

Wild-type and mutant recombinant human transforming growth factor β-induced protein (TGFBIp) were cloned, purified and crystallized. Preliminary X-ray crystallography data were obtained from wild-type TGFBIp.
Keywords: corneal dystrophy, recombinant protein
Abstract
Transforming growth factor β-induced protein (TGFBIp) has been linked to several corneal dystrophies as certain point mutations in the protein may give rise to a progressive accumulation of insoluble protein material in the human cornea. Little is known about the biological functions of this extracellular protein, which is expressed in various tissues throughout the human body. However, it has been found to interact with a number of extracellular matrix macromolecules such as collagens and proteoglycans. Structural information about TGFBIp might prove to be a valuable tool in the elucidation of its function and its role in corneal dystrophies caused by mutations in the TGFBI gene. A simple method for the purification of wild-type and mutant forms of recombinant human TGFBIp from human cells under native conditions is presented here. Moreover, the crystallization and preliminary X-ray analysis of TGFBIp are reported.
1. Introduction
Transforming growth factor β-induced protein (TGFBIp) is a 75 kDa 683-amino-acid residue protein (SwissProt sequence database access code Q15582) that is encoded by the TGFBI gene. TGFBIp is composed of an N-terminal secretion-signal sequence (residues 1–23), an N-terminal Cys-rich region (EMI) and four consecutive repeats that show some sequence similarity to Drosophila fasciclin-1 (FAS1; Clout & Hohenester, 2003 ▶). In addition, the ‘classic’ integrin-recognition motif, Arg-Gly-Asp (RGD), is located close to the C-terminus of TGFBIp (residues 642–644).
Size-exclusion chromatography and mass spectrometry show that TGFBIp exists as a monomer in solution, with no post-translational modifications, when purified from human corneas (Andersen et al., 2004 ▶). Detailed structural information is not known for full-length TGFBIp, but the three-dimensional structure of the Drosophila fasciclin-1 FAS1 domain has been solved (Clout et al., 2003 ▶) and revealed an overall globular shape containing seven β-strands and five α-helices.
Two amino-acid residue stretches within FAS1 domains (H1 and H2) are highly conserved in proteins from both eukaryotes and prokaryotes (Kawamoto et al., 1998 ▶). The sequence identity between the FAS1 domains of TGFBIp and fasciclin-1 is approximately 20% and accordingly it has been suggested that the overall fold might be similar (Clout & Hohenester, 2003 ▶). Apart from TGFBIp, only three other human proteins are known to contain FAS1 domains: periostin (Horiuchi et al., 1999 ▶; Takeshita et al., 1993 ▶), stabilin-1 and stabilin-2 (Adachi & Tsujimoto, 2002 ▶; Politz et al., 2002 ▶; Tamura et al., 2003 ▶). FAS1-containing proteins are known to be involved in cell adhesion and several copies of these domains are often found within each protein (Hortsch & Goodman, 1990 ▶), as is the case for TGFBIp. Extracellular TGFBIp is found in a wide range of tissues, including the cornea (Escribano et al., 1994 ▶; Rawe et al., 1997 ▶), skin (LeBaron et al., 1995 ▶), bone (Kitahama et al., 2000 ▶), tendon (Ferguson et al., 2003 ▶; Ohno et al., 2002 ▶), endometrium (Carson et al., 2002 ▶) and kidney (Lee et al., 2003 ▶). The biological function of TGFBIp remains unclear, but more than 30 mutations in the TGFBI gene have been shown to cause protein aggregates in an array of different corneal dystrophies (Klintworth, 2003 ▶; Kannabiran & Klintworth, 2006 ▶). These autosomal dominant diseases are characterized by a progressive abnormal accumulation of protein in the corneal stroma, which eventually leads to visual impairment (Klintworth, 2003 ▶). The resulting phenotypes can be divided into four groups: granular corneal dystrophies (GCDs), which are characterized by crystalloid amorphous aggregates, lattice corneal dystrophy (LCD) type 1 and its variants, with amyloid deposition in the cornea, a combination of GCD deposits with amyloid deposition (GCD type II) and Thiel–Behnke corneal dystrophy (TBCD), in which curly fibres are deposited in the superficial cornea (Klintworth, 2003 ▶). Although TGFBIp has been shown to co-localize with protein deposits in TGFBI-linked corneal dystrophies (Streeten et al., 1999 ▶), the composition of the protein aggregates remains to be characterized in detail.
