

Abstract
Two epoxide-containing peptidomimetics of the isopeptide, glutamyl-γ-glutamate, have been synthesized via a route that should be generally applicable to the synthesis of isopeptide analogs in which an oxirane replaces the scissile peptide bond. Enzymes that catalyze the hydrolysis of peptides and isopeptides are often susceptible to inactivation by electrophilic substrate analogs. In this research, an epoxide was installed as an electrophilic replacement of the scissile isopeptide bond. The C-terminal glutamyl mimic was accessed by the stereospecific synthesis of suitably substituted cyclopentenes as surrogates for either the l- or d- enantiomer. The enantiomeric cyclopentenes were further elaborated to incorporate an appended sulfone that was reacted with a suitably protected glutamyl-γ-semialdehyde in a Julia-Kocienski olefination reaction. This olefination afforded predominantly the desired E-olefin isosteres of l-glutamyl-γ-d-glutamate and l-glutamyl-γ-l-glutamate, following which peracid-mediated epoxidation and deprotection provided the epoxide-containing peptidomimetics, 4 and 5.
Introduction
Although the design and synthesis of mechanism-based inhibitors of proteases that act on proteins and peptides involving α-linked amino acids has been investigated extensively,1 similar efforts to identify specific inhibitors of enzyme-catalyzed isopeptide hydrolysis are much less common.2 The latter reaction, although less well studied, is of critical importance in areas such as the ubiquitination-deubiquitination cycle3 and folate polyglutamate homeostasis involving the formation and hydrolysis of γ-glutamyl peptides.4 The use of electrophilic substrate analogs as enzyme inactivators and probes of enzyme mechanism and structure has been at the forefront of medicinal and bioorganic chemistry research for decades. Thus, the development of “active-site-directed irreversible inhibitors”5,6 and subsequently, “mechanism-based enzyme inhibitors”7 has been a powerful tool in elucidating the mechanism of numerous enzyme-catalyzed reactions.
This approach has been applied to the design of selective inactivators of proteases that are postulated (and in many cases proven) to proceed via acyl enzyme intermediates; i.e., cysteine, serine, and threonine proteases.1 In most of these inhibitors, however, the pendant electrophilic moiety (e.g., halomethylketone, acyloxymethylketone, epoxysuccinates, α,β-epoxyketones, vinyl sulfones) is usually incorporated in place of the C-terminal carboxyl group of the peptide substrate. The well-studied ubiquitination pathway of protein degradation involves ATP-dependent attachment of ubiquitin, a small protein, to a targeted protein, followed by recycling of the ubiquitin via hydrolysis of the isopeptide bond between the C-terminal glycine residue of ubiquitin and the ε-amino group of a lysine residue of the targeted protein.3 Inhibition of the isopeptide hydrolase has been effected by the attachment of an electrophilic moiety to the C-terminus of ubiquitin.2 The structure of one ubiquitin C-terminal hydrolase, UCH-L3, has been determined by X-ray crystallography in the absence8 and presence of ubiquitin to which an electrophilic vinyl methyl ester was attached to the C-terminal glycine.9
γ-Glutamyl hydrolase (GH, EC 3.4.19.9), is a cysteine peptidase10 that catalyzes the hydrolysis of γ-glutamyl peptides11 which, together with the ATP-dependent ligase, folylpolyglutamate synthetase (FPGS, EC 6.3.2.17) controls folate homeostasis in living cells. Our interest in GH led us to consider the synthesis of electrophilic substrate analogs for use in further probing the catalytic mechanism as well as for developing probes for the localization of this enzyme in complex biological environments. Our initial focus was the synthesis of C-terminal epoxides due to the demonstrated selectivity of the epoxide moiety for cysteine proteases, as well as the many reported syntheses of C-terminal epoxides and related epoxysuccinyl peptides in the literature.1,12 The reaction catalyzed by GH (1a → 2a + 3a) is shown in Figure 1. Evaluation of a C-terminal epoxide derivative, 1b, as an inhibitor of GH revealed that 1b inhibited the enzyme but most, if not all of the inhibition could be attributed to GH-catalyzed hydrolysis (1b → 2a + 3b); i.e., 1b acts an alternate substrate.13 GH does not display product inhibition and, consistent with this fact, 2b, a compound lacking the isopeptide bond but also containing a C-terminal epoxide, is neither a substrate nor an inhibitor. As a result, subsequent synthetic efforts have been aimed at the synthesis of epoxide peptidomimetics in which the scissile isopeptide bond is replaced by an oxirane.
Figure 1.

Reaction catalyzed by γ-glutamyl hydrolase
There is some precedent for the replacement of a peptide bond by an oxirane and our research builds on those initial studies.14-18 These syntheses involved either peracid-mediated epoxidation of the corresponding E- or Z-olefins, or elaboration of the corresponding β-hydroxy selenide to the desired epoxide via an oxidative cyclization process. However, to the best of our knowledge, no such epoxide peptidomimetics of isopeptides have been synthesized. In this paper, using a peracid-mediated epoxidation of an olefin, we describe the synthesis of two diastereomeric epoxide peptidomimetics, 4 and 5, in which the scissile isopeptide bond is replaced by an oxirane.

Results & Discussion
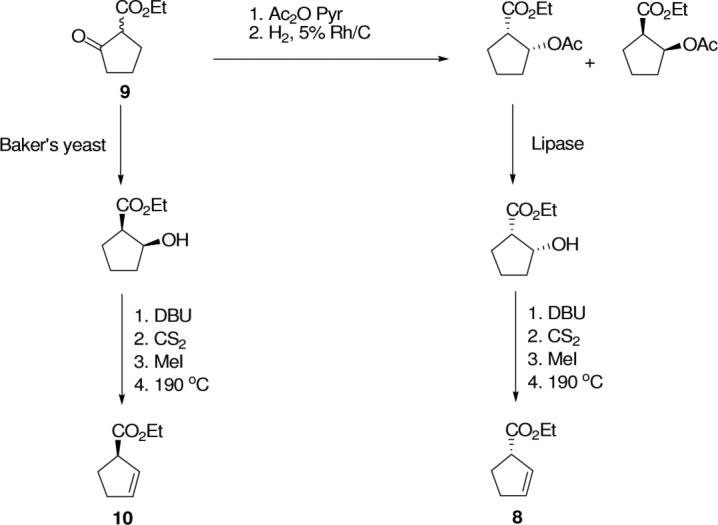
In previous research from our laboratory, the use of cyclopentene derivatives as glutarate surrogates in the synthesis of phosphinic acid-containing pseudopeptide inhibitors of FPGS has been described.19 In the present work, a similar approach has been pursued. Thus, the desired oxiranes are envisioned to be accessible from the corresponding E-olefin as shown in Figure 2 for the l-Glu-γ-l-Glu mimetic, 4, with the chiral centers derived ultimately from (N,N-Boc2)-l-glutamic acid γ-semialdehyde, 7, and (S)-1-(carboethoxy)cyclopent-2-ene, 8. The l-Glu-γ-d-Glu mimetic, 5, is also accessible via the same route from the enantiomer of 8 (ent-8). In developing a strategy for the synthesis of target olefin isosteres of the γ-glutamyl dipeptide (e.g., 6), and the corresponding internal epoxide derivatives (e.g., 4), the two important synthetic challenges in the molecules that need to be addressed are, (i) stereospecific generation of the chiral center which serves as a pseudo α-carbon in the C-terminal glutamic acid replacement, analogous to the 2S or 2R stereochemistry in the parent isopeptide, and (ii) selective formation of the E-olefin during synthesis of the olefinic dipeptide isostere. Stereospecific generation of either stereoisomer at C-1 of the cyclopent-2-ene derivative, 8 or ent-8, from ethyl 2-oxocyclopenane carboxylate, 9, has been achieved via lipase-mediated ester hydrolysis13,19-21, or a dynamic kinetic resolution,22 respectively, (Scheme 1) leading ultimately to the “l-Glu-γ-l-Glu” mimetic, 4, and the “l-Glu-γ-d-Glu” mimetic, 5. The desired E-olefin is most consistently achieved by the Julia-Kocienski reaction through the olefination of an aldehyde (e.g., 7).23,24 As outlined in Scheme 1, the critical stereoselective synthesis of ent-8 (10), via sequential dynamic kinetic resolution, xanthate formation, and pyrolysis to form the desired cyclopentene derivative25 proceeded in higher yield (65%, 5 steps)26 than the synthesis of 8 from 9 via acetylation, hydrogenation of the enol acetate, lipase-catalyzed resolution, and pyrolysis of the enantiomeric xanthate (21%, 7 steps).19 Accordingly, the synthesis of 5 from the more readily available enantiomer, 10, was investigated before the synthesis of 4 from 8 (Schemes 2-5).
Figure 2.
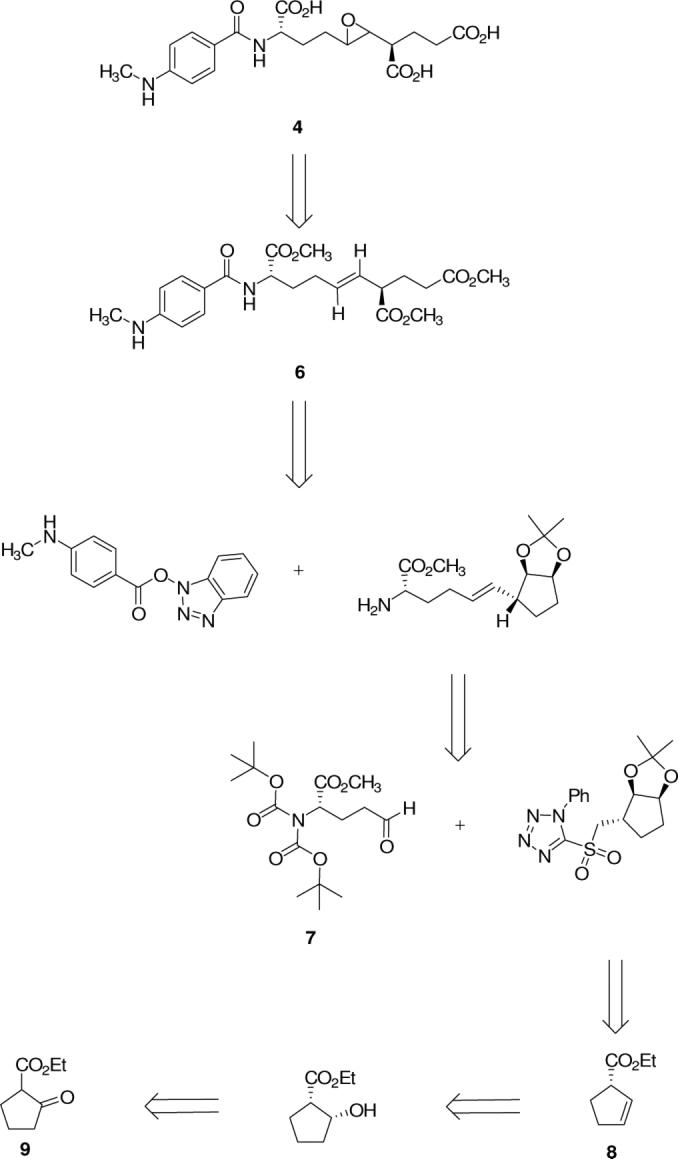
Synthesis of oxirane- and olefin-containing analogs of γ-glutamyl peptides: retrosynthetic analysis.
