

Two members of the class C family of bacterial nonspecific acid phosphatases have been cloned, expressed, purified and crystallized. One of the crystal forms exhibited epitaxial twinning.
Keywords: acid phosphatases, class C nonspecific acid phosphatases, Francisella tularensis, Pasteurella multocida, epitaxial twinning
Abstract
Class C nonspecific acid phosphatases are bacterial enzymes that are secreted across the cytoplasmic membrane and hydrolyze a variety of phosphomonoesters at acidic pH. These enzymes are of interest for the development of improved vaccines and clinical diagnostic methods. In one case, the category A pathogen Francisella tularensis, the class C phosphatase plays a role in bacterial fitness. Here, the cloning, expression, purification and crystallization methods for the class C acid phosphatases from F. tularensis and Pasteurella multocida are reported. Crystals of the F. tularensis enzyme diffracted to 2.0 Å resolution and belonged to space group C2221, with one enzyme molecule in the asymmetric unit. Crystals of the P. multocida enzyme diffracted to 1.85 Å resolution and belonged to space group C2, with three molecules in the asymmetric unit. Diffraction patterns from crystals of the P. multocida enzyme exhibited multiple interpenetrating reciprocal-space lattices, indicating epitaxial twinning. Despite this aberrance, autoindexing was robust and the data could be satisfactorily processed to 1.85 Å resolution using MOSFLM and SCALA.
1. Introduction
Acid phosphatases (EC 3.1.3.2) are ubiquitous and catalyze the transfer of phosphoryl groups from phosphomonoesters to water at acidic pH (Vincent et al., 1992 ▶). They play essential roles in the generation, acquisition and mobilization of inorganic phosphate, as well as critical roles in the phosphoryl relay systems involved in signal transduction pathways in both prokaryotes and eukaryotes.
Three classes of bacterial nonspecific acid phosphatases (NSAPs) have been identified (denoted A, B and C) based on subcellular localization and conserved amino-acid sequence motifs (Rossolini et al., 1998 ▶). The enzymes studied here belong to class C, which is characterized by the presence of four invariant aspartate residues known as the DDDD motif (in bold) contained within the bipartite signature motif (I/V)-(V/A/L)-D-(I/L)-D-E-T-(V/M)-L-X-(N/T)-X-X-Y near the N-terminus and (I/V)-(L/M)-X-X-G-D-(N/T)-L-X-D-F near the C-terminus (Thaller et al., 1998 ▶). Several class C enzymes have been purified and/or characterized, including those from Haemophilus influenzae [known as e (P4); Reilly et al., 1999 ▶; Reilly & Smith, 1999 ▶], Bacillus anthracis (Felts et al., 2006 ▶), Streptococcus equisimilis (Malke, 1998 ▶), Staphylococcus aureus (du Plessis et al., 2002 ▶), Helicobacter pylori (Reilly & Calcutt, 2004 ▶), Chryseobacterium meningosepticum (Passariello et al., 2003 ▶) and Clostridium perfringens (Reilly et al., 2009 ▶). Shared attributes include a polypeptide size of 25–30 kDa, a requirement for metal cation for catalytic activity and rather broad substrate specificity. Most have an N-terminal lipidated Cys that anchors them to the outer membrane of the bacterium. The S. aureus and C. perfringens enzymes appear to be exceptions in this respect.
The crystal structures of the class C NSAPs from H. influenzae (Felts et al., 2007 ▶; Ou et al., 2006 ▶) and B. anthracis (PDB code 2i33) have been determined. The structures show that class C NSAPs belong to the haloacid dehalogenase structural superfamily and that the conserved Asp residues of the DDDD motif participate in binding an active-site Mg2+ ion (Felts et al., 2007 ▶). Furthermore, the structure of the H. influenzae enzyme complexed with a tungstate-ion inhibitor suggests that the first Asp of the motif is the nucleophile that attacks the substrate P atom (Felts et al., 2007 ▶).
