

Abstract
Astrocytic, oligodendroglial and mixed gliomas are the commonest gliomas in adults. They have distinct phenotypes and clinical courses, but as they exist as a continuous histological spectrum differentiating them can be difficult. Co-deletions of total 1p and 19q are found in the majority of oligodendrogliomas and considered as a diagnostic marker and a prognostic indicator. The 1p status of astrocytomas has not yet been thoroughly examined. Using a chromosome 1 tile path array, we investigated 108 adult astrocytic tumours for copy number alterations. Total 1p deletions were rare (2%), however partial deletions involving 1p36 were frequently identified in anaplastic astrocytomas (22%) and glioblastomas (34%). Multivariate analysis showed that patients with total 1p deletions had significantly longer survival (p=0.005). In 9 glioblastomas homozygous deletions at 1p36 were identified. No somatic mutations were found among the 5 genes located in the homozygously deleted region. However, the CpG island of TNFRSF9 was hypermethylated in 19% of astrocytic tumours and 87% of glioma cell lines. TNFRSF9 expression was upregulated after demethylation of glioma cell lines. Akt3 amplifications were found in four glioblastomas. Our results indicate that 1p deletions are common anaplastic astrocytomas and glioblastomas but are distinct from the 1p abnormalities in oligodendrogliomas.
Keywords: Microarray, Array-CGH, Methylation, Astrocytoma, Oligodendroglioma
Introduction
Astrocytic and oligodendroglial tumours together with the mixed oligoastrocytomas account for more than 80% of all adult human gliomas (2005-2006 Statistical Report: Primary Brain Tumors in the United States Statistical Report, http://www.cbtrus.org/reports/reports.html). They are subdivided into several malignancy grades according to the World Health Organization (WHO) classification (Kleihues & Cavenee, 2000) and prognosis varies dependent on tumour type and malignancy grade. As these three tumour types form a continuous histological spectrum, differentiating them is sometimes difficult (Kros et al., 2007; Smith et al., 1999).
Classical astrocytic and oligodendroglial tumours have typical genotypes. Astrocytic tumours often show gains of chromosome 7 and/or deletions of chromosome 10, mutations of RB1 or PTEN, as well as amplification of EGFR, CDK4 or MDM2, which are rare in oligodendroglial tumours. The great majority of oligodendrogliomas have hemizygous losses of all of 1p (total 1p deletion), almost always combined with losses of all of 19q (total 19q deletion) (Ichimura et al., 2004; Reifenberger & Louis, 2003). Recent prospective control trials show that loss of one entire copy of 1p and 19q is a favourable prognostic indicator in both anaplastic oligodendrogliomas and anaplastic oligoastrocytomas (Cairncross et al., 2006; van den Bent et al., 2006). Attempts to find the gene(s) targeted by focusing on the smallest consistently deleted region found in a small subset of oligodendroglial tumours have been unsuccessful (Dong et al., 2004; Felsberg et al., 2004; Hashimoto et al., 2003; Iuchi et al., 2002; Nigro et al., 2001). Recent reports of an unbalanced t(1;19)(q10;p10) translocation as the mechanism of co-deletions of 1p/19q in oligodendrogliomas make such an approach less likely to be rewarding (Griffin et al., 2006; Jenkins et al., 2006).
The status of chromosome 1 in astrocytic tumours has not received so much attention. Metaphase CGH studies report rare deletions of 1p (Koschny et al., 2002), while Loss-of-Heterozygosity (LOH) analyses with limited numbers of markers indicate an overall incidence of 1p deletions of up to 20%, probably targeting 1p36 (Barbashina et al., 2005; Mueller et al., 2002; Schmidt et al., 2002; Smith et al., 1999; Ueki et al., 2002; von Deimling et al., 2000). However, the prognosis for patients with astrocytic tumours of any grade is much worse than that for similar grades of oligodendrogliomas or oligoastrocytomas regardless of therapy (MRC, 2001). A recent study of a series of astrocytic, oligodendrocytic and oligoastrocytic tumours of all grades suggested that the presence of a partial 1p loss was associated with poorer prognosis as compared with total 1p loss (Idbaih et al., 2005). Thus the presence or absence as well as the extent of chromosome 1 deletions in these tumours and the association of such genetic abnormalities with prognosis or response to treatment requires further study (Idbaih et al., 2005; Ino et al., 2000; Schmidt et al., 2002; Smith et al., 2000).
To date the techniques used in many studies of 1p in astrocytic tumours (i.e. LOH or FISH analysis) examining a limited numbers of loci cannot accurately identify the presence or the extent of all 1p losses. Here we provide a comprehensive analysis of chromosome 1 copy number alterations in adult astrocytic tumours using a chromosome 1 tile path array. We found that deletions of chromosome 1 are present in increasing frequency among anaplastic astrocytomas and glioblastomas. Unlike oligodendrogliomas, the majority of deletions are small and involving 1p36.22-p36.23. Furthermore, we identified novel overlapping homozygous deletions in 9 glioblastomas in this region.
Results
Chromosome 1 tile path array analysis of astrocytic tumours
A total of 108 of the astrocytic tumours (9 A, 22 AA, 77 GB) were examined by a chromosome 1 tile path array. Among these were tumours with allelic imbalance at 1p detected by MSA, or copy number reduction at SLC45A1/DNB5 detected by LightCycler analysis (qPCR) and a consecutive series of tumours collected during the year 1994 (5 A, 9 AA and 32 GB, used as an unbiased subset to estimate incidence). Twelve of the total series were examined by version 1 array alone, 65 by version 2 array alone and 31 by both. The interpretation of the data from both versions of the array gave similar findings for each of these 31 tumours (data not shown) confirming the validity of the normalization methods used. MSA and chromosome 1 array-CGH results showed good concordance, i.e., array-CGH consistently indicating either loss, gain or amplification where MSA showed allelic imbalance (Fig. 1).
Fig. 1.

Log2 ratio plots of chromosome 1 in two glioblastomas, GB219 and GB52. Insets show microsatellite data displaying allelic balance (D1S247 in GB219 and D1S2655 in GB52) and imbalance (D1S2667 in GB219 and GB52). Positions of clones that harbour the corresponding markers are indicated by grey spots. Hemizygous and homozygous deletions are indicated by arrows (see also Fig. 2). A grey arrow in GB52 indicates the region that appeared as copy number gain due to homology to a part of chromosome 7 which showed trisomy in this tumour (see Fig. 2 legend).
