

The site-specific unglycosylated mutant of metabotropic glutamate receptor 3 extracellular domain has been overexpressed, purified, and crystallized. A complete data set has been collected to 2.35 Å resolution.
Keywords: metabotropic glutamate receptors, extracellular domains
Abstract
Metabotropic glutamate receptors (mGluRs) are involved in the regulation of many physiological and pathological processes in the central nervous system. The extracellular domain (ECD) of mGluR subtype 3 (mGluR3) was produced using the baculovirus expression system and purified from the culture medium. However, the recombinant protein showed heterogeneity in molecular weight on SDS–PAGE analysis. It was found that the unglycosylation of Asn414 significantly reduced the heterogeneity. Consequently, three site-specifically unglycosylated mutant proteins of mGluR3 ECD, replacing Asn414 only or replacing Asn414 in combination with other glycosylation sites, were successfully crystallized in the presence of l-glutamate. Among them, crystals of the N414/439Q mutant diffracted X-rays to 2.35 Å resolution using synchrotron radiation. The crystal belonged to the monoclinic space group P21, with unit-cell parameters a = 84.0, b = 97.5, c = 108.1 Å, β = 93.0°. Assuming the presence of two protomers per crystallographic asymmetric unit, the Matthews coefficient V M was calculated to be 3.5 Å3 Da−1 and the solvent content was 65%.
1. Introduction
Glutamate is a major excitatory neurotransmitter in the central nervous system. There are two major categories of receptors that respond to glutamate: ionotropic glutamate receptors and metabotropic glutamate receptors (mGluRs). To date, eight subtypes of mGluR have been discovered. They are classified into three subgroups (groups I, II and III) according to sequence homology, associated second messenger systems and pharmacological properties (Nakanishi & Masu, 1994 ▶; Hollmann & Heinemann, 1994 ▶).
mGluRs are class C G-protein-coupled receptors (GPCRs) that have been implicated in major neurological disorders such as anxiety, depression, addiction, schizophrenia and pain (Holden, 2003 ▶). The various subtypes of the receptors exhibit distinct but partially overlapping distributions and functions in the brain. The group II mGluRs are located at both presynaptic and postsynaptic sites in the central nervous system (Tamaru et al., 2001 ▶). Three-dimensional structures of the receptor could provide valuable information for the design of group-specific drugs. For example, the subtype 3 receptor (mGluR3) of group II has been suggested to be a potential target for drug development against schizophrenia (Corti et al., 2007 ▶; Patil et al., 2007 ▶).
The receptor molecule consists of three domains: the N-terminal extracellular, transmembrane and C-terminal cytoplasmic domains (Pin et al., 2003 ▶). The N-terminal extracellular domain (ECD) is divided into ligand-binding and cysteine-rich subdomains. Crystal structures of the ligand-binding subdomain (LBD) of the homodimeric mGluR subtype 1 (mGluR1, group I) with or without ligands have been reported previously (Kunishima et al., 2000 ▶; Tsuchiya et al., 2002 ▶). The structures revealed that the ‘active’ form was stabilized by glutamate binding, whereas the ‘resting’ conformation was fixed by antagonist binding. However, the structure of the cysteine-rich (CR) subdomain, which intervenes between the LBD and the transmembrane domain, is missing. This structural information is indispensable for the investigation of the mechanism of signal transduction across membranes. The CR subdomain contains nine cysteine residues, which are strictly conserved among the class C GPCRs, with the exception of the GABAB receptor (Pin et al., 2003 ▶).
Here, we report the expression, purification, crystallization and preliminary X-ray crystallographic analysis of mGluR3 ECD (group II). The recombinant protein was expressed in insect cells using the baculovirus expression system and was purified from the culture supernatant. The ECD protein showed heterogeneity on SDS–PAGE analysis. Therefore, we designed site-specifically unglycosylated mutants in order to eliminate the heterogeneity. The purified mutant proteins were characterized by SDS–PAGE, gel-filtration chromatography and ligand-binding assay. One of the mutant proteins, N414/439Q, provided thin plate-shaped crystals that exhibited sufficient quality for crystal structure determination.
