nM in normal Krebs]. Ten minute exposure to the CCK-A receptor blocker, 1uM lorglumide in Krebs, was followed by a second exposure to CCK. The effect on calcium flux of the second CCK application was essentially blocked by lorglumide [Figure 4], thus identifying that the calcium response is mediated by type A CCK receptors.
Figure 4. Identification of signaling mechanisms used by CCK to activate vagal terminals.

To guard against unintentional pre-conditioning or other modulatory effects on these pre-labeled varicosities, each brainstem slice could only be used for one comparison of responsiveness to CCK. In all cases, the responsiveness of each varicosity to an initial exposure to CCK [100nM in normal Krebs] was determined. A minimum change of 20% change in magnitude of fluorescence [i.e., ΔF/F)%], was required for the varicosity to be considered responsive to CCK and included for additional study on underlying signaling mechanisms. This period was followed by a 10min exposure to one of these conditions: receptor blocker [lorglumide in Krebs], signal transduction blockers [e.g., U73122, H89, or chelerythrine], or specific channel blockers [e.g., nitrendipine, cadmium, or TTX]. At the end of this 10min exposure, the slice was again perfused with CCK [100nM]. For ease of comparison, data are expressed as a percentage of the initial calcium response to 100nM CCK equal to 100%. Thus, each varicosity served as its own control and minimized variability. Normalized changes in calcium flux, (ΔF/F)% to the second CCK exposure [i.e., post treatment] were compared across all groups via one-way ANOVA [F9,232 = 16; p<0.0001]; Dunnett’s post hoc tests for significance were made using CCK “time control” as point of comparison; * p<0.05. Lorglumide effectively blocked CCK action on previously responsive vagal afferent varicosities. CCK-induced calcium responses were significantly reduced by [refer to Figure 4 for all transduction data]: (a) blockade of IP3 production via U73122; (b) L-channel antagonist, nitrendipine; (c) PKA antagonist, H89; (d) PKC antagonist, chelerythrine; (e) sodium channel antagonist, tetrodotoxin [TTX], and (f) general calcium channel blocker, cadmium [Cd++]. Additionally, the combination of U73122 and nitrendipine had the additive effect of essentially eliminating the CCK-induced calcium response.
Normalized changes in calcium flux, (ΔF/F)%, to the second CCK exposure [i.e., post treatment] were compared across all groups via one-way ANOVA [F9,232 = 16; p<0.0001]; Dunnett’s post hoc tests for significance were made using CCK “time control” as point of comparison; * p<0.05. CCK-induced calcium responses were significantly reduced by [refer to Figure 4 for all transduction data]: (a) blockade of IP3 production via U73122; (b) L-channel antagonist, nitrendipine; (c) PKA antagonist, H89; (d) PKC antagonist, chelerythrine; (e) sodium channel antagonist, tetrodotoxin [TTX], and (f) general calcium channel blocker, cadmium [Cd++]. Additionally, the combination of U73122 and nitrendipine had the additive effect of essentially eliminating the CCK-induced calcium response.
Discussion
Several recent studies suggest that CCK effects to suppress feeding behavior and to alter autonomic control of digestion are mediated by direct action of the peptide on vagal afferent terminals in the brainstem [3, 5]. These results are supported by observations that CCK antagonists operating preferentially within the CNS are more effective than those excluded by the blood-brain diffusion barrier [44, 46]. Our results provide the first observations of the direct effects of CCK to increase calcium levels within identified vagal afferent terminals in the brainstem.
Recording experiments performed on cultured nodose ganglion cells have, thus far, provided the only direct mechanistic information available concerning CCK action on vagal afferent neurons [36, 57]. Those studies have provided valuable insight into the general mechanism of action of CCK to increase excitability. It is possible, however, that results obtained from cultured neurons may not accurately depict transduction events elsewhere in the cell [2, 35, 39, 54]. We hasten to add that recording terminal activity in an in vitro slice preparation may also not reflect the physiological situation of the intact animal.
Results from in vitro whole cell voltage and current clamp studies of NST neurons strongly imply that CCK can act directly on apposed vagal afferent terminals as well as on terminals from unknown inputs [3–5]. The essential effects of vagal input to NST neurons studied using these methods (i.e., the generation of glutamatergic post-synaptic currents) are not controversial. However, observations of the modulation of that process can be open to interpretation given that agonists and drugs that act on receptors and transduction processes in the presynaptic terminal may also act on the post-synaptic cell. Direct electrical recordings from vagal fibers and terminals with maximum cross-sections of 1 micron or less are improbable with present electrophysiological methods [8].