The chromosome 1 copy number status was determined at each clone based on the array-CGH results as either homozygous deletion, hemizygous deletion, normal copy number, copy number gain, or amplification, assuming that the tumours are near-diploid (Bigner et al., 1988). The results are summarized in Table 1 and the patterns of chromosome 1 alterations in individual tumours are schematically presented in Figure 2.
Table 1.
Summary of chromosome 1 status in 108 astrocytic tumours
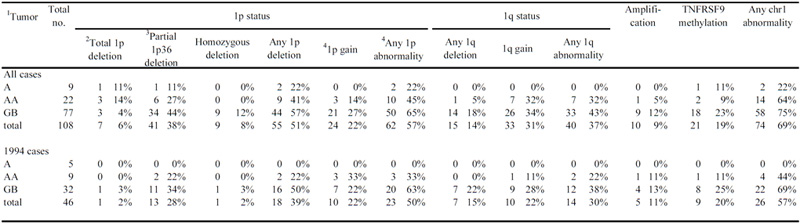
A, diffuse astrocytomas malignancy grade II; AA, anaplastic astrocytomas; GB, glioblastomas; 1994 cases, tumors operated within the calendar year of 1994 (see text)
the number of cases with hemizygous deletion of all 1p clones with or without other abnormalities
tumors that have deletion at 1p36 but not for the whole 1p
Gains limited to the homologous region to chromosome 7 were not included
Fig. 2.

A. A diagram mapping all copy number abnormalities on chromosome 1. Copy number status is schematically represented along chromosome 1 (1pter on the left to 1qter on the right) by colour-coded bars as indicated. Chromosomal band 1p36 is indicated at the top. Previously reported minimum regions of deletion by LOH analysis in various gliomas are schematically shown by grey bars (references, Hashimoto (Hashimoto et al., 2003), Iuchi (Iuchi et al., 2002), Felsberg (Felsberg et al., 2004), Barbashina (Barbashina et al., 2005), Nigro (Nigro et al., 2001), Smith (Smith et al., 1999), Dong (Dong et al., 2004)). The 1p36 region shown in more detail in Fig. 2B is boxed. A region homologous to chromosome 7 is indicated by a grey arrow all tumours with chromosome 7 gain (as judged by the 5Mb clone results) showed copy number gain in this region, most likely because of sequence homology. Codes for the 1994 tumours are in blue. B. A detailed view of copy number abnormalities at 1p36.21-33 for cases that had deletions within the region. Only deletions are shown. The boundaries of the largest homozygous deletion (GB84), telomeric break point at RP3-438L4 and centromeric at RP11-807G9, are indicated by dotted lines. For more precise borders of homozygous deletions see Fig. 3. Chromosomal bands are indicated below.
Chromosome 1 deletions in astrocytic tumours
In total, copy number abnormalities on chromosome 1 were seen in 74/108 tumours (Table 1). Deletions of any part of 1p were found in 55 tumours and of 1q in 15. Deletions of one entire copy of 1p (“total 1p deletion”) were only seen in 7 tumours (1 A, 3 AA, 3 GB, Table 1 and Fig. 2A). Careful re-evaluation of the histopathology of these cases revealed that two of the GBs contained small regions of oligodendroglioma-like cells. Both of these GBs also had total 19q deletions (1Mb array-CGH data, manuscript in preparation). The other five tumours, three of which also showed total 19q deletions (1 A, 2 AA), contained no significant oligodendroglial component in the tumour tissue submitted to histopathology. The commonest finding was partial deletions of 1p found in 48 tumours (1 A, 6 AA, 41 GB) and this involved 1p36 in 41 of these (1A, 6 AA, 34 GB, Table 1 and Fig. 2). The other 7 partial deletions of 1p affected different parts of 1p centromeric to 1p36. Furthermore, 9 homozygous deletions were identified in glioblastomas involving 1p36 (Figs. 1 through 3). Array-CGH analysis of patients’ constitutional DNA showed normal copy number in this region, indicating that the homozygous deletions were somatic changes.
Fig. 3.

A. Top; A schematic diagram of homozygous deletions (indicated by closed boxes). Middle; A schematic diagram of physical locations of the clones on the array, the genes in the region, the corresponding chromosomal bands and nucleotide position ruler (modified after Ensembl 33.35e, September 2005). Orientation of transcription is indicated by an arrow above corresponding genes. Bottom; A schematic diagram of exon positions of RERE and SLC45A1/DNB5 as well as the BAC clones. B-D. Multiplex PCR at an STS in RP11-185B14, RERE exon 23 and SLC45A1/DNB5 exon 8 (B), RERE exon 13 and 14 (C) and SLC45A1/DNB5 exon 1 (D). Xenografts are indicated by suffix X after the code corresponding to their parental primary tumours. The densitometrically calculated copy numbers are shown below the gel photographs (normal = 2, homozygous deletions (< 0.6) are indicated by bold text and underlining). c, control locus (WI-3306, an STS at 2q21). Arrows indicate the bands representing the target locus.
In the consecutive series of 46 tumours from 1994, the incidence of any deletion of 1p was 39% (18/46), the vast majority (13/46; 28%) being partial deletions involving 1p36 (0% in A, 22% in AA and 34% in GB) with one GB having a homozygous deletion of this region. Only a single GB showed total deletion of one copy of 1p. Deletions of any part of 1p were most common among GBs occurring in 16/32 GBs (50%) in this series while no deletions, or any other abnormalities of chromosome 1 were detected in the 5 As (Table 1).
Chromosome 1 status and prognosis
The potential influence of the 1p status on overall survival was studied on the 106 astrocytic tumour patients whose clinical data were available (Table 2). A univariate analysis using the log rank test showed that the tumours with total 1p deletion (1 A, 3 AA, 3 GB) had significantly longer overall survival (p=0.001, Table 2, Kaplan-Meier curve shown in Supplementary Fig. 1). Multivariate analysis using Cox’ regression showed that the 1p status predicts overall survival independent of age and WHO grade (p=0.005). Similar tendency was also observed among GB and AA patients, when analysed separately (Table 2).
Table 2.
