

The crystal structure of a hyperthermophilic d-tagatose 3-epimerase-related protein with a unique active-site architecture was determined.
Keywords: hyperthermophiles, Thermotoga maritima, ketohexose 3-epimerases, d-tagatose 3-epimerase
Abstract
The crystal structure of a d-tagatose 3-epimerase-related protein (TM0416p) encoded by the hypothetical open reading frame TM0416 in the genome of the hyperthermophilic bacterium Thermotoga maritima was determined at a resolution of 2.2 Å. The asymmetric unit contained two homologous subunits and a dimer was generated by twofold symmetry. The main-chain coordinates of the enzyme monomer proved to be similar to those of d-tagatose 3-epimerase from Pseudomonas cichorii and d-psicose 3-epimerase from Agrobacterium tumefaciens; however, TM0416p exhibited a unique solvent-accessible substrate-binding pocket that reflected the absence of an α-helix that covers the active-site cleft in the two aforementioned ketohexose 3-epimerases. In addition, the residues responsible for creating a hydrophobic environment around the substrate in TM0416p differ entirely from those in the other two enzymes. Collectively, these findings suggest that the substrate specificity of TM0416p is likely to differ substantially from those of other d-tagatose 3-epimerase family enzymes.
1. Introduction
d-Tagatose 3-epimerase (DTE) family enzymes catalyze C3 epimerization of various ketohexoses, including rare sugars that exhibit unique properties and may even exert beneficial health effects in humans (Livesey & Brown, 1996 ▶; Levin, 2002 ▶; Takata et al., 2005 ▶). Indeed, it is hoped that DTE-family enzymes can serve as useful catalysts for the commercially viable production of these otherwise rare sugars (Takeshita et al., 2000 ▶; Granström et al., 2004 ▶). To date, however, detailed descriptions of the structure and of the relationship between structure and enzymatic properties have been reported for only two DTE-family enzymes: DTE from Pseudomonas cichorii (Yoshida et al., 2007 ▶) and d-psicose 3-epimerase (DPE) from Agrobacterium tumefaciens (Kim et al., 2006 ▶).
In recent years, much effort has gone into the isolation and characterization of enzymes from hyperthermophiles, to a large extent because their greater thermostability represents a significant advantage over their counterparts from mesophiles. In particular, thermostable enzymes that are involved in carbohydrate metabolism are very attractive for industrial applications (Lee et al., 2004 ▶). Although genome analysis has enabled the identification of several DTE homologues from hyperthermophiles, the structures and true functions of these enzymes remain unclear. For example, TM0416p, which is encoded by a hypothetical open reading frame (ORF ID TM0416) in the genome of the hyperthermophilic bacterium Thermotoga maritima, is predicted to be a DTE homologue (Kim et al., 2006 ▶), but the structural and functional details of TM0416p have not yet been reported. In the present study, therefore, we determined the crystal structure of TM0416p at 2.2 Å resolution. This is the first description of the structure of a DTE-related protein from a hyperthermophile and we found the active-site architecture to be quite unique and to differ substantially from those of previously described DTE-family enzymes.