Scheme 1.
Scheme 2.

Scheme 5.

As shown in Scheme 2, dihydroxylation of the (1R)-β, γ-unsaturated ethyl ester derivative 10 was performed using the Upjohn procedure (OsO4/N-methylmorpholine-N-oxide)27 to afford the corresponding vicinal diols as an inseparable mixture of trans and cis diastereomers in 97% yield. To protect the free hydroxyl groups of the vicinal diols, reaction with 2, 2-dimethoxypropane/acetone/TsOH afforded the corresponding isopropylidene derivatives (93% yield, diastereomeric ratio (trans:cis) = ca 1.7:1 ), which were separated using flash chromatography to give 11 in 58% yield and the cis diastereomer, cis-11, in 35% yield. Stereochemistry of the diastereomers was confirmed by a nuclear Overhauser effect (nOe) between H-1 and H-2 that was observed only in the NMR spectra of the cis diastereomer. The ethyl ester functionality of the trans isomer was reduced using LiAlH4 at −78 °C to obtain the corresponding hydroxymethyl derivative in 95% yield. Mitsunobu coupling28 of this alcohol to 1-phenyl-1H-tetrazole-5-thiol using PPh3/diisopropyl-azodicarboxylate (DIAD) (0 °C → rt) furnished the sulfide product in 75% yield, which was followed by oxidation (H2O2, (NH4)6Mo7O24•4H2O, 0 °C) to give sulfone 12 in 87% yield.
The enantiomeric sulfone, 14, was obtained in an analogous manner from the enantiomeric cyclopentene derivative, 8. Thus, dihydroxylation of the β, γ-unsaturated ethyl ester derivative 8 using the Upjohn procedure27 afforded the corresponding vicinal diols as an inseparable mixture of trans and cis diastereomers in 78% yield. Subsequent protection of the vicinal diols yielded the corresponding isopropylidene derivatives (91% yield, diastereomeric ratio (trans:cis) = ca 1.2:1). Separation of the diastereomers was accomplished by flash chromatography to give 13 in 49% yield and cis-13 in 43% yield. The ethyl ester functionality of the trans isomer was reduced using LiAlH4 to obtain the corresponding hydroxymethyl derivative in 81% yield, followed by Mitsunobu coupling28 to 1-phenyl-1H-tetrazole-5-thiol to give the corresponding thioether in 92% yield. This product was oxidized to sulfone 14 in 90% yield.
As shown in Scheme 3, the cis diastereomer, cis-13, was converted to the corresponding sulfone, cis-14, by identical chemistry to that described above for the trans diastereomer. Crystallization of cis-14 enabled the determination of the absolute configuration of cis-14 by X-ray crystallography and confirmed the absolute stereochemistry of all substituents in this series of compounds (11−14) originally deduced based on 1H-NMR spectral data (see Supporting Information for details of the synthesis of cis-14 and also the crystal structure determination).13
Scheme 3.

Previous research in our laboratory has shown that compounds similar to 12 and 14 with the 2,3-isopropylidene substituent trans to a bromomethyl substituent at C-1 are more reactive in SN2 reactions than the cis diastereomers.19 In the current research, olefination reactions of sulfone 14 or cis-14 proceeded in similar yields. Since the stereochemical distinction between these diastereomers is lost in the oxidative cleavage of the masked cyclopentene moiety to the glutarate targets, it would be reasonable to proceed with the synthesis using a mixture of diastereomeric isopropylidenes; e.g., 14 and cis-14. However, the 1H NMR spectra of these mixtures are very complex, involving numerous diastereomeric and diastereotopic nuclei. In contrast, spectra of compounds derived from a single diastereomer; i.e., 12 or 14, are considerably simplified and more readily interpreted. Therefore, we chose to explore the olefination reactions primarily with a single diastereomer, usually the more readily available trans diastereomer.29 Subsequent investigations can be carried out with the mixture of diastereomers.
One of the important features of this reaction design is the preferred formation of a trans double bond using the Julia-Kocienski olefination.23,24 Initial investigation of the reaction on a small scale at low temperature indicated that the reaction did not go to completion and a large proportion of unreacted sulfone 14 was recovered along with the desired product.13 However, as depicted in Scheme 4, reaction of glutamyl semialdehyde derivative 730 and sulfone 12 under optimized conditions23,24 (KHMDS, −78 °C) provided the olefin product 15 in 69% yield. Analysis of 1H NMR spectral data showed predominant formation of the desired E-olefin (E:Z = 9:1; Jtrans = 15 Hz vs. Jcis = 11 Hz). Similarly, reaction of 7 and sulfone 14 under identical conditions provided the desired olefin product 18 in 71% yield. Analysis of the 1H NMR spectral data indicated that 18 was also mostly trans (E:Z = 4:1; Jtrans = 15 Hz vs. Jcis = 11 Hz).
Scheme 4.

The Julia-Kocienski reaction is known to be readily scalable and the olefination reactions described herein are amenable to scale-up in order to provide adequate quantities of 15 and 18 for further elaboration to the desired target compounds.
In order to remove the isopropylidene functionality, the Julia-Kocienski olefination product 15 was treated with 50% acetic acid to furnish the vicinal diol in 96% yield. Oxidative cleavage of the vicinal diol was performed with NaIO4 to give the dialdehyde as a crude product (92% yield). Without purification, the dialdehyde was oxidized under Kraus conditions (NaClO2/NaH2PO4/2-methyl-2-butene, 0 °C) to the diacid 16 (78% yield). Esterification of 16 (TMSCHN2, 0 °C → rt) gave the corresponding triester (85% yield), which on removal of the Boc groups gave the amine 17 (80% yield). A similar series of reactions converted 18 to 20. Thus, sequential hydrolysis to the vicinal diol (95%), oxidative cleavage to the crude dialdehyde (quantitative), further oxidation to diacid 19 under Kraus conditions (92%), esterification with TMSCHN2 (83%) and finally N-deprotection afforded 20 (84%).
Subsequent elaboration to the target epoxide peptidomimetics, 4 and 5, is shown in Scheme 5. As described in the Supporting Information, the 1-hydroxybenzotriazole (HOBt) ester of para-aminobenzoic acid, 21, was synthesized by DCC-mediated coupling of 4-(N-methyl)aminobenzoic acid (N-Me-pABA) and HOBt in 84% yield. This preactivated N-Me-pABA was coupled with olefin isostere 17 in the presence of Et3N to give 22 (71% yield). As observed in the synthesis of terminal epoxides, 1b and 2b (see Supporting Information),13 epoxidation of 22 without prior N-protection resulted in the oxidation of the secondary amine. To avoid this problem, the amine nitrogen atom of 22 was protected as a trifluoroacetamide (CF3CO)2O/Et3N, 0 °C, 70% yield). Reaction of the olefinic amide with mCPBA/NaHCO3 proceeded as expected, without facial selectivity, to yield the epoxide 23 (99% yield) as a mixture of trans epoxide diastereomers. TLC, NMR and MS analysis of the product confirmed the isomeric mixture was contaminant-free. Epoxide 23 was treated with LiOH, to give, after concomitant deprotection of the acids and the amine, the corresponding target compound 4. Formation of the desired product 4 was confirmed by NMR and MS analysis.33,34 Similarly, amine 20 was coupled with 21 to obtain 24 (68%). Formation of the N-Tfa derivative (76%) followed by epoxidation afforded epoxide 25 (97%) as a mixture of trans epoxide diastereomers. TLC, NMR and MS analysis of the product confirmed the isomeric mixture contained no impurities. Epoxide 25 was treated with LiOH to give the corresponding triacid 5, the structure of which was confirmed by NMR and mass spectral data.35 Biochemical studies on the effect of 4 and 5 on the reaction catalyzed by GH are in progress and will be reported in due course.
Experimental Section
General Experimental Procedures
See Supporting Information. The selectivity of the Julia-Kocienski reaction was consistent with literature precedent and provided primarily the E geometry for the newly formed olefin. For compounds 15 and 18, the diastereomeric ratio (E:Z) was determined by comparing the relative integration values of corresponding methine protons, the resonances of which do not overlap with any other signals. Independent decoupling of the methine protons for both the trans and cis olefin isomers within the mixture allowed the coupling between the vinyl protons to be measured at J = 15 Hz and J = 11 Hz for the trans and cis olefin respectively. The E;Z ratio determined for the products of the olefination reaction, 15 (9:1) and 18 (4:1), was assumed to be maintained during subsequent chemical transformations. Compounds 7,30 8,13,19-21 and 1022,25 were synthesized as described in the literature.