Median post-operative survival in astrocytic tumours
| All tumours | Glioblastomas | Anaplastic astrocytomas | ||||
|---|---|---|---|---|---|---|
| 1p status | (n=) | Survival in days | (n=) | Survival in days | (n=) | Survival in days |
| Normal 1p | (41) | 292 | (23) | 156 | (11) | 590 |
| Total 1p deletion | (7) | 4091 | (3) | 3321 | (3) | >5119 |
| 1p36 deletion | (40) | 301 | (33) | 299 | (6) | 406 |
| Other 1p alteration | (18) | 240 | (17) | 279 | (1) | 60 |
| 1Univariate analysis | p=0.001 | p=0.002 | p=0.021 | |||
| 2Multivariate analysis | p=0.005 | p=0.017 | p=0.06 | |||
Univariate analysis using log rank test.
Multivariate analysis including age and WHO grade using Cox’ Regression
Defining the commonly deleted region at 1p36
The boundaries of the 9 homozygous deletions were defined as shown in Fig. 3A. The largest homozygous deletion spanning a 2.9Mb region which included all the other 8 homozygous deletions was found in GB84 (Figs. 2B and 3A). This region was also overlapping with 36 of the tumours with hemizygous deletions with 1p36 (Fig. 2A). Five tumours with hemizygous deletions effecting 1p36 (GB63, GB152, AA14, AA29 and AA65, see Fig. 2) did not encompass this region.
Multiplex PCR at selected loci (an STS on RP11-185B14, RERE exons 12, 13, 14, 15 and 23, SLC45A1/DNB5 exons 1 and 8, TNFRSF9 exon 5) confirmed homozygous deletions at one or more of these loci in all 9 tumours (Fig 3A-D, Fig. 4A). However, a closer analysis showed that not all the homozygous deletions shared a single common region. GB219 showed homozygous deletion at exon 13 of RERE but not at exon 14 (Fig. 3C), defining the telomeric boundary of homozygous deletion between these loci (the region covered by clones RP11-126I6 and RP11-141M15, Fig. 3A). GB136 had homozygous deletion at exon 5 of TNFRSF9 but not at any of the exons examined at SLC45A1/DNB5 or RERE (Fig. 3B-C, Fig. 4A), defining the centromeric boundary as being telomeric to SLC45A1/DNB5. Thus, these results confirmed that homozygous deletions in GB219 and GB136 were not overlapping. Identical patterns of deletion were observed in xenografts of these tumours (GB219X and GB136X, Fig. 3B-D).
Fig. 4.

Analysis of TNFRSF9 alterations. A. Multiplex PCR at TNFRSF9 exon 5. The densitometrically calculated copy numbers are shown below the gel photographs (normal = 2, homozygous deletions (< 0.6) are indicated by bold text and underlining). c= control locus (WI-3306, an STS at 2q21). An arrow indicates the bands representing the target locus. B. MSP at the CpG island of TNFRSF9. The primer pair used for TNFRSF9 specifically amplifies bisulfite-modified methylated sequences. PCR products for ACTB using the primer pair that amplifies bisulfite-modified sequences regardless of methylation status are shown to indicate successful bisulfite modification. G-DNA, unmodified genomic DNA; Blood, modified DNA from WBC; SssI, SssI methylase treated DNA as positive control; Neg, no template. C. Bisulfite sequencing of TNFRSF9 in GB105. Upper panel is a diagram of the CpG island of TNFRSF9 (abscissa, nucleotide position; left ordinate, GC%; shaded area, CpG island). The positions of CpG dinucleotides in the island are indicated by vertical red bars. Frequency of methylation at each CpG dinucleotides in the 10 sequenced clones, each of which is presented in diagram form in the lower panel (closed circle, methylated; open circle, unmethylated), is superimposed in the upper panel (left ordinate, methylation%). The position of MSP primer pair (PC4317/PC4318) is indicated on the bottom of upper panel and the CpG dinucleotides recognized by the primers are indicated in boxes in the lower panel. D. mRNA expression levels of TNFRSF9 and RERE in glioma cell line U251 and breast carcinoma cell line MCF7 after treatment with 5′-Aza-2′-deoxycytidine for either 48 hours or 96 hours. Expression levels relative to that of the mock experiment are shown.
When these two regions were considered separately, the clones most frequently involved in the centromeric homozygous deletions were RP11-126I6, RP11-141M15, RP5-1115A15 and RP11-3J23 (8 tumours, Fig 3A). These clones contain SLC45A1/DNB5 and a part of RERE. The clones most frequently involved in the telomeric region were RP5-892F13, which was deleted in 7 tumours and harbours TNFRSF9, as well as RP11-478I22 and RP11-431K24 (no genes mapped) and RP4-633I8 (contained a part of RERE). These 3 genes, as well as two other genes, ERRFI1/MIG-6 and PARK7, located adjacent to TNFRSF9, were screened for presence of somatic mutations.
Mutation analysis of candidate genes
A total of 230 tumours (96 glioblastomas, 95 oligodendroglial tumours, 24 glioblastoma xenografts and 15 glioma cell lines) were screened for somatic mutations using either DGGE (primary tumours) or direct sequencing (xenografts and cell lines) of all coding sequences and intron sequences adjacent to the exons in RERE (24 exons, Accession No.NM_012102) and SLC45A1/DNB5 (9 exons, Accession No. XM_937695). Mutations of ERRFI1/MIG-6 (4 exons, NM_018948) were examined for 110 tumours (3 A, 12 AA, 56 GB, 24 GB xenografts and 15 glioma cell lines) and TNFRSF9 (8 exons, NM_001561) and PARK7 (7 exons, NM_007262) for 63 tumours (24 glioblastomas, 24 glioblastoma xenografts and 15 glioma cell lines) by DGGE. A number of nucleotide changes were identified in the coding regions or introns of RERE, SLC45A1/DNB5 and ERRFI1/MIG-6 (supplementary table 3), which were also observed in the patients’ constitutional DNA and therefore judged to be polymorphisms. In the cell line H4, a non-synonymous homozygous nucleotide change was identified at ERRFI1/MIG-6 (c.325G>A, D109N). Although no constitutional DNA for this cell line was available, this nucleotide change has been identified as a polymorphism in the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/index.html, refSNP ID: rs34781518) and therefore assumed as such. No nucleotide changes were identified in PARK7 or TNFRSF9.