There is emerging interest in class C NSAPs. Because of their localization to the bacterial outer membrane, they are potentially attractive candidates for vaccine development. In fact, significant progress has been made towards creating a vaccine against nontypeable H. influenzae using catalytically inactive mutants of recombinant e (P4) (Hotomi et al., 2005 ▶; Mason et al., 2004 ▶; Green et al., 2005 ▶). Moreover, the class C NSAP from the category A bioterrorism pathogen Francisella tularensis has been implicated in phagosomal escape and virulence (Mohapatra et al., 2008 ▶). Finally, detection of acid phosphatase activity is a useful diagnostic tool for the clinical identification of C. perfringens (Adcock & Saint, 2001 ▶; Eisgruber et al., 2003 ▶; Sartory et al., 2006 ▶). Presumably, these methods detect the Clostridium class C NSAP. By extension, it may be possible to develop analogous tools for the identification of other bacteria based on immunological and biochemical detection of an organism’s unique class C NSAP. Elucidation of the crystal structures of class C NSAPs will aid this effort.
To facilitate further investigations of this enzyme family, we have developed recombinant expression systems for the class C enzymes from F. tularensis (FtAcpC) and Pasteurella multocida (PmAcpC), the latter being a pathogen of significant agricultural importance (Harper et al., 2006 ▶). Methods for expressing, purifying and crystallizing these enzymes are described here, together with preliminary analyses of their X-ray diffraction data.
2. Methods and results
2.1. Cloning of the FtAcpC gene
The FtAcpC gene has been identified previously as one of five phosphatase genes present in the genome sequence of F. tularensis subsp. novicida (Mohapatra et al., 2008 ▶). The gene encodes a 247-residue protein that has 25% identity over 225 amino acids to H. influenzae e (P4), the archetype of class C NSAPs. Moreover, the sequence contains a bipartite motif that is characteristic of class C NSAPs. The motif for FtAcpC is ILDIDETALDNS at residues 73–84 followed by IAYFGDNIQDF at residues 202–212. Secretion across the cytoplasmic membrane is a defining characteristic of class C NSAPs, so the protein sequence was analyzed for potential signal peptides. Analysis using the SignalP 3.0 (Bendtsen et al., 2004 ▶), Signal-BLAST (Frank & Sippl, 2008 ▶) and PSORT 6.4 (Nakai & Kanehisa, 1991 ▶) servers suggests that the N-terminus contains a signal peptide of 20–23 residues in length, implying that the protein is expressed as a precursor polypeptide that is exported from the cytoplasm and cleaved to yield the mature enzyme. There is a cysteine at position 24 which could potentially be lipidated, as is predicted for most other class C NSAPs, but this remains to be determined. The sequence similarity to e (P4), the bipartite DDDD motif and the predicted export from the cytoplasm are consistent with classification of the enzyme into the class C NSAP family.
The FtAcpC gene (NCBI RefSeq No. YP_169641) was cloned from genomic DNA of F. tularensis strain SCHU S4 (Larsson et al., 2005 ▶). The gene was amplified by PCR and inserted into pET20b using NcoI and XhoI restriction sites. To avoid potential problems with membrane association, the N-terminal 24 residues were replaced by the pelB leader sequence followed by Met-Gly. Thus, the mature recombinant enzyme expressed in Escherichia coli is exported to the periplasmic space and is predicted to have N-terminal residues MGNSVNI and a C-terminal His6 tag.
2.2. Identification and cloning of the PmAcpC gene
A query of the complete genome sequence of P. multocida strain Pm70 (May et al., 2001 ▶) with class C NSAP genes identified an open reading frame, designated PM1064, which had the potential to be a class C NSAP. PM1064 encodes 272 residues having 59% global amino-acid sequence identity to e (P4). PmAcpC contains a classic 19-residue lipoprotein signal peptide at the N-terminus (Hayashi & Wu, 1990 ▶). The predicted signal peptide contains positively charged residues (Lys) at positions −18 and −15, followed by a hydrophobic region and ending in the lipoprotein box LLAA-C. Thus, the mature form of PmAcpC is predicted to be a 253-residue lipoprotein with Cys1 modified by lipidation. Furthermore, the predicted protein contains a class C bipartite sequence motif VVDLDETMIDNS at residues 61–72 of the mature protein and VLFVGDNLNDF at residues 175–185. The high sequence identity with e (P4), the bipartite DDDD motif and lipoprotein prediction are consistent with assignment of this protein as a class C NSAP.