2. Materials and methods
2.1. Preparation of mGluR3 ECD and LBD
The mGluR3 ECD (Met1–Ala575) and LBD (Met1–Ser507) proteins from Rattus norvegicus (PIR accession No. P31422) were cloned by a PCR method using a full-length mGluR3 cDNA as a template. Several PCR primers included an extra oligonucleotide sequence encoding amino-acid residues LVPRGSHHHHHHHH to attach a thrombin-cleavable His-tag next to the C-terminus of each protein. The recombinant baculovirus was constructed using the Bac-to-Bac Baculovirus Expression system (Invitrogen) according to the manufacturer’s protocol. In order to produce the target protein, Trichoplusia ni BTI-TN-5B1-4 (High Five, Invitrogen) cells were cultured in a monolayer using Express Five serum-free medium (Invitrogen) supplemented with 16.5 mM l-glutamine. Baculovirus infection was performed as described previously (Tsuji et al., 2000 ▶; Suzuki et al., 2004 ▶). The culture medium into which the target protein was successfully secreted was collected 4–5 d after inoculation.
The conditioned medium, including protease inhibitors (5 µM E-64 and 1 mM phenylmethylsulfonyl fluoride), was filtered using a 0.22 µm membrane. The filtrate was loaded onto Ni–NTA agarose (Qiagen) packed in an XK26 column (GE Healthcare) equilibrated with phosphate-buffered saline (PBS; 10 mM phosphate, 2.7 mM potassium chloride and 137 mM sodium chloride, pH 7.4). After washing with 40 mM imidazole in PBS, the protein bound to the beads was eluted with a linear gradient of 40–300 mM imidazole in PBS. Aliquots of the eluate were analyzed by SDS–PAGE (4–20% gel). Positively stained fractions were digested by thrombin (Sigma) to remove the C-terminal His-tag. The nontagged protein was extracted by collecting the flowthrough fractions of an Ni2+-charged HiTrap Chelating HP column (GE Healthcare). The pooled fractions were applied onto a HiTrap Q column (GE Healthcare) equilibrated with 20 mM Tris–HCl pH 8.8 and eluted with a linear NaCl gradient of 20–1000 mM in the Tris–HCl buffer. The eluate was concentrated to 5–7 mg ml−1 for crystallization using an Amicon Ultra centrifugal filter unit (30 kDa molecular-weight cutoff membrane, Millipore). The purity of the proteins was analyzed by SDS–PAGE (4–20% gel). In order to confirm the N-terminal amino acid of the secreted mGluR3 ECD, the purified proteins were subjected to automated Edman degradation using a Procise model 492 protein sequencer (Applied Biosystems).
2.2. Preparation of the site-specifically unglycosylated mutant proteins
To produce the site-specifically unglycosylated mutants of LBD, each asparagine residue in the potential N-glycosylation sites was genetically replaced with glutamine using a PCR-based method. The four mutant LBDs (N209Q, N292Q, N414Q and N439Q), secreted into the culture medium of High Five cells, were purified by Ni-affinity chromatography.
In the case of the unglycosylated mutants of ECD, the amino-acid substitution at the N-glycosylation sites was introduced independently or in combinations of two. The mutant proteins, N414Q, N292/414Q and N414/439Q, were also secreted into the culture supernatant of High Five cells infected with the corresponding baculovirus. The purification procedure was the same as that for wild-type ECD.
2.3. Analysis of the oligomerization state
The formation of the interprotomer disulfide bridge in the mGluR3 ECD proteins was analyzed by SDS–PAGE and Western blotting analysis. SDS–PAGE was performed with a 4–20% linear gradient polyacrylamide gel (Daiichi Pure Chemicals) under reduced and nonreduced conditions followed by silver or Coomassie Brilliant Blue staining. For Western blotting, an anti-penta-His antibody (Qiagen) was used as a primary antibody. The molecular-weight standards were purchased from Daiichi Pure Chemicals for SDS–PAGE and from BioRad for Western blotting.