2. Materials and methods
2.1. Overexpression and purification of recombinant protein
We initially carried out PCR with the following set of oligonucleotide primers to amplify a TM0416 gene fragment using the following primer pair: 5′-AAACATATGAAGCTATCTCTGGTGA-3′, containing a unique NdeI restriction site (bold) overlapping the 5′ initial codon, and 5′-GGGATCCTCATGTAAGTTTAATAATC-3′, containing a unique BamHI restriction site (bold) proximal to the 3′-end of the termination codon. Chromosomal DNA from T. maritima was obtained from the American Type Culture Collection (Manassas, Virginia, USA) and used as the template. The amplified 0.8 kbp fragment was digested with NdeI and BamHI and ligated into the expression vector pET-15b linearized with NdeI and BamHI, yielding pETM0416. Escherichia coli strain BL21 (DE3) Codon Plus RIL (Stratagene) was then transformed with pETM0416, after which the transformants were cultivated at 310 K in 0.5 l medium containing 6 g tryptone, 12 g yeast extract, 2.5 ml glycerol, 6.25 g K2HPO4, 1.9 g KH2PO4 and 50 µg ml−1 ampicillin until the optical density at 600 nm reached 0.6. Expression was then induced by adding 1 mM isopropyl β-d-1-thiogalactopyranoside to the medium and cultivation was continued for an additional 3 h at 310 K. The cells were then harvested by centrifugation, suspended in buffer (50 mM NaH2PO4 pH 8.0, 0.3 M NaCl, 10 mg ml−1 egg-white lysozyme and 1 mg ml−1 bovine pancreatic DNase I), incubated for 10 min at 310 K and lysed by sonication. The resultant lysate was centrifuged at 15 000g for 20 min, after which the supernatant was collected, heated for 10 min at 353 K and then clarified by centrifugation. This supernatant was then loaded onto a Protino Ni-IDA Resin column (MACHEREY-NAGEL) equilibrated with 50 mM NaH2PO4 pH 8.0 containing 0.3 M NaCl. After washing the column with the same buffer, the protein was eluted with 250 mM imidazole in the same buffer. The TM0416p-containing fractions were collected, concentrated by ultrafiltration and then subjected to gel filtration on a Superdex 200 26/60 column (GE Healthcare) equilibrated with 10 mM potassium phosphate buffer pH 7.0 containing 0.2 M NaCl, after which the resultant protein solution was dialyzed against 10 mM potassium phosphate buffer pH 7.0. The entire procedure was carried out at room temperature (∼298 K) and the TM0416p-containing fractions were checked by SDS–PAGE during each purification step. Based on the SDS–PAGE, the molecular mass of the enzyme was estimated to be about 32 kDa, which was consistent with the molecular mass calculated from the amino-acid sequence (32 627 Da for a total of 290 amino acids) including the His-tagged sequence.
2.2. Crystallization and data collection
Crystallization of TM0416p was performed using the sitting-drop vapour-diffusion method. 2 µl drops of protein solution (34 mg ml−1) were mixed with an equal volume of 20–30% PEG 200, 5% PEG 1000 and 0.1 M MES pH 6.0 and equilibrated against 0.1 ml reservoir solution. Colourless parallelepiped-shaped crystals appeared within 3 d at 293 K and reached maximum dimensions of 0.3 × 0.3 × 0.2 mm within one week. The crystals were found to belong to the triclinic space group P1, with unit-cell parameters a = 50.3, b = 55.2, c = 58.7 Å, α = 107.1, β = 102.7, γ = 91.5°. Diffraction data were collected to a resolution of 2.2 Å on an R-AXIS VII imaging-plate detector using a rotating copper-anode in-house generator (MicroMax007, Rigaku, Japan) operating at 40 kV and 20 mA. All measurements were carried out on crystals cryoprotected with 30%(v/v) PEG 200 and cooled to 100 K in a stream of nitrogen gas. The data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶).