The PmAcpC gene (GenBank accession No. FJ609981) was cloned from genomic DNA from a clinical isolate. The gene was amplified by PCR and cloned as an NcoI–XhoI fragment into pET20b. To produce soluble protein in E. coli, the N-terminal 20 residues were replaced by the pelB leader sequence followed by Met-Val. The mature recombinant enzyme is thus exported to the periplasmic space and is predicted to have N-terminal residues MVSNQQA and a C-terminal His6 tag.
2.3. Expression and purification of FtAcpC
Recombinant FtAcpC was expressed in E. coli BL21 (DE3). A starter culture was grown overnight at 310 K in 5 ml LB supplemented with 50 µg ml−1 ampicillin and 0.2%(w/v) glucose. The starter culture was diluted 1:500 into 35 ml LB containing 50 µg ml−1 ampicillin and 0.2%(w/v) glucose. The culture was incubated at 310 K with constant aeration supplied by orbital shaking at 250 rev min−1 until the OD600 reached 0.6. The cells were then centrifuged at 277 K (3600g for 10 min) and the resulting pellet was resuspended in 4 ml LB and diluted 1000-fold into 1 l LB containing 50 µg ml−1 ampicillin and 0.2%(w/v) glucose. The culture was incubated at 310 K with constant aeration (250 rev min−1) and induced with 0.4 mM IPTG when the OD600 reached 0.4. The incubation was continued at the lower temperature of 298 K and with a decreased shaking rate of 220 rev min−1 until the OD600 reached 1.0. The cells were harvested by centrifugation (3600g, 277 K), suspended in 50 mM Tris–HCl pH 8.4 and frozen.
The frozen cell suspension was thawed and the cells were disrupted in a Thermo 40K cell French Press adjusted to 69 MPa for two cycles at a low flow rate to maintain the cell pressure. Unbroken cells and cellular debris were removed by centrifugation at 31 000g for 20 min at 277 K. Remaining bacterial membranes were pelleted by centrifugation at 183 960g for 1 h at 277 K.
The supernatant from ultracentrifugation was filtered through a 0.2 µm Millipore filter and applied onto a Q Sepharose anion-exchange column with an ÄKTA FPLC chromatography system. The sample was loaded onto the column at 2 ml min−1 using a buffer of 50 mM Tris–HCl pH 8.4. A linear NaCl gradient was then applied, eluting FtAcpC in the range 60–100 mM NaCl. Fractions containing FtAcpC were identified using SDS–PAGE and phosphatase-activity assays, the latter consisting of a discontinuous colorimetric assay using p-nitrophenylphosphate as the substrate (Reilly et al., 2006 ▶). The pooled fractions were dialyzed overnight at 277 K against 20 mM phosphate buffer pH 7.0 containing 0.5 M NaCl in preparation for immobilized metal-ion affinity chromatography. The dialyzed sample was loaded onto an Ni2+-charged affinity column and eluted with 175–200 mM imidazole. Fractions having the highest purity level, based on SDS–PAGE, were pooled and dialyzed overnight into 50 mM sodium acetate buffer pH 6.0. Finally, the sample was concentrated to 10 mg ml−1 using a centrifugal concentrating device. Protein concentration was assessed using the BCA method (Pierce kit).