Methylation/expression analysis of candidate genes
Methylation status at the CpG islands (CGI) of the following genes in the homozygously deleted region, CAMTA1, VAMP3, PER3, ERRFI1/MIG-6, TNFRSF9, SLC45A1/DNB5, RERE, ENO1, H6PD and SPSB1/SSB1, were studied using MSP (for gene locations see Fig. 3A). No CGIs were identified for UTS2, CA6 and SLC2A7 (see Materials and Methods for definition of CGI used). Among them, methylation of the TNFRSF9 CGI was found in 21/108 (19%) of primary tumours (18/77 (23%) GB, 2/22 (9%) AA, 1/9 (11%) A) and 13/15 cell lines (87%, Table 1). The methylation of TNFRSF9 was further confirmed in two tumours (GB105 and GB152) by bisulfite-sequencing (Fig. 4C and see Materials and methods). No methylation of TNFRSF9 was found in DNA from three normal brain specimens or one specimen of peripheral white blood cells. Methylation of ENO1 was found in one GB and two GBs had methylation of SPSB1/SSB1. The other seven genes examined showed no evidence of methylation.
The expression levels of TNFRSF9 were examined by real-time PCR using a LightCycler in 21 astrocytic tumours with methylation of TNFRSF9 and 30 tumours without. No significant difference of expression levels between these two groups was observed (data not shown). To further investigate the association between the methylation status of TNFRSF9 and its expression, U251, a glioblastoma cell line with methylation of TNFRSF9, was treated with the demethylating agent 5-aza-2′-deoxycytidine (Fig. 4B). Following treatment for 4 days, there was a 15-fold increase in TNFRSF9 expression (Fig. 4D, top panel). MCF7, a breast cancer cell line that did not have methylation of TNFRSF9, showed no significant change in expression levels following the de-methylation treatment (Fig. 4D, bottom panel).
Chromosome 1 amplification in astrocytic tumours
Amplification was noted in 10 tumours (1 AA, 9 GB, see Table 1 and Figs. 2 and 5). Four glioblastomas, GB291, GB225, GB153 and GB263 had amplicons of various sizes, all including a common region at 1q44 between RP11-150L22 and RP11-350M10 (Fig. 5). This region contained four genes, SDCCAG8, AKT3, ZNF238 and NP_001013732 (Fig. 5B, lower panel). Further amplicons were found in GB197 at 1p36.21 between RP11-219C24 and RP5-1108H3, a region included in a larger amplicon in GB251 (between RP11-474O21 and RP11-227B14 with interruptions). The commonly targeted region in these two cases includes 5 genes (PRDM2, PRAMEF8, PRAMEF9, LRRC38 and PDPN). Yet another amplicon on 1p31.3-p32.1 in AA99 (between RP11-351G1 and RP11-776H12) was entirely included in a larger amplicon in GB168 (between RP5-994L8 and RP4-537K17). This region does not contain any known genes. Other amplified regions were found in GB149 (1p35.1; RP11-131M11 - RP1-149P10) and GB126 (1p34.2-p34.3; RP3-423B22 - RP4-562N20).
Fig. 5.

A. A log2 ratio plot of chromosome 1 in GB263. Homozygous deletion at 1p36.22-p36.23 (see also Fig. 3) and amplification at 1q44 are indicated by arrows. B. Superimposed log2 ratio plots of GB291, GB225, GB153 and GB263 around the amplicons at 1q44. A schematic diagram of physical locations of clones and genes in the minimal region of overlap, corresponding chromosomal bands and nucleotide position ruler are shown below the plot (modified after Ensembl 33.35e, September 2005).
Discussion
This study focused on profiling chromosome 1 alterations and precisely defining the minimum common regions of alterations in astrocytic tumours. Our results show that deletions at 1p36 are common abnormalities among astrocytic tumours. They are generally small and overlap with the 9 homozygous deletions we identified in glioblastomas. It occurred in increasing incidence with malignancy grade, suggesting that this region is involved in progression. In contrast, total deletions of 1p were rare in all grades of astrocytoma. Thus, deletions affecting 1p are common abnormalities in both astrocytic and oligodendroglial tumours, however the patterns of deletions are different. Moreover, total 1p deletions in this astrocytoma series had a significantly longer overall survival independent of age and malignancy grade as compared to the tumours with normal 1p status or other patterns of deletions (p=0.005 in Cox’s regression).
The association between 1p status and prognosis of astrocytic tumours has been controversial (He et al., 2001; Homma et al., 2006; Schmidt et al., 2002). Some of these studies examined a limited number of loci on 1p36, in which case the different types of 1p deletion cannot be distinguished. In line with our study, Idbaih et al compared total and partial 1p deletions in gliomas and found that total deletions of 1p, but not partial deletions, were associated with longer survival (Idbaih et al., 2005). Our results suggest that tumours with total 1p deletions and those with partial 1p36 deletions belong to biologically distinct classes of tumours, and must be clearly distinguished. This is particularly important when an assessment of 1p and 19q status is used in stratification or selection for clinical trials or recommended as a supplement to morphological diagnosis. Great care must thus be taken when choosing loci for conventional microsatellite or FISH analysis of 1p.
We identified 9 homozygous deletions at 1p36 in GBs. Detailed mapping revealed that homozygous deletions of GB219 and GB136 did not overlap, indicating that two separate regions were being targeted (Fig. 3). In the telomeric region defined by the homozygous deletion of GB136, RP5-892F13, RP11-478I22 and RP11-431K24 were most frequently deleted. TNFRSF9 located on RP5-892F13 was deleted in 7 tumours including GB263, which had an isolated homozygous deletion at this clone. No somatic mutations were found in TNFRSF9. However hypermethylation of the CGI of TNFRSF9 was identified in approximately 20% of primary astrocytic tumours and 87% of glioblastoma cell lines.