(3aS, 4R, 6aR)-ethyl 2, 2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxole-4-carboxylate (11)
A solution of N-methylmorpholine N-oxide (7.03 g, 60 mmol) and osmium tetroxide (3 mL 2.5 wt% solution, 300 μmol) in water (20 mL) was added to a cooled (0 °C) stirred solution of optically pure ethyl (cyclopent-2-ene)-1-carboxylate 10 (4.22 g, 30 mmol) in acetone/THF (15 mL/15 mL). The solution was warmed to rt and stirring was continued for 22h. TLC analysis showed the complete consumption of the starting olefin. Then the reaction mixture was diluted with DCM (70 mL) and washed with 10% Na2SO3 (40 mL) and saturated NH4Cl (40 mL). The combined aqueous layers were extracted with DCM (2 × 50 mL). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/Hexane 2:3) to provide an inseparable mixture of diastereomeric diols as colorless oil (5.08 g, 97% yield). 1H NMR (400 MHz; CDCl3) δH: 4.24−4.05 (m, 4H), 3.52 (bs, 0.3H), 2.98−2.84 (m, 2H), 2.63 (bs, 0.6H), 2.19−1.71 (m, 4H), 1.30−1.26 (m, 3H). 13C NMR (100 MHz, CDCl3) δC: 175.2, 77.3, 74.0, 73.8, 72.9, 61.0, 60.8, 48.3, 46.2, 30.8, 29.8, 24.0, 23.8, 14.2, 14.1. Rf 0.16 (EtOAc/Hexane 2:3). MS (ESI) m/z (relative intensity): 197.1 ([M+Na]+, 100).
The diastereomeric mixture of cis and trans diols (6.49 g, 37 mmol) and 2, 2-dimethoxypropane (100 mL) were dissolved in acetone (115 mL) and stirred for 5 min. TsOH•H2O (704 mg, 3.7 mmol) was then added portionwise and stirring was continued at rt for 18h. TLC analysis showed complete consumption of the starting diastereomeric diols. The reaction mixture was diluted with EtOAc (150 mL), washed with saturated NaHCO3 (50 mL), and then with saturated NaCl (150 mL). The aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue (7.48 g) was purified by flash chromatography (EtOAc/Hexane 1:9) to give compounds cis-11 (2.67 g, 35%) and 11 (4.47 g, 58%) as a colorless oil). Cis: 1H NMR (500 MHz; CDCl3) δH: 4.79 (t, J = 5.0 Hz, 1H), 4.66 (t, J = 5.5 Hz, 1H), 4.24−4.13 (m, 2H), 2.61−2.59 (m, 1H), 2.14−2.10 (m, 1H), 1.94−1.90 (m, 1H), 1.76−1.71 (m, 1H), 1.46−1.41 (m, 4H), 1.28−1.25 (m, 6H). 13C NMR (125 MHz, CDCl3) δC: 171.1, 109.9, 80.9, 80.4, 60.4, 50.1, 31.4, 25.8, 24.1, 24.0, 14.3. Rf 0.26 (EtOAc/Hexane 1:9). MS (ESI) m/z (relative intensity): 237.1 ([M+Na]+, 100). Trans: 1H NMR (500 MHz; CDCl3) δH: 4.83 (d, J = 5.5 Hz, 1H), 4.71 (t, J = 5.0 Hz, 1H), 4.12 (q, J = 7.5 Hz, 2H), 2.89 (d, J = 7.5 Hz, 1H), 2.11−2.08 (m, 1H), 1.92−1.75 (m, 3H), 1.45 (s, 3H), 1.32−1.24 (m, 6H). 13C NMR (125 MHz, CDCl3) δC: 174.1, 110.2, 83.2, 81.4, 60.9, 50.9, 31.8, 27.0, 26.7, 24.4, 14.5. Rf 0.41 (EtOAc/Hexane 1:9). MS (ESI) m/z (relative intensity): 237.1 ([M+Na]+, 100).
5-(((3aS,4R,6aR)-2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxol-4yl)methylthio)-1-phenyl-1H-tetrazole (12)
The optically pure bicyclic ethyl ester derivative 11 (2.97 g, 13.8 mmol) was dissolved in THF (30 mL). In a separate flask, LiAlH4 (0.73 g, 19.2 mmol) was suspended in THF (10 mL) in an atmosphere of argon and cooled to 0 °C. The THF solution of the ester was added dropwise to the LiAlH4 suspension at 0 °C. Vigorous bubbling was observed during the addition of the ester derivative. The reaction mixture was stirred for 4h at 0 °C in an atmosphere of argon and then quenched by dropwise addition of 10% NH4Cl solution (0.8 mL). The solution was allowed to warm to rt and stirring was continued for 60 min. The reaction mixture was filtered through Celite using EtOAc and the solvent was removed under reduced pressure to give the crude product as clear oil (2.4 g). The crude product was purified by flash chromatography (EtOAc/Hexane 3:7) to give the desired hydroxymethyl derivative as a colorless oil (2.27 g, 95%). 1H NMR (500 MHz; CDCl3) δH: 4.65 (t, J = 5.6 Hz, 1H), 4.47 (d, J = 5.5 Hz, 1H), 3.53−3.45 (m, 2H), 2.22−2.21 (m, 1H), 1.98−1.96 (m, 1H), 1.82−1.71 (m, 3H), 1.49−1.46 (m, 3H), 1.30 (s, 3H). 13C NMR (125 MHz, CDCl3) δC: 110.2, 83.5, 81.1, 63.6, 48.5, 31.4, 26.8, 25.6, 24.3. Rf 0.18 (EtOAc/Hexane 3:7). MS (ESI) m/z (relative intensity): 195.1 ([M+Na]+, 100).
The bicyclic hydroxymethyl derivative (890 mg, 5.1 mmol) was dissolved in THF (25 mL) at 0 °C in an atmosphere of argon followed by addition of Ph3P (2.0 g, 7.62 mmol) and 1-phenyl-1H-tetrazole-5-thiol (1.45 g, 8.13 mmol). In a separate flask, diisopropylazadicarboxylate, DIAD (2.08 g, 10.3 mmol) was dissolved in THF (10 mL) which gave a yellow solution. This DIAD solution was added dropwise to the reaction mixture at 0 °C. After addition of each drop of DIAD solution the reaction mixture became yellow, the color persisted for a second and then disappeared. When approximately half of the DIAD solution was added then the reaction mixture remained yellow. Stirring was continued at rt for 12 h. The TLC analysis showed the complete consumption of the starting material. The reaction mixture was diluted with Et2O (150 mL) and washed with saturated NaHCO3 solution (100 mL). The aqueous layer was extracted with Et2O (3 × 100 mL). The combined organic layer was washed with saturated NaCl solution (100 mL), dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to give a yellow oil which was purified by flash chromatography (EtOAc/Hexane 1:9) to give the thioether precursor of 12 as a yellow oil (1.5 g, 88%). 1H NMR (500 MHz; CDCl3) δH: 7.60−7.53 (m, 5H, Ar-H), 4.70 (t, J = 5.0 Hz, 1H), 4.43 (t, J = 5.5 Hz, 1H), 3.36 (dd, J = 13.1, 6.5 Hz, 1H), 3.27 (dd, J = 13.1, 7.0 Hz, 1H), 2.46−2.44 (m, 1H), 2.11−2.08 (m, 1H), 1.88−1.82 (m, 2H), 1.59−1.56 (m, 1H), 1.43 (s, 3H), 1.28 (s, 3H). 13C NMR (125 MHz, CDCl3) δC: 154.3, 133.9, 130.4, 130.0, 124.1, 110.4, 85.1, 80.8, 45.3, 35.1, 31.0, 28.2, 26.6, 24.3, 22.2. Rf 0.45 (EtOAc/Hexane 3:7). MS (ESI) m/z (relative intensity): 355.1 ([M+Na]+, 100).
(NH4)6Mo7O24•4H2O (165 mg, 133 μmol) was dissolved in 30% H2O2 (1.13 mL, 10 mmol) at 0 °C to give a yellow solution which was added dropwise to the ethanolic solution of the thioether precursor (222 mg, 667 μmol) in EtOH (10 mL) at 0 °C. The yellow color of the (NH4)6Mo7O24•4H2O solution disappeared after addition of each drop in the reaction mixture and a white precipitate was observed. The resultant suspension was stirred at rt for 24h, added to brine (40 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure to afford the crude product as an oil (590 mg). Purification of the crude product by flash chromatography (EtOAc/Hexane 3:7) gave the pure sulfoxide 12 as a clear, colorless oil (208 mg, 87%). 1H NMR (500 MHz; CDCl3) δH: 7.69−7.59 (m, 5H, Ar-H), 4.67 (t, J = 5.5 Hz, 1H), 4.49 (dd, J = 5.0, 2.0 Hz, 1H), 3.78 (dd, J = 15, 7.5 Hz, 1H), 3.67 (dd, J = 15.0, 7.0 Hz, 1H), 2.75−2.74 (m, 1H), 2.18−2.15 (m, 1H), 1.88−1.80 (m, 2H), 1.66−1.63 (m, 1H), 1.42 (s, 3H), 1.26 (s, 3H). 13C NMR (125 MHz, CDCl3) δC:154.1, 133.4, 131.9, 130.1, 125.5, 111.3, 85.3, 80.5, 57.9, 40.1, 31.2, 28.9, 26.9, 24.6. Rf 0.45 (EtOAc/Hexane 3:7). MS (ESI) m/z (relative intensity): 387.1 ([M+Na]+, 100).
(3aR, 4S, 6aS)-ethyl 2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxole-4-carboxylate (13)
A solution of N-methylmorpholine N-oxide (1.86 g, 15.87 mmol) and osmium tetroxide (0.80 mL 2.5 wt% solution, 79 μmol) in water (5 mL) was added to a cooled (0 °C) stirred solution of optically pure cyclopent-2-ene-ethylcarboxylate 8 (1.12 g, 7.99 mmol) in acetone/THF (3.5 mL/3 mL). Subsequent reaction conditions, workup, and purification employed identical reagents and solvents proportionately to that described in the synthesis of 11, again providing an inseparable mixture of diastereomeric diols as a colorless oil (1.08 g, 78% yield). 1H NMR (400 MHz; CDCl3) δH: 4.19−4.04 (m, 4H), 3.36 (bd, 0.2H), 2.90−2.80 (m, 1H), 2.26−2.10 (m, 1H), 2.00−1.90 (m, 1H), 1.82−1.68 (m, 2H), 1.30−1.26 (m, 3H). 13C NMR (125 MHz, CDCl3) δC: 175.9, 174.4, 77.2, 74.4, 74.3, 73.3, 61.2, 61.0, 48.8, 46.9, 30.6, 30.4, 24.6, 23.5, 14.5, 14.4. Rf 0.33 (EtOAc/Hexane 3:2). MS (ESI) m/z (relative intensity): 197.1 ([M+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C8H14O4Na [M+Na]+ 197.0790, found 197.0792.