The oligomerization state of the N414/439Q mutant protein was further investigated by size-exclusion chromatography and chemical cross-linking. The overall molecular size was estimated using a gel-filtration column (Superdex 200 PC 3.2/30, GE Healthcare) equilibrated with PBS (flow rate: 30 µl min−1). The cross-linking reaction of the protein was carried out in the presence of 1.2 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, Pierce) at 298 K for 30 or 120 min. The cross-linked proteins were analyzed by SDS–PAGE (4–20% gel) and silver staining.
2.4. Binding assay
The ligand-binding assays were performed using the polyethylene glycol (PEG) precipitation method (Tsuji et al., 2000 ▶; Suzuki et al., 2004 ▶) with minor modifications. (2S)-2-Amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl)propanoic acid (LY341495), which is a highly potent and selective antagonist for the group II mGluRs (mGluR2 and mGluR3), was used for the displacement assay. Briefly, 1.5 pmol 3H-labelled LY341495 ([3H]-LY341495, 1.35 TBq mmol−1, Tocris), 0.8 µg protein and various amounts of ligands were mixed in 150 µl binding buffer (40 mM HEPES pH 7.5, 2.5 mM CaCl2). After incubation at 277 K for 1 h, PEG 6000 and γ-globulin were added to the mixture to final concentrations of 15%(w/v) and 3 mg ml−1, respectively. Vortexing and centrifugation of the solution then precipitated the protein which bound the ligand. The precipitate was washed twice with 1 ml binding buffer containing 8%(w/v) PEG 6000 and was dissolved in 1 ml water. After the addition of 14 ml Clearsol II (Nacalai Tesque), the radioactivity was counted using a scintillation counter. The binding data were analyzed using Prism 3 (Graphpad Software). The displacement curve was fitted to a single-site binding model to estimate the K i value.
2.5. Crystallization
The initial crystallization trial was performed using the hanging-drop vapour-diffusion method in 24-well VDX greased plates (Hampton Research) with Grid Screen PEG 6000, Grid Screen Ammonium Sulfate and Crystal Screen Lite (Hampton Research). All of the drops were prepared by mixing 1 µl protein solution (5–7 mg ml−1 in 20 mM Tris–HCl buffer pH 8.8 including approximately 0.3 M NaCl) with 1 µl reservoir solution. Protein solution with 1 mM glutamate was also examined for co-crystallization. Each drop was equilibrated against 0.4 ml reservoir solution at a temperature of 293 K.
2.6. Data collection
The X-ray diffraction data were collected under a nitrogen-gas stream at 100 K and recorded on a CCD (charge-coupled device) detector using synchrotron radiation at SPring-8 (Hyogo, Japan). Protein crystals were cryoprotected by soaking them briefly in a buffer containing 26–28%(v/v) glycerol, 2.5–5.0%(w/v) PEG 6000, 0.2 M NH4H2PO4 pH 4.9–5.1 and 0.3 M NaCl prior to flash-cooling in a boiled-off N2 stream. The data-collection parameters are listed in Table 1 ▶.
Table 1. Preliminary crystallographic data and data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 84.0, b = 97.5, c = 108.1, β = 93.0 |
| No. of protomers per ASU | 2 |
| X-ray source | BL41XU, SPring-8 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.0000 |
| Oscillation range (°) | 1.0 |
| Crystal-to-detector distance (mm) | 320 |
| No. of frames | 180 |
| Resolution range (Å) | 100–2.35 (2.48–2.35) |
| Observed reflections | 269513 |
| Unique reflections | 72267 |
| Completeness (%) | 99.9 (100) |
| Multiplicity | 3.7 (3.8) |
| Mean I/σ(I) | 6.9 (1.4) |
| Rmerge† | 0.067 (0.520) |
R
merge = 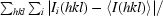
 , where 〈I(hkl)〉 is the mean intensity of symmetry-equivalent reflections.
, where 〈I(hkl)〉 is the mean intensity of symmetry-equivalent reflections.