2.4. Expression and purification of PmAcpC
Recombinant PmAcpC was expressed using the auto-induction method (Studier, 2005 ▶). A single colony of E. coli BL21 (DE3) containing the engineered plasmid was used to inoculate 5 ml LB containing ampicillin (50 µg ml−1) and incubated overnight at 310 K. The culture was pelleted at 3660g and suspended in fresh LB prior to inoculation of four 2.8 l flasks, each containing 500 ml TY broth supplemented with the ZYM-5052 media components and ampicillin (50 µg ml−1). The culture was grown with constant aeration at 310 K overnight.
The enzyme was purified using standard nondenaturing methods. All procedures were conducted at 277 K unless noted otherwise. Cultures from auto-induction were centrifuged at 3660g for 20 min and the pellet was suspended in 40 ml pH 7.0 buffer containing 20 mM sodium phosphate and 0.5 M NaCl. The suspension was stored at 253 K overnight and thawed the next day. A protease-inhibitor tablet (SigmaFAST) and DNase were added to the thawed suspension and mixed for approximately 30 min. The cells were disrupted and subjected to low-speed centrifugation and ultracentrifugation steps as described above for FtAcpC. The ultracentrifugation supernatant was retained and refrigerated.
The supernatant from ultracentrifugation was loaded at 2 ml min−1 onto a 5 ml Ni2+-charged HiTrap metal-chelate chromatography resin using an ÄKTAprime system. The resin was washed with 20 mM sodium phosphate, 0.5 M NaCl, 50 mM imidazole pH 7.0 until the A 280 returned to baseline. A 200 ml linear imidazole gradient (0.05–2.0 M imidazole in 20 mM sodium phosphate pH 7.0, 0.5 M NaCl) was applied and the eluate was collected in 2 ml fractions. PmAcpC eluted at approximately 200 mM imidazole. Fractions containing high phosphatase activity (p-nitrophenylphosphate substrate) were pooled and dialyzed overnight against 50 mM sodium acetate pH 6.0.
The dialyzed sample was loaded at a flow rate of 2.0 ml min−1 onto a 5 ml SP-Sepharose cation-exchange column that had been equilibrated with 50 mM sodium acetate pH 6.0. The column was washed with 50 mM sodium acetate pH 6.0 until the A 280 returned to baseline. The phosphatase was eluted from the column with a 200 ml linear NaCl gradient (0.0–1.0 M) and collected in 2 ml fractions. PmAcpC eluted in the range 0.44–0.53 M NaCl. Enzymatically active fractions that were judged to be highly pure by SDS–PAGE were pooled and dialyzed overnight against 20 mM sodium phosphate pH 7.0. The sample was then concentrated to 10 mg ml−1 (BCA assay) using a 10 000 molecular-weight cutoff Centricon filtering device.
2.5. Crystallization of FtAcpC and preliminary analysis of X-ray diffraction data
Crystallization trials were performed at 293 K using the sitting-drop method of vapor diffusion with Cryschem plates and reservoir volumes of 1 ml. Drops were formed by mixing 2 µl protein stock solution and 2 µl reservoir solution. Crystal screening trials using commercially available reagent kits revealed promising crystallization conditions consisting of 25–30%(w/v) PEG 3350, 0.1 M bis-tris pH 5.5–6.5 and 0.1–0.2 M of either sodium chloride or ammonium acetate. The latter salt proved to be the preferred additive and eventually the best crystals were grown using reservoir solutions consisting of 24–32%(w/v) PEG 3350, 0.1 M bis-tris pH 6.5 and 0.1 M ammonium acetate. The optimized crystals appeared as rectangular blocks with a maximum dimension of approximately 0.2 mm (Fig. 1 ▶). In preparation for low-temperature data collection, the crystals were soaked in 30%(w/v) PEG 3350, 0.1 M bis-tris pH 6.5, 0.1 M ammonium acetate and 20%(v/v) PEG 200. The cryoprotected crystals were picked up with Hampton loops and plunged into liquid nitrogen.
Figure 1.

A crystal of FtAcpC. The smallest dimension of the ruler corresponds to 20 µm.