The diastereomeric mixture of cis and trans diols (770 mg, 4.41 mmol) and 2, 2-dimethoxypropane (15.0 mL) were dissolved in acetone (10.0 mL) and stirred for 5 min. Then TsOH•H2O (22 mg, 116 μmol) was added portionwise and stirring was continued at rt for 30 min. Subsequent workup and purification employed identical reagents and solvents proportionately to that described in the synthesis of 11, providing compounds cis-13 (398 mg, 43%) and 13 (466 mg, 49%) as colorless oils. Cis: 1H NMR (400 MHz; CDCl3) δH: 4.79 (t, J = 5.7 Hz, 1H), 4.66 (t, J = 5.2 Hz, 1H), 4.25−4.11 (m, 2H), 2.63−2.57 (m, 1H), 2.18−2.06 (m, 1H), 1.94−1.89 (m, 1H), 1.77−1.70 (m, 1H), 1.49−1.41 (m, 4H), 1.28−1.25 (m, 6H). 13C NMR (125 MHz, CDCl3) δC: 171.4, 110.3, 81.2, 80.8, 60.8, 50.5, 31.8, 26.2, 24.5, 24.4, 14.6. Rf 0.25 (EtOAc/Hexane 1:9). MS (ESI) m/z (relative intensity) 237.2 [M+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C11H18NaO4 237.1103, found 237.1104. Trans: 1H NMR (400 MHz; CDCl3) δH: 4.83 (d, J = 5.6 Hz, 1H), 4.72 (t, J = 5.2 Hz, 1H), 4.12 (q, J = 7.6 Hz, 2H), 2.90 (d, J = 7.2 Hz, 1H), 2.15−2.05 (m, 1H), 1.93−1.70 (m, 3H), 1.44 (s, 3H), 1.31−1.23 (m, 6H). 13C NMR (125 MHz, CDCl3) δC: 174.0, 110.2, 83.2, 81.4, 60.9, 50.9, 31.8, 27.0, 26.0, 24.4, 14.5. Rf 0.39 (EtOAc/Hexane, 1:9). MS (ESI) m/z (relative intensity): 237.2 ([M+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C11H18O4Na 237.1103; found, 237.1102.
5-(((3aR,4S,6aS)-2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)methyl-thio)-1-phenyl-1H-tetrazole (14)
The optically pure ethyl ester-containing bicycle 13 (145 mg, 0.68 mmol) was dissolved in Et2O (1 mL). In a separate flask, LiAlH4 (36 mg, 0.95 mmol) was suspended in Et2O (4 mL) and cooled to 0 °C. The solution of 13 was transferred to the reaction flask via syringe along with an Et2O rinse. Vigorous bubbling was observed upon addition. The ice bath was removed and the reaction allowed to stir at rt for 1 h, at which point TLC analysis indicated the reaction was complete (hexanes-EtOAc 2:1; 13, Rf = 0.59, HCl then KMnO4, the desired hydroxymethyl derivative, Rf = 0.14, HCl then KMnO4). The reaction was worked up according to the Fieser and Fieser protocol.36 The mixture was cooled to 0 °C and H2O (36 μL), 15 % NaOH (36 μL), and H2O (105 μL) were added in that order. The mixture was stirred for 45 min during which time the grey solid turned to white solid. Filtration through Celite with EtOAc and removal of the solvent in vacuo produced 123 mg clear oil. Purification by flash chromatography (1 × 3 inch silica; hexanes-EtOAc 2:1 → 1:1) yielded pure hydroxymethyl derivative (114 mg, 98 %) as a clear, colorless oil: [α]22D = + 9.7 (c = 1.0, CHCl3). FTIR (film, NaCl) 3425, 1041 cm−1. 1H NMR (500 MHz) δ 4.60 (t, J = 5.5 Hz, 1 H), 4.40 (d, J = 5.8 Hz, 1 H), 3.42 (dd, J = 7.1, 10.8 Hz, 1 H), 3.38 (dd, J = 8.2, 10.8 Hz, 1 H), 2.15 (m, 1 H), 2.62 (bs, 1 H), 1.91 (m, 1 H), 1.76 (m, 1 H), 1.68 (m, 1 H), 1.43 (m, 1 H), 1.40 (s, 3 H), 1.25 (s, 3 H). 13C NMR (125 MHz) δ 110.0, 83.3, 81.0, 63.1, 48.2, 31.2, 26.6, 25.3, 24.2. MS (ESI) m/z (relative intensity): 227.2 ([M+Na+MeOH]+, 100), 195.2 ([M+Na]+, 78). HRMS-ESI (m/z): [M+Na]+ calcd for C9H16NaO3 195.0997, found 195.1001.
The 1-hydroxymethyl derivative (104 mg, 0.60 mmol) was dissolved in THF (1 mL) under argon before Ph3P (236 mg, 0.90 mmol) and 1-phenyl-1H-tetrazole-5-thiol (160 mg, 0.90 mmol) were added. In a separate flask, DIAD (213 μL, 1.08 mmol) was dissolved in THF (4 mL) to give a yellow solution. The reaction mixture was cooled to 0 °C and the DIAD solution was added dropwise. The yellow color persisted approximately one second after each drop was added before the solution returned to colorless. After approximately 75 % of the DIAD had been added, the yellow color remained. After 30 min of reaction, TLC analysis (hexanes-EtOAc 1:1) indicated the alcohol (Rf = 0.29) had been consumed and the sulfide (Rf = 0.55) had formed. The reaction was diluted with Et2O (50 mL) and washed with sat. NaHCO3 (20 mL). The aqueous layer was repeatedly extracted with Et2O (3 × 40 mL) and the combined organic layer was washed with brine (30 mL) and dried (Na2SO4). The solvent was removed by rotary evaporation to yield 816 mg of yellow oil that was purified by flash chromatography (Isco CombiFlash RETRIEVE with RediSep™ 10 g column; hexanes-EtOAc 5:1; Rf = 0.12) to afford 184 mg of clear, colorless oil that was pure sulfide (93 % yield). [α]24D = + 9.3 (c = 1.0, CHCl3). FTIR (film, NaCl) 1500, 1043 cm−1. 1H NMR (500 MHz) δ 7.51 (m, 5 H), 4.64 (t, J = 5.3 Hz, 1 H), 4.38 (d, J = 5.7 Hz, 1 H), 3.29 (dd, J = 13.0, 7.3 Hz, 1 H), 3.21 (dd, J = 13.0, 9.2 Hz, 1 H), 2.03 (m, 1 H), 2.40 (m, 1 H), 1.77 (m, 1 H), 1.51 (m, 1 H), 1.37 (s, 3 H), 1.21 (s, 3 H). 13C NMR (125 MHz) δ 154.1, 133.6, 130.1, 129.8, 123.8, 110.0, 84.8, 80.5, 45.0, 34.8, 30.7, 27.7, 26.4, 24.1. MS (ESI) m/z 355.1 (MNa+, 100). HRMS (ESI) calcd for C16H10N4NaO2S 355.1205 (MNa+), found 355.1209.
The sulfide (85 mg, 0.26 mmol) was dissolved in EtOH (95 %, 1 mL) and cooled to 0 °C. In a separate flask, (NH4)6Mo7O24•4H2O (63 mg, 0.051 mmol) was dissolved in H2O2 (30 %, 465 μL, 4.10 mmol) at 0 °C to give a yellow solution. The oxidant solution was added to the sulfide and the reaction monitored by TLC (hexanes-EtOAc 3:1; sulfide, Rf = 0.23, UV; 14, Rf = 0.23, UV; sulfoxide, Rf = 0.06, UV). The starting material and product have identical Rf values. Therefore, the reaction progress was evaluated based on the appearance and then disappearance of the sulfoxide intermediate. Additionally, reaction progression was indicated by the voluminous precipitate that formed during the last six hours of the 24 h reaction period. The mixture was diluted with brine (20 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried (Na2SO4), filtered, and the solvent was removed in vacuo to afford 113 mg clear oil. Flash chromatography (1 × 3 inch silica; hexanes-EtOAc 9:1 → 3:1) provided 14 (91 mg, 98 %) as a colorless oil. [α]24D = + 13.4 (c = 1.0, CHCl3). FTIR (film, NaCl) 1498, 1340, 1153, 1044 cm−1. 1H NMR (500 MHz) δ 7.66 (m, 2 H), 7.59 (m, 3 H), 4.66 (t, J = 5.4 Hz, 1 H), 4.48 (dd, J = 5.7, 1.6 Hz, 1 H), 3.77 (dd, J = 7.4, 14.6 Hz, 1 H), 3.66 (dd, J = 7.0, 14.6 Hz, 1 H), 2.72 (m, 1 H), 2.16 (m, 1 H), 1.87 (m, 1 H), 1.79 (m, 1 H), 1.62 (m, 1 H), 1.41 (s, 3 H), 1.24 (s, 3 H). 13C NMR (125 MHz) δ 153.7, 133.1, 131.6, 129.8, 125.2, 110.9, 85.0, 80.2, 57.5, 39.8, 30.9, 28.6, 26.5, 24.3. MS (ESI) m/z 387.1 (MNa+, 100). HRMS (ESI) calcd for C16H20N4NaO4S 387.1103 (MNa+), found 387.1102.