In the present study, we developed a mammalian expression system and a purification protocol to produce large amounts of wild-type recombinant human TGFBIp, as well as of the point mutants R124H, R124L and R124C that are known to cause GCD type II (Munier et al., 1997 ▶), GCD type III (Mashima et al., 1999 ▶) and LCD type I (Munier et al., 1997 ▶), respectively. In addition, the wild-type protein was crystallized and preliminary X-ray diffraction data were collected using synchrotron radiation.
2. Materials and methods
2.1. Materials
Unless otherwise stated, chemicals were purchased from Sigma–Aldrich (St Louis, Missouri, USA). SDS–PAGE molecular-weight standards were purchased from Bio-Rad Laboratories (Hercules, California, USA). Chromatographic protein purifications were performed using fast protein liquid chromatography (FPLC; GE Healthcare) or an ÄKTAprime (GE Healthcare) system.
2.2. Cloning and expression of TGFBIp
A cDNA clone encoding human TGFBIp (Invitrogen, Carlsbad, California, USA) was obtained from a human placenta cDNA library and directionally cloned into the NotI and EcoRV restriction sites of the pCMV-SPORT6 vector. High-level expression results from the constitutive CMV (cytomegalovirus) promoter. In order to produce the R124C, R124L and R124H mutants, three different nucleotide mutations were introduced (see Table 1 ▶). Each mutation was performed with two complementary mutation primers using the QuikChange XL site-directed mutagenesis kit from Stratagene (La Jolla, California, USA) and DpnI from New England Biolabs (Ipswich, Massachusetts, USA) according to the manufacturer’s instructions.
Table 1. Mutations introduced in TGFBIp.
Nucleotide numbering is according to entry NM_000358.
| Mutant | Primer sequence† | Changes introduced |
|---|---|---|
| R124C | CTCAGCTGTACACGGACTGCACGGAGAAGCTGAGG | C417 to T |
| R124H | CTCAGCTGTACACGGACCACACGGAGAAGCTGAGG | G418 to A |
| R124L | CTCAGCTGTACACGGACCTCACGGAGAAGCTGAGG | G418 to T |
Only the sequence of the forward primer is shown. The reverse mutation primer is complementary to the forward primer. Changes introduced are shown in bold italics.
The TGFBIp clones were used without further modification to express wild-type and mutant TGFBIp in the human embryonal kidney fibroblast cell line HEK293. Cells were grown to 60–70% confluency in 9 cm Petri dishes in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% bovine calf serum (BCS) for 24 h prior to transfection. To facilitate efficient transfection, the medium was supplemented with 25 µM chloroquin for 1 h before transfection by the calcium phosphate precipitation method (Luthman & Magnusson, 1983 ▶). The medium was changed 16 h after transfection and after another 8 h the cells were transferred to serum-free medium. The medium was harvested for protein purification from day 2 to day 4 after transfection.
2.3. Purification of wild-type and mutant TGFBIp
Cell-culture medium containing TGFBIp (900–1000 ml) was successively dialyzed against two volumes of 12 l buffer A (20 mM Tris–HCl pH 7.45) at 277 K and purified by affinity chromatography using a heparin Sepharose column (5 ml HiTrap Heparin HP column, GE Healthcare). The cell-culture medium was applied in batches of approximately 500 ml and eluted using a linear gradient of buffer A to buffer B (20 mM Tris–HCl pH 7.45 containing 1 M NaCl) at a flow rate of 2 ml min−1. The protein elution was monitored at 280 nm and 2 ml fractions were collected. Selected fractions were analyzed by reducing SDS–PAGE. Fractions containing TGFBIp were pooled and dialyzed against 2 × 2 l buffer A at 277 K. TGFBIp was purified from the resulting dialysate by anion-exchange chromatography (1 ml HiTrap Q HP or Mono Q, GE Healthcare) and eluted with a linear gradient from buffer A to buffer B. Selected fractions were analyzed by reducing SDS–PAGE and fractions containing TGFBIp were pooled and stored at 253 K.