3. Results and discussion
3.1. Preparation of the mGluR3 ECD proteins
The mGluR3 ECD proteins were expressed in High Five cells and successfully secreted into the culture medium. Our results contrast with the previous attempts by Yao et al. (2004 ▶) in which the mGluR3 ECD could not be secreted from HEK cells. The proteins were purified by a combination of Ni-affinity and anion-exchange chromatography. On analysis by Western blotting, the His-tagged wild-type ECD was heterogeneous (Fig. 1 ▶ a, lane 1). At least two bands were present in close vicinity to each other under reduced conditions. The N-terminal amino-acid sequence of the ECD protein was uniform as confirmed by Edman degradation. The C-terminal portion was also intact because of the presence of the His-tag detected by the primary antibody. Considering these facts, the multiple bands appeared as a result of modification of the ECD protein.
Figure 1.

(a) Western blotting analysis of the mGluR3 ECD proteins. The samples were loaded in the presence (lanes 1 and 2) and absence (lanes 3 and 4) of DTT. The positions of the molecular-weight markers are shown on the left (kDa). Lanes 1 and 3, mGluR3 ECD wild type with a His-tag; lanes 2 and 4, N414Q mutant with a His-tag. (b) SDS–PAGE analysis of the wild-type and mutant LBDs. These proteins were analyzed under reduced conditions. The gel was stained with silver nitrate. Lane 1, wild type; lane 2, N209Q; lane 3, N292Q; lane 4, N414Q; lane 5, N439Q. (c) The site-specifically unglycosylated mutant proteins of ECD without a His-tag were analyzed by SDS–PAGE. The purified samples were loaded in the presence (lanes 1–3) and absence (lanes 4–6) of DTT. The gel was stained with Coomassie Brilliant Blue. The positions of the molecular-weight markers are shown on the left (kDa). Lanes 1 and 4, N414Q; lanes 2 and 5, N292/414Q; lanes 3 and 6, N414/439Q.
We hypothesized that the attachment of variable sugar chains was the reason for the heterogeneity. In order to eliminate the glycosylation sites, we constructed several mutant proteins of mGluR3 LBD. Four asparagine residues (Asn209, Asn292, Asn414 and Asn439) were found to be putative N-glycosylation sites (-Asn-X-Thr/Ser-) in the mGluR3 LBD sequence. In fact, Asn414 was responsible for the heterogeneity. Whereas the N414Q mutant protein yielded a single band, multiple bands were observed for the other mutants (Fig. 1 ▶ b). In addition, the N209Q mutation caused an insignificant alteration in the SDS–PAGE analysis, suggesting that the oligosaccharide moiety at Asn209 might be small.
We then designed mutant ECD proteins with unglycosylated residues at site 414. In addition to this site, two other sites (292 and 439) were also considered because the oligosaccharides attached to these sites would be large. In total, three types of mutant protein (N414Q, N292/414Q and N414/439Q) were examined. These mutant proteins were secreted and purified in a similar manner to the wild type. As shown in Fig. 1 ▶(c), all of the mutants were confirmed as a single band on SDS–PAGE.
3.2. Characterization of the N414/439Q mutant of ECD
The N414/439Q protein was the sole mutant for which the crystal gave reasonable X-ray diffraction (see §3.3). Thus, we characterized this mutant protein as a strong candidate for structure determination.
The N-terminal amino acids of the wild-type and N414/439Q proteins were confirmed using a protein sequencer. The predicted N-terminus of the secreted protein was at Leu23, according to the PIR database (accession No. P31422). However, Asp25 was the first amino acid of the two proteins. As a result, the theoretical molecular weight and isoelectric point of the N414/439Q protein with the additional four residues (LVPR) at the C-terminus were 63 047 and 5.5, respectively, as calculated using ProtParam (Gasteiger et al., 2005 ▶). The former value is consistent with the result of SDS–PAGE analysis under reduced conditions (Fig. 1 ▶ c, lane 3).
Next, we investigated the oligomerization state of the N414/439Q protein. LBDs of mGluR1 and mGluR7 formed homodimers in which the two protomers were connected by a disulfide bond (Tsuji et al., 2000 ▶; Muto et al., 2007a ▶). SDS–PAGE analysis of the mGluR3 ECD protein indicated that a band appeared at a position corresponding to twice the molecular weight under nonreduced conditions (Fig. 1 ▶ c, lanes 1–3). The corresponding mutant LBD proteins also gave similar results (data not shown). Size-exclusion chromatography showed that the N414/439Q protein eluted as an oligomer rather than a monomer (Fig. 2 ▶ a). These results indicated that the N414/439Q protein of mGluR3 could form an interprotomer disulfide bond, as demonstrated for the LBDs of mGluR1 and mGluR7.