(S,E)-methyl 2-(bis(tert-butoxycarbonyl)amino)-6-((3aS,4S,6aR)-2,2-dimethyltetra-hydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)hex-5-enoate (15)
KHMDS in toluene (164 μL 0.5M, 82 μmol) was added dropwise to a solution of the sulfone 12 (25 mg, 68 μmol) in THF (3 mL) in an atmosphere of argon at −78 °C. The resultant yellow solution was stirred for 60 min followed by the dropwise addition of a solution of methyl (S)-5-oxo-2-di-tert-butoxycarbonylaminopentaneoate, 7, (43 mg, 123 μmol) in THF (2 mL). Approximately 15 min after the addition of the aldehyde, the yellow color became lighter. The reaction mixture was stirred for 1 h at −78 °C, warmed to rt, and stirred for an additional 20 h. The reaction mixture was quenched with satd. NH4Cl solution (20 μL), stirred for 5 min, diluted with ether (10 mL), the layers were separated and the organic layer was washed with water (10 mL). The aqueous layer was extracted with ether (3 × 15 mL), the combined organic layer was dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure to afford a colorless oil which was purified by flash chromatography (EtOAc/Hexane 1:9) to give the olefin derivative 15 as an oil (23 mg, 69%). Diastereomeric ratio (E:Z) = 9:1. 1H NMR (500 MHz; CDCl3) δH: 5.47−5.39 (m, 1H), 5.37−5.31 (m, 0.9H), 5.23 (t, J = 10Hz, 0.1H), 4.90−4.83 (m, 1H), 4.66−4.62 (m, 1H), 4.40−4.38 (m, 0.9H), 4.29−4.26 (m, 0.1H), 3.71 (s, 3H), 2.93−2.90 (m, 0.1H), 2.66−2.64 (m, 0.9H), 2.20−2.13 (m, 1H), 2.06−1.72 (m, 4H), 1.79−1.72 (m, 2H), 1.53−1.41 (m, 22H), 1.29 (s, 3H). 13C NMR (75 MHz, CDCl3) δC: 171.4, 152.2, 131.2, 129.3, 109.6, 85.9, 83.1, 57.5, 52.2, 47.6, 31.2, 29.8, 29.3, 28.2, 28.0, 26.4, 24.1. Rf 0.73 (EtOAc/Hexane 3:7). MS (ESI) m/z (relative intensity): 506.2 ([M+Na]+, 100). HRMS-ESI (m/z) calcd for C25H41NO8Na [M+Na]+ 506.2730, found 506.2731.
(S)-2-((S,E)-5-(bis(tert-butoxycarbonyl)amino)-6-methoxy-6-oxohex-1-enyl)pentane-dioic acid (16)
Acetonide 15 (17 mg, 35 μmol) was dissolved in 50% AcOH (15 mL) at 0 °C, brought to rt, and then the solution was stirred at rt for 15 h. The solvent was removed under reduced pressure. The crude reaction product (16 mg) was purified by flash chromatography (EtOAc/Hexane 2:3) to afford the corresponding vicinal diol as a white solid (15 mg, 96%). Diol: 1H NMR (500 MHz; CDCl3) δH: 5.52−5.46 (m, 1H), 5.33−5.29 (m, 1H), 4.92−4.85 (m, 1H), 4.12−4.07 (m, 1H), 3.71 (s, 3H), 3.61−3.54 (m, 1H), 3.24 (bs, 0.8H), 2.88−2.82 (m, 0.3H), 2.61−2.48 (m, 2H), 2.27−1.87 (m, 6H), 1.70−1.63 (m, 1H), 1.50 (s, 18H), 1.32−1.24 (m, 1H). 13C NMR (125 MHz, CDCl3) δC: 171.5, 152.4, 133.6, 133.1, 130.4, 130.3, 83.3, 83.2, 79.6, 78.7, 72.6, 72.4, 57.6, 56.9, 52.2, 47.2, 42.1, 30.4, 30.4, 29.8, 29.1, 28.8, 28.0, 27.4, 27.0. Rf 0.19 (EtOAc/Hexane 2:3). MS (ESI) m/z (relative intensity): 466.2 ([M+Na]+, 100).
The vicinal diol (19 mg, 42 μmol) was dissolved in THF/H2O (v/v, 9/1, 3 mL) at rt and stirred. NaIO4 (47 mg, 219 μmol) was added to the above solution and stirring was continued for 20 hours during which time a white precipitate was observed in the reaction mixture. TLC analysis showed the complete consumption of the starting material and formation of desired product. The solvent was removed under reduced pressure, the crude product was dissolved in DCM (12 mL), and washed with water (10 mL). The aqueous layer was extracted with DCM (3 × 10 mL). The combined organic layer was dried over Na2SO4, the solvent was removed under reduced pressure, and dried under vacuum to obtain the desired α,γ-dialdehyde (17 mg). 1H NMR (500 MHz; CDCl3) δH: 9.76 (bt, 1H), 9.54 (d, J = 1.5 Hz, 1H), 5.69−5.63 (m, 1H), 5.31−5.27 (m, 1H), 4.88−4.85 (m, 1H), 3.71 (s, 3H), 3.01−2.96 (m, 1H), 2.50 (t, J = 7 Hz, 2H), 2.17−1.23 (m, 24H). 13C NMR (125 MHz, CDCl3) δC: 201.4, 200.8, 171.2, 152.2, 152.1, 135.9, 124.3, 83.2, 57.3, 55.0, 52.2, 40.9, 29.6, 29.4, 28.0, 20.7. MS (ESI) m/z (relative intensity): 496.1 ([M+Na+MeOH]+, 100).
The α,γ-dialdehyde (17 mg, 38 μmol) was dissolved in THF (2 mL) at 0 °C followed by addition of 2-methyl-2-butene (0.9 mL 2M). In a separate vial NaClO2 (18 mg, 199 μmol), NaH2PO4 (5 mg, 41 μmol), and 2-methyl-2-butene (1.0 mL 2M) were dissolved in t-BuOH/H2O (v/v, 7/3, 4 mL) at 0 °C. This solution was added dropwise to the reaction mixture at 0 °C, and the stirring was continued at 0 °C for 3 h. Excess NaClO2 was quenched by addition of 2-propanol (1.0 mL). Solvents were removed under reduced pressure and the crude product was purified by flash chromatography (EtOAc/Hexane 2:3, few drops of AcOH) to give the desired olefinated glutaric acid 16 as an oil (14 mg, 78%, two steps). 1H NMR (500 MHz; CDCl3) δH: 5.63−5.57 (m, 1H), 5.50−5.39 (m, 1H), 4.88−4.84 (m, 1H), 4.04 (bs, 2H), 3.72 (bs, 3H), 3.37−3.36 (m, 1H), 3.01−2.97 (m, 1H), 2.32 (t, J = 8 Hz, 2H), 2.23−1.79 (m, 6H), 1.63−1.46 (m, 18H). 13C NMR (125 MHz, CDCl3) δC: 176.6, 175.9, 171.7, 152.3, 132.7, 131.8, 128.4, 83.9, 83.8, 58.0, 57.8, 52.4, 52.4, 31.7, 30.2, 29.8, 29.4, 28.0, 28.0, 27.4, 27.6, 27.4, 24.6. Rf 0.14 (EtOAC/Hexane 2:3). MS (ESI) m/z (relative intensity): 496.1 ([M+Na]+, 100).
(3S,8S,E)-trimethyl 8-aminooct-4-ene-1,3,8-tricarboxylate (17)
The diacid 16 (39 mg, 82 μmol) was dissolved in MeOH (4 mL), cooled to 0 °C, followed by dropwise addition of Me3SiCHN2 (4.0 mL 2M solution in diethyl ether) under continuous stirring. Initially when Me3SiCHN2 was added, the solution became yellow and then turned colorless. When all the Me3SiCHN2 was added the solution remained yellow. Stirring was continued at rt for 16 h, after which the reaction was quenched by addition of a few drops of HOAc . Solvent was removed under reduced pressure and the crude product (46 mg) was purified by flash chromatography (EtOAc/Hexane 2:3) to give the corresponding trimethyl ester derivative as an oil (35 mg, 85%). 1H NMR (500 MHz; CDCl3) δH: 5.60−5.55 (m, 1H), 5.45−5.34 (m, 1H), 4.87−4.83 (d, J = 8.5 Hz, 1H), 3.71−3.66 (m, 9H), 3.03−2.98 (m, 1H), 2.38−1.80 (m, 8H), 1.49 (s, 18H). 13C NMR (125 MHz, CDCl3) δC: 174.1, 173.4, 171.2, 152.1, 133.0, 127.6, 83.2, 83.1, 57.6, 52.2, 51.9, 51.6, 48.2, 31.4, 29.6, 29.1, 28.0, 27.2. Rf 0.66 (EtOAc/Hexane 1:1). MS (ESI) m/z (relative intensity): 424.2 ([M-Boc+Na]+, 100).
The trimethyl ester (44 mg, 87 μmol) was dissolved in THF (4 mL), cooled to 0 °C, followed by addition of Et3SiH (2 mL) under stirring condition. Then CF3COOH (6 mL) was added dropwise at 0 °C and stirring was continued at rt for 16 h. Solvent was removed under reduced pressure and the crude product was purified by flash chromatography (EtOAc/Hexane 4:1, ca. 2% Et3N) to give the free amine derivative 17 as an oil (21 mg, 80%). 1H NMR (500 MHz; CDCl3) δH: 5.59−5.53 (m, 1H), 5.46−5.35 (m, 1H), 3.72 (s, 3H), 3.66−3.64 (m, 6H), 3.45−3.43 (m, 1H), 3.04−2.99 (m, 1H), 2.33−2.29 (m, 2H), 2.17−2.02 (m, 3H), 1.88−1.78 (m, 2H), 1.65−1.59 (m, 1H). 13C NMR (125 MHz, CDCl3) δC: 176.4, 174.2, 173.4, 133.0, 133.3, 127.7, 127.5, 53.8, 52.01, 51.95, 51.91, 51.6, 48.2, 42.9, 34.2, 31.4, 31.2, 28.6, 27.4, 27.2, 23.8. Rf 0.17 (EtOAc/Haxane 4:1). MS (ESI) m/z (relative intensity): 302.2 ([M+H]+, 100), 324.2 [M+Na]+.