All chromatographic steps were performed at room temperature (298 K) and all samples and buffers were filtered using 0.22 µm filtration units prior to chromatography.
2.4. SDS–PAGE
SDS–PAGE was performed using 5–15% gradient gels or 10% uniform gels (10 cm × 10 cm × 1.5 mm) using the glycine/2-amino-2-methyl-1,3-propanediol/HCl system (Bury, 1981 ▶). Unless otherwise stated, samples were diluted/dissolved in 50 mM Tris–HCl, 150 mM NaCl pH 7.45. Protein was visualized using Coomassie Brilliant Blue.
2.5. Mass-spectrometric analysis
To determine the molecular weight of purified TGFBIp, the protein was desalted and concentrated using custom-made micro-columns packed with Poros R1 in GelLoader pipette tips. The resin (1–3 mm in height) was equilibrated by flushing 10 µl 5% formic acid through the column and the sample was subsequently loaded and washed in the same buffer. The desalted sample was then eluted directly onto the MALDI target using 20 g l−1 sinapic acid dissolved in a solution containing 70% acetonitrile and 0.1% trifluoroacetic acid (TFA) in water. The MALDI–TOF MS analysis was performed on a Voyager-DE STR (Applied Biosystems, Foster City, California, USA) controlled by Data Explorer software (v.3.4.0.0 and v.4.4; Applied Biosystems). Operations were performed in linear and positive polarity mode. The instrument was externally calibrated prior to use. The spectrum was viewed in MoverZ (Genomic Solutions, Holliston, Massachusetts, USA).
2.6. N-terminal amino-acid sequence analysis
Purified TGFBIp was mixed with 0.1% TFA and applied onto a ProSorb membrane (Applied Biosystems) activated with methanol. The membrane was then washed twice with 1 ml 0.1% TFA and analyzed by automated Edman degradation in an Applied Biosystems model 477A/120A protein-sequencing system.
2.7. Crystallization of TGFBIp
Frozen protein samples were slowly thawed on ice and then pooled and concentrated to a final concentration of 3.9 mg ml−1 using a Centricon YM-30 (Amicon, Houston, Texas, USA). The concentrated protein was subjected to final purification by gel-filtration chromatography on a Superdex 200 column (GE Healthcare) equilibrated with buffer C (20 mM Tris, 50 mM NaCl pH 7.45). The sample purity was assessed by 10% SDS–PAGE. Pure and homogeneous samples were pooled and concentrated again with the same device to a final concentration of 3.3 mg ml−1. A standard set of commercially available crystallization screens from Hampton Research (Aliso Viejo, California, USA) were assayed using the sitting-drop (200 and 300 nl) vapour-diffusion method at 277 and 293 K at the Automated Crystallization Platform of the Barcelona Science Park–Molecular Biology Institute of Barcelona employing a Tecan robot and a Cartesian nanodrop dispenser robot and two-well MRC crystallization plates (Wilden/Innovadyne). The assayed volume ratios of protein solution to precipitant were 1:1 and 2:1.
3. Results and discussion
TGFBIp is involved in various inheritable corneal dystrophies, in which point mutations in the protein cause impaired vision owing to gradual accumulation of protein material in the cornea. By determining the three-dimensional structure of TGFBIp, it will be possible to predict structural motifs and binding sites for protein–protein interactions, which will provide insight into the general function of TGFBIp in the extracellular matrix. Knowing the structure of wild-type TGFBIp might also allow us to predict the structural changes caused by mutations in TGFBIp, which could provide valuable insight into aggregation mechanisms.