FtAcpC crystals were analyzed on Advanced Light Source beamline 4.2.2 using a NOIR-1 CCD detector. Autoindexing calculations with d*TREK (Pflugrath, 1999 ▶) and MOSFLM (Leslie, 1992 ▶, 2006 ▶) indicated a C-centered orthorhombic lattice with unit-cell parameters a = 59.2, b = 124.2, c = 62.2 Å. Using the method of Matthews (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶), the asymmetric unit is predicted to contain one FtAcpC molecule and 46% solvent. A data set consisting of 115 frames was collected with a crystal-to-detector distance of 150 mm, an oscillation width of 1.0° per frame and an exposure time of 5 s per frame. The data set was integrated with MOSFLM through the iMosflm graphical interface and scaled to 2.0 Å resolution with SCALA (Evans, 2006 ▶) using the CCP4i interface (Potterton et al., 2003 ▶). Data-processing statistics are listed in Table 1 ▶.
Table 1. Data-processing statistics for FtAcpC.
Values in parentheses are for the outermost resolution shell.
| Space group | C2221 |
| Wavelength (Å) | 0.97909 |
| Unit-cell parameters (Å) | a = 59.2, b = 124.2, c = 62.2 |
| Protein molecules in ASU | 1 |
| VM (Å3 Da−1) | 2.3 |
| Solvent content (%) | 46 |
| Resolution (Å) | 27.8–2.00 (2.11–2.00) |
| Total observations | 70800 |
| Unique reflections | 15842 |
| Redundancy | 4.5 (4.0) |
| Completeness (%) | 99.8 (99.1) |
| Mean I/σ(I) | 16.3 (3.8) |
| Rmerge† | 0.073 (0.470) |
| Rmerge† in low-resolution bin | 0.034 |
R
merge = 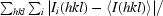
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
2.6. Crystallization of PmAcpC and preliminary analysis of X-ray diffraction data
Crystallization trials were performed at 293 K using the sitting-drop method (Cryschem plates, 1 ml reservoir volume) with drops formed by mixing 5 µl protein stock solution and 5 µl reservoir solution. Initial crystal screening trials revealed several promising conditions with PEG as the precipitating agent. The crystals invariably appeared as clusters of needles and although extensive optimization trials reduced the degree of fusion, it was not possible to grow truly single crystals. For example, the best crystals, which were grown using a reservoir solution of 20%(w/v) PEG 3350, 0.2 M ammonium citrate dibasic and 10%(v/v) n-propanol, still exhibited significant clustering and needle-like morphology (Fig. 2 ▶). In preparation for low-temperature data collection, the crystals were soaked in 25%(w/v) PEG 3350, 0.2 M ammonium citrate dibasic and 25%(v/v) PEG 200. The clusters were teased apart with Hampton loops in an attempt to isolate single crystals for data collection. The cryoprotected crystals were then plunged into liquid nitrogen.
Figure 2.

Crystals of PmAcpC. Note that the crystals grow as clusters of needles. The thickest clusters are approximately 20–40 µm thick.
X-ray diffraction data were collected on Advanced Light Source beamline 4.2.2 using a NOIR-1 CCD detector. The diffraction images clearly exhibited multiple interpenetrating reciprocal-space lattices (Fig. 3 ▶ a), particularly at low resolution (Fig. 3 ▶ b). The interpenetrating reciprocal lattices are symptomatic of epitaxial twinning and arise from multiple real-space lattices in the sample that are not perfectly aligned (Yeates & Fam, 1999 ▶; Chandra et al., 1999 ▶). The crystals diffracted to surprisingly high resolution despite being rather thin. As shown in Fig. 3 ▶(c), reflections extending beyond 1.84 Å resolution were observed. Despite the epitaxial twinning, autoindexing using either d*TREK (Pflugrath, 1999 ▶) or MOSFLM was robust and consistently indicated C2 as the space group, with unit-cell parameters a = 80.0, b = 106.1, c = 89.7 Å, β = 93.1°. The assumption of three PmAcpC molecules in the asymmetric unit implies 45% solvent content and V M = 2.2 Å3 Da−1 (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶).