(S,E)-methyl 2-(bis(tert-butoxycarbonyl)amino)-6-((3aR,4R,6aS)-2,2-dimethyltetra-hydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)hex-5-enoate (18)
Sulfone 14 (11 mg, 30 μmol) was dissolved in THF (0.5 mL) under an atmosphere of argon and cooled to −78 °C under stirring condition. KHMDS solution (80 μL 0.5M, 40 μmol) was added dropwise to the sulfone at −78 °C and stirring continued for 15 min. The reaction mixture was then warmed to −50 °C and stirring was continued for 1 h, after which it was again cooled to −78 °C. A solution of methyl (S)-2-di-tert-butoxycarbonylaminopentaneoate, 7, (20 mg, 57 μmol) in THF (1.3 mL) was added dropwise into the reaction mixture and stirring was continued at −78 °C for 3 h. The reaction solution was warmed to rt and stirring was continued for 12 h during time which a white precipitate was observed. The reaction was quenched with saturated NH4Cl solution (200 μL). The aqueous layer was extracted with ether (3 × 15 mL). The combined organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to afford a colorless oil which was purified by flash chromatography (EtOAc/Hexane 1:9). Olefin derivative 18 was obtained as an oil (11 mg, 71%). Diastereomeric ratio (E:Z) = 4:1. 1H NMR (500 MHz; CDCl3) δH: 5.47−5.31 (m, 1.8H), 5.23 (t, J = 11 Hz, 0.2H), 4.90−4.83 (m, 1H), 4.67−4.62 (m, 1H), 4.38 (d, J = 5.5 Hz, 0.8H), 4.28 (d, J = 5.5 Hz, 0.2H), 3.71 (s, 3H), 2.93−2.90 (m, 0.2H), 2.70−2.62 (m, 0.8H), 2.26−1.70 (m, 8H), 1.64−1.40 (m, 21H), 1.30−1.18 (m, 3H). 13C NMR (100 MHz, CDCl3) δC: 171.3, 152.1, 131.1, 130.7, 129.8, 129.2, 109.5, 86.6, 85.8, 83.0, 80.7, 80.6, 57.6, 57.4, 52.1, 47.4, 43.3, 31.4, 31.2, 29.9, 29.8, 29.6, 29.4, 29.3, 28.0, 27.9, 26.4, 24.3, 24.0. Rf 0.73 (EtOAc/Hexane 3:7). MS (ESI) m/z (relative intensity): 506.2 ([M+Na]+, 100). HRMS-ESI (m/z) calcd for C25H41NO8Na [M+Na]+ 506.2730, found 506.2731.
(R)-2-((S,E)-5-(bis(tert-butoxycarbonyl)amino)-6-methoxy-6-oxohex-1-enyl)pentane-dioic acid (19)
Acetonide 18 (75 mg, 155 μmol) was dissolved in 50% AcOH (40 mL) at rt, and then the solution was stirred at rt for 11 h. The solvent was removed under reduced pressure. The crude reaction product (72 mg) was purified by flash chromatography (EtOAc/Hexane 2:3) to afford the corresponding vicinal diol as an oil (57 mg, 83%). Diol: 1H NMR (400 MHz; CDCl3) δH: 5.50−5.42 (m, 1H), 5.36−5.30 (m, 1H), 4.96−4.92 (m, 0.8H), 4.84−4.81 (m, 0.2H), 4.14−4.06 (m, 1H), 3.71 (s, 3H), 3.63−3.59 (m, 1H), 3.35 (bd, 0.8H), 2.91−2.82 (m, 0.3H), 2.61 (bs, 0.7H), 2.54−2.45 (m, 1H), 2.34−1.64 (m, 7H), 1.50 (bs, 18H), 1.39−1.19 (m, 1H). 13C NMR (100 MHz, CDCl3) δC: 171.4, 152.1, 133.4, 133.3, 130.7, 130.3, 83.5, 83.4, 79.8, 78.8, 72.6, 72.6, 57.4, 57.2, 52.2, 52.2, 47.3, 42.1, 30.3, 30.3, 29.8, 29.5, 29.3, 28.0, 28.0, 27.5, 27.2, 24.4. Rf 0.19 (EtOAc/Hexane 2:3). MS (ESI) m/z (relative intensity): 466.2 ([M+Na]+, 100).
The vicinal diol (96 mg, 216 μmol) was dissolved in THF/H2O (v/v, 9/1, 3 mL) at rt and NaIO4 (232 mg, 1.08 mmol) was added with stirring. After 1 h 30 min, additional NaIO4 (231 mg, 1.07 mmol) was added and stirring was continued for 45 min during which time a white precipitate was observed in the reaction mixture. TLC analysis showed the complete consumption of the starting material and formation of desired product. The solvent was removed under reduced pressure, the crude product was dissolved in DCM (25 mL), and washed with water (20 mL). The aqueous layer was extracted with DCM (3 × 20 mL). The combined organic layer was dried over Na2SO4, the solvent was removed under reduced pressure, and dried under vacuum to obtain the crude α,γ-dialdehyde as an oil (93 mg). 1H NMR (300 MHz; CDCl3) δH: 9.77 (bt, 1H), 9.54 (d, J = 1.5 Hz, 1H), 5.68−5.60 (m, 1H), 5.32−5.21 (m, 1H), 4.88−4.81 (m, 1H), 3.71 (s, 3H), 3.40−3.30 (m, 0.2H), 3.01−2.96 (m, 0.8H), 2.60−1.23 (m, 18H). 13C NMR (100 MHz, CDCl3) δC: 201.4, 200.7, 171.2, 152.1, 135.9, 124.9, 83.2, 57.3, 55.0, 52.2, 40.8, 29.5, 29.4, 28.2, 27.9, 20.7. MS (ESI) m/z (relative intensity): 496.1 ([M+Na+MeOH]+, 100). HRMS-ESI (m/z): [M+Na+MeOH]+ calcd for C23H39NO9Na 496.2523, found 464.2260 [M+Na]+.
The crude α,γ-dialdehyde (93 mg, 0.21 mmol) was dissolved in THF (4 mL) at 0 °C followed by addition of 2-methyl-2-butene (3 mL 2M). In a separate vial NaClO2 (196 mg, 2.16 mmol), NaH2PO4 (52 mg, 0.43 mmol), and 2-methyl-2-butene (1.0 mL 2M) were dissolved in t-BuOH/H2O (v/v, 1/1, 2 mL) at 0 °C. This solution was added dropwise to the reaction mixture at 0 °C, and the stirring was continued at 0 °C. After 2 h a solution of NaClO2 (98 mg, 1.08 mmol), NaH2PO4 (25 mg, 0.20 mmol), 2-methyl-2-butene (0.8 mL 2M) in t-BuOH/H2O (v/v, 1/1, 1 mL) was added to the reaction mixture. Stirring was continued at 0 °C for an additional 1 h 15 min. Excess NaClO2 was quenched by addition of 2-propanol (4.0 mL) and stirring the reaction for 30 min. Solvents were removed under reduced pressure and the crude product was purified by flash chromatography (EtOAc/Hexane 1:1, ca. 2% AcOH) to give the desired olefinated glutaric acid 19 as an oil (94 mg, 94%, two steps). 1H NMR (500 MHz; CDCl3) δH: 5.63−5.58 (m, 1H), 5.53−5.44 (m, J = 15.3 Hz, 1H), 4.86−4.83 (m, 1H), 3.71 (s, 3H), 3.43−3.39 (m, 0.2H), 3.07−3.03 (m, 0.8H), 2.51−1.82 (m, 8H), 1.53 (s, 18H). 13C NMR (100 MHz, CDCl3) δC: 179.4, 178.7, 171.3, 152.2, 133.2, 127.4, 83.5, 83.3, 57.5, 52.2, 48.1, 31.6, 29.4, 29.1, 28.0, 27.4. Rf 0.14 (EtOAC/Hexane 2:3). MS (ESI) m/z (relative intensity): 496.1 ([M+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C22H35NO10Na 496.2159, found 496.2159.
(3R,8S,E)-trimethyl 8-aminooct-4-ene-1,3,8-tricarboxylate (20)
The diacid 19 (91 mg, 192 μmol) was dissolved in MeOH (4 mL), cooled to 0 °C, followed by dropwise addition of Me3SiCHN2 (5.0 mL 2M solution in diethyl ether) under continuous stirring. Initially when Me3SiCHN2 was added, the solution became yellow and then turned colorless. When all the Me3SiCHN2 was added the solution remained yellow. Stirring was continued at rt for 17 h, after which the reaction was quenched by addition of a few drops of HOAc. Solvent was removed under reduced pressure and the crude product (107 mg) was purified by flash chromatography (EtOAc/Hexane 2:30) to give the corresponding trimethyl ester derivative as an oil (80 mg, 83%). 1H NMR (500 MHz; CDCl3) δH: 5.62−5.53 (m, 1H), 5.45−5.35 (m, 1H), 4.88−4.83 (m, 1H), 3.71−3.66 (s, 9H), 3.39−3.34 (m, 0.2H), 3.03−2.98 (m, 0.8H), 2.38−1.80 (m, 8H), 1.50 (s, 18H). 13C NMR (100 MHz, CDCl3) δC: 174.2, 173.4, 171.3, 152.2, 133.0, 127.7, 83.2, 83.1, 57.5, 52.2, 51.9, 51.6, 48.2, 31.4, 29.6, 29.6, 29.1, 28.0, 27.2. Rf 0.66 (EtOAc/Hexane 1:1). MS (ESI) (relative intensity): m/z 424.2 ([M-Boc+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C24H39NO10Na 524.2472, found 524.2485.
The trimethyl ester (44 mg, 87 μmol) was dissolved in THF (4 mL), cooled to 0 °C, followed by addition of Et3SiH (2 mL) with stirring, after which excess CF3COOH was added dropwise at 0 °C and stirring was continued at rt for 16 h. Solvent was removed under reduced pressure and the crude product was purified by flash chromatography (EtOAc/Hexane 4:1, ca. 2% Et3N) to give the free amine derivative 20 as an oil (22 mg, 84%). 1H NMR (500 MHz; CDCl3) δH: 5.59−5.53 (m, 1H), 5.46−5.5.35 (m, 1H), 3.73−3.67 (s, 9H), 3.45−3.42 (m, 1H), 3.04−2.99 (m, 1H), 2.33−2.17 (m, 2H), 2.16−2.02 (m, 3H), 1.88−1.79 (m, 2H), 1.65−1.57 (m, 1H). 13C NMR (100 MHz, CDCl3) δC: 176.4, 174.2, 173.4, 133.1, 127.7, 53.8, 52.0, 51.9, 51.6, 48.2, 42.9, 34.2, 31.4, 29.7, 28.6, 27.2, 23.8. Rf 0.17 (EtOAc/Haxane 4:1). MS (ESI) m/z (relative intensity): 302.2 ([M+H]+, 100).