It is commonly known that proteins with a high propensity to aggregate can be difficult to purify under native conditions. The Arg124 mutants analyzed in this study have all been found to accumulate in the corneas of patients (Korvatska et al., 2000 ▶), indicating aggregation of the proteins in vivo.
However, the conditions used in our purification did not appear to cause the mutant protein to aggregate. Future experiments could therefore involve probing different cornea-specific factors, such as collagens, proteoglycans and glucosaminoglycans, for their ability to induce TGFBIp aggregation and fibrillate in vitro using thioflavin T fluorescence assays (Naiki et al., 1989 ▶).
3.1. Expression and purification of recombinant TGFBIp
Recombinant TGFBIp was expressed in HEK293 cells following transfection with the pCMV-SPORT6 vector containing full-length TGFBIp cDNA. Since TGFBIp is an extracellular protein, it accumulates in the cell medium, which was collected over a 3 d period and stored at 253 K until purification was initiated.
Heparin-affinity chromatography was used as a first step in the purification of recombinant TGFBIp (Ferguson et al., 2003 ▶). Dialyzed cell medium was applied onto a heparin Sepharose column and proteins were separated using a 0–1 M gradient of NaCl. SDS–PAGE analysis of the resulting fractions showed that TGFBIp eluted in fractions 23–27, corresponding to a salt concentration of about 250 mM (Fig. 1 ▶ a). Fractions containing TGFBIp were pooled and dialyzed for further purification. In the second purification step, proteins were separated using ion-exchange chromatography. The TGFBIp pool was applied onto the column, separated by a salt gradient as described above and analyzed by SDS–PAGE (Fig. 1 ▶ b). TGFBIp eluted as a broad peak between approximately 200 and 250 mM salt (fractions 22–26). These fractions were pooled and stored at 253 K. N-terminal amino-acid sequencing revealed the expected N-terminus at residue 24 (Gly-Pro-Ala-Lys…) and the molecular weight was determined to be 69 066 Da by mass spectrometry. These results indicate that purified TGFBIp is truncated between residues 655 and 656, which corresponds to a previously reported truncation in human tissue (Andersen et al., 2004 ▶). SDS–PAGE analysis of the final Arg124 mutant purification pools is shown in Fig. 2 ▶.
Figure 1.

Purification of rhTGFBIp using heparin-affinity chromatography. (a) Reducing SDS–PAGE of selected fractions separated by heparin-affinity chromatography. TGFBIp migrates as a clear band at approximately 65 kDa. Fractions 23–27 were pooled for further purification. (b) Reducing SDS–PAGE of selected fractions from anion-exchange chromatography. TGFBIp eluted around fraction No. 23 with a purity of approximately 95% (by visual estimation). Fractions 22–27 were pooled and used for crystallization. Fraction numbers are indicated at the top of the gels and molecular weights (in kDa) are indicated at the side.
Figure 2.