Using in vitro calcium imaging methods, we find that vagal afferent terminals in the NST are activated by CCK in a concentration-dependent manner. These processes are minimally sensitive to concentrations of CCK at 10–100pM and robustly responsive above 1nM. Not surprisingly, we find that the selective CCK-A antagonist, lorglumide, blocks CCK effects to elevate calcium in central vagal afferent terminals. This validates the view that important physiological effects of CCK on the afferent vagus are mediated by the CCK-A receptor [15, 36, 57].
Electrophysiological recordings of nodose ganglion cells as well as calcium imaging studies performed on other cell types, have implicated two potential intracellular transduction mechanisms by which CCK may elevate cytoplasmic calcium in vagal afferents. The first appears to involve an activation of a transmembrane ion flux, most likely through an L-type calcium channel [36]. The second [derived from data collected in other cell types] involves activation of intracellular calcium release from stores in the endoplasmic reticulum [64]. Our results suggest that, for the central vagal afferent terminal, both mechanisms are triggered in parallel. That is, while nitrendipine or U73122, alone, significantly reduce the effects of CCK to elevate terminal calcium, the combination of these two antagonists completely block CCK effects.
The effects of CCK to elevate calcium in vagal terminals also appear to be sensitive to blockade of PKA by H89. A well recognized effect of PKA activation in excitable cells is the activation of L-type calcium channels [25, 60]. A less direct effect of PKA activation is the modulation of the sensitivity of the PLC mechanism responsible for releasing calcium from ER stores [20, 52]. It is likely that the CCK-A receptor in the vagal afferent terminal activates both a PKA-dependent mechanism [L-channel] and a PLC-dependent mechanism [ER calcium release]. This conclusion is consistent with data that have shown that the CCK-A receptor possesses activation domains for both Gs and Gq proteins which activate PKA and PLC mechanisms, respectively [55, 66, 67]. However, the present studies on vagal afferent fibers suggest that the PLC mechanism may be dependent on intact PKA signaling given that nearly all calcium signaling was blocked by H89. Reports in other models suggest that PLC activity may, indeed, be gated by active PKA signaling pathways [11, 61]. The dominant nature of PKA influences in regulating CCK signaling in the afferent vagus is supported by behavioral studies by Sutton et al [58, 59]. These studies demonstrated that long-term changes in NST activity related to CCK-induced suppression of feeding behavior are based on activation of extracellular signal related kinase [ERK] and cAMP response element binding protein [CREB]; both of these transduction elements are activated by PKA [21].
While the dual activation of L-calcium channels and ER calcium release will certainly elevate intracellular calcium, the ability of CCK to cause these effects also can be blocked by tetrodotoxin [TTX] and by the generalized blockade of calcium conductances by cadmium-replacement in the media. These observations suggest that CCK-mediated increase of calcium in vagal afferent fibers is, ultimately, dependent on sodium channel-mediated spiking depolarizations that are sensitive to TTX [30]. Spiking depolarizations, in turn, activate voltage-sensitive transmembrane calcium conductances [32]. This sequence of events may provide the mechanism to explain the erratic oscillatory calcium spiking elicited by CCK in central vagal afferents [see Figure 1]. One transduction mechanism that can connect membrane depolarization with the activation of both PKA and PLC involves PKC.
PKC is activated by increases in intracellular calcium and by the production of DAG by PLC [21]. PKC, in turn, is a potent inhibitor of transient potassium conductances [e.g., AP4 sensitive] and a weak inhibitor of resting potassium conductances. Inhibition of these potassium conductances will allow TTX-sensitive action potential-like discharges in synaptosomes with coincident calcium spikes in terminals and an increase in transmitter release, particularly glutamate [6, 14]. Note that our present studies demonstrated that elevations in calcium in vagal terminals could be significantly reduced by: (a) chelerythrine; PKC inhibition, (b) Cd++; calcium channel inhibition, or (c) TTX; sodium channel inhibition.