(3S,8S,E)-trimethyl 8-(4-(methylamino)benzamido)oct-4-ene-1,3,8-tricarboxylate (22)
Et3N (0.12 mL) was added to a stirred solution of the N-methyl p-aminobenzoic acid HOBt ester, 21, (24 mg, 86 μmol) and amine derivative 17 (26 mg, 86 μmol) in THF (4 mL) at 0 °C in an atmosphere of argon. The reaction was warmed to rt and stirring was continued for 26 h, after which time additional 21 (24 mg) and Et3N (0.2 mL) were added and stirring continued for 24 h. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (EtOAc/Hexane 1:1) to give pure product 22 as an oil (27 mg, 73%). 1H NMR (400 MHz; CDCl3) δH: 7.71−7.65 (m, 2H), 6.64−6.51 (m, 3H), 5.61−5.52 (m, 1H), 5.45−5.33 (m, 1H), 4.84−4.79 (m, 1H), 4.16 (bs, 1H), 3.77−3.63 (s, 9H), 3.40−3.34 (m, 0.2H), 3.03−2.97 (m, 0.8H), 2.87 (s, 3H), 2.32−1.80 (m, 8H). 13C NMR (100 MHz, CDCl3) δC: 174.1, 173.4, 173.3, 166.8, 152.1, 132.7, 132.6, 128.9, 128.8, 127.8, 127.6, 121.7, 111.3, 52.4, 52.4, 52.0, 52.0, 51.9, 51.6, 48.2, 43.0, 32.3, 32.2, 31.4, 31.2, 30.2, 28.3, 27.3, 27.1. Rf 0.61 (EtOAc/Hexane 4:1). MS (ESI) m/z (relative intensity): 457.2 ([M+Na]+, 100).
(2S)-dimethyl 2-(3-((S)-4-methoxy-4-oxo-3-(4-(2,2,2-trifluoro-N-methylacetamido)-benzamido)butyl)oxiran-2-yl)pentanedioate (23)
The N-Me-pAB-olefinic isostere 22 (27 mg, 62 μmol) was dissolved in THF (2 mL) in an atmosphere of argon, cooled to −70 °C and stirred. Then Et3N (500 μL, 3.6 mmol) was added to the above solution followed by dropwise addition of (CF3CO)2O (40 μL, 290 μmol) at −70 °C. Stirring was continued for 1.5 h after which the reaction temperature was allowed to rise to 0 °C. When TLC analysis showed the complete consumption of the starting material (10 h), solvent was removed and the crude product was purified by flash chromatography (EtOAc/Hexane 1:1) to give pure N-Tfa derivative as an oil (20 mg, 62%). 1H NMR (500 MHz; CDCl3) δH: 7.96−7.87 (m, 2H), 7.35−7.34 (m, 2H), 6.72−6.71 (bd, 0.8H), 5.61−5.54 (m, 1H), 5.47−5.35 (m, 1H), 4.85−4.79 (m, 1H), 3.80−3.79 (s, 3H), 3.67−3.62 (m, 6H), 3.37 (bs, 3H), 3.04−2.99 (m, 1H), 2.32−1.80 (m, 8H). 13C NMR (125 MHz, CDCl3) δC: 174.0, 173.3, 172.8, 165.7, 134.5, 132.3, 132.2, 131.9, 128.7, 128.6, 128.3, 128.2, 127.8, 127.7, 52.6, 52.4, 52.3, 52.2, 52.1, 52.0, 51.7, 48.13, 43.06, 39.6, 32.0, 31.7, 31.4, 31.2, 28.3, 27.3, 27.1, 23.5. Rf 0.44 (EtOAc/Hexane 3:2). MS(ESI) m/z (relative intensity): 553.17 ([M+Na]+, 100).
The N-Tfa-N-Me-pAB-olefin isostere, prepared as described directly above (15 mg, 11 μmol), was dissolved in DCM (2 mL), and cooled to 0 °C under stirring condition. Then NaHCO3 (7 mg, 84 μmol) was added followed by mCPBA (12 mg, 70 μmol) at 0 °C. Stirring was continued at rt for 24 h. TLC and MS analysis showed the complete consumption of the starting material and formation of the product. The reaction mixture was filtered through Celite and Na2SO4 using a Pasteur pipette and the crude product was purified by flash chromatography (EtOAc/Hexane 7:3, 1% Et3N) to give the desired epoxide 23 as an oil (12 mg, 80%). 1H NMR (500 MHz; CDCl3) δH: 7.97−7.88 (m, 2H), 7.35 (bs, 2H), 4.91−4.82 (m, 1H), 3.81−3.62 (s, 9H), 3.37 (s, 3H), 3.18−2.83 (m, 3H), 2.48−1.83 (m, 8H). 13C NMR (125 MHz, CDCl3) δC: 172.9, 172.8, 172.4, 172.3, 165.8, 156.6, 143.6, 134.3, 128.65, 128.58, 127.6, 58.7, 58.5, 58.4, 57.6, 57.3, 57.1, 57.0, 56.1, 52.7, 52.2, 52.1, 51.77, 51.68, 47.18, 47.15, 39.6, 31.4, 31.3, 29.7, 29.3, 28.7, 27.6, 25.0, 23.9. 19F NMR (376 MHz, CDCl3) δF: −67.01, −70.04, −75.68. Rf 0.51 (EtOAc/Hexane 4:1). MS (ESI) m/z 569.1 (relative intensity): ([M+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C24H29F3N2O9Na 569.1723, found 569.1723.
(3R,8S,E)-trimethyl 8-(4-(methylamino)benzamido)oct-4-ene-1,3,8-tricarboxylate (24)
Et3N (1.5 mL) was added to a stirred solution of the N-methyl p-aminobenzoic acid HOBt ester, 21, (59 mg, 219 μmol) and amine derivative 20 (22 mg, 73 μmol) in THF (1.5 mL) at 0 °C in an atmosphere of argon. The reaction was warmed to rt and stirring was continued for 48 h. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (EtOAc/Hexane 1:1) to give pure product 24 as an oil (21 mg, 68%). 1H NMR (500 MHz; CDCl3) δH: 7.72−7.66 (m, 2H), 6.58−6.56 (m, 2H), 6.50−6.49 (bd, 1H), 5.57−5.50 (m, 1H), 5.50−5.30 (m, 1H), 4.90−4.80 (m, 1H), 3.77−3.66 (s, 9H), 3.42−3.40 (m, 0.2H), 3.02−2.98 (m, 0.9H), 2.88 (s, 3H), 2.33−1.80 (m, 8H). 13C NMR (100 MHz, CDCl3) δC: 174.1, 173.4, 173.0, 166.8, 152.1, 132.7, 132.6, 128.9, 128.8, 127.9, 127.8, 121.7, 111.4, 111.3, 52.44, 52.39, 51.97, 51.93, 51.90, 51.70, 51.63, 48.2, 32.3, 31.4, 31.1, 30.3, 30.0, 29.7, 28.3, 27.24, 27.15. Rf 0.61 (EtOAc/Hexane 4:1). MS (ESI) m/z (relative intensity): 457.2 ([M+Na]+, 100). HRMS-ESI m/z: [M+Na]+ calcd for C22H30N2O7Na 457.1951, found, 457.1942.
(2R)-dimethyl 2-(3-((S)-4-methoxy-4-oxo-3-(4-(2,2,2-trifluoro-N-methylacetamido)-benzamido)butyl)oxiran-2-yl)pentanedioate (25)
The N-Me-pAB-olefinic isostere 24 (21 mg, 47 μmol) was dissolved in THF (3.5 mL) in an atmosphere of argon, cooled to 0 °C and stirred. Then Et3N (68 μL, 470 μmol)) was added to the above solution followed by dropwise addition of (CF3CO)2O (67 μL, 470 μmol) at 0 °C. Stirring was continued for 10 h. TLC analysis showed the complete consumption of the starting material. Solvent was removed and the crude product was purified by flash chromatography (EtOAc/Hexane 1:1) to give pure N-Tfa derivative as an oil (19 mg, 76%) 1H NMR (500 MHz; CDCl3) δH: 7.97 (d, J = 8.5 Hz, 0.4H), 7.90−7.88 (m, 1.7H), 7.35 (d, J=7.5 Hz, 2H), 7.10 (bd, 0.2H), 6.73 (bd, 0.9H), 5.64−5.54 (m, 1H), 5.48−5.36 (m, 1H), 4.85−4.75 (m, 1H), 3.80−3.63 (s, 9H), 3.37 (bs, 3H), 3.04−2.99 (m, 1H), 2.35−1.80 (m, 8H). 13C NMR (100 MHz, CDCl3) δC: 174.1, 173.3, 172.79, 172.76, 165.7, 132.3, 132.2, 132.1, 128.8, 128.6, 128.3, 128.2, 127.1, 127.6, 64.2, 52.7, 52.6, 52.3, 52.2, 52.05, 51.95, 51.73, 51.67, 48.1, 42.7, 39.6, 32.0, 31.7, 31.4, 31.0, 28.3, 27.24, 27.15, 23.8. Rf 0.44 (EtOAc/Hexane 3:2). MS (ESI) m/z (relative intensity): 553.1 ([M+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C24H29F3N2O8Na 553.1774, found 553.1774.
The N-Tfa-N-Me-pAB-olefin isostere, prepared as described directly above (18 mg, 33 μmol), was dissolved in DCM (3 mL), and cooled to 0 °C under stirring condition. Then NaHCO3 (18 mg, 132 μmol) was added followed by mCPBA (19 mg, 85 μmol) at 0 °C. The reaction was allowed to warm to rt. Stirring was continued at rt for 48 h. TLC and MS analysis showed the complete consumption of the starting material and formation of the product. The solvent was removed under reduced pressure. The residue was dissolved in EtOAc (20 mL) and washed with NaHCO3 solution (3 × 10 mL) and water (2 × 10 mL). The organic layer was dried over Na2SO4 and the crude product was purified by flash chromatography (EtOAc/Hexane 3:2, 1% Et3N) to give the desired epoxide 25 as an oil (18 mg, 97%). 1H NMR (500 MHz; CDCl3) δH: 7.96−7.89 (m, 2H), 7.35−7.30 (m, 2H), 7.08−7.05 (m, 0.4H), 6.87 (bd, 0.7H), 6.59 (bd, 0.2H), 4.90−4.83 (m, 1H), 3.81−3.58 (s, 9H), 3.37 (s, 3H), 3.19−2.83 (m, 3H), 2.47−1.44 (m, 8H). 13C NMR (100 MHz, CDCl3) δC: 173.0, 172.9, 172.5, 172.4, 165.8, 128.8, 128.6, 127.7, 59.0, 58.7, 58.4, 57.7, 57.34, 57.26, 57.1, 57.0, 52.8, 52.2, 51.8, 51.7, 47.19, 47.16, 43.5, 39.6, 31.4, 31.3, 30.6, 28.8, 28.6, 27.6, 25.0, 24.3, 23.9. 19F NMR (376 MHz, CDCl3) δF: −67.02, −70.05. Rf 0.51 (EtOAc/Hexane 4:1). MS (ESI) m/z (relative intensity): 569.1 ([M+Na]+, 100). HRMS-ESI (m/z): [M+Na]+ calcd for C24H29F3N2O9Na 569.1723, found 569.1711.