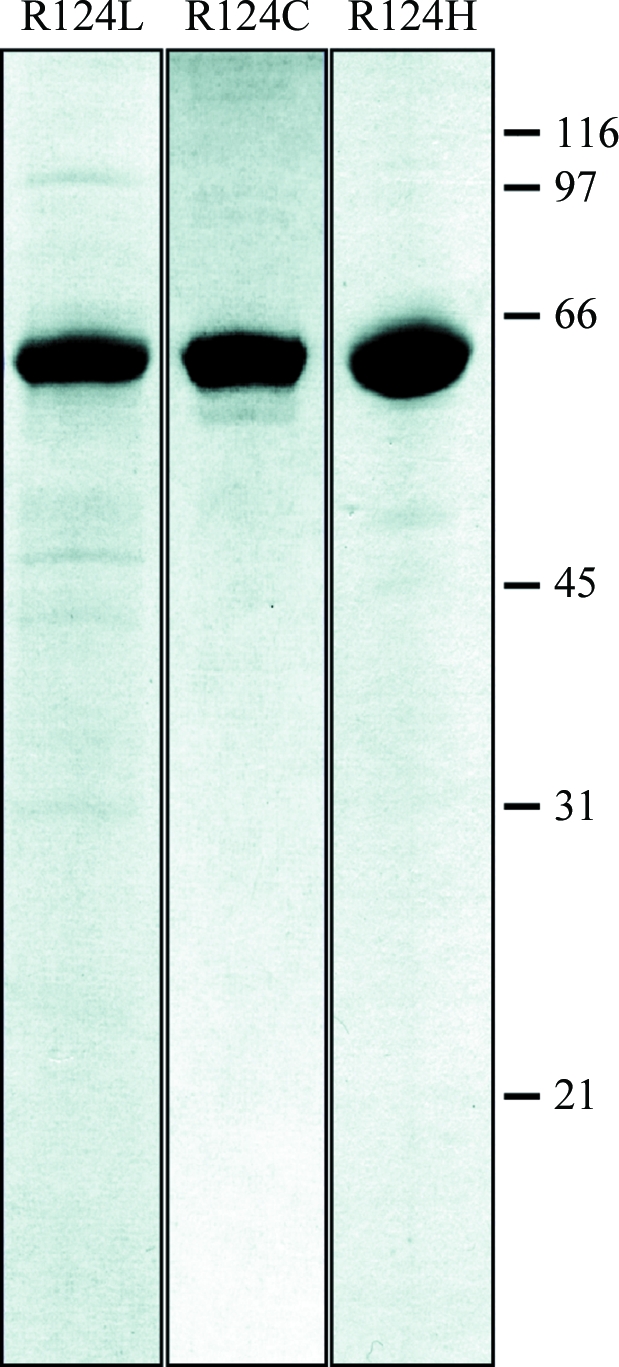
SDS–PAGE of purified TGFBIp mutants. The R124L, R124H and R124C mutants were purified as for wild-type TGFBIp. Final pools were separated by SDS–PAGE and visualized using Coomassie Brilliant Blue. molecular weights (in kDa) are indicated on the right. The purity was estimated to be greater than 90%.
The average yield from the purifications was approximately 1.2 mg of pure TGFBIp per litre of expression medium. For crystallization experiments, wild-type recombinant TGFBIp was concentrated to about 3.9 mg ml−1 and homogeneity of the protein was ensured by gel filtration. The final protein pool was concentrated to 3.3 mg ml−1.
3.2. Crystallization and preliminary X-ray diffraction data analysis.
Hexagonal crystals with maximal dimensions of 10 × 10 × 60 µm that were likely to belong to space group P61 (or enantiomorph) were obtained. These crystals resulted from drops containing 0.2 µl protein solution (3.3 mg ml−1 in buffer C) and 0.1 µl precipitating agent (0.1 M sodium acetate, 4% PEG 4000 pH 4.6) after 5 d at 293 K (Fig. 3 ▶ a) and are expected to contain two molecules in the asymmetric unit (V M = 2.5 Å3 Da−1; 51% solvent content; Matthews, 1968 ▶). Assessment of protein identity was performed by tryptic digestion and subsequent mass spectrometry of carefully washed and dissolved crystals (data not shown). Prior to diffraction analysis, a cryocooling protocol was established in order to protect crystals from radiation damage during data collection. This protocol consisted of initial stabilization in harvesting solution (4.8% PEG 4000, 0.1 M sodium acetate pH 4.6) followed by soaking in cryoprotective solution (4.8% PEG 4000, 0.1 M sodium acetate, 20% glycerol pH 4.6) just prior to flash-vitrification of the crystals in liquid nitrogen. Diffraction data (see Fig. 3 ▶ b) were collected at 100 K from a single crystal using a ADSC Q315R CCD detector on beamline ID29 of the European Synchrotron Radiation Facility (Grenoble, France). Data were processed to 3.25 Å resolution and scaled with XDS (Kabsch, 2001 ▶) and SCALA (Evans, 1993 ▶) from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶; see Table 2 ▶ for statistics of data collection and processing). Refinement of crystallization conditions to obtain larger and better diffracting crystals is currently under way.