TNFRSF9 encodes the CD137 (ILA / 4-1BB) protein, a member of tumour necrosis factor receptor family (reviewed in (Watts, 2005)). TNFRSF9/CD137 is expressed on the surface of activated T-cells. Antigen presenting cells express its ligand as a cell surface trans-membrane protein (TNFSF9/4-1BBL). TNFRSF9/CD137 mediated signals promote cell survival and the secretion of various chemokines by T-cells. The role of TNFRSF9/CD137 in non-immune cells is not clear. It is interesting that TNFRSF9 is homozygously deleted, or hemizygously deleted with methylation of the retained allele in a subset of astrocytic tumours and glioma cell lines, while no such methylation was found in normal brain tissues. Although no significant difference of TNFRSF9 expression levels was observed between primary tumours with TNFRSF9 methylation and those without, a strong association between the methylation status of TNFRSF9 and its mRNA expression was demonstrated by demethylating cultured glioblastoma cells with 5′-aza-2′-deoxycytidine (Fig. 4D). The lack of correlation between expression levels and methylation status in primary tumour tissue may be due the presence of non-neoplastic cells such as macrophages that express high levels of this receptor (Melero et al., 1997) and that are commonly present in high numbers in glioblastomas. TNFRSF9 was the only gene found frequently altered among the genes recognised so far in the homozygously deleted region. TNFRSF9 was not methylated in GB219 or GB176, in which TNFRSF9 was not involved in the homozygous deletions, excluding the possibility that TNFRSF9 is the sole target of all 1p36 deletion cases (Fig. 3A).
RERE was identified as a gene coding for a protein homologous to the atrophin-1 protein, in which abnormal glutamine expansion causes dentatorubral-pallidoluysian atrophy (DRPLA), a rare hereditary neurodegenerative disorder (Yanagisawa et al., 2000). Ectopically expressed RERE has been shown to induce apoptosis in a caspase-3-dependent manner (Waerner et al., 2001), suggesting that RERE may be involved in promoting apoptosis and is therefore a potential TSG. All but one homozygous deletion (GB136) involved at least a part of RERE, however no somatic mutations or promoter hypermethylation were identified in RERE after screening a large number of gliomas. Thus the significance of RERE homozygous deletions in glioblastomas remains obscure.
The possibility still exists that other gene(s) or regulatory sequences in this region may be being targeted. Several non-coding RNA genes have been mapped in this region. The presence of novel, as yet unrecognised gene(s) cannot be excluded. The lack of recurrent breakpoints for gains/deletions (see Fig. 2) indicates that a recurrent translocation with a resultant a novel fusion gene is unlikely.
Hemizygous 1p36 deletions also occurred outside the homozygously deleted region defined above (e.g., GB63, GB152 and AA14, see Fig. 2B). These deletions overlap with minimally deleted regions reported previously (Barbashina et al., 2005; Dong et al., 2004). Barbashina et al. defined the minimal region of deletion around CAMTA1. A limited analysis of this gene identified no mutations (Barbashina et al., 2005). CAMTA1 was also included in the homozygously deleted regions in 5GBs of our series (Fig. 3A). TP73, a homologue of TP53 involved in regulation of cell proliferation and apoptosis, is located on 1p36.32, telomeric to the homozygously deleted region but was included in a number of hemizygous deletions (Fig. 2B). No somatic mutations of TP73 have been identified so far (Alonso et al., 2001), however hypermethylation of the promoter region has been reported in a subset of gliomas (Watanabe et al., 2002). Bagchi et al identified CHD5 (Chromodomain helicase DNA binding domain 5) as a candidate tumour suppressor gene using mice genetically engineered to harbour alterations in the chromosomal region corresponding to human 1p36 (Bagchi et al., 2007). Mouse Chd5 was shown to positively regulate the p19Arf/p53 pathways and mouse embryonic fibroblasts deficient in Chd5 formed tumours in nude mice following transplantation. Human gliomas with CHD5 deletions showed low expression levels. CHD5 maps telomeric to but not within the homozygously deleted region we identified (Fig. 2B). So far no somatic mutation of CHD5 has been reported in human tumours but this requires further investigation. Interestingly, among the other genes mapped to this region, neither Camta1 nor Tp73 showed the ability to modulate proliferation in the study of Bagchi et al, while Tnfrsf9 was not examined.
We identified 10 novel amplifications on chromosome 1. Most notably, 4 glioblastomas had amplifications encompassing AKT3. Akt3 (PKBγ) is one of three isoforms of phosphoinositide-dependent serine-threonine protein kinases, Akt (Protein Kinase B). Akt plays a central role in the PI3K-Akt-mTOR signalling pathway, which is involved in regulating many cellular functions including cell proliferation, growth survival, cell motility and angiogenesis (Bellacosa et al., 2005). Deregulation of this pathway such as mutations of PTEN is common in glioblastomas. Components of this pathway are frequently altered in diverse tumour types. Amplification involving the AKT3 locus has been particularly observed in hepatitis C virus-associated hepatocellular carcinomas (Hashimoto et al., 2004), although previous reports on glioblastomas have not found this (Knobbe & Reifenberger, 2003). As our tumour series was not selected for alterations of this region (see Results), the frequency of amplification would be about 5% (4/77). Interestingly, two of the four GBs in our series with AKT3 amplification also had PTEN mutations ((Schmidt et al., 1999) and unpublished data). PTEN mutation and AKT3 amplification/overexpression may have a synergetic effect. A precise amplicon mapping with correlation to mRNA expression is underway.
The data presented here revealed that chromosome 1 status in gliomas, particularly in astrocytic tumours, is much more complex than previously thought. A thorough assessment of 1p status across different glioma types, correlation of the findings with clinical features, the identification of what is being targeted as well as an understanding of the biological consequences of the abnormalities will establish their clinical significance and improve our understanding of the molecular mechanisms underlying the progression of the astrocytic gliomas.
Materials and methods
All chromosome 1 array clones, positions of copy number variations and log2-ratio are shown in supplementary table 1. All primers, PCR and gel conditions are shown in supplementary table 2.
Tumour materials and nucleic acid extraction
A total of 265 primary astrocytic tumours including 22 astrocytoma malignancy grade II (prefixed as A), 61 anaplastic astrocytoma malignancy grade III (AA) and 182 glioblastoma malignancy grade IV (GB), as well as 24 glioblastoma xenografts (established from 24 glioblastomas included in the above series) and 15 glioma cell lines were included in the study. In addition, 95 oligodendroglial tumours were included in the mutation analysis of selected genes. The histopathological diagnosis of the astrocytic tumours was reviewed by one neuropathologist (V.P.C) and made according to the WHO classification (Kleihues & Cavenee, 2000). Collection, handling and DNA/RNA extraction of tumour tissues and the patients’ blood samples were as described previously (Ichimura et al., 2000). cDNA was generated using total RNA that had been treated with RNase-free DNaseI as published (Liu et al., 2005). The study was approved by the Ethical Committee of the Karolinska Hospital (No. 91:16) and Cambridge Local Research Ethics Committee, Cambridge, UK (Ref. LREC 03/115).