Figure 3.

Diffraction image collected from a PmAcpC crystal. (a) Entire resolution range. (b) Enlarged view of the low-resolution region. The circle corresponds to 3.69 Å resolution. (c) Enlarged view of the high-resolution data.
A data set consisting of 360 frames was collected with an oscillation width of 0.5° per frame and a crystal-to-detector distance of 140 mm. Integration was performed with MOSFLM through the iMosflm interface and scaling calculations were performed with SCALA via CCP4i. The data could be processed satisfactorily to 1.85 Å resolution, as shown in Table 2 ▶. Note, however, that R merge is 0.065 in the low-resolution bin, which is rather high. Presumably, this suboptimal result is a consequence of the presence of multiple reciprocal-space lattices, which are particularly evident at low resolution (Fig. 3 ▶ b). For comparison, R merge is 0.034 in the low-resolution bin for the FtAcpC data (Table 1 ▶).
Table 2. Data-processing statistics for PmAcpC.
Values in parentheses are for the outermost resolution shell.
| Space group | C2 |
| Wavelength (Å) | 1.00000 |
| Unit-cell parameters (Å, °) | a = 80.0, b = 106.1, c = 89.7, β = 93.1 |
| Protein molecules in ASU | 3 |
| VM (Å3 Da−1) | 2.2 |
| Solvent content (%) | 45 |
| Resolution (Å) | 27.5–1.85 (1.95–1.85) |
| Total observations | 215684 |
| Unique reflections | 62294 |
| Redundancy | 3.5 (2.5) |
| Completeness (%) | 97.8 (86.7) |
| Mean I/σ(I) | 10.4 (3.1) |
| Rmerge† | 0.094 (0.272) |
| Rmerge† in low-resolution bin | 0.065 |
R
merge =
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
Acknowledgments
We thank Dr Jay Nix of ALS beamline 4.2.2 for assistance with data collection and Dr Fran Nano for providing the F. tularensis DNA used for cloning. HS was supported by a pre-doctoral fellowship from National Institutes of Health grant DK071510. This research was supported by National Institutes of Health grant U54-AI057160 to the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (to JJT and TJR), the University of Missouri Research Board (to JJT and TJR) and the Program for Prevention of Animal Infectious Diseases USDA ARS 58-1940-5-519 (to TJR and MJC). Part of this work was performed at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Adcock, P. W. & Saint, C. P. (2001). Appl. Environ. Microbiol.67, 4382–4384. [DOI] [PMC free article] [PubMed]
- Bendtsen, J. D., Nielsen, H., von Heijne, G. & Brunak, S. (2004). J. Mol. Biol.340, 783–795. [DOI] [PubMed]
- Chandra, N., Acharya, K. R. & Moody, P. C. E. (1999). Acta Cryst. D55, 1750–1758. [DOI] [PubMed]
- Eisgruber, H., Geppert, P., Sperner, B. & Stolle, A. (2003). Int. J. Food Microbiol.82, 81–86. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Felts, R. L., Ou, Z., Reilly, T. J. & Tanner, J. J. (2007). Biochemistry, 46, 11110–11119. [DOI] [PubMed]
- Felts, R. L., Reilly, T. J., Calcutt, M. J. & Tanner, J. J. (2006). Acta Cryst. F62, 705–708. [DOI] [PMC free article] [PubMed]
- Frank, K. & Sippl, M. J. (2008). Bioinformatics, 24, 2172–2176. [DOI] [PubMed]
- Green, B. A., Baranyi, E., Reilly, T. J., Smith, A. L. & Zlotnick, G. W. (2005). Infect. Immun.73, 4454–4457. [DOI] [PMC free article] [PubMed]