(2S)-2-(3-((S)-3-carboxy-3-(4-(methylamino)benzamido)propyl)oxiran-2-yl)pentanetrioic acid (4)
The epoxide derivative 23 (3 mg, 5.49 μmol) was dissolved in MeOH (2 mL) under stirring condition and LiOH•H2O (0.21 mL 0.1M solution, 22 μmol) was added dropwise at rt. The reaction mixture was continuously stirred at rt, during which time, additional LiOH was added in portions while the reaction progress was monitored carefully by MS analysis, which confirmed formation of desired tricarboxylate derivative 4. After 7 days, solvent was removed under reduced pressure and high vacuum to provide the desired product as a solid (5 mg, quantitative yield). 1H NMR (600 MHz; CD3OD) δH: 7.69−7.66 (m, 2H), 6.59−6.57 (m, 2H), 4.47−4.40 (m, 1H), 2.80 (s, 3H), 2.65−1.59 (m, 8H). 13C NMR (100 MHz, CDCl3) δC: 182.8, 182.7, 179.64, 179.60, 176.0, 169.7, 169.6, 161.7, 154.5, 141.1, 134.0, 130.1, 122.4, 122.2, 119.4, 117.4, 115.5, 112.2, 112.0, 69.2, 56.8, 56.7, 56.5, 52.7, 50.5, 50.02, 49.98, 45.1, 39.0, 37.4, 31.0, 30.3, 30.1. HRMS-ESI(m/z): [M-H]− cald for C19H23N2O8 407.1460, found 407.1469).
(2R)-2-(3-((S)-3-carboxy-3-(4-(methylamino)benzamido)propyl)oxiran-2-yl)pentanetrioic acid (5)
The epoxide derivative 25 (2 mg, 3.66 μmol) was dissolved in MeOH (2 mL) under stirring condition and LiOH•H2O (0.14 mL 0.1M solution, 14 μmol) was added dropwise at rt. The reaction mixture was continuously stirred at rt, during which time, additional LiOH was added in portions while the reaction progress was monitored carefully by MS analysis, which confirmed formation of desired tricarboxylate derivative 5. After 7 days, solvent was removed under reduced pressure and high vacuum to afford the desired product as a solid (3 mg, quantitative yield). 1H NMR (600 MHz; CD3OD) δH: 7.70−7.66 (m, 2H), 6.58 (d, J = 8.4 Hz, 2H), 4.48−4.40 (m, 1H), 2.80 (s, 3H), 2.63−1.59 (m, 8 H). 13C NMR (100 MHz, CDCl3) δC: 182.8, 182.7, 176.9, 169.7, 161.7, 154.5, 141.1, 138.0, 130.1, 122.4, 112.2, 112.0, 69.2, 56.7, 52.8, 50.5, 50.02, 49.97, 39.0, 37.4, 34.8, 31.0, 30.3, 30.1. HRMS-ESI(m/z): [M-H]− calcd for C19H23N2O8 407.1460, found 407.1454.
Supplementary Material
Acknowledgments
This research was supported by a grant from the National Cancer Institute (CA28097). We thank Mr. Jeff Kampf for determining the crystal structure of cis-14.
Footnotes
Supporting Information Available: General Experimental Procedures, synthesis of 5-(((3aS,4S,6aR)-2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)methyl-sulfonyl)-1-phenyl-1H-tetrazole (cis-14), ORTEP plot of compound cis-14 and X-Ray crystallographic data, synthesis of HOBt ester of 4-(N-Methyl)aminobenzoic acid (21), synthesis of N-Tfa, methyl ester precursors, S3 and S6, of terminal epoxide derivatives, 2b and 1b, 1H NMR and 13C NMR spectra for all compounds described in Experimental Section. This material is available free of charge via the Internet at http:pubs.acs.org.
References
- 1.Powers JC, Asgian JL, Ekici OD, James KE. Chem. Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 2.Love KR, Catic A, Schlieker C, Ploegh HL. Nature Chem. Biol. 2007;3:697–705. doi: 10.1038/nchembio.2007.43. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson KD. Cell Dev. Biol. 2000;11:141–148. doi: 10.1006/scdb.2000.0164. [DOI] [PubMed] [Google Scholar]
- 4.Schneider E, Ryan TJ. Clin. Chim. Acta. 2006;374:25–32. doi: 10.1016/j.cca.2006.05.044. [DOI] [PubMed] [Google Scholar]
- 5.Baker BR. Design of active-site-directed irreversible enzyme inhibitors; the organic chemistry of the enzymic active-site. Wiley; New York: 1967. [Google Scholar]
- 6.Baker BR. Ann. Rev. Pharmacol. 1970;10:35–50. doi: 10.1146/annurev.pa.10.040170.000343. [DOI] [PubMed] [Google Scholar]
- 7.Silverman RB. Mechanism-based enzyme inactivation: chemistry and enzymology. CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- 8.Johnston SC, Larsen CN, Cook WJ, Wilkinson KD, Hill CP. EMBO J. 1997;16:3787–3796. doi: 10.1093/emboj/16.13.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misaghi S, Galardy PJ, Meester WJN, Ovaa H, Ploegh HL, Gaudet R. J. Biol. Chem. 2005;280:1512–1520. doi: 10.1074/jbc.M410770200. [DOI] [PubMed] [Google Scholar]
- 10.Chave KJ, Galivan J, Ryan TJ. Biochem. J. 1999;343:551–555. [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander JP, Ryan TJ, Ballou DP, Coward JK. Biochemistry. 2008;47:1228–1239. doi: 10.1021/bi701607v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albeck A. Drug Dev. Res. 2000;50:425–434. [Google Scholar]
- 13.Alexander MD. Ph.D. Thesis. The University of Michigan; 2004. [Google Scholar]
- 14.Jenmalm A, Berts W, Li YL, Luthman K, Csoregh I, Hacksell U. J. Org. Chem. 1994;59:1139–1148. [Google Scholar]
- 15.Mann A, Quaranta L, Reginato G, Taddei M. Tetrahedron Lett. 1996;37:2651–2654. [Google Scholar]
- 16.Perlman N, Livneh M, Albeck A. Tetrahedron. 2000;56:1505–1516. [Google Scholar]
- 17.Demarcus M, Ganadu ML, Mura GM, Porcheddu A, Quaranta L, Reginato G, Taddei M. J. Org. Chem. 2001;66:697–706. doi: 10.1021/jo000961w. [DOI] [PubMed] [Google Scholar]
- 18.Wiktelius D, Berts W, Jensen AJ, Gullbo J, Saitton S, Csoregh I, Luthman K. Tetrahedron. 2006;62:3600–3609. [Google Scholar]
- 19.Bartley DM, Coward JK. J. Org. Chem. 2005;70:6757–6774. doi: 10.1021/jo0507439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rozzell JD. Tetrahedron Lett. 1982;23:1767–1770. [Google Scholar]
- 21.Xie ZF, Suemune H, Sakai K. J. Chem. Soc. Chem. Comm. 1987:838–839. [Google Scholar]
- 22.Wahhab A, Tavares DF, Rauk A. Can. J. Chem. 1990;68:1559–1563. [Google Scholar]
- 23.Blakemore PR. J. Chem. Soc. Perkin Trans 1. 2002:2563–2585. [Google Scholar]
- 24.Plesniak K, Zarecki A, Wicha J. Top. Curr. Chem. 2007;275:163–250. doi: 10.1007/128_049. [DOI] [PubMed] [Google Scholar]
- 25.Maeda K, Inouye Y. Bull. Chem. Soc. Jpn. 1994;67:2880–2882. [Google Scholar]
- 26.Bartley DM, Coward JK. J. Org. Chem. 2006;71:372–374. doi: 10.1021/jo051854a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanrheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976:1973–1976. [Google Scholar]
- 28.Mitsunobu O. Synthesis-Stuttgart. 1981:1–28. [Google Scholar]
- 29.The trans diastereomers of the isopropylidene precursors, 11 and 13, are predicted to have a lower dipole moment, with associated decreased polarity, than the corresponding cis diastereomers. As a result, the trans diastereomers (higher Rf) were readily separated from the more polar cis diastereomers in high purity by flash chromatography.
- 30.Padron JM, Kokotos G, Martin T, Markidis T, Gibbons WA, Martin VS. Tetrahedron Asymm. 1998;9:3381–3394. [Google Scholar]
- 31.Kraus GA, Taschner MJ. J. Org. Chem. 1980;45:1175–1176. [Google Scholar]
- 32.Santhanam B, Worfent MA, Moore JN, Boons GJ. Chem. Eur. J. 2004;10:4798–4807. doi: 10.1002/chem.200400376. [DOI] [PubMed] [Google Scholar]
- 33.The use of a stereorandom epoxidation method was chosen in order to obtain the target compounds as “proof of concept” irreversible inhibitors of GH. Following biochemical evaluation of 4 and 5, the use of chiral epoxidation methods34 will be pursued in order to optimize both efficacy and selectivity.
- 34.Frohn M, Shi Y. Synthesis-Stuttgart. 2000:1979–2000. [Google Scholar]
- 35.All of the reactions depicted in Schemes 4 and 5 were performed multiple times on less than 100 mg scale with consistent results. However, all of the chemical transformations utilized here were chosen for their generality and are known to work well on significantly larger scale. Thus, we expect the synthetic route described here to be readily amenable to gram scale without modification.
- 36.Fieser LF, Fieser M. Reagents for organic synthesis. Vol. 1. John Wiley & Sons, Inc.; New York: 1967. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.