Microsatellite analysis
The following chromosome 1 microsatellite markers were used; (1p) D1S468, D1S2667, D1S228, D1S247, D1S1598, D1S220, D1S464, D1S418, (1q) D1S2655, D1S2703. Microsatellite analysis (MSA) was performed as previously described (Ichimura et al., 1998).
Chromosome 1 tile path array
Two versions of chromosome 1 tile path array were used. The first version has been previously described (Buckley et al., 2005). The second version consisted of the same set of clones with an additional 113 chromosome 1 clones added to fill some gaps in the first version. In total, 2222 clones (1437 BACs, 778 PACs, 7 cosmids) covering 97.9% of the sequenced regions of chromosome 1 were included for the analysis (NCBI build 35, (Gregory et al., 2006)). The second version also included a set of 538 BACs that were distributed over all autosomes with an average midpoint interval of 5.2Mb (5Mb clone set). The majority of the tumours (96 tumours) were studied using the second version, of which 31 were examined using both to confirm reproducibility. The construction, hybridization and analysis of the chromosome 1 tile path array versions 1 and 2 were carried out according to the published protocol with minor modifications (Fiegler et al., 2003; Ichimura et al., 2006). For normalization, the median of the ratio of test/reference spot intensity of all clones on either 1p or 1q was used as the normalization factor in the first version of array. The chromosomal arm on which most microsatellite markers showed allelic balance was chosen for normalization. In the second version, the median of test/reference spot intensity ratios of all autosomal clones in the 5-Mb clone set was used as a normalization factor independent of the prior knowledge of chromosome 1 allelic status.
Multiplex PCR, DGGE and sequencing
Multiplex PCR was performed and the copy number (normal value = 2) at each target locus was calculated as described (Schmidt et al., 1999). PCR products on an agarose gel were quantified using a Labworks software and GelDoc-It Imaging System (UVP, Cambridge, UK). Denaturing Gradient Gel Electrophoresis (DGGE) and sequencing were optimized and performed as described (Ichimura et al., 2000). Primer design and sequencing analysis were carried out using MacVector v8.1 (MacVector, Inc. Cambridge, UK) or Accelrys Gene 2.0 (Accelrys, Cambridge, UK).
Methylation analysis
Bisulfite-modification of genomic DNA and methylation-specific PCR (MSP) was performed according to the published protocol with minor modifications (Herman et al., 1996). CpG islands (CGI) for the genes at 1p36.22-p36.23 (CAMTA1, VAMP3, PER3, ERRFI1/MIG-6, TNFRSF9, SLC45A1/DNB5, RERE, ENO1, H6PD and SPSB1/SSB1) were identified using the Methprimer software (http://www.urogene.org/methprimer/index.html) with the following criteria: Island size > 100bp, G+C content > 50.0%, Observed to Expected ratio of CpG dinucleotides > 0.60. Primers for methylation-specific PCR (MSP) were designed within the defined CGIs to specifically amplify bisulfite-modified methylated sequences. Bisulfite-modified DNA was amplified using a primer pair for ACTB that amplified bisulfite-modified sequences regardless of their original methylation status to confirm the efficiency of modification (Harden et al., 2003). Unmodified genomic DNA, bisulfite-modified normal blood DNA, SssI methylase-treated and bisulfite-modified normal blood DNA, and no template control were included in every PCR run as a control.
Primers for bisulfite-sequencing of TNFRSF9 were designed to amplify bisulfite-modified sequences of the entire CGI regardless of the methylation status. The PCR products were then subcloned into a pCR4-TOPO vector according to the manufacturer’s recommendations (Invitrogen, Paisley, UK) and a minimum of 10 clones from each construct were sequenced.
Real-time quantitative PCR
Gene copy number change at exon 6 of SLC45A1/DNB5 and mRNA expression of TNFRSF9 and RERE were assessed by real-time PCR using a FastStart DNA Master SYBR Green I® with LightCycler® (Roche Diagnostics, Mannheim, Germany). WI-3306 (STS at 2q21) and the 18S ribosomal RNA gene were used as an internal reference for copy number and expression analysis respectively. Samples were amplified in duplicate using a LightCycler version 3.5 software according to the manufacturer’s recommendations (Roche Diagnostics, Mannheim, Germany). A consistent calibrator (mixture of various genomic DNA or cDNA) was included in each run for normalization. The PCR products of the target or reference genes, directly or subcloned into a pCR4-TOPO vector, were used in a dilution series to determine relative standard curves for efficiency correction. After the amplification, the Crossing Points for individual samples were automatically determined using the Second Derivative Maximum Method using LightCycler version 3.5. The PCR efficiency-corrected and normalized target/reference ratio was then determined using a RelQuant software (Roche Diagnostics, Mannheim, Germany). For SLC45A1/DNB5, a gene dosage was calculated as a relative target/reference ratio of each tumour DNA to that of control DNA (average of five normal tissue DNA).
5-aza-2′-deoxycytidine treatment
For each treatment group, 2 × 105 cells (U251 or MCF7) were plated in a T25 flask in triplicate. After 24 hours, cells were cultivated with the medium containing 1 μM of 5-aza-2′-deoxycytidine for either 48 hours or 96 hours. The medium containing the reagent was changed every 24 hours. After the treatment, cells were cultivated for further 48 hours without 5-aza-2′-deoxycytidine, RNA extracted and expression levels quantified using real-time PCR as described above.
Statistical analysis
The log rank test was used for survival analysis, assessing the potential prognostic value of the 1p status. Cox’ regression analysis was performed to adjust for age and WHO grade. All statistical analysis were performed using Statistical Package for the Social Sciences ver 14.0 (SPSS).
Supplementary Material
{kind=link}
Acknowledgements
We would like to thank the Mapping Core, Map Finishing and Microarray Facility groups of the Wellcome Trust Sanger Institute, Hinxton, UK, for initial clone supply and verification, Rachel Cooper for technical assistance in the version 1 chromosome 1 array production, the Centre for Microarray resources in the Department of Pathology, University of Cambridge, for printing of the arrays, and Bo Nilsson for his assistance in statistical analysis.