2.3. Phasing and refinement
A heavy-atom derivative was prepared by soaking the crystals for 18 h in mother liquor containing 1 mM ethylmercurithiosalicylic acid sodium salt (thimerosal). Phase calculation was carried out using the single isomorphous replacement with anomalous scattering (SIRAS) method with SOLVE (Terwilliger & Berendzen, 1999 ▶). The SIRAS map at 2.2 Å was subjected to maximum-likelihood density modification followed by autotracing using RESOLVE (Terwilliger, 1999 ▶). The model was built using XtalView (McRee, 1999 ▶) and refinement to a resolution of 2.2 Å was carried out using REFMAC5 (Murshudov et al., 1997 ▶) and CNS (Brünger et al., 1998 ▶). After several cycles of inspection of the 2F o − F c and F o − F c density maps, the model was rebuilt. The R factor and R free values for the final model were 19.9% and 24.3%, respectively (Table 1 ▶). The N-terminal region, including a His-tagged sequence (amino acids −19 to 0 in subunit A and −19 to −1 in subunit B), amino-acid residues 12–18 in both subunits and the C-terminal region (amino acids 267–270 in subunit A and 269–270 in subunit B) were disordered and not visible in the electron-density map. Because the side chain of residue 0 in subunit B had poor electron density, this residue was represented by Ala. The model geometry was analyzed using PROCHECK (Laskowski et al., 1993 ▶) and the final structure showed good geometry, with no Ramachandran outliers. Molecular-graphics figures were created using PyMOL (http://www.pymol.sourceforge.net/).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution data shell
| Native | Thimerosal | |
|---|---|---|
| Data collection | ||
| Wavelength () | 1.5418 | 1.5418 |
| Temperature (K) | 100 | 100 |
| Space group | P1 | P1 |
| Unit-cell parameters (, ) | a = 50.3, b = 55.2, c = 58.7, = 107.1, = 102.7, = 91.5 | a = 50.3, b = 55.3, c = 58.8, = 107.3, = 102.3, = 92.0 |
| Resolution range () | 502.2 (2.252.2) | 502.2 (2.252.2) |
| No. of measured reflections | 235335 | 229457 |
| No. of unique reflections | 29676 | 29687 |
| Redundancy | 7.9 | 7.7 |
| Completeness (%) | 96.0 (93.1) | 95.8 (91.2) |
| R merge † (%) | 3.9 (9.4) | 6.5 (17.0) |
| I/(I) | 24.1 (21.7) | 16.1 (9.5) |
| Refinement | ||
| Resolution range () | 37.62.2 | |
| R/R free ‡ (%) | 19.9/24.3 | |
| No. of protein atoms | 4151 | |
| No. of water molecules | 232 | |
| R.m.s.d. | ||
| Bond lengths () | 0.006 | |
| Bond angles () | 1.2 | |
| Average B factors (2) | 20.6 | |
| Ramachandran statistics (%) | ||
| Most favoured | 92.6 | |
| Additionally allowed | 7.2 | |
| Generously allowed | 0.2 | |
| Disallowed | 0 |
R
merge = 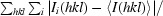
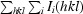 , where Ii(hkl) is the scaled intensity of the ith observation of reflection hkl and I(hkl) is the mean value; the summation is over all measurements.
, where Ii(hkl) is the scaled intensity of the ith observation of reflection hkl and I(hkl) is the mean value; the summation is over all measurements.
R free was calculated with randomly selected reflections (10%).
3. Results and discussion
3.1. Overall structure and structural homologues
TM0416p assembled as a dimer (Fig. 1 ▶ a) and the asymmetric unit consisted of two homologous subunits related by noncrystallographic twofold symmetry. Since the dimer is formed with a relatively small subunit interface, the homodimeric structure of this protein is not well supported by the structural evidence. Each monomer folded into an (α/β)8-barrel carrying four additional helical segments α2′, α3′, α4′ and α6′, which were inserted after β2, β3, β4 and β6, respectively (Fig. 1 ▶ b). When we submitted our model of the TM0416p monomer to the DALI server (Holm & Sander, 1998 ▶), seeking proteins with similar structures (as of 7 November 2008), the two proteins with the highest structural similarity were DTE from P. cichorii (PDB codes 2qul, 2qun, 2qum and 2ou4, with r.m.s.d.s between 2.0 and 2.1 Å) and DPE from A. tumefaciens (PDB codes 2hk1 and 2hk0, with r.m.s.d.s between 2.0 and 2.1 Å), as expected. The main-chain coordinates of the TM0416p monomer were similar to those of P. cichorii DTE and A. tumefaciens DPE, although we found a clear topological difference between TM0416p and the other two enzymes: the α8′ helix, which covers the active-site cleft in P. cichorii DTE and A. tumefaciens DPE, was absent in TM0416p (Fig. 2 ▶ a). Consequently, the substrate-binding pocket of TM0416p appears to be uniquely solvent-accessible (Fig. 2 ▶ b).