Figure 2.

(a) Size-exclusion chromatography of the N414/439Q mutant protein of mGluR3 ECD. The elution positions of the marker proteins are shown by arrowheads with molecular weights in kDa. The estimated molecular weight is 205 kDa. (b) Chemical cross-linking of the N414/439Q protein. After reaction with EDC, the aliquots were analyzed by SDS–PAGE under reduced conditions. Lane 1, without EDC; lane 2, with EDC for 30 min; lane 3, with EDC for 2 h. The arrowheads indicate the positions of the native/cross-linked proteins.
We also confirmed oligomer formation of mGluR3 ECD by a cross-linking experiment. Fig. 2 ▶(b) displays the results of a cross-linking experiment using the N414/439Q protein. The cross-linked protein appeared at positions corresponding to the molecular weight of the homodimer (126 kDa) and homotetramer (252 kDa). These two bands intensified when the reaction time was extended from 30 min to 2 h. These results suggested the possibility of forming a homodimer as well as a homotetramer, although it is uncertain whether the formation of the tetramer has biological significance. We speculate that the dimer–dimer interaction for tetramerization was too weak to be detected by size-exclusion chromatographic analysis.
In addition, we investigated the ligand-binding ability of the N414/439Q mutant of ECD using the PEG-precipitation method (Tsuji et al., 2000 ▶; Suzuki et al., 2004 ▶) with [3H]-LY341495. The K i values estimated from the binding data (Fig. 3 ▶) are summarized in Table 2 ▶. The rank order of the ligand potency was consistent with a previous report (Wright et al., 2001 ▶). This finding indicated that the N414/439Q protein produced by insect cells folded correctly and that the modification of the protein did not influence its ligand-binding ability.
Figure 3.

Displacement of [3H]-LY341495 binding to the mGluR3 N414/439Q ECD mutant by the ligands. The data are plotted with the following symbols: red squares, LY341495; yellow triangles, DCGIV [(2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine]; green inverted triangles, l-glutamate; crosses, 2R,4R-APDC [(2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate)]; blue diamonds, 1S,3S-ACPD [(1S,3S)-1-aminocyclopentane-1,3-dicarboxylic acid]; purple circles, 1S,3R-ACPD [(1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid]. Each point shows the mean ± standard deviation. The continuous lines indicate the curves representing the results of the corresponding sigmoidal fitting.
Table 2. K i values for various ligands to inhibit the binding of [3H]-LY341495 to the N414/439Q mutant of mGluR3 ECD.
| Ligand | Ki (µM) | 95% confidence interval (µM) |
|---|---|---|
| LY341495 | 0.120 | 0.0909–0.159 |
| DCGIV | 0.784 | 0.658–0.934 |
| L-Glutamate | 1.36 | 1.12–1.65 |
| 2R,4R-APDC | 24.1 | 18.9–30.8 |
| 1S,3S-ACPD | 110 | 95.7–126 |
| 1S,3R-ACPD | 146 | 114–187 |
3.3. Crystallization
The three mutant proteins of mGluR3 ECD were crystallized by the hanging-drop vapour-diffusion method at 293 K. Crystals of the three mutant proteins were obtained using PEG as a precipitant. Both the N414Q and N292/414Q proteins with glutamic acid yielded crystals of stacked laminar shape (Fig. 2 ▶ a) that diffracted X-rays to very limited resolution. In contrast, crystals of the N414/439Q protein grew into thin isolated plates in the presence of l-glutamate (Fig. 4 ▶ b). These crystals appeared to be suitable for structural analysis and the X-ray diffraction reached 2.35 Å resolution. Our results demonstrated that the elimination of the sugar chains on Asn414 and Asn439 significantly improved the quality of the crystal.
Figure 4.

Crystals of the mGluR3 ECD mutants. (a) The N414Q mutant. The maximum dimensions are 0.15 × 0.1 × 0.005 mm. (b) The N414/439Q mutant. The maximum dimensions are 0.25 × 0.1 × 0.03 mm.