Together, these observations suggest a mechanism by which CCK activates vagal afferent fibers [summarized in the schematic of Figure 5]. Activation of the CCK-A receptor causes the co-activation of PKA and PLC through the CCK-A Gs and Gq intracellular domain, respectively. PKA causes an initial L-channel dependent increase in calcium in the terminal and applies an essential positive bias on the activity of PLC. The resultant increase in terminal calcium, plus the PLC-mediated production of DAG activates PKC, a potent inhibitor of potassium conductances that normally restrain autonomous terminal spiking by maintaining a resting potential below a level that would increase spontaneous sodium channel activity. The resulting local depolarizations will open voltage gated calcium channels. Thus, CCK can induce erratic, spiking, calcium oscillations in vagal afferent terminals.
Figure 5. CCK activated transduction mechanisms in vagal afferent varicosities.
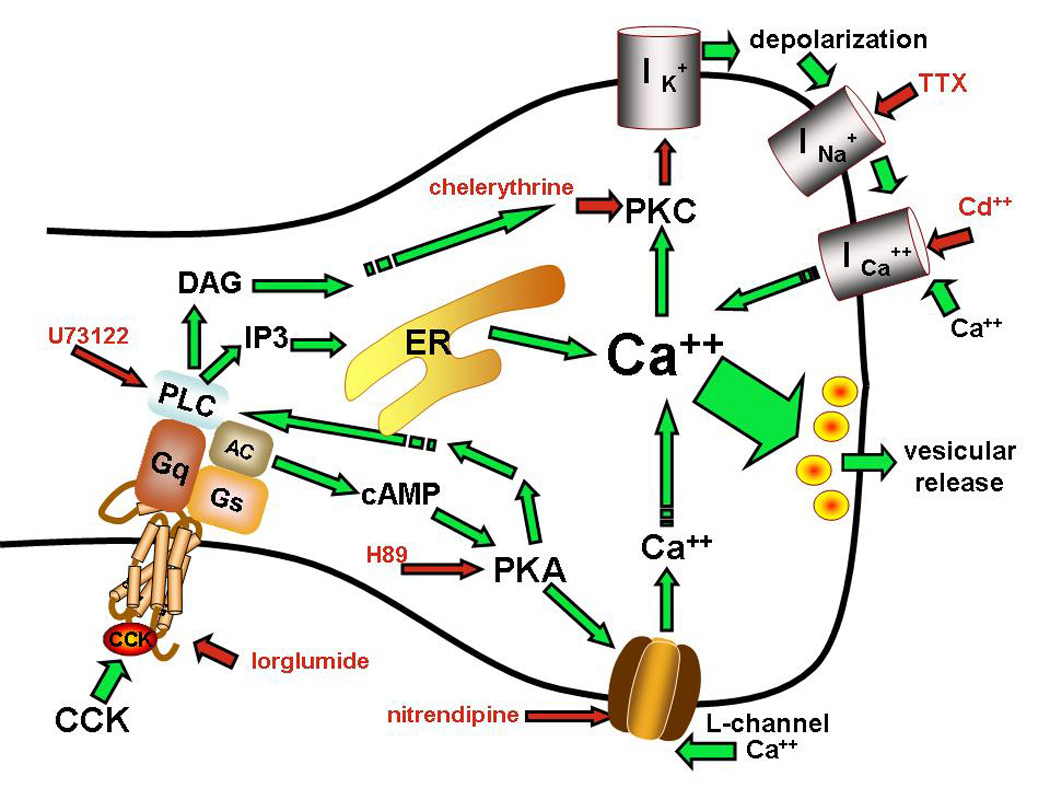
The combination of observations from our vagal afferent terminal imaging studies and those of previous investigators using other methods suggest that CCK activates vagal afferents by activating protein kinase A [PKA], phospholipase C [PLC], and protein kinase C [PKC] transduction pathways in an interdependent manner. Activation of vagal afferent terminal varicosities by CCK is dependent on intact CCK-A receptor signaling [blocked by lorglumide, a selective CCK-A antagonist]. CCK-A receptor causes [through adenyl cyclase] the co-activation of PKA and PLC through the CCK-A Gs and Gq intracellular domain, respectively. PKA causes an initial L-channel-dependent increase in terminal calcium and applies an essential positive bias on the activity of PLC. The resultant increase in terminal calcium, plus the PLC-mediated production of DAG activates PKC, a potent inhibitor of potassium conductances that ordinarily restrains autonomous sodium channel-mediated terminal spiking. Spiking terminal depolarizations permitted by PKC-potassium channel inhibition cause the opening of voltage gated calcium channels in the terminal membrane, leading to the erratic spiking calcium oscillations we observe after exposing the preparation to CCK [see Figure 1].