- Harper, M., Boyce, J. D. & Adler, B. (2006). FEMS Microbiol. Lett.265, 1–10. [DOI] [PubMed]
- Hayashi, S. & Wu, H. C. (1990). J. Bioenerg. Biomembr.22, 451–471. [DOI] [PubMed]
- Hotomi, M., Ikeda, Y., Suzumoto, M., Yamauchi, K., Green, B. A., Zlotnick, G., Billal, D. S., Shimada, J., Fujihara, K. & Yamanaka, N. (2005). Vaccine, 23, 1294–1300. [DOI] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci.12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Larsson, P. et al. (2005). Nature Genet.37, 153–159. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4–ESF/EACBM Newsl. Protein Crystallogr.26
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Malke, H. (1998). Appl. Environ. Microbiol.64, 2439–2442. [DOI] [PMC free article] [PubMed]
- Mason, K. W., Zhu, D., Scheuer, C. A., McMichael, J. C., Zlotnick, G. W. & Green, B. A. (2004). Vaccine, 22, 3449–3456. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- May, B. J., Zhang, Q., Li, L. L., Paustian, M. L., Whittam, T. S. & Kapur, V. (2001). Proc. Natl Acad. Sci. USA, 98, 3460–3465. [DOI] [PMC free article] [PubMed]
- Mohapatra, N. P., Soni, S., Reilly, T. J., Liu, J., Klose, K. E. & Gunn, J. S. (2008). Infect. Immun.76, 3690–3699. [DOI] [PMC free article] [PubMed]
- Nakai, K. & Kanehisa, M. (1991). Proteins, 11, 95–110. [DOI] [PubMed]
- Ou, Z., Felts, R. L., Reilly, T. J., Nix, J. C. & Tanner, J. J. (2006). Acta Cryst. F62, 464–466. [DOI] [PMC free article] [PubMed]
- Passariello, C., Schippa, S., Iori, P., Berlutti, F., Thaller, M. C. & Rossolini, G. M. (2003). Biochim. Biophys. Acta, 1648, 203–209. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Plessis, E. M. du, Theron, J., Joubert, L., Lotter, T. & Watson, T. G. (2002). Syst. Appl. Microbiol.25, 21–30. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Reilly, T. J. & Calcutt, M. J. (2004). Protein Expr. Purif.33, 48–56. [DOI] [PubMed]
- Reilly, T. J., Chance, D. L., Calcutt, M. J., Tanner, J. J., Felts, R. L., Waller, S. C., Henzl, M. T., Mawhinney, T. P., Ganjam, I. K. & Fales, W. H. (2009). Submitted. [DOI] [PMC free article] [PubMed]
- Reilly, T. J., Chance, D. L. & Smith, A. L. (1999). J. Bacteriol.181, 6797–6805. [DOI] [PMC free article] [PubMed]
- Reilly, T. J., Felts, R. L., Henzl, M. T., Calcutt, M. J. & Tanner, J. J. (2006). Protein Expr. Purif.45, 132–141. [DOI] [PubMed]
- Reilly, T. J. & Smith, A. L. (1999). Protein Expr. Purif.17, 401–409. [DOI] [PubMed]
- Rossolini, G. M., Schippa, S., Riccio, M. L., Berlutti, F., Macaskie, L. E. & Thaller, M. C. (1998). Cell. Mol. Life Sci.54, 833–850. [DOI] [PMC free article] [PubMed]
- Sartory, D. P., Waldock, R., Davies, C. E. & Field, A. M. (2006). Lett. Appl. Microbiol.42, 418–424. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif.41, 207–234. [DOI] [PubMed]
- Thaller, M. C., Schippa, S. & Rossolini, G. M. (1998). Protein Sci.7, 1647–1652. [DOI] [PMC free article] [PubMed]
- Vincent, J. B., Crowder, M. W. & Averill, B. A. (1992). Trends Biochem. Sci.17, 105–110. [DOI] [PubMed]
- Yeates, T. O. & Fam, B. C. (1999). Structure Fold. Des.7, R25–R29. [DOI] [PubMed]