Crystals of the N414/439Q protein were obtained from seven conditions: A4 and B4 of Grid Screen PEG 6000, and 9, 15, 22, 40 and 41 of Crystal Screen Lite. All conditions contained 5–15%(w/v) PEG as a precipitant in the pH range 5.6–8.5. However, these crystals were too thin and fragile to handle for X-ray data collection. Subsequent refinement of the buffer conditions gave optimal crystallization conditions consisting of 2.5–5.0%(w/v) PEG 6000 and 0.2 M NH4H2PO4 pH 4.9–5.1. The protein solution also included 1 mM l-glutamate. The crystals appeared within a few days and the fully grown crystals had dimensions of 0.25 × 0.1 × 0.03 mm (Fig. 4 ▶ b).
3.4. Data collection and molecular-replacement model
For X-ray data collection, single crystals of the N414/439Q protein picked up from a droplet using a nylon loop (Hampton Research) were transferred into a cryoprotectant solution [26–28%(v/v) glycerol, 2.5–5.0%(w/v) PEG 6000, 0.2 M NH4H2PO4 pH 4.9–5.1 and 0.3 M NaCl] and placed directly into a cold nitrogen-gas stream at 100 K. Diffraction data were obtained in the resolution range 100–2.35 Å (Fig. 5 ▶) and were processed with MOSFLM (Leslie, 2006 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶). The crystals belonged to the monoclinic space group P21, with unit-cell parameters a = 84.0, b = 97.5, c = 108.1 Å, β = 93.0°. The asymmetric unit contained two protomers with a solvent content of 65% and a crystal volume per protein weight V M of 3.5 Å3 Da−1. These two values are within the ranges for ordinary protein crystals (Matthews, 1968 ▶). The preliminary crystallographic data are summarized in Table 1 ▶.
Figure 5.

Diffraction pattern of the crystal of the mGluR3 ECD N414/439Q mutant. (a) A typical diffraction image obtained using the oscillation method. The concentric circles indicate resolution ranges of 8.7, 4.4, 2.9 and 2.2 Å. (b) An enlarged view of the region enclosed by a dotted line in (a).
Primary phasing of the diffraction data was performed by the molecular-replacement method using the program AMoRe (Navaza, 1994 ▶) with the atomic coordinates of mGluR1 LBD (PDB code 1ewk; Kunishima et al., 2000 ▶) as the search probe. The initial electron-density map clearly indicated the positions of the CR subdomains, which were absent in the probe. The subsequent structure determination and biological significance of the structure have been described elsewhere (Muto et al., 2007b ▶).
We crystallized the unglycosylated mutant of mGluR3 ECD. The wild-type ECD protein contains four potential N-glycosylation sites: Asn209, Asn292, Asn414 and Asn439. Because glycosylation of these residues produces heterogeneity, mutation at these sites is important to obtain a form of mGluR3 ECD suitable for crystallization. In fact, only the N414/439Q mutant gave suitable crystals for structure determination. The flexibility and heterogeneity of N-glycans attached to protein molecules can hinder crystallization because these properties are unfavourable for the establishment of rigid crystal packing. Several reports involving protein unglycosylation for crystallization have been published, such as nonglycosylated Equ c 1 produced by Escherichia coli (Grégoire et al., 1999 ▶), site-specific unglycosylated mutants of BST-1/CD157 (Yamamoto-Katayama et al., 2001 ▶) and CD58 with Endo H-treated N-glycans (Butters et al., 1999 ▶). Nevertheless, N-glycosylation of protein molecules is important for their secretion into extracellular space (Muto et al., 2000 ▶). A trade-off between crystallizability and amount of secretion is one of the troublesome aspects in crystallizing glycoproteins. Although we adopted a trial-and-error approach, a theoretical strategy for the elimination of glycosylation sites is awaited.