Financial support: Cancer Research UK, The Jacqueline Seroussi Memorial Foundation for Cancer Research, Samantha Dickson Brain Tumour Trust, CAMPOD, the Ludwig Institute for Cancer Research and the Wellcome Trust.
Abbreviations
- CGH
comparative genomic hybridisation
- TSG
tumour suppressor gene
- MSA
microsatellite analysis
- PAC
P1-derived artificial chromosome
- BAC
bacterial artificial chromosome
- DGGE
denaturing gradient gel electrophoresis
- CGI
CpG island
- MSP
methylation-specific PCR
Footnotes
The raw data for array-CGH can be retrieved from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/, Accession No. GSE7428).
References
- Alonso ME, Bello MJ, Lomas J, Gonzalez-Gomez P, Arjona D, De Campos JM, et al. Absence of mutation of the p73 gene in astrocytic neoplasms. Int J Oncol. 2001;19:609–612. doi: 10.3892/ijo.19.3.609. [DOI] [PubMed] [Google Scholar]
- Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, et al. CHD5 Is a Tumor Suppressor at Human 1p36. Cell. 2007;128:459–475. doi: 10.1016/j.cell.2006.11.052. [DOI] [PubMed] [Google Scholar]
- Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M. Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin Cancer Res. 2005;11:1119–1128. [PubMed] [Google Scholar]
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- Bigner SH, Mark J, Burger PC, Mahaley MS, Jr., Bullard DE, Muhlbaier LH, et al. Specific chromosomal abnormalities in malignant human gliomas. Cancer Res. 1988;48:405–411. [PubMed] [Google Scholar]
- Buckley PG, Jarbo C, Menzel U, Mathiesen T, Scott C, Gregory SG, et al. Comprehensive DNA copy number profiling of meningioma using a chromosome 1 tiling path microarray identifies novel candidate tumor suppressor loci. Cancer Res. 2005;65:2653–2661. doi: 10.1158/0008-5472.CAN-04-3651. [DOI] [PubMed] [Google Scholar]
- Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707–2714. doi: 10.1200/JCO.2005.04.3414. [DOI] [PubMed] [Google Scholar]
- Dong Z, Pang JS, Ng MH, Poon WS, Zhou L, Ng HK. Identification of two contiguous minimally deleted regions on chromosome 1p36.31-p36.32 in oligodendroglial tumours. Br J Cancer. 2004;91:1105–1111. doi: 10.1038/sj.bjc.6602093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsberg J, Erkwoh A, Sabel MC, Kirsch L, Fimmers R, Blaschke B, et al. Oligodendroglial tumors: refinement of candidate regions on chromosome arm 1p and correlation of 1p/19q status with survival. Brain Pathol. 2004;14:121–130. doi: 10.1111/j.1750-3639.2004.tb00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiegler H, Carr P, Douglas EJ, Burford DC, Hunt S, Scott CE, et al. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer. 2003;36:361–374. doi: 10.1002/gcc.10155. [DOI] [PubMed] [Google Scholar]
- Gregory SG, Barlow KF, McLay KE, Kaul R, Swarbreck D, Dunham A, et al. The DNA sequence and biological annotation of human chromosome 1. Nature. 2006;441:315–321. doi: 10.1038/nature04727. [DOI] [PubMed] [Google Scholar]
- Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD, et al. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol. 2006;65:988–994. doi: 10.1097/01.jnen.0000235122.98052.8f. [DOI] [PubMed] [Google Scholar]
- Harden SV, Guo Z, Epstein JI, Sidransky D. Quantitative GSTP1 methylation clearly distinguishes benign prostatic tissue and limited prostate adenocarcinoma. J Urol. 2003;169:1138–1142. doi: 10.1097/01.ju.0000049627.90307.4d. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Mori N, Tamesa T, Okada T, Kawauchi S, Oga A, et al. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Mod Pathol. 2004;17:617–622. doi: 10.1038/modpathol.3800107. [DOI] [PubMed] [Google Scholar]
- Hashimoto N, Murakami M, Takahashi Y, Fujimoto M, Inazawa J, Mineura K. Correlation between genetic alteration and long-term clinical outcome of patients with oligodendroglial tumors, with identification of a consistent region of deletion on chromosome arm 1p. Cancer. 2003;97:2254–2261. doi: 10.1002/cncr.11322. [DOI] [PubMed] [Google Scholar]
- He J, Mokhtari K, Sanson M, Marie Y, Kujas M, Huguet S, et al. Glioblastomas with an oligodendroglial component: a pathological and molecular study. J Neuropathol Exp Neurol. 2001;60:863–871. doi: 10.1093/jnen/60.9.863. [DOI] [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma T, Fukushima T, Vaccarella S, Yonekawa Y, Di Patre PL, Franceschi S, et al. Correlation among pathology, genotype, and patient outcomes in glioblastoma. J Neuropathol Exp Neurol. 2006;65:846–854. doi: 10.1097/01.jnen.0000235118.75182.94. [DOI] [PubMed] [Google Scholar]
- Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res. 2000;60:417–424. [PubMed] [Google Scholar]
- Ichimura K, Mungall AJ, Fiegler H, Pearson DM, Dunham I, Carter NP, et al. Small regions of overlapping deletions on 6q26 in human astrocytic tumours identified using chromosome 6 tile path array-CGH. Oncogene. 2006;25:1261–1271. doi: 10.1038/sj.onc.1209156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura K, Ohgaki H, Kleihues P, Collins VP. Molecular pathogenesis of astrocytic tumours. J Neurooncol. 2004;70:137–160. doi: 10.1007/s11060-004-2747-2. [DOI] [PubMed] [Google Scholar]
- Ichimura K, Schmidt EE, Miyakawa A, Goike HM, Collins VP. Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Genes Chromosomes Cancer. 1998;22:9–15. doi: 10.1002/(sici)1098-2264(199805)22:1<9::aid-gcc2>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Idbaih A, Marie Y, Pierron G, Brennetot C, Hoang-Xuan K, Kujas M, et al. Two types of chromosome 1p losses with opposite significance in gliomas. Ann Neurol. 2005;58:483–487. doi: 10.1002/ana.20607. [DOI] [PubMed] [Google Scholar]