Red arrows indicate sites of drug inhibition; green arrows indicate positive interactions.
Fibers and varicosities can be distinguished in the live cell imaging preparation, at least at a gross morphological level. Figure 1, for example, shows the effects of CCK on varicosities in the inset. Note that CCK causes considerable brightening of the varicosity but not much in the attached branching axon. Previous work has associated the endoplasmic reticulum [ER; source of much of the calcium produced by the action of CCK] with the varicosities and axon terminal endings, whereas the axon fibers appear not to possess much ER [50]. Further, the L-type calcium channels are associated with axon terminals and in this role are critical to the regulation of presynaptic transmission plasticity [38, 51]. Thus, most of the transduction machinery that CCK operates on is localized to varicosities and axon terminals as opposed to the axon itself and the varicosity would emerge as the most likely locus of initial CCK action. Our results suggest that the ultimate action of CCK is to induce spiking activity in the terminal ending and there is no reason to doubt that these spikes would be propagated throughout the afferent vagal axon. To determine if CCK acts primarily on varicosities, followed by propagation of spikes through the rest of the fiber, one would need to observe the effects of CCK on vagal fibers with an extremely high-speed camera; one capable of taking 1 image per millisecond. This speed would allow one to track the spike propagation. Such camera systems are only now becoming available.
Not unlike the electrophysiological studies on cultured nodose ganglion cells [36, 57], our live cell imaging studies indicate that CCK elevates intracellular calcium in vagal afferent terminals, ultimately, by admitting it from the extracellular space. However, our imaging methods show that this final effect is first mediated by a complex transduction chain that relies on the regulation of intracellular calcium levels as a critical link. It would be quite interesting to test the hypothesis that this same interdependent mechanism is responsible for activation of the peripheral end of sensory vagal afferent fibers by the paracrine action of CCK.
The broader question of the mechanism of action of CCK to produce satiety is still controversial. The role of the vagus in transducing the CCK signal and relaying it to brainstem regions involved in the control of digestion and feeding behavior is well accepted, though the details are still debated. In particular, it appears that neither peripheral nor central vagal afferents are likely to have a large response to concentration levels of CCK found in the general circulation. Several papers cite evidence that the amount of CCK appearing in the plasma after nutrient stimulation of the duodenum is not sufficient to induce satiety [9, 45]. This suggests that CCK released into the duodenal extracellular milieu and the local hepatic portal circulation can activate vagal afferent fibers in the vicinity of release, where the concentration of the peptide is higher than in the general circulation. Indeed, reports by Calingasan [12] and Cox [17] have shown that local injections of CCK into the arterial circulation of the proximal gut are effective in producing satiety. The minimum amounts of exogenous CCK necessary are higher than that found in the general circulation after a meal [i.e., 30–70pM vs 5–10pM] [45]. However, this observation fits well with a study showing that intraduodenal HCl-induced CCK concentrations are about 5-fold higher in portal plasma as opposed to plasma in the general circulation [13]. Thus, these observations suggest that vagal afferents which produce satiety are sensitive to local duodenal-portal concentrations of CCK, but not to those lower concentrations seen in the general circulation. The present results suggest that the central vagal afferents within the NST are at least minimally sensitive to the same concentrations of CCK as those afferents in the duodenum that are involved in producing satiety.
This provokes the question of the physiological and behavioral significance of CCK-sensitive vagal afferents within the NST. While they are sensitive to the similar concentrations of CCK as is found in the portal circulation, these levels of CCK are not typically found in the general circulation, except when CCK is injected exogenously, or, when CCK is released by tumors in the digestive tract [1]. CCK released into the NST by central CCK-ergic efferents would probably be more effective and the more likely source. CCK is virtually ubiquitous in the brain and there are numerous possibilities for CCK-ergic projections to the NST that could affect changes in the way that vagal afferents control autonomic and behavioral functions [41]. Potential sources of putative CCK-ergic terminations in the NST [7, 16, 28] include CCK neurons in the reticular formation [33], paraventricular hypothalamus [34], ventral tegmental area, cortex, and amygdala as well as the NST itself [7, 28, 29]. However, neither the exact pathways nor the physiological significance of any CCK afferent projection to the NST has yet been established.