Acknowledgments
The authors would like to thank Drs N. Shimizu and T. Oyama for help during X-ray diffraction data collection. We are very grateful to Ms R. Sata, Ms H. Kishi and Mr T. Tomura for assistance with technical support. We thank Dr S. Nakanishi for a gift of mGluR3 cDNA. The synchrotron X-ray experiment at SPring-8 was performed with the approval of the Program Advisory Committee of the Japan Synchrotron Radiation Institute (Proposal No. 2005A0339-NL1-np). This work was partly supported by a research grant endorsed by the New Energy and Industrial Technology Development Organization (NEDO).
References
- Butters, T. D., Sparks, L. M., Harlos, K., Ikemizu, S., Stuart, D. I., Jones, E. Y. & Davis, S. J. (1999). Protein Sci.8, 1696–1701. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Corti, C., Crepaldi, L., Mion, S., Roth, A. L., Xuereb, J. H. & Ferraguti, F. (2007). Biol. Psychiatry, 62, 747–755. [DOI] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Grégoire, C., Tavares, G. A., Lorenzo, H. K., Dandeu, J.-P., David, B. & Alzari, P. M. (1999). Acta Cryst. D55, 880–882. [DOI] [PubMed]
- Holden, C. (2003). Science, 300, 1866–1868. [DOI] [PubMed]
- Hollmann, M. & Heinemann, S. (1994). Annu. Rev. Neurosci.17, 31–108. [DOI] [PubMed]
- Kunishima, N., Shimada, Y., Tsuji, Y., Sato, T., Yamamoto, M., Kumasaka, T., Nakanishi, S., Jingami, H. & Morikawa, K. (2000). Nature (London), 407, 971–977. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Muto, T., Feese, M. D., Shimada, Y., Kudou, Y., Okamoto, T., Ozawa, T., Tahara, T., Ohashi, H., Ogami, K., Kato, T., Miyazaki, H. & Kuroki, R. (2000). J. Biol. Chem.275, 12090–12094. [DOI] [PubMed]
- Muto, T., Tsuchiya, D., Morikawa, K. & Jingami, H. (2007a). Acta Cryst. F63, 627–630. [DOI] [PMC free article] [PubMed]
- Muto, T., Tsuchiya, D., Morikawa, K. & Jingami, H. (2007b). Proc. Natl Acad. Sci. USA, 104, 3759–3764. [DOI] [PMC free article] [PubMed]
- Nakanishi, S. & Masu, M. (1994). Annu. Rev. Biophys. Biomol. Struct.23, 319–348. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Patil, S. T. et al. (2007). Nature Med.13, 1102–1107. [DOI] [PubMed]
- Pin, J.-P., Galvez, T. & Prézeau, L. (2003). Pharmacol. Ther.98, 325–354. [DOI] [PubMed]
- Suzuki, Y., Moriyoshi, E., Tsuchiya, D. & Jingami, H. (2004). J. Biol. Chem.279, 35526–35534. [DOI] [PubMed]
- Tamaru, Y., Nomura, S., Mizuno, N. & Shigemoto, R. (2001). Neuroscience, 106, 481–503. [DOI] [PubMed]
- Tsuchiya, D., Kunishima, N., Kamiya, N., Jingami, H. & Morikawa, K. (2002). Proc. Natl Acad. Sci. USA, 99, 2660–2665. [DOI] [PMC free article] [PubMed]
- Tsuji, Y., Shimada, Y., Takeshita, T., Kajimura, N., Nomura, S., Sekiyama, N., Otomo, J., Usukura, J., Nakanishi, S. & Jingami, H. (2000). J. Biol. Chem.275, 28144–28151. [DOI] [PubMed]
- Wright, R. A., Arnold, M. B., Wheeler, W. J., Ornstein, P. L. & Schoepp, D. D. (2001). J. Pharmacol. Exp. Ther.298, 453–460. [PubMed]
- Yamamoto-Katayama, S., Sato, A., Ariyoshi, M., Suyama, M., Ishihara, K., Hirano, T., Nakamura, H., Morikawa, K. & Jingami, H. (2001). Biochem. J.357, 385–392. [DOI] [PMC free article] [PubMed]
- Yao, Y., Koo, J. C., Wells, J. W. & Hampson, D. R. (2004). Biochem. Biophys. Res. Commun.319, 622–628. [DOI] [PubMed]