Figure 3.

TGFBIp crystals and preliminary diffraction. (a) Hexagonal P6 crystals obtained using 0.1 M sodium acetate, 4% PEG 4000 pH 4.6 as precipitating agent solution. Diffraction data were collected from the encircled crystal. (b) Diffraction pattern obtained on beamline ID29 at the ESRF synchrotron (Grenoble, France).
Table 2. Preliminary crystallographic statistics of data collection and processing.
Values in parentheses are for the outermost resolution shell.
| Space group | P61 or P65 |
| Unit-cell parameters (Å) | a = b = 114.8, c = 181.1 |
| Wavelength (Å) | 0.9762 |
| No. of measurements/unique reflections | 261330/19822 |
| Resolution range (Å) | 49.69–3.25 (3.42–3.25) |
| Completeness (%) | 99.9 (100.0) |
| Rmerge† | 0.114 (0.405) |
| Rr.i.m.† | 0.122 (0.432) |
| Rp.i.m.† | 0.042 (0.148) |
| Average intensity over standard deviation {〈[〈I〉/σ(〈I〉)]〉} | 18.1 (5.8) |
| Average multiplicity | 8.3 (8.3) |
R
merge = 
 , R
r.i.m. =
, R
r.i.m. = 
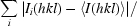 and R
p.i.m. =
and R
p.i.m. = 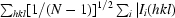 −
−  , where I
i(hkl) is the ith intensity measurement, N is the number of observations of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average intensity. R
r.i.m. (or R
meas) and R
p.i.m. are improved multiplicity-weighted indicators of the quality of the data, the redundancy-independent merging R factor and the precision-indicating merging R factor, the latter computed after averaging multiple measurements (Weiss, 2001 ▶; Evans, 2006 ▶).
, where I
i(hkl) is the ith intensity measurement, N is the number of observations of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average intensity. R
r.i.m. (or R
meas) and R
p.i.m. are improved multiplicity-weighted indicators of the quality of the data, the redundancy-independent merging R factor and the precision-indicating merging R factor, the latter computed after averaging multiple measurements (Weiss, 2001 ▶; Evans, 2006 ▶).
4. Conclusions
In recent years there has been increasing interest in the field of protein aggregation and fibrillation. This is a consequence of the recently recognized involvement of protein fibrils in several, mainly neurodegenerative, diseases such as Alzheimer’s disease, Huntington’s disease and Parkinson’s disease (Rochet, 2007 ▶). The main goal of these studies is to gain insight into the mechanisms of fibrillation and subsequently to produce therapeutic agents that are capable of dissolving the aggregates. However, much remains to be learned about the mechanisms that lead to fibrillation.
In the present study, we have successfully expressed and purified recombinant human wild-type TGFBIp as well as three naturally occurring TGFBIp mutants (R124L, R124C and R124H). Purification strategies for recombinant TGFBIp have previously been reported (Hanssen et al., 2003 ▶; Morand et al., 2003 ▶; Ohno et al., 1999 ▶; Skonier et al., 1994 ▶; Yuan et al., 2004 ▶), but this is the first report of milligram-level expression and purification of untagged recombinant human TGFBIp expressed in human cells under nondenaturing conditions. The simple two-step purification procedure resulted in very high purity, enabling crystallization of TGFBIp. TGFBIp showed a high propensity to form crystals and needle-shaped crystals were observed in the nano-drop screening procedure after only 5 d at 293 K. These crystals were found to diffract to 3.25 Å resolution.