One point that needs to be addressed in this vein concerns the use of subdiaphragmatic vagotomy as a "proof" of peripheral versus central site of action for CCK. While peripheral vagal nerve section may leave vagal afferent and efferent neurons generally intact from a morphological perspective, this trauma to the peripheral vagus produces (after a delay of several days) profound functional and microstructural changes that lead to reduced excitability and frank degeneration [27, 35, 62]. So, peripheral vagotomy not only destroys peripheral vagal sensory-motor function but it also seriously damages the remaining central components. It is highly unlikely that the "surviving" central components of the vagus are as sensitive to CCK after a peripheral vagotomy.
The present report provides a mechanism for CCK action on central vagal terminals, though it does not address the source of CCK that could activate central vagal afferent varicosities in the intact animal. Clearly, a program of research on the specific role of circulating versus CNS pathway-specific CCK in the regulation of the NST is critical to understanding the role of this peptide in homeostasis.
Acknowledgments
This work was supported by NIH grants DK56373, DK 52142, and HD 47643.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aguilera A, Codoceo R, Selgas R, Garcia P, Picornell M, Diaz C, Sanchez C, Bajo MA. Anorexigen (TNF-alpha, cholecystokinin) and orexigen (neuropeptide Y) plasma levels in peritoneal dialysis (PD) patients: their relationship with nutritional parameters. Nephrol Dial Transplant. 1998;13:1476–1483. doi: 10.1093/ndt/13.6.1476. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez FJ, Villalba RM, Carr PA, Grandes P, Somohano PM. Differential distribution of metabotropic glutamate receptors 1a, 1b, and 5 in the rat spinal cord. J Comp Neurol. 2000;422:464–487. doi: 10.1002/1096-9861(20000703)422:3<464::aid-cne11>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 3.Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neuroscience. 2005;25:3578–3585. doi: 10.1523/JNEUROSCI.4177-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baptista V, Browning KN, Travagli RA. Effects of cholecystokinin-8s in the nucleus tractus solitarius of vagally deafferented rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1092–R1100. doi: 10.1152/ajpregu.00517.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baptista V, Zheng ZL, Coleman FH, Rogers RC, Travagli RA. Cholecystokinin octapeptide increases spontaneous glutamatergic synaptic transmission to neurons of the nucleus tractus solitarius centralis. J Neurophysiol. 2005;94:2763–2771. doi: 10.1152/jn.00351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrie AP, Nicholls DG, Sanchez-Prieto J, Sihra TS. An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes. J Neurochem. 1991;57:1398–1404. doi: 10.1111/j.1471-4159.1991.tb08306.x. [DOI] [PubMed] [Google Scholar]
- 7.Berk ML, Smith SE, Karten HJ. Nucleus of the solitary tract and dorsal motor nucleus of the vagus nerve of the pigeon: localization of peptide and 5-hydroxytryptamine immunoreactive fibers. J Comp Neurol. 1993;338:521–548. doi: 10.1002/cne.903380404. [DOI] [PubMed] [Google Scholar]
- 8.Boyer S, Ruel J, Puel JL, Chabbert C. A procedure to label inner ear afferent nerve endings for calcium imaging. Brain Res Brain Res Protoc. 2004;13:91–98. doi: 10.1016/j.brainresprot.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Brenner L, Yox DP, Ritter RC. Suppression of sham feeding by intraintestinal nutrients is not correlated with plasma cholecystokinin elevation. Am J Physiol. 1993;264:R972–R976. doi: 10.1152/ajpregu.1993.264.5.R972. [DOI] [PubMed] [Google Scholar]
- 10.Broberger C, Holmberg K, Shi TJ, Dockray G, Hokfelt T. Expression and regulation of cholecystokinin and cholecystokinin receptors in rat nodose and dorsal root ganglia. Brain Res. 2001;903:128–140. doi: 10.1016/s0006-8993(01)02468-4. [DOI] [PubMed] [Google Scholar]