Figure 1.

(a) Ribbon representation of the TM0416p dimer, with subunits shown in different colours. (b) Subunit structure of TM0416p. α-Helices (numbered from α1 to α8) and β-strands (numbered from β1 to β8) are indicated in red and yellow, respectively.
Figure 2.

(a) Superposition of the monomeric structures of TM0416p (green), P. cichorii DTE (magenta) and A. tumefaciens DPE (cyan). The bound d-fructose molecule in P. cichorii DTE is shown as a stick model in yellow. (b) Representation of TM0416p in (a) as a surface model (grey).
3.2. Active site
The crystal structure of P. cichorii DTE in complex with Mn2+ and d-fructose (PDB code 2qun) has been determined (Yoshida et al., 2007 ▶). In this structure, d-fructose (the C4 epimer of d-tagatose) was held in the active-site cavity as an open form. Superposition of this structure onto the structure of TM0416p (1.3 Å r.m.s.d. for the Cα atoms of 200 residues) enabled us to compare the amino-acid residues involved in Mn2+ and substrate binding (Fig. 3 ▶ a). The Mn2+ ion is coordinated by Glu152, Asp185, His211 and Glu246 in P. cichorii DTE and these four residues are strictly conserved in TM0416p as Glu149, Asp182, His208 and Glu243, respectively. In addition, Glu158, His188 and Arg217 in P. cichorii DTE, which are responsible for the interaction between the enzyme and O1, O2 and O3 of d-fructose, are also conserved in TM0416p as Glu155, His185 and Arg214, respectively. In our model, O1 of the fructose is within hydrogen-bonding distance of the side chains of Glu155, His185 and Arg214. The Mn2+ ion is within 2.6 Å of the side chains of Glu149, Asp182, His208 and Glu243 and within 2.9 Å of O2 and O3. In addition, O2 and O3 are within hydrogen-bonding distance of the side chains of His185 and Glu149, respectively. In contrast, the amino-acid residues providing a hydrophobic environment around the substrate are completely different in the two enzymes. In P. cichorii DTE, Phe7, Trp15, Cys66, Leu108, Trp113 and Phe248 create a hydrophobic pocket around the 4-, 5- and 6-positions of d-fructose. Of these residues, Phe7, Cys66, Leu108, Trp113 and Phe248 are replaced by Val6, Gly68, Gly112, Leu113 and Leu245, respectively, in TM0416p. TM0416p does not contain a residue corresponding to Trp15.
Figure 3.

Stereographic close-up of the substrate-binding site. (a) The structure of P. cichorii DTE (magenta and red labels) is superimposed on that of TM0416p (green and black labels). The d-fructose molecule is shown as a stick model in yellow. Mn2+ is shown in orange. (b) Comparison of the active-site structures in P. cichorii DTE (magenta and red labels) and A. tumefaciens DPE (cyan and black labels). The d-fructose molecules are shown as stick models in yellow and green for P. cichorii DTE and A. tumefaciens DPE, respectively. Mn2+ ions are shown in orange and grey for P. cichorii DTE and A. tumefaciens DPE, respectively.