- Ino Y, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, Jhung S, et al. Long survival and therapeutic responses in patients with histologically disparate high-grade gliomas demonstrating chromosome 1p loss. J Neurosurg. 2000;92:983–990. doi: 10.3171/jns.2000.92.6.0983. [DOI] [PubMed] [Google Scholar]
- Iuchi T, Namba H, Iwadate Y, Shishikura T, Kageyama H, Nakamura Y, et al. Identification of the small interstitial deletion at chromosome band 1p34-p35 and its association with poor outcome in oligodendroglial tumors. Genes Chromosomes Cancer. 2002;35:170–175. doi: 10.1002/gcc.10080. [DOI] [PubMed] [Google Scholar]
- Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Cavenee WK. Pathology and Genetics of Tumours of the Nervous System. IARC Press; Lyon: 2000. [Google Scholar]
- Knobbe CB, Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003;13:507–518. doi: 10.1111/j.1750-3639.2003.tb00481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschny R, Koschny T, Froster UG, Krupp W, Zuber MA. Comparative genomic hybridization in glioma: a meta-analysis of 509 cases. Cancer Genet Cytogenet. 2002;135:147–159. doi: 10.1016/s0165-4608(01)00650-1. [DOI] [PubMed] [Google Scholar]
- Kros JM, Gorlia T, Kouwenhoven MC, Zheng PP, Collins VP, Figarella-Branger D, et al. Panel Review of Anaplastic Oligodendroglioma From European Organization for Research and Treatment of Cancer Trial 26951: Assessment of Consensus in Diagnosis, Influence of 1p/19q Loss, and Correlations With Outcome. J Neuropathol Exp Neurol. 2007;66:545–551. doi: 10.1097/01.jnen.0000263869.84188.72. [DOI] [PubMed] [Google Scholar]
- Liu L, Backlund LM, Nilsson BR, Grander D, Ichimura K, Goike HM, et al. Clinical significance of EGFR amplification and the aberrant EGFRvIII transcript in conventionally treated astrocytic gliomas. J Mol Med. 2005;83:917–926. doi: 10.1007/s00109-005-0700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- MRC (The Medical Research Council Brain Tumour Working Party) Randomized trial of procarbazine, lomustine, and vincristine in the adjuvant treatment of high-grade astrocytoma: a Medical Research Council trial. J Clin Oncol. 2001;19:509–518. doi: 10.1200/JCO.2001.19.2.509. [DOI] [PubMed] [Google Scholar]
- Mueller W, Hartmann C, Hoffmann A, Lanksch W, Kiwit J, Tonn J, et al. Genetic signature of oligoastrocytomas correlates with tumor location and denotes distinct molecular subsets. Am J Pathol. 2002;161:313–319. doi: 10.1016/S0002-9440(10)64183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro JM, Takahashi MA, Ginzinger DG, Law M, Passe S, Jenkins RB, et al. Detection of 1p and 19q loss in oligodendroglioma by quantitative microsatellite analysis, a real-time quantitative polymerase chain reaction assay. Am J Pathol. 2001;158:1253–1262. doi: 10.1016/S0002-9440(10)64076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifenberger G, Louis DN. Oligodendroglioma: toward molecular definitions in diagnostic neuro-oncology. J Neuropathol Exp Neurol. 2003;62:111–126. doi: 10.1093/jnen/62.2.111. [DOI] [PubMed] [Google Scholar]
- Schmidt EE, Ichimura K, Goike HM, Moshref A, Liu L, Collins VP. Mutational profile of the PTEN gene in primary human astrocytic tumors and cultivated xenografts. J Neuropathol Exp Neurol. 1999;58:1170–1183. doi: 10.1097/00005072-199911000-00007. [DOI] [PubMed] [Google Scholar]
- Schmidt MC, Antweiler S, Urban N, Mueller W, Kuklik A, Meyer-Puttlitz B, et al. Impact of genotype and morphology on the prognosis of glioblastoma. J Neuropathol Exp Neurol. 2002;61:321–328. doi: 10.1093/jnen/61.4.321. [DOI] [PubMed] [Google Scholar]
- Smith JS, Alderete B, Minn Y, Borell TJ, Perry A, Mohapatra G, et al. Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene. 1999;18:4144–4152. doi: 10.1038/sj.onc.1202759. [DOI] [PubMed] [Google Scholar]
- Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J, Hosek SM, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol. 2000;18:636–645. doi: 10.1200/JCO.2000.18.3.636. [DOI] [PubMed] [Google Scholar]
- Ueki K, Nishikawa R, Nakazato Y, Hirose T, Hirato J, Funada N, et al. Correlation of histology and molecular genetic analysis of 1p, 19q, 10q, TP53, EGFR, CDK4, and CDKN2A in 91 astrocytic and oligodendroglial tumors. Clin Cancer Res. 2002;8:196–201. [PubMed] [Google Scholar]
- van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715–2722. doi: 10.1200/JCO.2005.04.6078. [DOI] [PubMed] [Google Scholar]
- von Deimling A, Fimmers R, Schmidt MC, Bender B, Fassbender F, Nagel J, et al. Comprehensive allelotype and genetic anaysis of 466 human nervous system tumors. J Neuropathol Exp Neurol. 2000;59:544–558. doi: 10.1093/jnen/59.6.544. [DOI] [PubMed] [Google Scholar]
- Waerner T, Gardellin P, Pfizenmaier K, Weith A, Kraut N. Human RERE is localized to nuclear promyelocytic leukemia oncogenic domains and enhances apoptosis. Cell Growth Differ. 2001;12:201–210. [PubMed] [Google Scholar]
- Watanabe T, Huang H, Nakamura M, Wischhusen J, Weller M, Kleihues P, et al. Methylation of the p73 gene in gliomas. Acta Neuropathol (Berl) 2002;104:357–362. doi: 10.1007/s00401-002-0549-1. [DOI] [PubMed] [Google Scholar]
- Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Bundo M, Miyashita T, Okamura-Oho Y, Tadokoro K, Tokunaga K, et al. Protein binding of a DRPLA family through arginine-glutamic acid dipeptide repeats is enhanced by extended polyglutamine. Hum Mol Genet. 2000;9:1433–1442. doi: 10.1093/hmg/9.9.1433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.