- 11.Bugrim AE. Regulation of Ca2+ release by cAMP-dependent protein kinase. A mechanism for agonist-specific calcium signaling? Cell Calcium. 1999;25:219–226. doi: 10.1054/ceca.1999.0027. [DOI] [PubMed] [Google Scholar]
- 12.Calingasan N, Ritter S, Ritter R, Brenner L. Low-dose near-celiac arterial cholecystokinin suppresses food intake in rats. Am J Physiol. 1992;263:R572–R577. doi: 10.1152/ajpregu.1992.263.3.R572. [DOI] [PubMed] [Google Scholar]
- 13.Cantor P, Rehfeld JF. Cholecystokinin in pig plasma: release of components devoid of a bioactive COOH-terminus. Am J Physiol. 1989;256:G53–G61. doi: 10.1152/ajpgi.1989.256.1.G53. [DOI] [PubMed] [Google Scholar]
- 14.Coffey ET, Sihra TS, Nicholls DG. Protein kinase C and the regulation of glutamate exocytosis from cerebrocortical synaptosomes. J Biol Chem. 1993;268:21060–21065. [PubMed] [Google Scholar]
- 15.Corp ES, McQuade J, Moran TH, Smith GP. Characterization of type A and type B CCK receptor binding sites in rat vagus nerve. Brain Res. 1993;623:161–166. doi: 10.1016/0006-8993(93)90024-h. [DOI] [PubMed] [Google Scholar]
- 16.Cotter JR, Laemle LK. Cholecystokinin (CCK)-like immunoreactivity in the brain of the little brown bat (Myotis lucifugus) J Hirnforsch. 1990;31:87–97. [PubMed] [Google Scholar]
- 17.Cox JE. Cholecystokinin satiety involves CCKA receptors perfused by the superior pancreaticoduodenal artery. Am J Physiol. 1998;274:R1390–R1396. doi: 10.1152/ajpregu.1998.274.5.R1390. [DOI] [PubMed] [Google Scholar]
- 18.Cox JE, Randich A. CCK-8 activates hepatic vagal C-fiber afferents. Brain Res. 1997;776:189–194. doi: 10.1016/s0006-8993(97)01036-6. [DOI] [PubMed] [Google Scholar]
- 19.
- 20.Evellin S, Nolte J, Tysack K, vom Dorp F, Thiel M, Weernink PA, Jakobs KH, Webb EJ, Lomasney JW, Schmidt M. Stimulation of phospholipase C-epsilon by the M3 muscarinic acetylcholine receptor mediated by cyclic AMP and the GTPase Rap2B. J Biol Chem. 2002;277:16805–16813. doi: 10.1074/jbc.M112024200. [DOI] [PubMed] [Google Scholar]
- 21.
- 22.
- 23.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- 24.Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg. 2002;94:313–318. doi: 10.1097/00000539-200202000-00015. table of contents. [DOI] [PubMed] [Google Scholar]
- 25.Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. Embo J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 27.Hermann GE, Hebert SL, Van Meter MJ, Holmes GM, Rogers RC. TNF(alpha)-p55 receptors: medullary brainstem immunocytochemical localization in normal and vagus nerve-transected rats. Brain Res. 2004;1004:156–166. doi: 10.1016/j.brainres.2003.11.078. [DOI] [PubMed] [Google Scholar]
- 28.Howes KA, Newton BW, Maley BE. Cholecystokinin octapeptide immunoreactivity in the nucleus tractus solitarius of the cat. Peptides. 1989;10:73–78. doi: 10.1016/0196-9781(89)90079-x. [DOI] [PubMed] [Google Scholar]
- 29.Ingram SM, Krause RG, 2nd, Baldino F, Jr., Skeen LC, Lewis ME. Neuronal localization of cholecystokinin mRNA in the rat brain by using in situ hybridization histochemistry. J Comp Neurol. 1989;287:260–272. doi: 10.1002/cne.902870209. [DOI] [PubMed] [Google Scholar]
- 30.Jin YH, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals in nucleus tractus solitarius. J Neurosci. 2004;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- 32.Katz B, Miledi R. The release of acetylcholine from nerve endings by graded electric pulses. Proc R Soc Lond B Biol Sci. 1967;167:23–38. doi: 10.1098/rspb.1967.0011. [DOI] [PubMed] [Google Scholar]
- 33.King JS, Bishop GA. Distribution and brainstem origin of cholecystokinin-like immunoreactivity in the opossum cerebellum. J Comp Neurol. 1990;298:373–384. [Google Scholar]
- 34.Kiss JZ, Williams TH, Palkovits M. Distribution and projections of cholecystokinin-immunoreactive neurons in the hypothalamic paraventricular nucleus of rat. J Comp Neurol. 1984;227:173–181. doi: 10.1002/cne.902270204. [DOI] [PubMed] [Google Scholar]