Acknowledgments
We would like to thank Anne Gylling for expert technical assistance. We also thank Professor Peter Højrup for performing mass-spectrometric analyses. This work was supported by National Eye Institute Grant R01 EY 12712, the Danish National Research Foundation, the Danish Natural Science Research Council, the Danish Association for Prevention of Eye Diseases and Blindness, the Synoptik Foundation, Aarhus University Research Foundation and the Danish Medical Research Council. Further support was provided by BIO2006-02668, PSE-010000-2007-1 and the CONSOLIDER-INGENIO 2010 Project ‘La Factoría de Cristalización’ (CSD2006-00015) from Spanish public agencies, FP6 Strep Project LSHG-2006-018830 ‘CAMP’ and FP7 Collaborative Project 223101 ‘AntiPathoGN’ from the European Union and 2005SGR00280 from the National Catalan Government.
References
- Adachi, H. & Tsujimoto, M. (2002). J. Biol. Chem.277, 34264–34270. [DOI] [PubMed]
- Andersen, R. B., Karring, H., Moller-Pedersen, T., Valnickova, Z., Thogersen, I. B., Hedegaard, C. J., Kristensen, T., Klintworth, G. K. & Enghild, J. J. (2004). Biochemistry, 43, 16374–16384. [DOI] [PubMed]
- Bury, A. F. (1981). J. Chromatogr. A, 213, 491–500.
- Carson, D. D., Lagow, E., Thathiah, A., Al-Shami, R., Farach-Carson, M. C., Vernon, M., Yuan, L., Fritz, M. A. & Lessey, B. (2002). Mol. Hum. Reprod.8, 871–879. [DOI] [PubMed]
- Clout, N. J. & Hohenester, E. (2003). Mol. Vis.9, 440–448. [PubMed]
- Clout, N. J., Tisi, D. & Hohenester, E. (2003). Structure, 11, 197–203. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Escribano, J., Hernando, N., Ghosh, S., Crabb, J. & Coca-Prados, M. (1994). J. Cell. Physiol.160, 511–521. [DOI] [PubMed]
- Evans, P. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122. Warrington: Daresbury Laboratory.
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Ferguson, J. W., Thoma, B. S., Mikesh, M. F., Kramer, R. H., Bennett, K. L., Purchio, A., Bellard, B. J. & LeBaron, R. G. (2003). Cell Tissue Res.313, 93–105. [DOI] [PubMed]
- Hanssen, E., Reinboth, B. & Gibson, M. A. (2003). J. Biol. Chem.278, 24334–24341. [DOI] [PubMed]
- Horiuchi, K., Amizuka, N., Takeshita, S., Takamatsu, H., Katsuura, M., Ozawa, H., Toyama, Y., Bonewald, L. F. & Kudo, A. (1999). J. Bone Miner. Res.14, 1239–1249. [DOI] [PubMed]
- Hortsch, M. & Goodman, C. S. (1990). J. Biol. Chem.265, 15104–15109. [PubMed]
- Kabsch, W. (2001). International Tables for Crystallography, Vol. F, edited by M. G. Rossmann & E. Arnold, pp. 730–734. Dordrecht: Kluwer Academic Publishers.