The crystal structure of A. tumefaciens DPE in complex with Mn2+ and d-fructose (PDB code 2hk1) has also been reported (Kim et al., 2006 ▶). Comparison of the substrate-binding structure of P. cichorii DTE with that of A. tumefaciens DPE revealed that the catalytic mechanisms for epimerization at the C3 position of the substrate are basically the same (Yoshida et al., 2007 ▶): the metal ion plays a pivotal role in catalysis by anchoring the bound d-fructose, while Glu152 and Glu246 (the residue numbers for P. cichorii DTE are given) carry out deprotonation/protonation at the C3 position. With respect to P. cichorii DTE, it has been proposed that a C3–O3 proton-exchange mechanism regulates the ionization state of the two Glu residues during the epimerization reaction (Yoshida et al., 2007 ▶). The key residues involved in the catalytic reaction are highly conserved between the two enzymes, but the residues that create a hydrophobic pocket around the 4-, 5- and 6-positions of d-fructose are less well conserved (Fig. 3 ▶ b). Trp15, Trp113 and Phe248 in P. cichorii DTE are conserved in A. tumefaciens DPE as Trp13, Trp112 and Phe246, respectively, but Phe7, Cys66 and Leu108 are replaced by Tyr6, Gly65 and Ala107, respectively. Because P. cichorii DTE exhibits a broader substrate specificity than A. tumefaciens DPE, it has been proposed that the structural differences in the hydrophobic pocket might affect substrate recognition at the 4-, 5- and 6-positions and that P. cichorii DTE only loosely recognizes substrates in this region (Yoshida et al., 2007 ▶).
In view of the crystal structure of TM0416p, the strict conservation of the key residues involved in catalysis supports the idea that TM0416p catalyzes the epimerization reaction in a manner similar to P. cichorii DTE and A. tumefaciens DPE: the two Glu residues, Glu149 and Glu243 in TM0416p, may be involved in deprotonation/protonation of the substrate. However, the clear difference observed in the hydrophobic pocket around the substrate as well as the presence of a unique solvent-accessible active site suggests that the substrate specificity of TM0416p differs substantially from those of other DTE-family enzymes. However, at this stage the true function of TM0416p remains unclear. Based on a whole-genome cDNA microarray analysis, it has been proposed that TM0416p is involved in myo-inositol degradation during exopolysaccharide production in T. maritima (Johnson et al., 2005 ▶). Our results may provide critical information for probing the putative substrate of TM0416p.
Supplementary Material
PDB reference: d-tagatose 3-epimerase-related protein, 2zvr, r2zvrsf
References
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Granström, T. B., Takata, G., Tokuda, M. & Izumori, K. (2004). J. Biosci. Bioeng. 97, 89–94. [DOI] [PubMed]
- Holm, L. & Sander, C. (1998). Nucleic Acids Res. 26, 316–319. [DOI] [PMC free article] [PubMed]
- Johnson, M. R., Montero, C. I., Conners, S. B., Shockley, K. R., Bridger, S. L. & Kelly, R. M. (2005). Mol. Microbiol. 55, 664–674. [DOI] [PubMed]
- Kim, K., Kim, H. J., Oh, D. K., Cha, S. S. & Rhee, S. (2006). J. Mol. Biol. 361, 920–931. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lee, D. W., Jang, H. J., Choe, E. A., Kim, B. C., Lee, S. J., Kim, S. B., Hong, Y. H. & Pyun, Y. R. (2004). Appl. Environ. Microbiol. 70, 1397–1404. [DOI] [PMC free article] [PubMed]
- Levin, G. V. (2002). J. Med. Food, 5, 23–36. [DOI] [PubMed]
- Livesey, G. & Brown, J. C. (1996). J. Nutr. 126, 1601–1609. [DOI] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Takata, M. K., Yamaguchi, F., Nakanose, K., Watanabe, Y., Hatano, N., Tsukamoto, I., Nagata, M., Izumori, K. & Tokuda, M. (2005). J. Biosci. Bioeng. 100, 511–516. [DOI] [PubMed]
- Takeshita, K., Suga, A., Takada, G. & Izumori, K. (2000). J. Biosci. Bioeng. 90, 453–455. [DOI] [PubMed]
- Terwilliger, T. C. (1999). Acta Cryst. D55, 1863–1871. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Yoshida, H., Yamada, M., Nishitani, T., Takada, G., Izumori, K. & Kamitori, S. (2007). J. Mol. Biol. 374, 443–453. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: d-tagatose 3-epimerase-related protein, 2zvr, r2zvrsf