- 35.Lancaster E, Oh EJ, Weinreich D. Vagotomy decreases excitability in primary vagal afferent somata. J Neurophysiol. 2001;85:247–253. doi: 10.1152/jn.2001.85.1.247. [DOI] [PubMed] [Google Scholar]
- 36.Lankisch TO, Tsunoda Y, Lu Y, Owyang C. Characterization of CCK(A) receptor affinity states and Ca(2+) signal transduction in vagal nodose ganglia. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1002–G1008. doi: 10.1152/ajpgi.00313.2001. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Hao Y, Owyang C. High-affinity CCK-A receptors on the vagus nerve mediate CCK-stimulated pancreatic secretion in rats. Am J Physiol. 1997;273:G679–G685. doi: 10.1152/ajpgi.1997.273.3.G679. [DOI] [PubMed] [Google Scholar]
- 38.Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- 39.Mateos JM, Azkue J, Benitez R, Sarria R, Losada J, Conquet F, Ferraguti F, Kuhn R, Knopfel T, Grandes P. Immunocytochemical localization of the mGluR1b metabotropic glutamate receptor in the rat hypothalamus. J Comp Neurol. 1998;390:225–233. doi: 10.1002/(sici)1096-9861(19980112)390:2<225::aid-cne5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 40.Moran TH, Kornbluh R, Moore K, Schwartz GJ. Cholecystokinin inhibits gastric emptying and contracts the pyloric sphincter in rats by interacting with low affinity CCK receptor sites. Regul Pept. 1994;52:165–172. doi: 10.1016/0167-0115(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 41.Moran TH, Schwartz GJ. Neurobiology of cholecystokinin. Crit Rev Neurobiol. 1994;9:1–28. [PubMed] [Google Scholar]
- 42.Peters JH, Karpiel AB, Ritter RC, Simasko SM. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology. 2004;145:3652–3657. doi: 10.1210/en.2004-0221. [DOI] [PubMed] [Google Scholar]
- 43.Peters JH, Simasko SM, Ritter RC. Modulation of vagal afferent excitation and reduction of food intake by leptin and cholecystokinin. Physiol Behav. 2006;89:477–485. doi: 10.1016/j.physbeh.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 44.Reidelberger RD, Hernandez J, Fritzsch B, Hulce M. Abdominal vagal mediation of the satiety effects of CCK in rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1005–R1012. doi: 10.1152/ajpregu.00646.2003. [DOI] [PubMed] [Google Scholar]
- 45.Reidelberger RD, Kalogeris TJ, Solomon TE. Plasma CCK levels after food intake and infusion of CCK analogues that inhibit feeding in dogs. Am J Physiol. 1989;256:R1148–R1154. doi: 10.1152/ajpregu.1989.256.5.R1148. [DOI] [PubMed] [Google Scholar]
- 46.Reidelberger RD, Varga G, Liehr RM, Castellanos DA, Rosenquist GL, Wong HC, Walsh JH. Cholecystokinin suppresses food intake by a nonendocrine mechanism in rats. Am J Physiol. 1994;267:R901–R908. doi: 10.1152/ajpregu.1994.267.4.R901. [DOI] [PubMed] [Google Scholar]
- 47.Ritter RC, Ladenheim EE. Capsaicin pretreatment attenuates suppression of food intake by cholecystokinin. Am J Physiol. 1985;248:R501–R504. doi: 10.1152/ajpregu.1985.248.4.R501. [DOI] [PubMed] [Google Scholar]
- 48.Rogers RC, Nasse JS, Hermann GE. Live-cell imaging methods for the study of vagal afferents within the nucleus of the solitary tract. J Neurosci Methods. 2006;150:47–58. doi: 10.1016/j.jneumeth.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 49.Rogers RC, Van Meter MJ, Hermann GE. Tumor necrosis factor potentiates central vagal afferent signaling by modulating ryanodine channels. J Neurosci. 2006;26:12642–12646. doi: 10.1523/JNEUROSCI.3530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ronde P, Dougherty JJ, Nichols RA. Functional IP3- and ryanodine-sensitive calcium stores in presynaptic varicosities of NG108-15 (rodent neuroblastoma x glioma hybrid) cells. J Physiol. 2000;529(Pt 2):307–319. doi: 10.1111/j.1469-7793.2000.00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sand O, Chen BM, Grinnell AD. Contribution of L-type Ca(2+) channels to evoked transmitter release in cultured Xenopus nerve-muscle synapses. J Physiol. 