- Kannabiran, C. & Klintworth, G. K. (2006). Hum. Mutat.27, 615–625. [DOI] [PubMed]
- Kawamoto, T., Noshiro, M., Shen, M., Nakamasu, K., Hashimoto, K., Kawashima-Ohya, Y., Gotoh, O. & Kato, Y. (1998). Biochim. Biophys. Acta, 1395, 288–292. [DOI] [PubMed]
- Kitahama, S., Gibson, M. A., Hatzinikolas, G., Hay, S., Kuliwaba, J. L., Evdokiou, A., Atkins, G. J. & Findlay, D. M. (2000). Bone, 27, 61–67. [DOI] [PubMed]
- Klintworth, G. K. (2003). Front Biosci.8, d687–d713. [DOI] [PubMed]
- Korvatska, E., Henry, H., Mashima, Y., Yamada, M., Bachmann, C., Munier, F. L. & Schorderet, D. F. (2000). J. Biol. Chem.275, 11465–11469. [DOI] [PubMed]
- LeBaron, R. G., Bezverkov, K. I., Zimber, M. P., Pavelec, R., Skonier, J. & Purchio, A. F. (1995). J. Invest. Dermatol.104, 844–849. [DOI] [PubMed]
- Lee, S. H., Bae, J. S., Park, S. H., Lee, B. H., Park, R. W., Choi, J. Y., Park, J. Y., Ha, S. W., Kim, Y. L., Kwon, T. H. & Kim, I. S. (2003). Kidney Int.64, 1012–1021. [DOI] [PubMed]
- Luthman, H. & Magnusson, G. (1983). Nucleic Acids Res.11, 1295–1308. [DOI] [PMC free article] [PubMed]
- Mashima, Y., Nakamura, Y., Noda, K., Konishi, M., Yamada, M., Kudoh, J. & Shimizu, N. (1999). Arch. Ophthalmol.117, 90–93. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Morand, S., Buchillier, V., Maurer, F., Bonny, C., Arsenijevic, Y., Munier, F. L. & Schorderet, D. F. (2003). Invest. Ophthalmol. Vis. Sci.44, 2973–2979. [DOI] [PubMed]
- Munier, F. L., Korvatska, E., Djemai, A., Le Paslier, D., Zografos, L., Pescia, G. & Schorderet, D. F. (1997). Nature Genet.15, 247–251. [DOI] [PubMed]
- Naiki, H., Higuchi, K., Hosokawa, M. & Takeda, T. (1989). Anal. Biochem.177, 244–249. [DOI] [PubMed]
- Ohno, S., Doi, T., Fujimoto, K., Ijuin, C., Tanaka, N., Tanimoto, K., Honda, K., Nakahara, M., Kato, Y. & Tanne, K. (2002). J. Dent. Res.81, 822–825. [DOI] [PubMed]
- Ohno, S., Noshiro, M., Makihira, S., Kawamoto, T., Shen, M., Yan, W., Kawashima-Ohya, Y., Fujimoto, K., Tanne, K. & Kato, Y. (1999). Biochim. Biophys. Acta, 1451, 196–205. [DOI] [PubMed]
- Politz, O., Gratchev, A., McCourt, P. A., Schledzewski, K., Guillot, P., Johansson, S., Svineng, G., Franke, P., Kannicht, C., Kzhyshkowska, J., Longati, P., Velten, F. W., Johansson, S. & Goerdt, S. (2002). Biochem. J.362, 155–164. [DOI] [PMC free article] [PubMed]
- Rawe, I. M., Zhan, Q., Burrows, R., Bennett, K. & Cintron, C. (1997). Invest. Ophthalmol. Vis. Sci.38, 893–900. [PubMed]
- Rochet, J. C. (2007). Expert Rev. Mol. Med.9, 1–34. [DOI] [PubMed]
- Skonier, J., Bennett, K., Rothwell, V., Kosowski, S., Plowman, G., Wallace, P., Edelhoff, S., Disteche, C., Neubauer, M., Marquardt, H., Rodgers, J. & Purchio, A. F. (1994). DNA Cell Biol.13, 571–584. [DOI] [PubMed]
- Streeten, B. W., Qi, Y., Klintworth, G. K., Eagle, R. C. Jr, Strauss, J. A. & Bennett, K. (1999). Arch. Ophthalmol.117, 67–75. [DOI] [PubMed]
- Takeshita, S., Kikuno, R., Tezuka, K. & Amann, E. (1993). Biochem. J.294, 271–278. [DOI] [PMC free article] [PubMed]
- Tamura, Y., Adachi, H., Osuga, J., Ohashi, K., Yahagi, N., Sekiya, M., Okazaki, H., Tomita, S., Iizuka, Y., Shimano, H., Nagai, R., Kimura, S., Tsujimoto, M. & Ishibashi, S. (2003). J. Biol. Chem.278, 12613–12617. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.
- Yuan, C., Reuland, J. M., Lee, L. & Huang, A. J. (2004). Protein Expr. Purif.35, 39–45. [DOI] [PubMed]