2001;536:21–33. doi: 10.1111/j.1469-7793.2001.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt M, Evellin S, Weernink PA, von Dorp F, Rehmann H, Lomasney JW, Jakobs KH. A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol. 2001;3:1020–1024. doi: 10.1038/ncb1101-1020. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz GJ, Moran TH. CCK elicits and modulates vagal afferent activity arising from gastric and duodenal sites. Ann N Y Acad Sci. 1994;713:121–128. doi: 10.1111/j.1749-6632.1994.tb44058.x. [DOI] [PubMed] [Google Scholar]
- 54.Scott BS, Edwards BA. Electric membrane properties of adult mouse DRG neurons and the effect of culture duration. J Neurobiol. 1980;11:291–301. doi: 10.1002/neu.480110307. [DOI] [PubMed] [Google Scholar]
- 55.Sethi T, Herget T, Wu SV, Walsh JH, Rozengurt E. CCKA and CCKB receptors are expressed in small cell lung cancer lines and mediate Ca2+ mobilization and clonal growth. Cancer Res. 1993;53:5208–5213. [PubMed] [Google Scholar]
- 56.Simasko SM, Ritter RC. Cholecystokinin activates both A- and C-type vagal afferent neurons. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1204–G1213. doi: 10.1152/ajpgi.00132.2003. [DOI] [PubMed] [Google Scholar]
- 57.Simasko SM, Wiens J, Karpiel A, Covasa M, Ritter RC. Cholecystokinin increases cytosolic calcium in a subpopulation of cultured vagal afferent neurons. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1303–R1313. doi: 10.1152/ajpregu.00050.2002. [DOI] [PubMed] [Google Scholar]
- 58.Sutton GM, Duos B, Patterson LM, Berthoud HR. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology. 2005;146:3739–3747. doi: 10.1210/en.2005-0562. [DOI] [PubMed] [Google Scholar]
- 59.Sutton GM, Patterson LM, Berthoud H-R. Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. The Journal Of Neuroscience. 2004;24:10240–10247. doi: 10.1523/JNEUROSCI.2764-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Urbano FJ, Depetris RS, Uchitel OD. Coupling of L-type calcium channels to neurotransmitter release at mouse motor nerve terminals. Pflugers Arch. 2001;441:824–831. doi: 10.1007/s004240000489. [DOI] [PubMed] [Google Scholar]
- 61.vom Dorp F, Sari AY, Sanders H, Keiper M, Oude Weernink PA, Jakobs KH, Schmidt M. Inhibition of phospholipase C-epsilon by Gi-coupled receptors. Cell Signal. 2004;16:921–928. doi: 10.1016/j.cellsig.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 62.Wang XY, Wong WC, Ling EA. NADPH-diaphorase activity in the nodose ganglion of normal and vagotomized guinea-pigs. Cell Tissue Res. 1996;285:141–147. doi: 10.1007/s004410050629. [DOI] [PubMed] [Google Scholar]
- 63.Wank SA. G protein-coupled receptors in gastrointestinal physiology. I. CCK receptors: an exemplary family. Am J Physiol. 1998;274:G607–G613. doi: 10.1152/ajpgi.1998.274.4.g607. [DOI] [PubMed] [Google Scholar]
- 64.Williams JA. Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu Rev Physiol. 2001;63:77–97. doi: 10.1146/annurev.physiol.63.1.77. [DOI] [PubMed] [Google Scholar]
- 65.Wu SV, Yang M, Avedian D, Birnbaumer M, Walsh JH. Single amino acid substitution of serine82 to asparagine in first intracellular loop of human cholecystokinin (CCK)-B receptor confers full cyclic AMP responses to CCK and gastrin. Mol Pharmacol. 1999;55:795–803. [PubMed] [Google Scholar]
- 66.Wu V, Yang M, McRoberts JA, Ren J, Seensalu R, Zeng N, Dagrag M, Birnbaumer M, Walsh JH. First intracellular loop of the human cholecystokinin-A receptor is essential for cyclic AMP signaling in transfected HEK-293 cells. J Biol Chem. 1997;272:9037–9042. doi: 10.1074/jbc.272.14.9037. [DOI] [PubMed] [Google Scholar]
- 67.Yule DI, Tseng MJ, Williams JA, Logdson CD. A cloned CCK-A receptor transduces multiple signals in response to full and partial agonists. Am J Physiol. 1993;265:G999–G1004. doi: 10.1152/ajpgi.1993.265.5.G999. [DOI] [PubMed] [Google Scholar]